Auto-inhibition of Human Dicer by its Internal Helicase Domain (original) (raw)
. Author manuscript; available in PMC: 2010 Aug 24.
Published in final edited form as: J Mol Biol. 2008 May 8;380(1):237–243. doi: 10.1016/j.jmb.2008.05.005
SUMMARY
Dicer, a member of the Ribonuclease III family of enzymes, processes double-stranded RNA substrates into ~21-27 nucleotide products that trigger sequence-directed gene silencing by RNA interference (RNAi). Although the mechanism of RNA recognition and length-specific cleavage by Dicer has been established, the way in which dicing activity is regulated is unclear. Here we show that the N-terminal domain of human Dicer, which is homologous to DExD/H-box helicases, substantially attenuates the rate of substrate cleavage. Deletion or mutation of this domain activates human Dicer in both single- and multiple-turnover assays. The catalytic efficiency (_k_cat/_K_m) of the deletion construct is increased by 65-fold over that exhibited by the intact enzyme. Kinetic analysis shows that this activation is almost entirely due to an enhancement in _k_cat. Modest stimulation of catalysis by the full-length Dicer enzyme was observed in the presence of the TAR-RNA binding protein (TRBP), which physically interacts with the DExD/H-box domain. These results suggest that the DExD/H-box domain likely disrupts the functionality of the Dicer active site until a structural rearrangement occurs, perhaps upon assembly with its molecular partners.
Keywords: Ribonuclease, Dicer, RNAi, helicase
INTRODUCTION
Dicer is a large multi-domain enzyme responsible for cytoplasmic production of both microRNAs (miRNAs) and short interfering RNAs (siRNAs) during sequence-directed gene regulation by RNAi. As a member of the Ribonuclease III family of proteins, Dicer recognizes the 5′ and 3′ helical ends of double-stranded RNA substrates and cleaves a specific distance away to produce 21-27 nucleotide products (reviewed in1; 2; 3). Dicer helps these mi- and siRNAs to load onto Argonaute proteins together with other protein components of the RNA-induced silencing complex (RISC). Once bound to target mRNAs, miRNAs typically regulate protein expression by controlling the level of translation, whereas siRNAs direct cleavage and subsequent degradation of complementary mRNAs.
Although the mechanism of double-stranded RNA recognition and length-specific RNA cleavage by Dicer has been established, the way in which this activity is regulated remains unclear. Biochemical and structural studies showed that the PAZ domain of Dicer binds the 3′ 2-nt overhanging end of a duplex RNA substrate, positioning it for cleavage by Dicer’s two RNase III-homologous domains at a distance of ~25 base pairs from the end4; 5; 6. Although a natural Dicer from Giardia containing just these functional domains is capable of robust dicing and can complement the lack of a functional Dicer in S. pombe6, most Dicers include numerous additional sequences. In particular, Dicer typically contains a large N-terminal domain homologous to DExD/H-box helicases. Although the presence of this domain correlates with a requirement for ATP by invertebrate Dicers7; 8; 9; 10, siRNA production by mammalian Dicer is ATP-independent11; 12.
To investigate the role of the DExD/H-box domain as well as other regions of the human Dicer enzyme, we overexpressed and purified five Dicer variants containing either domain deletions or point mutations. We demonstrate that Dicer is capable of multiple-turnover catalysis and that it behaves as a classic Michaelis-Menten enzyme. Surprisingly, deletion or mutation of the DExD/H-box domain led to substantially enhanced activity of Dicer when assayed for cleavage of a perfect-duplex substrate, with slight stimulation of hairpin substrate cleavage. Kinetic analysis shows that this rate enhancement is largely a _k_cat effect, indicating that the DExD/H-box domain inhibits catalysis rather than affecting RNA binding affinity. The protein TRBP, which binds directly to the DExD/H-box domain, modestly stimulates cleavage activity of wild-type Dicer. Our data suggest that the DExD/H-box domain functions as an intramolecular structural switch to maintain Dicer in a low-activity state until the enzyme assembles with its protein partners, and it may help to distinguish perfect-duplex versus hairpin substrates.
