The Localization of APOBEC3H Variants in HIV-1 Virions Determines Their Antiviral Activity (original) (raw)
Abstract
Several members of the human APOBEC3 family of cytidine deaminases can potently restrict retroviruses such as HIV-1. The single-domain APOBEC3H (A3H) is encoded by four haplotypes, of which only A3H haplotype II-RDD (hapII-RDD) restricts HIV-1 efficiently. The goal of this study was to elucidate the mechanisms underlying the differences in antiviral activity among A3H haplotypes. The naturally occurring A3H hapI-GKE and hapII-RDD variants differ at three amino acid positions. A panel of six site-directed mutants containing combinations of the three variable residues was used to determine A3H protein expression, requirements of A3H virion incorporation, and A3H-Gag interactions. The catalytic activity of each A3H protein was assessed directly by using an Escherichia coli mutator assay. We found that the incorporation efficiencies of A3H variants into HIV-1 virions were comparable despite major differences in cellular expression. An assessment of the enzymes' catalytic activities showed that the deaminase activity of each A3H variant correlated with protein expression, suggesting similar enzymatic efficiencies. Surprisingly, virion incorporation experiments using Gag deletion mutants demonstrated that A3H haplotypes interacted with different Gag regions. A3H hapII-RDD associated with nucleocapsid in an RNA-dependent manner, whereas A3H hapI-GKE associated with the C-terminal part of matrix and the N-terminal capsid domain. Our results show that the A3H hapII-RDD interaction with nucleocapsid is critical for its antiviral activity and that the inability of A3H hapI-GKE to interact with nucleocapsid underlies its limited antiviral potential. Thus, the antiviral activity of A3H haplotypes is determined by its incorporation into the viral core, in proximity to the reverse transcription complex.
The human apolipoprotein B mRNA-editing catalytic polypeptide 3 (APOBEC3 [A3]) family consists of seven cytidine deaminases (A3A to A3H), some of which can potently restrict retroviruses such as human immunodeficiency virus type 1 (HIV-1) (5, 13, 19, 22, 43). They each encode one or two cytidine deaminase domains. The double-deaminase-domain APOBEC3 proteins A3B, A3DE, A3F, and A3G exhibit antiviral activity against HIV-1 (14, 16, 36, 48). Although the single-domain A3A and A3C members do not restrict HIV-1, they do efficiently block the replication of endogenous retrotransposons and simian immunodeficiency virus (SIV), respectively (8, 45). APOBEC3 proteins exert antiviral activity by editing and nonediting mechanisms (6, 7). Mutagenesis is achieved by deaminating cytidines into uridines of the minus DNA strand during reverse transcription (21, 27, 29, 36).
The single-domain A3H is the only APOBEC3 molecule that contains a Z3-type catalytic domain (12). It was initially thought to have limited antiretroviral activity (14, 33). However, several recent studies convincingly showed that A3H protein variants encoding a single-nucleotide polymorphism (SNP) cluster (composed of G105R, K121D, and E178D, named haplotype II-RDD [hapII-RDD]) display potent activities against HIV-1 and LINE-1 retrotransposons (15, 20, 32, 39). The active A3H variant is highly prevalent in populations of African descent (32).
We recently showed that A3H was not only polymorphic in sequence but also subjected to alternative splicing (20). The arginine (R) or glycine (G) at position 105 was the main determinant for the cellular expression and antiretroviral activity of A3H haplotypes (20, 32). A3H hapI-GKE resists HIV-1 Vif-mediated degradation, whereas A3H hapII-RDD is differentially sensitive to Vif from different HIV-1 strains (20, 32). The aspartic acid (D) at position 121 of hapII-RDD was previously identified as the main determinant for resistance to HIV-1 Vif (28, 47).
A3H transcripts were initially detected in various human tissues such as peripheral blood mononuclear cells, liver, skin, ovary, and testis by PCR (33). Moreover, two recent studies confirmed A3H induction by alpha interferon and A3H expression using quantitative real-time PCR approaches in cell populations susceptible to HIV-1 infection (e.g., dendritic cells, macrophages, and CD4 T cells) (25, 34).
APOBEC3 molecules confer antiviral activity during viral reverse transcription in the subsequent cycle of infection by APOBEC3-containing progeny viral particles. Thus, APOBEC3 packaging into egressing viral particles is critical for its anti-HIV-1 activity. Our knowledge of virion incorporation is based mainly on studies with A3G (3, 10, 35, 46) and A3F (41), which both bind the nucleocapsid (NC) part of the Gag protein during virion assembly. The interaction between A3G and NC is dependent on RNA, but whether host RNA, viral RNA, or both are involved remains somewhat controversial (4, 9, 24, 38, 40). Both single-domain A3A and A3C are efficiently packaged into virions yet fail to restrict HIV-1 (11, 45). Interestingly, A3A and A3C packaging is NC independent, suggesting that they are targeted away from the reverse transcription complex (2, 18, 42).
Taken together, the cellular expression and the antiviral activity of A3H hapI-GKE are strongly reduced compared to those of A3H hapII-RDD (15, 20, 32, 39). Such pronounced differences between natural variants of the same APOBEC3 representative have been described only for A3H variants, yet the correlates of their antiviral function remain poorly understood.
In this study we investigated the molecular mechanism underlying the differences in antiviral activities between A3H haplotypes. We determined that the efficiencies of incorporation into HIV-1 virions and deaminase catalytic activities were comparable between A3H hapI-GKE and hapII-RDD. However, both haplotypes differed qualitatively in their associations with HIV-1 Gag and were therefore packaged into different subcompartments of egressing virions. Our data indicate that the localization of A3 molecules in the HIV-1 virion is an important, previously underappreciated, determinant of antiviral activity.
MATERIALS AND METHODS
Plasmids.
The replication-competent full-length molecular clone NL4-3 ΔVif mutant was provided by the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health NIH Reagent Program (17).
The mammalian expression vector pTR600 containing A3H hapI-GKE, A3H hapII-RDD, the six site-directed A3H mutants, or A3G was described previously (20).
