PUTTING TUMOURS IN CONTEXT (original) (raw)
. Author manuscript; available in PMC: 2010 Nov 8.
Published in final edited form as: Nat Rev Cancer. 2001 Oct;1(1):46–54. doi: 10.1038/35094059
Abstract
The interactions between cancer cells and their micro- and macroenvironment create a context that promotes tumour growth and protects it from immune attack. The functional association of cancer cells with their surrounding tissues forms a new ‘organ’ that changes as malignancy progresses. Investigation of this process might provide new insights into the mechanisms of tumorigenesis and could also lead to new therapeutic targets.
Under normal conditions, ORGANS are made up of TISSUES that exchange information with other cell types via cell–cell contact, cytokines and the EXTRACELLULAR MATRIX (ECM). The ECM, which is produced by collaboration between STROMAL fibroblasts and EPITHELIAL cells, provides structural scaffolding for cells, as well as contextual information. The endothelial vasculature provides nutrients and oxygen, and cells of the immune system combat pathogens and remove apoptotic cells. Epithelial cells associate into intact, polarized sheets. These tissues communicate through a complex network of interactions: physically, through direct contact or through the intervening ECM, and biochemically, through both soluble and insoluble signalling molecules. In combination, these interactions provide the information that is necessary to maintain cellular differentiation and to create complex tissue structures.
Occasionally, the intercellular signals that define the normal context become disrupted. Alterations in epithelial tissues can lead to movement of epithelial sheets and proliferation — for example, after activation of mesenchymal fibroblasts due to wounding. Normally, these conditions are temporary and reversible, but when inflammation is sustained, an escalating feedback loop ensues. Under persistent inflammatory conditions, continual upregulation of enzymes such as matrix metalloproteinases (MMPs) by stromal fibroblasts can disrupt the ECM, and invading immune cells can overproduce factors that promote abnormal proliferation.
As this process progresses, the normal organization of the organ is replaced by a functional disorder (FIG. 1). If there are pre-existing epithelial cells within this changing context that possess tumorigenic potential, they can start to proliferate. Alternatively, the abnormal interactions might lead to genomic instability within the epithelial cells and the acquisition of tumorigenic potential. The proliferating cancer cells can then interact with their microenvironment and enhance the abnormal interactions. At this point, the tumour has become its own organ, with a distinct context that now defines all its cellular responses. Here, we will examine how the mechanisms that contribute to the normal context also act to suppress developing tumours, how disruption of this context initiates and supports the process of tumorigenicity, and how some cells with a tumorigenic genotype can become phenotypically normal if the context is appropriately manipulated.
Figure 1. Normal versus malignant breast tumours.

a. The normal mammary gland shows a highly structured and segregated architecture. Ducts are formed by a double layer of cells: luminal epithelial cells surrounded by a layer of myoepithelial cells, enclosed by the basement membrane. Stromal fibroblasts secrete a collagenous extracellular matrix (ECM), and blood vessels are centrally located and well defined. b. Lobular breast carcinoma is less organized. Tumour angiogenesis produces poorly defined blood vessels, and carcinoma cells intermingle with all the stromal elements.
An innate anticancer mechanism
An important feature of the normal stromal context is the generation and maintenance of epithelial-cell polarity. Epithelial cells receive a variety of orientational cues from the environment that help them establish cellular apical and basal surfaces and to maintain the differentiated state. Loss of polarity has been shown to lead to increased cell proliferation and tumorigenesis (BOX 1). The basal surface of epithelial cells associates with the BASEMENT MEMBRANE, a specialized form of ECM that provides both structural support and polarization signals to epithelia. The basement membrane is a dynamic structure. Changes in its composition lead to changes in cell shape and behaviour1, altered binding affinity or cellular distribution of cell-surface receptors2, and cellular responses to soluble molecules3. Depending on the composition and physical characteristics of the basement membrane, different soluble factors can have completely different cellular effects, such as inducing cell proliferation, growth arrest, differentiation or apoptosis4.
Box 1 . Epithelial cell polarity and tumorigenesis in Drosophila.