RESULTS AND DISCUSSION
Dicer’s DExD/H-box domain inhibits single-turnover dsRNA cleavage rates
To investigate dsRNA recognition and cleavage by human Dicer, the wild-type (hDcr) (accession no. NP_803187) and five mutant forms of recombinant hDcr were prepared (Fig. 1A). Specifically, a point mutation of lysine to alanine at position 70 (K70A) in the ATP-binding motif (hWalker) and a deletion of amino acids 1-604 spanning the entire DExD/H-box domain (Δhelicase) were created to analyze the functional contributions of the DExD/H-box domain. To explore the role of the C-terminal double-stranded RNA binding domain (dsRBD) and the _d_omain of _u_nknown _f_unction (DUF283), hDcr variants lacking these domains were prepared (ΔdsRBD1844-1922 and ΔDUF630-709, respectively). A more substantially truncated form of hDcr (2DD) was prepared lacking both the DExD/H-box and the dsRBD domains. Each Dicer variant was produced using a baculovirus expression system, purified by Ni2+-affinity chromatography and concentrated after size exclusion chromatography following removing the N-terminal His6 tag. In each case, 0.5-1.0 mg of purified recombinant protein was obtained routinely from one liter of cell culture (Fig. 1B). Activity assays showed that all five hDcr variants are able to cleave double-stranded RNA substrates (Fig. 1C).
Figure 1. Domain structure and expression of human Dicer (hDcr).
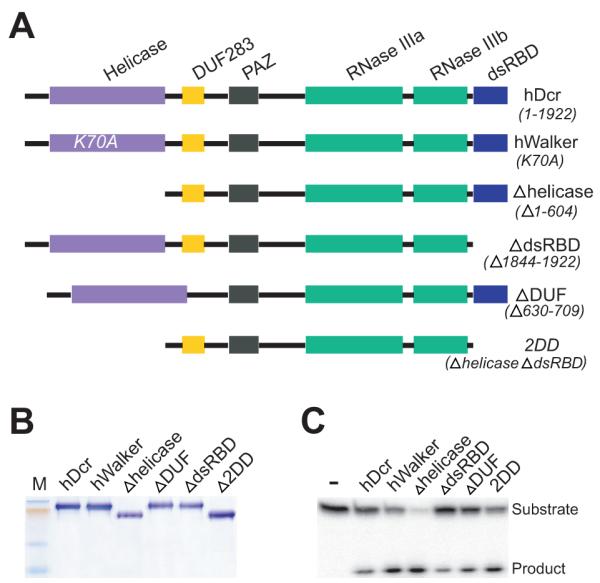
A, Domain structure of hDcr variants; B, 10% SDS-PAGE analysis of recombinant hDcr proteins; C, Single time-point dsRNase activity assays (60 min., 37□, 60 nM protein, 2 nM 32P-labeled 37ab RNA); M, protein size marker.
To assess the contributions of various hDcr domains to dicing activity, we first tested the catalytic activity of each hDcr variant under single-turnover conditions using two different substrates. One was a double-stranded RNA containing 2-nucleotide 3′ overhangs on either end (37ab), while the other was the human pre-hlet-7 hairpin RNA (Fig. 2A). In the presence of excess protein, the wild-type hDcr catalyzed cleavage of the 37ab substrate to yield 22-nt. products at an initial rate of 0.19 fmol min−1 (Fig. 2B left panel, C). Deletion of either the dsRBD or the DUF domain significantly reduced the observed cleavage rate. These results are consistent with previous data showing that deletion of the dsRBD resulted in decreases of 1.9- and 2.5-fold in the cleavage rates for dsRNA and hairpin RNA, respectively 5. Surprisingly, however, deletion or mutation of the DExD/H-box domain significantly enhanced the cleavage rate relative to that observed for the wild-type enzyme (Fig. 2B left panel, Fig. 2C). Deletion of the DExD/H-box domain had the most pronounced effect, with an ~8-fold faster rate of dicing for the 37ab dsRNA substrate. More subtle cleavage rate differences were observed for the pre-hlet-7 substrate, although in general this substrate was cleaved more rapidly than 37ab (Fig. 2B right panel, Fig. 2C).
Figure 2. Single-turnover activity of hDcr proteins.