For bacterial expression, the A3H variants were amplified from pTR600 plasmids by using Pfu polymerase (Stratagene) and the following primers (Integrated DNA Technologies): 5′-CTA GAG ATC TAT GGA TTA CAA GGA TGA CGA (the BglII site is underlined) and 5′-GAT CGA ATT CTT CAG GAC TGC TTT ATC CTC TC-3′ (the EcoRI restriction site is underlined). The purified fragments were ligated into bacterial expression plasmid pTrcHis2B (Invitrogen). The integrity of cloned inserts was verified by sequencing.
The full-length Gag and Gag deletion constructs for virus-like particle (VLP) production and glutathione _S_-transferase (GST)-fused Gag constructs in pCRV1 were described previously (46).
Culture of cell lines.
HEK 293T and TZM-bl reporter cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin-streptomycin. TZM-bl cells were provided by the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health NIH Reagent Program (44).
Transfection of HEK 293T cells.
The pTR600-FLAG-A3H-hapI-GKE and pTR600-FLAG-A3H-hapII-RDD expression vectors, the site-directed mutants, pTR600-FLAG-A3G, and the empty pTR600 plasmid (all 100 ng) were cotransfected with either NL4-3 ΔVif (500 ng) or pCRV1-Gag constructs (500 ng). The transfections were performed in a 24-well format using 4 μg/ml polyethylenimine (PEI; Polysciences, Inc.). For all transfections, the culture medium was replenished after 12 h. Supernatants were harvested 36 to 48 h after transfection, clarified by centrifugation, and used to infect TZM-bl reporter cells.
Western blotting of HEK 293T cell lysates.
Transfected cells were lysed in a solution containing 1% sodium dodecyl sulfate (SDS), 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 5 mM EDTA. Five microliters of 4× lithium dodecyl sulfate (LDS) sample buffer (NuPAGE; Invitrogen) and 2 μl of sample reducing agent (NuPAGE; Invitrogen) were added to 13 μl of the lysate and heated for an additional 10 min at 70°C. Proteins were separated on 10% SDS-polyacrylamide gels (Invitrogen), transferred onto polyvinylidene difluoride (PVDF) membranes (Pierce), and probed with anti-FLAG M2 monoclonal antibody (Sigma) and with anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Sigma) to ensure equal protein loading. Membranes were subsequently incubated with horseradish peroxidase-conjugated secondary antibodies (Sigma), developed with SuperSignal West Pico (Pierce), and detected by using the Fujifilm Intelligent Lightbox LAS-3000 instrument and Image Reader LAS-3000 software. Signals were detected at supersensitive settings with automatic settings.
Assessment of viral infectivity.
TZM-bl reporter cells, which carry an HIV-1 Tat-responsive beta-galactosidase indicator gene under the transcriptional control of the HIV-1 long terminal repeat (LTR), were used to assess the infectivity of viral stocks produced by transfection in the presence and absence of the different FLAG-APOBEC3H variants or FLAG-APOBEC3G. TZM-bl cells were infected in triplicate with 20 μl of cell-free viral supernatants in 96-well plates. Beta-galactosidase activity was quantified 48 h after infection by using chemiluminescent substrate (Tropix; Perkin-Elmer), as previously described (20). The data from three independent experiments were used to calculate average values and standard deviations.
Protease treatment of HIV-1 virions.
HIV-1 virions containing A3H or A3G and supernatants of A3G- and A3H-expressing cells (no HIV-1) were concentrated through a 20% sucrose cushion by centrifugation at 20,000 × g for 3 h at 4°C. Pellets were dissolved in either 20 μl of phosphate-buffered saline (PBS) alone, PBS containing proteinase K (100 μg/ml), PBS containing 0.5% Triton X (Promega), or a combination of both proteinase K and Triton X. After a 30-min incubation at 37°C, proteinase K was inactivated by the addition of phenylmethylsulfonyl fluoride (PMSF) (10 mM; Boehringer Mannheim Biochemicals) and LDS sample buffer (NuPAGE; Invitrogen). Virion and cell lysates were analyzed by Western blotting. A3H and A3G were detected with anti-FLAG antibodies (Sigma), and p24 and gp120 were detected by probing the membrane with diluted serum from an HIV-1-infected person.
Confocal microscopy of A3H.
HEK 293T cells were transfected with N-terminally FLAG-tagged A3H hapI-GKE and A3H hapII-GKE expression plasmids, fixed 24 h later with 3% formaldehyde, permeabilized with 0.2% Triton X, and stained with anti-FLAG (1:4,000, mouse; Sigma) primary antibodies. Red Alexa Fluor 594 mouse (Invitrogen) was used as a secondary antibody. DNA was stained with Hoechst dye (1:1,000). Images were collected with a Leica TCS-SP5 confocal microscope at a magnification of ×100. Representative fields are shown for A3H hapI-GKE and A3H hapII-RDD. The gain-offset settings were selected in such a manner as to avoid a saturation of the A3H hapII-RDD signals and were increased for A3H hapI-GKE to correct for low expression. Images were acquired by using MetaMorph 6.2 imaging software.
Bacterial mutator assay.
Escherichia coli BW310 (lacking uracil DNA glycosylase, obtained from the E. coli Genetic Stock Center, Yale University) cells were transformed with bacterial expression plasmid pTrcHis2B (Invitrogen) containing each of the APOBEC3 panels via electroporation and grown on LB agar (Fisher Scientific) plates containing ampicillin (100 ng/ml; Fisher BioReagents) at 37°C. Eight colonies were picked for each construct from the transformed E. coli BW310 plates and grown in 2 ml LB medium containing ampicillin (100 ng/ml) and IPTG (isopropyl-β-d-thiogalactopyranoside) (1 mM; Fisher Scientific) at 37°C. The optical density of each culture grown overnight was measured at an absorbance of 600 nm (optical density at 600 nm [OD600]). One hundred microliters was plated onto an LB agar plate containing rifampin (100 μg/ml; Fisher BioReagents) to select for rifampin resistance. Plates were grown at 37°C overnight, and rifampin-resistant colonies were counted and corrected for by dividing the number of viable cells using their respective OD600 values. Data from one representative experiment of two independent experiments are shown in Fig. 4A. The relative deaminase activity shown in Fig. 4B reflects two independent experiments, for which the average activity of all 11 constructs was set to 100%. Each mutant's activity was expressed relative to the reference points of 24 for A3H hapI-GKE and 325 for A3H hapII-RDD.
FIG. 4.