Loss of apicobasal polarity in carcinomas has often been viewed as a secondary consequence of oncogenic transformation134, but recent investigations of Drosophila mutants have shown that loss of polarity determinants can directly lead to increased proliferation and development of tumours. In a genetic screen for mutations that cause aberrant epithelial structures, Bilder, Li and Perrimon135 identified scribble (Scrib) as a gene that is required for proper localization of apical proteins and the components of the adherens junctions. Absence of Scrib is associated with the loss of epithelial polarity and neoplastic transformation, a phenotype previously observed in two other Drosophila mutants: discs large (Dlg) and lethal giant larvae (Lgl); subsequent investigations showed that all three proteins act in the same pathway136. Although many details are not yet resolved, Scrib and Dlg seem to colocalize at the septate junction, a structure similar to the mammalian tight junction, where they direct the polarized sorting of a specific population of Lgl-containing vesicles. It is loss of this directional-sorting mechanism that suppresses normal growth-control mechanisms.
Two mechanisms have been proposed to account for the increased proliferation. First, it is possible that misdirection of cell-surface signal receptors could lead to inappropriate receptor synergy; in this regard, it is noteworthy that ERBIN, a mammalian protein with homology to Scrib, has been shown to interact with the epidermal growth factor receptor (EGFR) family member ERBB2 (HER2)137. Alternatively, mislocalization of the components of the adherens junctions might release contact inhibition, leading to hyperproliferation. At present, these mechanisms cannot be distinguished, and it is possible that both contribute to neoplastic transformation. Further investigations of molecules that maintain the tissue-specific functions of polarized epithelia will doubtless reveal more about their complex relationship with tumorigenesis.
Epithelial cells maintain physical contact with their neighbours through a combination of ADHERENS JUNCTIONS, GAP JUNCTIONS, TIGHT JUNCTIONS and DESMOSOMES (FIG. 2). Of these, adherens junctions have been a particular focus of studies into the signals that generate epithelial-cell polarity, but more recent investigations have revealed mechanisms by which gap junctions, tight junctions and desmosomes also contribute to the formation of polarized epithelial tissues5–8.
Figure 2. Mechanisms of cell–cell and cell–ECM interactions.

Integrin and non-integrin cell-surface receptors form attachments with the actin filaments in the cytoskeleton, and are able to sense elements of the extracellular matrix (ECM) to promote growth-factor activation. Tight junctions act as a barrier to the diffusion of solutes through the intercellular space and act as a boundary between the apical and basolateral plasma-membrane domains. Adherens junctions, which consist of extracellular E-cadherin dimers connected to cytoplasmic α- and β-catenin molecules, are anchored to actin filaments. Gap junctions provide a communication mechanism by allowing solutes and small signalling molecules to pass between adjacent cells. Desmosomes serve as anchoring points for INTERMEDIATE FILAMENTS and also provide signalling information.
Adherens junctions are contacts between adjacent epithelial cells and are anchored to the cytoskeleton. Cadherins (such as E-CADHERIN) traverse the membrane, associating with cadherins on adjacent cells in a calcium-dependent manner. On the cytoplasmic face, β-catenin connects to the cadherin tail and associates with α-catenin, which in turn binds to actin. Loss or alteration of these components leads to premalignant phenotypes and even tumorigenesis9–11. Of these components, E-cadherin has been a particular target of study, as this molecule is lost in many types of tumour9, and its restoration can suppress cellular transformation. Decreased E-cadherin function is a component of EPITHELIAL–MESENCHYMAL TRANSITION, invasive tumour growth and metastasis11–13. Loss of E-cadherin can be accompanied by increased expression of alternate cadherin isoforms that promote inappropriate survival signals and enhance the malignant phenotype14. However, as all cellular responses are tissue- and context-dependent, there can be no universal generalizations, as shown by the fact that E-cadherin gain of function is an early step in ovarian carcinoma15.
Gap junctions are channel-forming complexes that allow passive diffusion of small signalling molecules between neighbouring cells5. The particular composition of CONNEXIN subunits within a gap junction determines the type of molecule that can be transported16. Much remains to be learned about how the specific combination of connexins facilitates tissue interactions, but it is clear, again, that generalizations should be avoided, as the expression patterns (and probably the function) of connexins are tissue dependent and change during tumour progression17,18. For example, some breast cancer cells upregulate connexin 32 (Cx32)19, but loss of Cx32 contributes to hepatocellular carcinoma20,21; Cx43 inhibits tumorigenicity of lung, cervical and bladder carcinoma cells22–24, but has no effect on squamous cell carcinomas25; and other connexins can facilitate cell adhesion during metastasis26.