A, Substrates and dicing reactions. Left panels, perfect-duplex dsRNA substrate, 37ab; right panels, hairpin pre-miRNA substrate, pre-hlet-7; asterisks (*) indicate 5′-end labeled with 32P. hDcr generates two products from RNA 37ab, 22-nt (P1) or 15-nt (P2), and one product (P) from pre-hlet-7. Relative rates of P1 and P2 production were the same for all hDcrs tested and were combined to give the total cleavage product for 37ab. B, Single-turnover reaction of hDcrs (60 nM) with 2 nM (3000 c.p.m.) duplex RNA 37ab (left panel) or pre-hlet-7 (right panel); values are the average from two independent experiments. Data were fit to the equation S_=(a-b)exp(−_k_obsd_t)+b, where S is the fraction of dsRNA cleaved at each time point, a is the fraction of dsRNA at the beginning of the reaction, b is the fraction of dsRNA at the reaction plateau (t-->∞), and _k_obsd is the observed rate constant; _k_obsd values (fmol/min) for the 37ab substrate were as follows: hDcr (wildtype), 0.32; hWalker, 0.8; Δhelicase (Δhel), 2; ΔDUF, 0.0034; ΔRBD, 0.2; 2DD, 0.46; _k_obsd values (fmol/min) for the pre-hlet-7 substrate were as follows: hDcr (wildtype), 3; hWalker, 2; Δhelicase, 2.2; ΔDUF, 2.6; ΔRBD, 0.74; 2DD, 0.74; C, summary of initial reaction rates of hDcr variants calculated for 20% substrate cleavage; S, RNA substrate; FL, wild-type hDicer; hW, hWalker mutant; dhel, Δhelicase mutant; dDUF, ΔDUF mutant; dRBD, ΔRBD mutant.
Different efficiencies of hDcr-catalyzed pre-miRNA versus generic dsRNA cleavage were observed previously11, implying that miRNAs and siRNAs may be recognized or processed differently in vivo. The lack of pronounced stimulation of pre-hlet-7 cleavage by hDcr lacking the DExD/H-box domain is consistent with the finding that deletion of the DExD/H-box domain only slightly increases pre-miRNA processing activity13; 14. Taken together, our data show that the activity of hDcr is substrate-dependent, and that its DExD/H-box domain inhibits the rate of cleavage, particularly for a perfect-duplex dsRNA substrate.
Dicer’s DExD/H-box domain does not significantly alter substrate binding affinity
The substrate-dependent differences in dicing activity observed for human Dicer variants might reflect differences in substrate recognition. To test this possibility, we measured the dissociation constants of recombinant hDcr complexes with the perfect duplex (37ab) or the pre-hlet-7 hairpin RNA. Under conditions in which dicing is inhibited (via magnesium ion chelation), the affinities of four of the mutant hDcr enzymes for either substrate are within three-fold of those measured for the wild-type enzyme (75 nM and 30 nM, respectively) (Fig. 3). Similar _K_d values were obtained using a 64-nt duplex RNA (data not shown) and are comparable to those measured previously11; 15. A catalytically inactive hDcr variant showed similar affinities for each RNA in the presence of 10 mM Mg2+ (data not shown), confirming that there are no significant effects of magnesium on RNA affinity11; 12. Notably, deletion of the dsRBD does not significantly affect RNA affinity. This observation contrasts with a previous report that a segment of the dsRBD alone binds to dsRNA5 (Fig. 3A, B). Furthermore, although the DUF283 domain has been suggested to be a dsRNA-binding domain16, our results do not support its role in enhancing Dicer’s affinity for RNA. The protein lacking both the DExD/H-box domain and the dsRBD (2DD) binds ~3-4 fold more weakly to either substrate relative to full-length Dicer (Fig. 3A-C). It is possible that this severely truncated protein is generally destabilized, or that the DExD/H-box domain plays a small but measurable role in substrate binding.
Figure 3. Duplex RNA/hDcr dissociation constants.

Equilibrium filter binding assays for hDcr proteins complexed with 37ab RNA (A) or pre-hlet-7 RNA (B). Values are averages from two independent assays. C, summary of dissociation constants (Kd, nM).
DExD/H-box domain deletion enhances the _k_cat values for Dicer
The lack of substantial differences in hDcr-substrate _K_d values, particularly for the DExD/H-box deletion protein, suggests that the enhanced rate of single turnover by this enzyme is reflected primarily in the _k_cat value. To test this, we measured the rate of 37ab substrate cleavage under steady-state conditions as a function of substrate concentration. Plots of initial reaction rate (_v_o) versus substrate concentration show that both the wild-type Dicer and the DExD/H-box deletion enzyme kinetics obey classical Michaelis-Menten formalism (Fig. 4A, B). The kinetic constants are given in Fig. 4C.