Direct assessment of the deaminase activities of the A3H variants using a bacterial mutator assay. (A) Bacterial expression plasmids encoding the different A3H variants were electroporated into E. coli BW310. Eight colonies of each construct were picked and cultured overnight in the presence of IPTG. Bacteria were plated on rifampin plates, and Rif-resistant colonies were counted the next day. Bacterial concentrations were measured by the OD600 in a spectrometer and used to correct the number of counted Rif-resistant colonies. (B) The deaminase activities and the respective expression levels of two independent experiments were used. The deaminase mutants were excluded from the correlation between deaminase activity and protein expression (Spearman correlation of 0.949; P < 0.01).
Assessment of bacterial expression of A3H variants by Western blotting.
Single colonies were picked from the original transformed E. coli BW310 plates, grown overnight (37°C) in 1 ml LB medium containing ampicillin (100 ng/ml), and induced the next day by the addition of 3 ml LB medium containing IPTG (4 mM; Fisher Scientific) and ampicillin (400 ng/ml; Fisher BioReagents) for 2 h at 37°C. Five hundred microliters of the bacterial culture was pelleted and lysed by the addition of 500 μl 1% SDS lysis buffer. Ponceau protein staining (DiaSys) was used to confirm equal sample loading and efficient transfer. Protein transfer and detection were identical to the procedures for the HEK 293T cell lysates described above. Protein expression was quantified by using the Fujifilm Intelligent Lightbox LAS-3000 instrument and Image Reader LAS-3000 software. Signals were detected at supersensitive settings with automatic settings. Only the nonsaturated signals were quantified, and background was subtracted by using ImageGauge 4.0 software and used to calculate protein expression levels. The relative expression levels shown in Fig. 4B reflect two independent experiments for which the average expression of all 11 constructs was set to 100%. Each mutant's expression was expressed relative to the reference points of 8 for A3H hapI-GKE and 331 for A3H hapII-RDD.
Incorporation of A3H and A3G into virions and virus-like particles.
HEK 293T cells transfected in 24-well plates with 100 ng pTR600-FLAG-A3H-hapI-GKE, pTR600-FLAG-A3H-hapII-RDD expression vectors, the site-directed mutants, pTR600-FLAG-A3G, or the empty pTR600 plasmid were cotransfected with pCRV1-Gag constructs (500 ng). The Gag expression plasmids were provided by P. Bieniasz (46). The transfections were performed in a 24-well format by using 4 μg/ml PEI (Polysciences, Inc.). The culture medium was replenished after 12 h. Supernatants were harvested 48 h after transfection and clarified by centrifugation. Virions/VLPs were concentrated through a 20% sucrose cushion by centrifugation at 20,000 × g for 3 h at 4°C. Pellets were dissolved in 1% SDS lysis buffer and subsequently analyzed by Western blotting as described above (37). Gag proteins were detected by probing the membrane with diluted serum from an HIV-1-infected person.
Gag-GST pulldown of A3H and A3G.
Five hundred nanograms of the GST-tagged Gag constructs was cotransfected with 100 ng A3H hapII-RDD or A3G plasmid. The GST-Gag expression plasmids were provided by P. Bieniasz (46). Two days posttransfection, the cells were washed twice with ice-cold PBS and lysed in 500 μl mild lysis buffer (1% Triton in PBS supplemented with EDTA-free protease inhibitor cocktail [Roche]). The cleared lysates were incubated with glutathione-Sepharose 4B beads (GE Healthcare) at 4°C for 3 h. In the case of A3H hapII-RDD and A3G in combination with GST-Gag and GST-NC, samples were split, and RNase A (Invitrogen) was added to half of the sample before incubation. The beads were washed five times with wash buffer (0.1% Triton in PBS), and the proteins were eluted by boiling in LDS buffer (Invitrogen). The proteins were analyzed by Western blotting as described above and probed with anti-FLAG M2 monoclonal antibody (Sigma), anti-GST antibody (Amersham), and anti-GAPDH (Sigma).
RESULTS
A3H hapI-GKE and hapII-RDD are equally packaged into HIV-1 virions.
We first set out to determine if different A3H expression levels were the underlying cause of differences in virion incorporation efficiencies. We used a panel of site-directed mutants containing the naturally occurring amino acid substitutions at positions 105, 121, and 178 in different combinations in the most common A3H splice variant, SV-183 (Fig. 1) (20). A3G served as a control. Viral stocks were produced in the presence of a small amount of A3 expression plasmids (100 ng) to better distinguish the differences in antiviral activity and to avoid a saturation of A3H packaging into virions (data not shown).
FIG. 1.

Schematic overview of the A3H variants used in this study. A3H hapI-GKE and A3H hapII-RDD differ at amino acid positions 105, 121, and 178. The panel of site-directed mutants containing all possible combinations of both haplotypes was used to assess the effect of individual residues on deaminase activity/antiviral activity. The A3H variants used represent the most common splice variant (SV183), are 184 residues long, and contain a FLAG tag at their N terminus (described in reference 20). G, glycine; K, lysine; E, glutamic acid; R, arginine; D, aspartic acid.
The cotransfection of the A3H expression panel with the infectious HIV-1 NL4-3 ΔVif molecular clone revealed poor protein expression in the lysates of the producer cell for hapI-GKE, whereas hapII-RDD and A3G were efficiently expressed (Fig. 2 A), which is in agreement with data from previous studies (20, 32). The expression of the A3H site-directed mutants indicated that an arginine at position 105 (105R) was the sole determinant for efficient expression.
FIG. 2.