Changing interactions between adjacent tissues might also affect tumour development (FIG. 3). For example, in the normal human mammary gland (FIG. 1), the ductallobular system is composed of an inner layer of luminal epithelial cells, which line the duct and produce milk during lactation, and an outer layer of myoepithelial cells, which express a number of proteases during tissue remodelling to pave the way for emerging ductules. This double-layered structure is separated from the INTERSTITIAL MATRIX by an intact basement membrane27. Breast cancer arises mainly in the luminal epithelial compartment, but myoepithelial cells also express molecules that have been shown to suppress transformation of luminal epithelial cells in vivo 27 (TABLE 1). These proteins have been named ‘class II tumour suppressors’28 and production of these proteins allows myoepithelial cells to act as tumour suppressors in the breast27,29.
Figure 3. Differences in stroma between tumours.

Interstitial stromal cells of normal and breast tumour tissues differ in levels of smooth muscle differentiation. a. Normal interstital stroma(s) does not express smooth muscle actin (red) or b. smooth muscle myosin (green), indicating that smooth muscle differentiation has not taken place, although these cells do produce blood vessels (bv). c–f. Tumour tissues, by contrast, express high levels of smooth muscle actin (c,e). These images also show, however, that not all tumour stroma are similar. The tumour stroma shown in (c) and (d) expresses smooth muscle actin (c) but not smooth muscle myosin (d). In the tumour shown in e and f, the stromal cells express high levels of both actin and smooth muscle myosin. Adapted from REF. 142.
Table 1.
Myoepithelial proteins that suppress luminal tumours
| Protein | Function | Reference |
|---|---|---|
| α-smooth muscle actin | Cell structure | 143 |
| Cytokeratin 5 | Cell structure | 144 |
| α6-integrin | ECM receptor | 145 |
| Caveolin-1 | Cell-surface molecule | 146 |
| Connexin 43 | Gap-junction component | 147 |
| Maspin | Protease inhibitor | 148 |
| TIMP-1 | Protease inhibitor | 29 |
| Relaxin | Hormone | 149 |
| Activin | Hormone | 150 |
In combination, these mechanisms create a dynamic equilibrium that helps cells to maintain a normal, differentiated phenotype. This equilibrium might attenuate the consequences of genetic mutations, as consideration of the frequency of spontaneous mutations indicates that many epithelial cells should possess oncogene-activating mutations, yet cells continue to function normally30,31. Analyses of normal epithelial tissue adjacent to tumours have shown that similar patterns of mutations can be found in both, indicating that malignant cells can exist within normal tissues but be restrained by normal contextual cues32–34.
Activated stroma as a carcinogen
Whereas normal stroma can delay or prevent tumorigenesis, abnormal stromal components can promote tumour growth (FIG 4). Acquired or inherited mutations that alter stromal-cell function can release the suppression placed on context-inhibited malignant cells. Literature that spans more than a century has shown that inflammation associated with tissue wounding can produce tumours (REFS 35–38 and references therein) (BOX 2). . Barcellos-Hoff and colleagues39 have shown that irradiation of the mammarygland stromal component promotes the tumorigenic potential of non-irradiated epithelial cells. These investigators had previously shown that even low levels of irradiation lead to remodelling of the ECM in breast tissue and activation of latent transforming growth factor-β (TGF-β), which affects tissue and organ function40. Moinfar et al.41 examined genetic alterations in tumour-associated stroma from several independent cases of mammary carcinoma, and found chromosomal rearrangements that were not present in the malignant carcinoma cells. These results indicate that characteristic mutations that affect stromal cells might have contributed to the formation of the epithelial tumours. Moreover, studies of a subset of inherited cancer-susceptibility syndromes42,43 also indicate that alterations in stromal cells can contribute to tumorigenesis. So, aberrations in stroma can both precede and stimulate the development of epithelial cancers44,45.
Figure 4. The tumour microenvironment assay.