Figure 4. Steady-state kinetic analysis of wild-type and Δhelicase hDcr constructs.

Plots of initial velocity versus substrate concentration: A, hDcr; and B, Δhelicase. C, summary of kinetic values.
The _K_m and _k_cat values, determined from nonlinear least squares fits of _v_o versus substrate concentration, show that the _K_m values for full-length hDcr and Δhelicase (18.8 and 11.6 nM, respectively) are close, while the _k_cat value is increased ~40-fold as a result of the DExD/H-box deletion (Fig. 4C). As the _K_m and _K_d values are only minimally affected by the presence of the DExD/H-box domain, it is likely that the substrate-binding site is equally accessible in each case. The DExD/H-box domain must therefore partially interfere with catalysis per se, possibly by stabilizing a conformational isoform that is relieved by DExD/H-box domain deletion or disruption. We note that the Dicer construct containing a point mutation in the ATP-binding region of the DExD/H-box domain, hWalker (Fig. 1A), shows significantly increased association with the hydrophobic reagent 1-anilino-naphthalene-8-sulfonate (ANS) relative to that observed for wild-type Dicer (Fig. S1). Because ANS binds more strongly to partially unfolded or “molten-globule” proteins17, this finding is consistent with the conclusion that structural destabilization or rearrangement of the DExD/H-box domain triggers catalytic activation of human Dicer.
Association with TRBP stimulates full-length Dicer catalytic activity
Earlier immunoprecipitation experiments showed that the DExD/H-box domain of Dicer is critical for interaction with human partner proteins TRBP and PACT13 and with the fly protein Loquacious (Loqs)14. In each case these partner proteins share homology with known RNA binding motifs, and have been thought to enhance the activity and/or specificity of Dicer for its substrates. We used size exclusion chromatography to test whether a direct physical interaction can be observed between purified hDcr and TRBP. Consistent with prior immunoprecipitation data, we found that wild-type human Dicer spontaneously forms a complex with TRBP, while Dicer lacking the DExD/H-box domain (Δhelicase) does not (Fig. 5A). Thus, the helicase motif of human Dicer is necessary for the association of TRBP with Dicer. No other factors are required.
Figure 5. Dicer-TRBP interaction and kinetics.

A, size exclusion chromatography of the hDcr-TRBP complex formed by incubation of hDcr (molecular weight 219 kDa; 2.5 nmol) and excess TRBP (molecular weight 39 kDa; 9 nmol) in 20 μl dicing buffer for 60 min. on ice; chromatogram peak C, hDcr-TRBP complex; chromatogram peak T, TRBP; chromatogram peak hD, hDcr. SDS-PAGE gel analysis of fractions are shown below each chromatogram. B, multiple-turnover assay for 37ab cleavage using 100 nM dsRNA and 5 nM hDcr or chromatographically pure hDcr-TRBP; the data shown are representative of two independent experiments.
We wondered whether TRBP binding to the DExD/H-box domain might stimulate hDcr activity and thus serve as a trigger to activate dicing by the complex. To test this possibility, the dsRNase activity of the hDcr-TRBP complex purified by size-exclusion chromatography was compared to that of wild-type hDcr or the DExD/H-box deletion protein in a multiple-turnover assay (Fig. 5B). Interestingly, we find that, although not as dramatic as DExD/H-box deletion, association of TRBP with full-length hDcr increases the cleavage rate by approximately two-fold. Consistent with this finding, reduction of TRBP protein levels in human cells led to decreased pre-miRNA processing18. Because the _K_d values of wild-type hDcr-dsRNA and hDcr-TRBP-dsRNA complexes are similar (75 vs. 48 nM, data not shown), the enhanced cleavage rate possibly results from conformational changes induced in hDcr upon association with TRBP.
Taken together, these observations suggest that human Dicer undergoes a conformational rearrangement upon interaction with partner proteins that activates dicing, particularly for perfect-duplex substrates. The stimulatory effect of DExD/H-box domain deletion on hDcr activity is at least superficially similar to that observed upon limited treatment of human Dicer with Proteinase K11; 12. Structural rearrangement of hDcr is perhaps mimicked by either cleaving or mutating Dicer in regions involved in the conformational change. In agreement with earlier observations, we found no evidence that ATP binding or hydrolysis accompanies RNA binding or dicing by any of the proteins tested in this study (data not shown). It remains possible that the DExD/H-box domain displays ATP or NTP-dependent behavior under conditions not yet identified, perhaps in conjunction with other interaction partners. Our data show that independent of such activity, the DExD/H-box domain functions as both a protein interaction domain and a structural switch that controls Dicer activity.