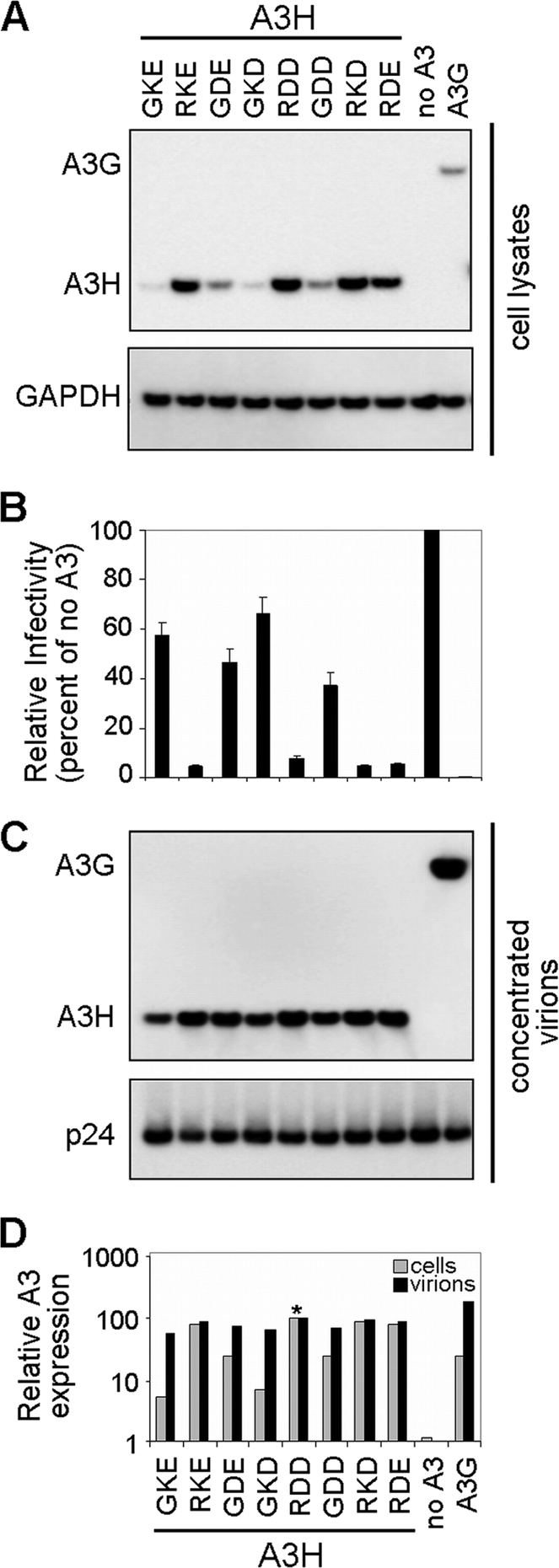
Expression, antiviral activity, and virion incorporation of A3H variants (hapI-GKE, hapII-RDD, and mutants) and A3G. (A) HIV-1 ΔVif viruses (500 ng) were produced by transfection in the presence of eight different APOBEC3H variants (100 ng), empty pTR600 plasmid (100 ng), or APOBEC3G (100 ng). A3H and A3G expression levels (anti-FLAG) were assessed by Western blotting of transfected HEK 293T cell lysates. Detection of GAPDH served as a protein loading control. Data from one representative experiment of three are shown. (B) Viral infectivity was assessed by infection of TZM-bl reporter cells. Results were normalized by using the no-APOBEC3 controls as a reference and are plotted as the percent relative mean infectivity. TZM-bl infections were performed in triplicates, and results from three independent transfection experiments are shown. (C) Viral stocks were concentrated through a 20% sucrose cushion and separated by SDS-PAGE. Virion incorporation of A3H and A3G was detected by Western blotting using an anti-FLAG monoclonal antibody. HIV-1 p24 was probed by using anti-HIV serum. (D) A3H and A3G cellular expression and virion incorporation were quantified by using the Fujifilm Intelligent Lightbox LAS-3000 instrument and Image Reader LAS-3000 software. Signals were normalized by setting both A3H hapII-RDD expression levels at 100% (indicated by an asterisk).
We infected TZM-bl reporter cells with virions produced in the presence of the different A3H and A3G constructs to compare infectivity levels (Fig. 2B). At the chosen DNA concentrations, we observed poor restriction for A3H hapI-GKE (∼40% restriction), whereas A3H hapII-RDD reduced HIV-1 infectivity by more than 7-fold (92% restriction). All A3H variants containing an arginine at position 105 potently restricted HIV-1 (95% restriction), while a glutamine at this position displayed infectivity levels comparable to those of A3H hapI-GKE (35 to 60% restriction). These observations are again in excellent agreement with data from previous reports from our laboratory and others (20, 32).
We used the viral stocks produced under the conditions described above and with the HIV-1 restriction patterns shown in Fig. 2B to determine the packaging efficiencies of A3H variants. Culture supernatants were concentrated through a 20% sucrose cushion, and the amounts of packaged A3H and A3G were assessed by Western blotting (Fig. 2C). Surprisingly, despite major differences in cellular A3H expression levels, the efficiencies of A3H incorporation into virions were comparable between A3H haplotypes (Fig. 2C). Quantifications of cellular expression and virion incorporation of the constructs (Fig. 2D) showed a 20-fold difference in cellular expression levels between A3H hapI-GKE and A3H hapII-RDD (the level of A3H hapI-GKE was 5.7% of that of A3H hapII-RDD), whereas the difference in viral packaging efficiency was less than 2-fold (the A3H hapI-GKE packaging efficiency was 58% of that of A3H hapII-RDD). Taken together, these findings indicate that (i) cellular A3H protein expression is a poor predictor of the level of A3H encapsidation and (ii) differences in antiviral activities of A3H hapI-GKE and A3H hapII-RDD cannot be explained simply by packaging differences but must have another underlying mechanism.
A3H is specifically packaged into HIV-1 virions.
To verify whether A3H variants were specifically incorporated into HIV-1 virions, we cotransfected A3H hapI-GKE, A3H hapII-RDD, and A3G with and without full-length HIV-1 expressing plasmids. In the presence of HIV-1, both A3H hapI-GKE and A3H hapII-RDD were packaged to comparable levels (Fig. 3 A). When the A3H haplotypes and A3G were expressed alone, no proteins were detected in the concentrated cell culture medium, indicating that HIV-1 virions were required. Furthermore, A3H variants, A3G, and Gag p24 proteins were not affected by treating the concentrated virions with proteinase K to degrade proteins that copurify with virions in an unspecific manner, whereas viral gp120, which resides on the outside of the viral membrane, was efficiently degraded. The further addition of Triton X (0.5%), which disrupts the viral envelope membrane, resulted in the complete degradation of A3H, A3G, p24, and gp120. Taken together, this indicates that both A3H haplotypes are specifically packaged within HIV-1 virions.
FIG. 3.