a. Primary breast carcinoma cells form spherical colonies when cultured in three-dimensional collagen type I. b. Co-cultivation with stromal cells, however, causes the tumour cells to spread and become invasive. The degree of tumour growth increases with the density of the stromal cells. Staining of the coculture assay (c) and of tumour (d) with anti-vimentin antibody reveals the structural similarities of stromal cells in the presence or absence of cancer cells. (Reproduced with permission from REF. 142 © (1995) American Society for Clinical Investigation.)
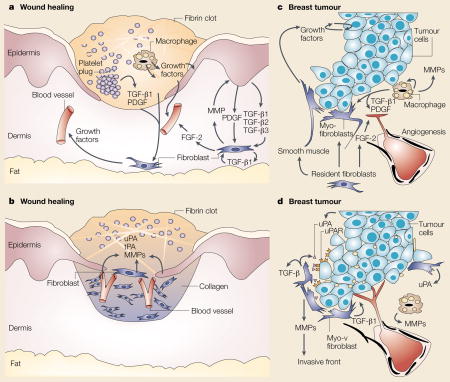
Box 2 . Comparison between wound healing and tumour development.
Wound healing and tumour development are dynamic, progressive processes that involve the interaction of several tissue types138, and comparison of the two reveals many mechanistic similarities. a | Immediate reaction to wounding. Tissue injury leads to activation of platelets that form a haemostatic plug and also release vasoactive mediators to increase vascular permeability and to enable the influx of serum fibrinogen to generate the fibrin clot. Platelets produce chemotactic factors, including transforming growth factor-β (TGF-β) and platelet-derived growth factor (PDGF). These factors initiate the formation of granulation tissue by activating fibroblasts to produce matrix metalloproteinases (MMPs) and a number of growth factors, such as fibroblast growth factor-2 (FGF-2). These factors degrade dermal extracellular matrix, stimulate infiltration of macrophages and promote the development of new blood vessels. These interactions are potentiated by reciprocal signalling between the epidermis and dermal fibroblasts through growth factors, MMPs, and members of the TGF-β family. b | Reformation of the epithelial sheet. The complex reaction to wounding reduces epithelial adhesiveness and increases epithelial-cell mobility to re-form an intact sheet of tissue over the wound. Production of MMPs and proteolytic enzymes such as uroplasminogen activator (uPA) and tissue plasminogen activator (tPA) facilitates this re-epithelialization. Blood vessels can then enter the fibrin clot as epidermal cells resurface the wound. The lateral migration of the epidermal cells is followed by a reversion to the normal, non-motile phenotype, including regeneration of a basement membrane and resynthesis of HEMIDESMOSOMES. Following re-epithelialization, a new basement membrane is produced and many of the fibroblasts take on a myofibroblast phenotype to facilitate wound contraction. c | Reciprocal activation mechanisms in early tumours. Building on a rich, but inconclusive, literature spanning nearly a century (reviewed in REF. 35), Dvorak proposed that tumours activate some of the normal wound-healing responses139. Although developing tumours do not disrupt the vascular tissue in the same way as in wounding, many of the processes occur in parallel. Tumour cells (blue) produce many of the same growth factors that activate the adjacent stromal tissues in wounding or fibrosis37,140,141. Activated fibroblasts and infiltrating immune cells (macrophage) secrete MMPs and cytokines such as TGF-β FGF-2, and PDGF. These factors potentiate tumour growth, stimulate angiogenesis, and induce fibroblasts to undergo differentiation into myofibroblasts and into smooth muscle. d | Expression of proteases at the invasive front. Tumour cells, myofibroblasts and activated macrophages increase production of MMPs and uPA at the invasive front to stimulate angiogenesis and proliferation. Production of TGF-β also promotes tumour growth. uPAR, uroplasminogen receptor.
Matrix metalloproteinases
MMPs can degrade ECM and are involved in promoting the inflammatory response, normal tissue remodelling, wound healing and angiogenesis46. These enzymes, however, also have an important function in malignancy (BOX 2). The sustained presence of these proteinases in the tumour environment, produced both by the activated cells and by the cancer cells, leads to destruction of normal ECM. Degradation of ECM stimulates both proliferative and apoptotic mechanisms, which can lead to the selection of apoptosis-resistant carcinoma cells and enhanced invasive potential47,48. In the tumour context, direct association of MMPs with specific ECM receptors provides spatial control of MMP activity and directional signals to the invading tumour cells49.