MATERIALS AND METHODS
RNA substrates
A 73 nucleotide (nt) human let-7 hairpin RNA (pre-hlet-7) was transcribed in vitro by T7 RNA polymerase from a construct containing a double ribozyme system to ensure homogeneous 5′ and 3′ ends19. All other RNA substrates were synthesized by IDT (Integrated DNA Technologies, Inc, Coralville, IA). All RNAs were purified by 16% urea-PAGE. For both filter binding and dicing assays, the purified RNA substrates were 5′-end labeled with 32P using T4 polynucleotide kinase (New England Biolabs, Inc. Beverly, MA). The sequences of RNA substrates used in this study are: pre-hlet-7, 5′-UGAGGUAGUAGGUUGUAUAGUUUUAGGGUCACACCCACCACUGGGAGAUAACUAUACAAUCUACUGUCUUACC-3′; 37a, 5′-UGAGGUAGUAGGUUGUAUAGUUUGAAAGUUCACGAUU-3′; 37b, 5′-UCGUGAACUUUCAAACUAUACAACCUACUACCUCAUU-3′.
Generation of human Dicers
To generate recombinant hDcr proteins, we first produced wild-type and Δhelicase (deletion of DExD/H-box domain) hDcr cDNAs (accession number NP_803187) by PCR with the primer sets hDcr-F/hDcr-R and helicase-F/hDcr-R, respectively (see below). The PCR products were cloned into a pFastBac plasmid (Invitrogen) using Sfo I and Xho I restriction sites. All other hDcr constructs were generated by PCR using QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene) with primer set DUF-F/DUF-R to delete DUF283, and with primer set dsRBD-F/dsRBD-R to delete the dsRBD domain (see sequences below). To generate a double-deletion of the DExD/H-box and dsRBD domains, Δhelicase pFastBac plasmid was used as the template in a PCR reaction with primer set dsRBD-F/dsRBD-R. For generation of hWalker (containing a point mutation in the ATP-binding motif in the DExD/H-box domain), PCR was performed with the primers hW-F and hW-R in the presence of wild-type hDcr pFastBac plasmid. The corresponding recombinant Bacmid DNAs were obtained by transforming pFastBac plasmids into competent DH10Bac E. coli cells (Invitrogen). Bacmid DNAs were transfected into Sf9 cells with FuGene Transfection Reagent (Roche Applied Science) for generating baculovirus, which is used to produce recombinant hDcr proteins in Sf9 cells. The hDcr proteins were purified by Ni2+-affinity chromatography, followed by TEV protease cleavage to remove the His6 tag and gel filtration chromatography using HiLoad 16/60 Superdex 200 (GE Healthcare). The PCR oligos are shown below (Sfo I and Xho I sites are underlined):
- hDcr-F: 5′-GGG_GGCGCC_ATGAAAAGCCCTGCTTTGCAACCCCTCAGCATGGCAG-3′
- hDcr-R: 5′-CCC_CTCGAG_TCAGCTATTGGGAACCTGAGGTTGATTAGC-3′
- helicase-F: 5′-GGG_GGCGCC_ATGGATGATGATGACGTTTTCCCACCATATGTGTTG-3′
- DUF-F: 5′-CGAGTCACAATCAACACGGACCATTTGATGCCAGTTGGGAAAGAG-3′
- DUF-R: 5′-CCCAACTGGCATCAAATGGTCCGTGTTGATTGTGACTCGTGGACC-3′
- dsRBD-F: 5′-GAAAAGTTTTCTGCAAATAATCAACCTCAGGTTCCCAATAGCTG-3′
- dsRBD-R: 5′-GGGAACCTGAGGTTGATTATTTGCAGAAAACTTTTCTATTAGTGGC-3′
- hW-F: 5′-AACACTGGCTCAGGGGCGACATTTATTGCAGTAC-3′helicase
- hW-R: 5′-GTACTGCAATAAATGTCGCCCCTGAGCCAGTGTT-3′
Filter binding assays
Dicer protein, prepared by serial dilution, was incubated in a buffer containing 20 mM Tris-HCl (pH 7.5), 25 mM NaCl, 5 mM EDTA, 1 mM dithiothreitol (DTT), 1% glycerol and ~0.5-1 nM (1500 c.p.m.) of 5′-end 32P-labeled duplex RNA substrate (one strand was labeled) at room temperature for 60 min in a 30 μl total volume. Following incubation, a 25 μl aliquot of each reaction was applied to a dot-blot apparatus equipped with three membranes: Tuffryn, Protran and Nytran (from top to bottom). After drying, the bound (on Protran) or free (on Nytran) RNAs were quantified by a Phosphorimager (GE Healthcare). Percent bound RNA, calculated as the ratio of radioactivity detected on the Protran membrane over the total input radioactivity, was plotted as a function of protein concentration. Kd was determined by global fitting to the equation: _k_obsd= (_k_max × [Dicer]) (_K_1/2 + [Dicer])−1, where _k_obsd is the observed rate constant at a given protein concentration, _k_max is the maximal rate constant with saturating protein, and _K_1/2 (or _K_d) is the protein concentration that provides half the maximal rate. Curve fitting was conducted with KaleidaGraph (Synergy Software, Reading, PA).