Specificity of A3H incorporation and subcellular localization. (A) HIV-1 virions (500 ng) were produced by transfection in the presence of A3H hapI-GKE, A3H hapII-RDD, A3G, or an empty pTR600 plasmid (100 ng). Virions were concentrated through a 20% sucrose cushion and dissolved in PBS alone, PBS with proteinase K, PBS with Triton X, and PBS with both proteinase K and Triton X. Virions were incubated at 37°C for 30 min. A3H and A3G expression levels (anti-FLAG) were assessed by Western blotting. HIV-1 p24 and gp120 were probed by using anti-HIV serum. A band corresponding to proteinase K is indicated by an asterisk. (B) Cellular A3H and A3G expression levels (anti-FLAG) were assessed by Western blotting. Detection of GAPDH served as a protein loading control. (C) 293T cells were transfected with N-terminally FLAG-tagged A3H hapI-GKE and A3H hapII-GKE, and the subcellular localization was assessed by confocal microscopy. Two different imaging settings were used since a higher offset was needed to capture the poorly expressed A3H hapI-GKE. Representative images are shown at a ×100 magnification. DNA was stained with Hoechst dye to visualize the nuclei.
Cytoplasmic localization of A3H hapI-GKE and hapII-RDD.
Next, we tested whether A3H haplotypes differ in their subcellular localizations. Results from previous studies addressing the subcellular localization of A3H were variable, possibly dependent on the cell type investigated and the position of the tag needed for detection (26, 30, 32, 49). We transfected N-terminally FLAG-tagged A3H hapI-GKE and A3H hapII-RDD in HEK 293T cells and detected the FLAG signal 24 h later by confocal microscopy. For better comparison with A3H hapII-RDD, we increased the red offset signal for imaging of the poorly expressed A3H hapI-GKE (Fig. 3C). This shows that the subcellular localizations of both haplotypes are similar and mostly cytoplasmic.
The catalytic activities of A3H variants are comparable.
Previous work from us and other laboratories showed that A3H deaminates cytidines in HIV-1 proviral DNA. The number of proviral G-to-A mutations correlated with the efficiency of viral restriction, supporting DNA editing as the mechanism underlying A3H restriction (20, 33). Since virion packaging appeared to be comparable between the two A3H haplotypes, we hypothesize that the SNPs might directly affect catalytic deaminase activity and thereby antiviral activity. We used an E. coli mutator assay (23, 33) to directly assess the enzymatic activities of the eight A3H variants. The mutator assay is based on the principle that mutations in the bacterial RNA polymerase gene rpoB lead to rifampin (Rif) resistance in E. coli strain BW310. The number of Rif-resistant colonies induced by a given deaminase directly reflects its DNA mutator activity.
We inserted the different A3H variants into the bacterial expression plasmid pTrcHis2B and assessed their catalytic activities as described previously (23). A deaminase active-site inactivating mutation (E56A) in both the A3H hapI-GKE and A3H hapII-RDD backbones was included as a negative control (20).
Figure 4 A shows the number of Rif-resistant colonies observed for A3H hapI-GKE, A3H hapII-RDD, as well as the mutants. A >10-fold difference in the numbers of Rif-resistant colonies was observed between A3H haplotypes I and II. Constructs encoding the G105R variant in all cases conferred higher deaminase activities than the G105 variants (compare RKE, RDD, RKD, and RDE with GKE, GDE, GKD, and GDD in Fig. 4A). Interestingly, the pattern of deaminase activity resembled the pattern of antiviral activity shown in Fig. 2B. The number of resistant colonies generated by the catalytic E56A mutants in each haplotype backbone was comparable to the level of mutations generated by the empty vector alone. Based on data from previous reports, we anticipated equal levels of expression of the A3H constructs in bacteria (33), but in our hands, expression levels were different between variants (Fig. 4B). The expression patterns in E. coli BW310 were comparable to the expression levels observed for mammalian HEK 293T cells (compare Fig. 4B and 2A).
To determine whether a correlation between deaminase activity, measured by the number of Rif-resistant colonies, and A3H protein expression existed, expression levels were plotted against deaminase activities. We observed a strong direct correlation (Spearman correlation of 0.949; P < 0.01) between expression and deaminase activity for hapI-GKE, hapII-RDD, the site-directed mutants, and the empty plasmid (Fig. 4B). The deaminase mutants of hapI-GKE and hapII-RDD were clearly outliers, in agreement with their lack of catalytic activity. A3H mutations that would directly influence deaminase activity without affecting expression would deviate from the regression line toward the deaminase mutants (decreased catalytic activity) or the upper left corner (increased catalytic activity). The fact that this was not the case for any of the A3H mutants tested strongly indicates that the different residues in A3H variants affected the protein expression levels but not the deaminase activity itself.
A3H hapII-RDD interacts with the NC region of Gag in an RNA-dependent manner.
Since virion incorporations (Fig. 2C) and catalytic activities (Fig. 4) of the A3H variants appeared equal, differential virion packaging requirements for A3H haplotypes could determine the variance in their antiviral activities. A3G contains two catalytic domains, of which the N-terminal domain mediates RNA binding and packaging and the C-terminal domain is catalytically active (31). This implies that A3H hapII-RDD must combine the different functions in a single domain.
We tested the abilities of A3H hapII-RDD and A3G to interact with several GST-fused Gag constructs (46), including full-length Gag, matrix-capsid (MA-CA), capsid (CA), NC, and GST alone. Pulldown assays indicated that A3H hapII-RDD interacted with either full-length Gag or NC. The interaction required an RNA bridge since the interaction was lost in the presence of RNase (Fig. 5, left). A similar requirement of NC and RNA was observed for A3G (Fig. 5, right), which is in agreement with data from previous studies (3, 10, 35, 46). We used only A3H hapII-RDD and A3G in these experiments since hapI-GKE could not be detected in the cell lysates, likely due to the mild lysis buffer that excluded hapI-GKE from the soluble protein fraction that was needed for the pulldown (data not shown). Taken together, these findings indicate that A3H hapII-RDD, similarly to A3G, interacts with NC in an RNA-dependent manner.
FIG. 5.

Gag interactions with A3H hapII-RDD and A3G. A3H hapII-RDD (100 ng) and A3G (100 ng) were cotransfected with GST-fused Gag constructs (500 ng). After 2 days, cells lysates were subjected to pulldown with anti-GST-Sepharose beads. In addition, the lysates containing A3H or A3G with Gag or NC were incubated with RNase A. A3H-Gag interactions were analyzed by Western blotting using anti-FLAG- and -GST-specific antibodies. Data from one representative experiment of three independent experiments are shown.