Stromelysin-1 (SL-1, also known as MMP-3), is an MMP that is involved in both mammary-gland development and breast cancer50,51. Cellular context determines the response of mammary epithelial cells to SL-1 treatment: when grown in basement-membrane gels, mammary epithelial cells undergo growth arrest and become functionally differentiated; subsequent treatment of these cells with SL-1 causes apoptosis52. However, when cultured on two-dimensional matrices and allowed to continuously proliferate, mammary epithelial cells react to treatment with SL-1 by undergoing an epithelial–mesenchymal transition and becoming tumorigenic50.
In transgenic mice that express SL-1 in mammary luminal epithelial cells, the mammary glands show morphogenesis defects and contain pre-neoplastic lesions44,53,54 that eventually lead to full malignancies50,54. Here, the causative mechanism seems to be that SL-1 — expressed ectopically at low levels in the epithelial cells — is subsequently produced at much higher levels by the stromal fibroblasts44, showing that a moderate disruption contributes to a self-sustaining tumorigenic state. Similar reciprocal feedback mechanisms have been observed in transgenic mice with altered expression of MMP-7 (REF. 55), MMP-11 (REF. 56) and MT1-MMP57.
Immune function in the tumour context
Immune surveillance is the mechanism by which the immune system targets and destroys developing malignancies. Investigations of transgenic mice with deficient responses to interferon-γ (IFN-γ), a cytokine that has been shown to be required for migration of T cells to tumour sites58,59, have led to increased interest in the mechanisms by which immune cells target tumours60,61. Although T cells seem to be the main effectors of immune surveillance62, the innate immune system (which includes natural killer cells, macrophages, monocytes and mast cells) is also involved63–65.
Malignant cells evade immunosuppression by downregulating intrinsic immunogenicity66,67. The tumour vasculature contributes to this process by preventing extravasation of the antitumour T cells, while continuing to allow the passage of innate immune cells68. Studies by Gloria Heppner and colleagues (REF. 69, and references therein) showed that natural killer cells actually provided positive signals for progression of preneoplastic mammary lesions. This initially controversial concept has received support from recent investigations of carcinomas of the skin70,71, pancreas72 and mammary gland73, showing that innate immune cells promote tumorigenesis by producing MMPs, inducing the stroma to produce MMPs and by activating latent MMPs that are present in the ECM74–76. The resultant increase in proteolytic activity potentiates tumour progression by further degrading ECM, activating tumour-associated fibroblasts and enhancing angiogenesis70,72.
Macrophage migration inhibitory factor (MIF) is another immunomodulator that is associated with tumour progression. This cytokine has been shown to be overexpressed by tumour cells77, contributing to neoangiogenesis and to epithelial cell proliferation77, as well as suppressing immune surveillance78. MIF might also contribute to the genomic instability within tumours, as MIF suppresses p53 function79, potentially leading to the attenuation of normal apoptosis and growth-arrest mechanisms and allowing for the accumulation of additional oncogenic mutations80. This might be one of the mechanisms by which persistent inflammation can increase the risk of cancer81.
Tumour-cell plasticity
One manifestation of the distinct tumour context is that cells from a given malignant tissue are not limited to that tissue’s normal panoply of physiological processes. The classic work of Beatrice Mintz (discussed below) is a prime example of this, but a more recent example can be found in ‘vasculogenic mimicry’, a process in which aggressive tumours can augment normal angiogenesis by forming hollow channels that connect to the existing vascular system. These vessels are believed to transport blood into the depths of the tumour82,83. This concept has now been well-characterized84–87 and could represent a general component of tumour development88. To produce more selective antiangiogenic therapies, it might be necessary to combine detailed examinations of vasculogenic mimicry with existing models of tumour angiogenesis.
Haematological tumours
More than 80% of human cancers are derived from the epithelium, but the role of context in the development and maintenance of cancer also seems to apply to tumours of haematological origin. Although most immune cells spend much of their lifespan in the circulatory system, key aspects of immune-cell development involve cell–cell and cell–ECM interactions within the stroma of the bone marrow, the thymus and the lymph nodes89,90. In haematopoietic cells, as in epithelial cells, these interactions control cell shape, adhesion and migration91.