Dicing assays
dsRNA substrates were 5′-end labeled with (γ-32P)ATP, annealed and incubated with hDcr (amounts indicated in figure legends) at 37□ for the specified time in a 10 μl vollume (unless otherwise indicated) containing 20 mM Tris-HCl (pH 6.5), 1.5 mM MgCl2, 25 mM NaCl, 1 mM DTT and 1% glycerol. Reactions were stopped by adding 1.2 volumes of loading buffer (95% formamide, 18 mM EDTA, 0.025% SDS, 0.1% xylene cyanol and 0.1% bromophenol blue). After heating at 75□ for 10 min, the samples were analyzed by electrophoresis through a 15% polyacrylamide-7M urea gel run in TBE buffer and quantified using a Phosphorimager.
Kinetic analysis of Dicers
Single-turnover experiments were performed in 90 μl reaction mixtures containing 60 nM protein and 2 nM radiolabeled duplex RNA substrate. 10 μl aliquots were removed and added to 12 μl of RNA loading buffer after 0, 0.5, 1, 2, 2.5, 5, 10, 20, 40, and 80 min of incubation at 37□. Each aliquot was fractionated by 15% urea-polyacrylamide gel electrophoresis and quantitated with a Phosphorimager. Plots were prepared using KaleidaGraph. Multiple-turnover experiments were performed in 90 μl reaction mixtures containing 5 nM protein and variable amounts of duplex RNA ranging from one-third to five times the _K_d values (25, 50, 75, 100, 150, 225, and 375 nM, respectively). Aliquots were taken after 0, 0.5, 1, 2, 2.5, 5, 10, 20, 40, and 80 min of incubation at 37□, and analyzed by gel electrophoresis. After quantitation, initial rates were determined by linear regression (from 0 to 10 min) using Excel. _K_m was determined by KaleidaGraph global fitting to the equation _vo_=(_V_max × S)/(_K_m + S), where vo is the initial rate of reaction and S is the RNA concentration.
Supplementary Material
01
Figure S1. ANS binding assay. 5 μM ANS (1 -anilino-naphthalene-8-sulfonate) and 1 μM hDcr were incubated for 30 min at room temperature in a 70 μl reaction volume containing dicing buffer; fluorescence intensity (425 to 545 nm) was measured following excitation at 460 nm using a FluoroMax-3 (Jobin Yvon Inc).
ACKNOWLEDGEMENTS
We thank Kaihong Zhou for expert technical assistance and other members of the Doudna lab for helpful discussions. This work was supported by a grant from the NIH to J.A.D. J.F.K. is supported by a grant from the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare that they have no competing financial interests.