A3H haplotypes require different regions of Gag for virion packaging.
Since SNPs in the different A3H haplotypes could change the interaction with virion components, Gag requirements for A3H hapI-GKE and hapII-RDD encapsidation into Gag virus-like particles (VLPs) were determined. For this experiment we took advantage of the fact that A3H hapI-GKE was efficiently packaged into HIV-1 virions, although it was poorly expressed (Fig. 2C). We used a set of HIV-1 Gag constructs, including full-length Gag, a matrix construct with a deletion of residues 10 to 110 (ΔMA), a matrix-capsid construct with a deletion of residues 10 to 277 (ΔMA-CA), and a nucleocapsid deletion construct (ΔNC) in which NC is replaced by a leucine zipper (1). All these Gag constructs assemble into VLPs upon transfection and were previously used to determine A3G virion incorporation (46).
VLPs were produced by the cotransfection of the panel of eight A3H variants and A3G with the Gag-expressing plasmids. The VLPs were pelleted through a 20% sucrose cushion and analyzed by Western blotting. The cellular expression level of the A3H variants in the presence of the Gag constructs was comparable to the expression levels seen in the presence of HIV-1 (compare Fig. 6 with 2A).
FIG. 6.

Gag determinants for A3H packaging into virus-like particles. The different A3H variants (100 ng) and A3G (100 ng) were cotransfected with full-length and progressive Gag deletion expression plasmids (500 ng). Supernatants containing VLPs were clarified and pelleted through a 20% sucrose cushion and analyzed by Western blotting. Anti-FLAG antibodies detected A3H and A3G, while anti-HIV-1 serum was used to detect HIV-1 Gag. Cell lysates were probed in parallel with anti-FLAG and anti-GAPDH. One representative experiment of three independent experiments is shown.
In agreement with our results with full-length HIV-1 (Fig. 2C), Gag VLPs packaged all A3H variants with equal efficiencies. Comparable packaging efficiencies of A3H variants were also observed for the VLPs lacking the majority of matrix. This observation indicates that matrix residues 11 to 110 are not required for A3H packaging. Further deletion of the N-terminal domain of capsid in the matrix-capsid construct with a deletion of residues 10 to 277 showed a clear difference in A3H virion incorporation. HapII-RDD and G105R variants remained efficiently incorporated, whereas hapI-GKE packaging was strongly reduced. In contrast, the ΔNC construct displayed the opposite effect. A3H hapII-RDD and the 105R-containing mutants were poorly packaged, whereas hapI-GKE and the G105 variants were efficiently encapsidated. A3G incorporation was abolished only when NC was replaced by a leucine zipper, indicating that NC is required for packaging, as reported previously (35, 46).
These findings indicate that A3H hapI-GKE is packaged into the virion through an association with the matrix-capsid region of Gag, whereas A3H hapII-RDD, similarly to A3G, required Gag-NC for virion incorporation.
DISCUSSION
There are four haplotypes of APOBEC3H, of which A3H hapI-GKE and A3H hapII-RDD are the most common (20, 32, 39). These two A3H variants differ by only three amino acid substitutions, but the cellular expression and antiviral activity of A3H hapII-RDD are superior to those of A3H hapI-GKE. Possible explanations for this difference in activity are that a minimal level of expression of A3H hapI-GKE leads to lower levels of packaging into virions or that the catalytic activities of the A3H variant deaminases are directly affected. We show in this study that the levels of A3H virion incorporation are the same despite dramatic differences in cellular expression levels among A3H variants (Fig. 2). It was shown previously that the reduced expression levels of A3H hapI-GKE resulted from increased protein degradation by a nonproteosomal degradation pathway (33), which together suggests that A3H hapI-GKE must be packaged into virions before it is degraded.
A3H antiviral activity correlated with the protein expression level in the producer cell and not with the amount of A3H packaged into virions. Of note, we observed similar A3H virion incorporation efficiencies with full-length HIV-1 and VLPs, which do not undergo maturation. These findings contrast with previous observations that the virion incorporation of A3H hapI-GKE and A3H hapII-RDD into full-length HIV-1 correlated with cellular A3H expression (28, 47). This discrepancy may be explained by a rupture of the virions due to high-velocity ultracentrifugation, thereby losing A3H hapI-GKE in the soluble fraction of the preparation.
By using a well-established bacterial mutator assay (23), intrinsic variation in catalytic activities among A3H variants was excluded (Fig. 4). We observed a direct correlation between expression and deaminase activity despite differences in expression in E. coli, indicating that the enzymatic activities of the different A3H variants were intrinsically similar. The lack of A3H hapI-GKE stability in both bacterial and mammalian cells suggests that a well-conserved mechanism is responsible for A3H hapI-GKE destabilization.
A3H packaging into virions with distinct Gag domain deletions revealed that A3H hapI-GKE interacts with a 166-amino-acid (aa)-long region mapping to the matrix-capsid junction, whereas hapII-RDD required NC for its virion incorporation. It was shown previously that A3B, A3DE, A3G, and A3F associate with Gag-NC in an RNA-dependent manner and efficiently restrict HIV-1 (14-16, 46). This indicates a necessity for APOBEC molecules to interact with Gag-NC in order to exert potent antiviral activity. We now show that this also holds true for A3H hapII-RDD. In contrast, the single-domain APOBECs such as A3A, A3C, and A3H hapI-GKE are packaged efficiently into HIV-1 virions but fail to interact with NC and therefore lack antiviral activity (2, 18, 42).
Our results collectively suggest that APOBEC3 localization within the virion is essential for antiviral activity. A requirement for the presence of APOBEC in the viral core in close proximity to the RNase complex could be derived from the fact that APOBECs deaminate single-stranded DNA during reverse transcription (Fig. 7). This argument is supported by data from experiments showing that redirecting A3A to the viral core by fusion to VPR or to APOBEC3G resulted in HIV-1 restriction (2, 18). This finding underscores the importance of correct intravirion localization for antiviral activity.
FIG. 7.