Accordingly, defects in the function of bone-marrow stromal cells can cause a predisposition to cancer, such as in cases of Shwachman–Diamond syndrome, an inherited preleukaemic disorder that is caused by a faulty bone-marrow microenvironment92. As with tumours that are derived from the epithelium, haematological tumour cells interact with their stromal microenvironment through cell-surface receptors93–95. These interactions lead to increased production of MMPs96,97, altered expression of ECM receptors98,99 and increased angiogenesis100,101. The interactions between haematological tumour cells and the tumour stroma are, therefore, a significant component of tumour growth and resistance to anticancer therapeutics102–106.
Restoring the normal context
Although an abnormal context can contribute to tumorigenesis and tumour progression, there is no compelling evidence that this process, once initiated, is irreversible. The possibility that reintroduction of the normal context could suppress the transformed phenotype was first suggested by the work of Mintz and Illmensee, who showed that TERATOCARCINOMA CELLS, even after prolonged passage, were still capable of differentiating and generating normal mice107. This seminal observation indicated that maintenance of a normal context could lead to inhibition or even reversion of tumours in situ. In another example, Rous sarcoma virus — one of the most potent oncogenic viruses — is not tumorigenic in the early embryo108, but when the embryonic cells that host the virus cells are put in culture, they become transformed109.
In co-culture assays, normal stromal cells inhibit the progression to epithelial malignancy110. Norbert Fusenig and colleagues have developed an assay to model the natural tissue context of the stratified skin epithelium111. Using this system, they were able to suppress early stages of neoplastic progression of malignant keratinocytes by introducing an excess of normal keratinocytes112.
An assay involving a three-dimensional (3D) basement membrane113 has been used to investigate the response of a series of human breast-tumour cell lines at different stages of progression, cultured within a physiological context114. Although the non-malignant cells are similar in appearance to the malignant cells when cultured on plastic substrata, the phenotypic differences are striking when the cells are cultured in a reconstituted basement membrane (rBM)115. Under these conditions, the non-malignant cells undergo growth arrest and form a polarized, alveolar structure, whereas the malignant cells proliferate and form amorphous structures. Analysis of ECM and growth-factor receptors in the non-malignant and malignant cell types indicates that the malignant cells overexpress INTEGRINS and epidermal growth factor receptor (EGFR). Addition of anti-β1-integrin antibodies to the malignant cells, when cultured in 3D rBM, downregulated EGFR expression, restored cellular organization, and decreased overall tumorigenicity115. This observation led to the discovery of a bidirectional cross-modulation of integrin and EGFR signalling that exists only when cells are cultured in 3D116. Furthermore, the tumorigenic phenotype of the malignant cells was reversed by treatment with EGFR-inhibitory antibodies, mitogen-activated protein kinase (MAPK) pathway inhibitors, or phosphatidylinositol 3-kinase (PI3K) pathway inhibitors116,117. Inhibiting several different signalling pathways restores even an aggressive breast-cancer cell line to a normal phenotype117.
Therefore, assays in which tumour cells are cultured in physiological conditions can be used to identify combinations of signalling inhibitors with the potential to reverse the progression of a broad range of tumours. The success of agents that are designed to inhibit other signal transduction pathways, such as herceptin (which blocks signalling by the EGFR ERBB2 (HER2) in breast cancer cells) and STI-571 (which inhibits BCR–ABL kinase activity in chronic myelogenous leukaemia cells) indicates that this might be a valid approach118,119. It is clear, however, that relapses occur and many patients do not respond. These agents were designed to target one particular oncoprotein, so it might be necessary — in cases of more complex cancers — to target both the tumour and its context, using combinations of drugs.
Targeting the tumour organ
The efficacy of targeting the tumour organ can be found in recent strategies for treating hepatocellular carcinoma. This cancer type is accompanied by a fibrotic stromal reaction in which HEPATIC STELLATE CELLS show increased proliferation, fibrogenesis and matrix degradation, as well as reduced retinoid production and cytokine release120 — physiological responses often found in tumour tissues. Recent clinical studies indicate that chemotherapy for hepatocarcinoma could be more effective if therapies to target the underlying liver fibrosis were also employed120,121. As fibrotic breast disease is also associated with a predisposition to breast cancer122, and environmentally induced fibrotic disorders of the lung can increase incidence of lung cancer123, targeting the tumour environment might also increase the treatment effectiveness for these types of cancer.