REFERENCES
- 1.Carmell MA, Hannon GJ. RNase III enzymes and the initiation of gene silencing. Nature Struct. Mol. Biol. 2004;11:214–8. doi: 10.1038/nsmb729. [DOI] [PubMed] [Google Scholar]
- 2.Murchison EP, Hannon GJ. miRNAs on the move: miRNA biogenesis and the RNAi machinery. Current Opinion Cell Biol. 2004;16:223–9. doi: 10.1016/j.ceb.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 3.MacRae IJ, Doudna JA. Ribonuclease revisited: structural insights into ribonuclease III family enzymes. Current Opinion Struct. Biol. 2007;17:138–45. doi: 10.1016/j.sbi.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Song JJ, Liu J, Tolia NH, Schneiderman J, Smith SK, Martienssen RA, Hannon GJ, Joshua-Tor L. The crystal structure of the Argonaute2 PAZ domain reveals an RNA binding motif in RNAi effector complexes. Nature Struct. Mol. Biol. 2003;10:1026–32. doi: 10.1038/nsb1016. [DOI] [PubMed] [Google Scholar]
- 5.Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W. Single processing center models for human Dicer and bacterial RNase III. Cell. 2004;118:57–68. doi: 10.1016/j.cell.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 6.Macrae IJ, Zhou K, Li F, Repic A, Brooks AN, Cande WZ, Adams PD, Doudna JA. Structural basis for double-stranded RNA processing by Dicer. Science. 2006;311:195–8. doi: 10.1126/science.1121638. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–6. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 8.Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterk RH. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001;15:2654–9. doi: 10.1101/gad.927801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Q, Rand TA, Kalidas S, Du F, Kim HE, Smith DP, Wang X. R2D2, a bridge between the initiation and effector steps of the Drosophila RNAi pathway. Science. 2003;301:1921–5. doi: 10.1126/science.1088710. [DOI] [PubMed] [Google Scholar]
- 10.Nykanen A, Haley B, Zamore PD. ATP requirements and small interfering RNA structure in the RNA interference pathway. Cell. 2001;107:309–21. doi: 10.1016/s0092-8674(01)00547-5. [DOI] [PubMed] [Google Scholar]
- 11.Provost P, Dishart D, Doucet J, Frendewey D, Samuelsson B, Radmark O. Ribonuclease activity and RNA binding of recombinant human Dicer. EMBO J. 2002;21:5864–74. doi: 10.1093/emboj/cdf578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H, Kolb FA, Brondani V, Billy E, Filipowicz W. Human Dicer preferentially cleaves dsRNAs at their termini without a requirement for ATP. EMBO J. 2002;21:5875–85. doi: 10.1093/emboj/cdf582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee Y, Hur I, Park SY, Kim YK, Suh MR, Kim VN. The role of PACT in the RNA silencing pathway. EMBO J. 2006;25:522–32. doi: 10.1038/sj.emboj.7600942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ye X, Paroo Z, Liu Q. Functional anatomy of the Drosophila microRNA-generating enzyme. J. Biol. Chem. 2007;282:28373–8. doi: 10.1074/jbc.M705208200. [DOI] [PubMed] [Google Scholar]
- 15.Vermeulen A, Behlen L, Reynolds A, Wolfson A, Marshall WS, Karpilow J, Khvorova A. The contributions of dsRNA structure to Dicer specificity and efficiency. RNA. 2005;11:674–82. doi: 10.1261/rna.7272305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dlakic M. DUF283 domain of Dicer proteins has a double-stranded RNA-binding fold. Bioinformatics. 2006;22:2711–4. doi: 10.1093/bioinformatics/btl468. [DOI] [PubMed] [Google Scholar]
- 17.Semisotnov GV, Rodionova NA, Razgulyaev OI, Uversky VN, Gripas AF, Gilmanshin RI. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 1991;31:119–28. doi: 10.1002/bip.360310111. [DOI] [PubMed] [Google Scholar]
- 18.Haase AD, Jaskiewicz L, Zhang H, Laine S, Sack R, Gatignol A, Filipowicz W. TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with Dicer and functions in RNA silencing. EMBO Rep. 2005;6:961–7. doi: 10.1038/sj.embor.7400509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferre-D’Amare AR, Doudna JA. Use of cis- and trans-ribozymes to remove 5′ and 3′ heterogeneities from milligrams of in vitro transcribed RNA. Nucleic Acids Res. 1996;24:977–8. doi: 10.1093/nar/24.5.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01
Figure S1. ANS binding assay. 5 μM ANS (1 -anilino-naphthalene-8-sulfonate) and 1 μM hDcr were incubated for 30 min at room temperature in a 70 μl reaction volume containing dicing buffer; fluorescence intensity (425 to 545 nm) was measured following excitation at 460 nm using a FluoroMax-3 (Jobin Yvon Inc).