Model of A3H haplotype I and II localization in HIV-1 virions. Shown is a model of immature (left) and mature (right) HIV-1 virions. The immature virion contains full-length Gag proteins, of which the outside layer of the virion is formed by the matrix (MA) domain, followed by the capsid (CA) protein, which consists of an N-terminal (square) domain and a C-terminal (circle) domain. The NC part of Gag lines the center of the virion. A3H hapI-GKE interacts with the C-terminal part of MA and the N-terminal domain of CA, whereas A3H hapII-RDD interacts with NC. During virion maturation, the Gag protein is cleaved into the respective subunits. MA now lines the outside of the virion, whereas CA forms a condensed cone-shaped core, of which the N-terminal domain of CA is facing outwards. The C-terminal domain of CA covers the inside of the core. The core contains the dimeric genomic viral RNA and NC. A3H hapI-GKE localizes outside the core, and A3H hapII-RDD is present in the viral core.
The failure of A3H hapI-GKE to interact with NC explains its reduced antiviral activity, but deleting MA-CA does not result in the retargeting of A3H hapI-GKE to NC. This finding indicates that the specific interaction of A3H hapI-GKE with MA-CA is likely not a virus-mediated mechanism to reduce its antiviral activity.
So far, A3H hapII-RDD is the only single-domain APOBEC to efficiently restrict HIV-1. This observation suggests that the molecular requirements that permit A3H variants to be incorporated and exert their antiviral function are unique and determined solely by the amino acid at position 105. These properties set the A3H hapII-RDD variant apart from all other single-deaminase-domain A3 enzymes, which are catalytically active yet fail to restrict HIV-1. In summary, correct virion incorporation mediates the antiviral activities of A3H haplotypes.
Acknowledgments
We thank members of the Simon laboratory and G. A. Versteeg for helpful discussions.
This work was supported by NIH/NIAID grant R01 AI064001 (V.S.), the Alexandrine and Alexander L. Sinsheimer Fund (V.S.), and the NIGMS T32GM007280-33 Mount Sinai Medical Scientist Training Program (C.W.S.).
Footnotes
▿
Published ahead of print on 2 June 2010.
REFERENCES
- 1.Accola, M. A., B. Strack, and H. G. Gottlinger. 2000. Efficient particle production by minimal Gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1 capsid-p2 and a late assembly domain. J. Virol. 74**:**5395-5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguiar, R. S., N. Lovsin, A. Tanuri, and B. M. Peterlin. 2008. Vpr.A3A chimera inhibits HIV replication. J. Biol. Chem. 283**:**2518-2525. [DOI] [PubMed] [Google Scholar]
- 3.Alce, T. M., and W. Popik. 2004. APOBEC3G is incorporated into virus-like particles by a direct interaction with HIV-1 Gag nucleocapsid protein. J. Biol. Chem. 279**:**34083-34086. [DOI] [PubMed] [Google Scholar]
- 4.Bach, D., S. Peddi, B. Mangeat, A. Lakkaraju, K. Strub, and D. Trono. 2008. Characterization of APOBEC3G binding to 7SL RNA. Retrovirology 5**:**54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bieniasz, P. D. 2003. Restriction factors: a defense against retroviral infection. Trends Microbiol. 11**:**286-291. [DOI] [PubMed] [Google Scholar]
- 6.Bishop, K. N., R. K. Holmes, A. M. Sheehy, N. O. Davidson, S. J. Cho, and M. H. Malim. 2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 14**:**1392-1396. [DOI] [PubMed] [Google Scholar]
- 7.Bishop, K. N., M. Verma, E. Y. Kim, S. M. Wolinsky, and M. H. Malim. 2008. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 4**:**e1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bogerd, H., H. Wiegand, B. Doehle, K. Lueders, and B. Cullen. 2006. APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 34**:**89-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bogerd, H. P., and B. R. Cullen. 2008. Single-stranded RNA facilitates nucleocapsid: APOBEC3G complex formation. RNA 14**:**1228-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cen, S. 2004. The interaction between HIV-1 Gag and APOBEC3G. J. Biol. Chem. 279**:**33177-33184. [DOI] [PubMed] [Google Scholar]
- 11.Chen, H., C. E. Lilley, Q. Yu, D. V. Lee, J. Chou, I. Narvaiza, N. R. Landau, and M. D. Weitzman. 2006. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 16**:**480-485. [DOI] [PubMed] [Google Scholar]
- 12.Conticello, S. G., C. J. Thomas, S. K. Petersen-Mahrt, and M. S. Neuberger. 2005. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol. Biol. Evol. 22**:**367-377. [DOI] [PubMed] [Google Scholar]
- 13.Cullen, B. R. 2006. Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J. Virol. 80**:**1067-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dang, Y., X. Wang, W. J. Esselman, and Y. H. Zheng. 2006. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J. Virol. 80**:**10522-10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dang, Y., L. M. Siew, X. Wang, Y. Han, R. Lampen, and Y. H. Zheng. 2008. Human cytidine deaminase APOBEC3H restricts HIV-1 replication. J. Biol. Chem. 283**:**11606-11614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doehle, B. P., A. Schafer, and B. R. Cullen. 2005. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology 339**:**281-288. [DOI] [PubMed] [Google Scholar]
- 17.Gibbs, J. S., D. A. Regier, and R. C. Desrosiers. 1994. Construction and in vitro properties of HIV-1 mutants with deletions in “nonessential” genes. AIDS Res. Hum. Retroviruses 10**:**343-350. [DOI] [PubMed] [Google Scholar]
- 18.Goila-Gaur, R., M. A. Khan, E. Miyagi, S. Kao, and K. Strebel. 2007. Targeting APOBEC3A to the viral nucleoprotein complex confers antiviral activity. Retrovirology 4**:**61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goila-Gaur, R., and K. Strebel. 2008. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology 5**:**51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harari, A., M. Ooms, L. C. Mulder, and V. Simon. 2009. Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J. Virol. 83**:**295-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris, R. S., K. N. Bishop, A. M. Sheehy, H. M. Craig, S. K. Petersen-Mahrt, I. N. Watt, M. S. Neuberger, and M. H. Malim. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113**:**803-809. [DOI] [PubMed] [Google Scholar]
- 22.Harris, R. S., and M. T. Liddament. 2004. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 4**:**868-877. [DOI] [PubMed] [Google Scholar]
- 23.Harris, R. S., S. K. Petersen-Mahrt, and M. S. Neuberger. 2002. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 10**:**1247-1253. [DOI] [PubMed] [Google Scholar]
- 24.Khan, M. A., S. Kao, E. Miyagi, H. Takeuchi, R. Goila-Gaur, S. Opi, C. L. Gipson, T. G. Parslow, H. Ly, and K. Strebel. 2005. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J. Virol. 79**:**5870-5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koning, F. A., E. N. Newman, E. Y. Kim, K. J. Kunstman, S. M. Wolinsky, and M. H. Malim. 2009. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 83**:**9474-9485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LaRue, R. S., S. R. Jonsson, K. A. Silverstein, M. Lajoie, D. Bertrand, et al. 2008. The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol. Biol. 9**:**104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lecossier, D. 2003. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science 300**:**1112. [DOI] [PubMed] [Google Scholar]
- 28.Li, M. M., L. I. Wu, and M. Emerman. 2010. The range of human APOBEC3H sensitivity to lentiviral Vif proteins. J. Virol. 84**:**88-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mangeat, B. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424**:**99-103. [DOI] [PubMed] [Google Scholar]
- 30.Muckenfuss, H., M. Hamdorf, U. Held, M. Perkovic, J. Lower, K. Cichutek, E. Flory, G. G. Schumann, and C. Munk. 2006. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J. Biol. Chem. 281**:**22161-22172. [DOI] [PubMed] [Google Scholar]
- 31.Navarro, F., B. Bollman, H. Chen, R. Konig, Q. Yu, K. Chiles, and N. R. Landau. 2005. Complementary function of the two catalytic domains of APOBEC3G. Virology 333**:**374-386. [DOI] [PubMed] [Google Scholar]
- 32.OhAinle, M., J. A. Kerns, M. M. H. Li, H. S. Malik, and M. Emerman. 2008. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 4**:**249-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.OhAinle, M., J. A. Kerns, H. S. Malik, and M. Emerman. 2006. Adaptive evolution and antiviral activity of the conserved mammalian cytidine deaminase APOBEC3H. J. Virol. 80**:**3853-3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Refsland, E. W., M. D. Stenglein, K. Shindo, J. S. Albin, W. L. Brown, and R. S. Harris. 22 March 2010, posting date. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. doi: 10.1093/nar/gkq174. [DOI] [PMC free article] [PubMed]
- 35.Schafer, A., H. P. Bogerd, and B. R. Cullen. 2004. Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology 328**:**163-168. [DOI] [PubMed] [Google Scholar]
- 36.Sheehy, A., N. Gaddis, J. Choi, and M. Malim. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418**:**646-650. [DOI] [PubMed] [Google Scholar]
- 37.Simon, V., N. Padte, D. Murray, J. Vanderhoeven, T. Wrin, N. Parkin, M. Di Mascio, and M. Markowitz. 2003. Infectivity and replication capacity of drug-resistant human immunodeficiency virus type 1 variants isolated during primary infection. J. Virol. 77**:**7736-7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Svarovskaia, E. S., H. Xu, J. L. Mbisa, R. Barr, R. J. Gorelick, A. Ono, E. O. Freed, W. S. Hu, and V. K. Pathak. 2004. Human apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (APOBEC3G) is incorporated into HIV-1 virions through interactions with viral and nonviral RNAs. J. Biol. Chem. 279**:**35822-35828. [DOI] [PubMed] [Google Scholar]
- 39.Tan, L., P. T. N. Sarkis, T. Wang, C. Tian, and X.-F. Yu. 2008. Sole copy of Z2-type human cytidine deaminase APOBEC3H has inhibitory activity against retrotransposons and HIV-1. FASEB J. 1**:**279-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang, T., C. Tian, W. Zhang, K. Luo, P. T. Sarkis, L. Yu, B. Liu, Y. Yu, and X. F. Yu. 2007. 7SL RNA mediates virion packaging of the antiviral cytidine deaminase APOBEC3G. J. Virol. 81**:**13112-13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang, T., C. Tian, W. Zhang, P. T. Sarkis, and X. F. Yu. 2008. Interaction with 7SL RNA but not with HIV-1 genomic RNA or P bodies is required for APOBEC3F virion packaging. J. Mol. Biol. 375**:**1098-1112. [DOI] [PubMed] [Google Scholar]
- 42.Wang, T., W. Zhang, C. Tian, B. Liu, Y. Yu, L. Ding, P. Spearman, and X. F. Yu. 2008. Distinct viral determinants for the packaging of human cytidine deaminases APOBEC3G and APOBEC3C. Virology 377**:**71-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wedekind, J. E., G. S. Dance, M. P. Sowden, and H. C. Smith. 2003. Messenger RNA editing in mammals: new members of the APOBEC family seeking roles in the family business. Trends Genet. 19**:**207-216. [DOI] [PubMed] [Google Scholar]
- 44.Wei, X., J. M. Decker, H. Liu, Z. Zhang, R. B. Arani, J. M. Kilby, M. S. Saag, X. Wu, G. M. Shaw, and J. C. Kappes. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46**:**1896-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu, Q., D. Chen, R. Konig, R. Mariani, D. Unutmaz, and N. R. Landau. 2004. APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J. Biol. Chem. 279**:**53379-53386. [DOI] [PubMed] [Google Scholar]
- 46.Zennou, V., D. Perez-Caballero, H. Gottlinger, and P. D. Bieniasz. 2004. APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J. Virol. 78**:**12058-12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhen, A., T. Wang, K. Zhao, Y. Xiong, and X. F. Yu. 2010. A single amino acid difference in human APOBEC3H variants determines HIV-1 Vif sensitivity. J. Virol. 84**:**1902-1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng, Y. H., D. Irwin, T. Kurosu, K. Tokunaga, T. Sata, and B. M. Peterlin. 2004. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J. Virol. 78**:**6073-6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zielonka, J., I. G. Bravo, D. Marino, E. Conrad, M. Perkovic, M. Battenberg, K. Cichutek, and C. Munk. 2009. Restriction of equine infectious anemia virus by equine APOBEC3 cytidine deaminases. J. Virol. 83**:**7547-7559. [DOI] [PMC free article] [PubMed] [Google Scholar]