Antagonism of the developing tumour context also offers potential for cancer prevention therapies124,125. In the best-characterized example of this approach so far, chronic suppression of inflammation through use of non-steroidal anti-inflammatory drugs (NSAIDs) has been shown to lower the incidence of colon and breast cancer125,126. This antitumour activity seems to occur through inhibition of cyclooxygenase-2 (COX2)127–129, an enzyme that is involved in the synthesis of pro-inflammatory prostaglandins (see the review by Rajnish Gupta and Raymond DuBois on pp. 11–21 in this issue). The demonstration of the role of COX2 in tumorigenesis serves as a remarkable example of how the several tissue types can collaborate to promote tumour progression, as fibroblasts, immune system cells and cells involved in neoangiogenesis are all part of this pathway130.
The requirement of tumours for a vascular supply has also produced a diverse group of angiogenesis inhibitors that are currently undergoing evaluation in the clinic131. Similarly, the role of MMPs in tumorigenesis, tumour invasion and metastasis has prompted clinical testing of MMP inhibitors. Although the results with patients suffering from advanced stages of cancer have shown no clinical efficacy, recent data indicate that MMP inhibitors could be more successful when used in early-stage cancer or in conjunction with traditional treatment methods132,133.
So, agents that target the tumour microenvironment represent an important new direction for cancer therapy. Just as the normal context creates a dynamic equilibrium to maintain normal tissue function, so the tumour context contains many overlapping mechanisms to maintain its functional disorder and to evade anticancer therapies. Therefore, it is likely that combinations of the next-generation therapeutic agents, targeting specific molecular targets, will be required not only to inhibit and destroy the tumour cells, but also to normalize the tumour microenvironment. Gaining a better understanding of the complexities of the tumour context will improve our prospects for developing effective cancer treatments. Dormant metastases are not the only sheep in wolves’ clothing — the altered microenvironment of the tumour is itself a powerful and insidious carcinogen that needs to be targeted.
Acknowledgments
The research performed in the laboratory of M.J.B. and summarized in this review was supported by the United States Department of Energy (DOE), the Office of Biological and Environmental Research, and by the NCI. D.R. was supported by a Distinguished Hollaender Postdoctoral Fellowship from the DOE.
ORGAN
An anatomically discrete collection of tissues, integrated to perform specific functions
TISSUE
A relatively homogenous structure, composed of an organized collection of cells of similar morphology and function
EXTRACELLULAR MATRIX (ECM)
A complex, three-dimensional network of very large macromolecules that provides contextual information and an architectural scaffold for cellular adhesion and migration
STROMA
Organ compartment serving as the connective tissue framework includes fibroblasts; immune defence cells and fat cells
EPITHELIUM
A diverse group of tissues that covers or lines nearly all body surfaces, cavities and tubes, functioning as interfaces between different biological compartments. Epithelial layers provide physical protection and containment, and also mediate organ-specific transport properties
BASEMENT MEMBRANE
A specialized form of ECM that consists of laminins, collagen IV, nidogen (entactin), proteoglycans and a number of other glycoproteins that separates epithelia from underlying supporting tissues. Different organs have different compositions of basement membrane
ADHERENS JUNCTION
A physical junction that links apicolaterally localized belts of actin in adjacent epithelial cells
GAP JUNCTION
An aqueous channel that interconnects the cytoplasms of adjacent cells and allows direct exchange of small cytoplasmic components. It is created by the association of two hemichannels, each a hexamer of connexin subunits
TIGHT JUNCTION
A component of cell–cell adhesion in epithelial and endothelial cell sheets. Acts as a mediator of the diffusion of solutes through the intercellular space. Also acts as a boundary between the apical and basal plasma-membrane domains
DESMOSOME
An adhesive junction that anchors intermediate filaments between adjoining cells
E-CADHERIN
The main adhesion receptor in adherens junctions. Mediates Ca2+-dependent interactions between adjacent epithelial cells and regulates cell proliferation. It also sequesters the transcriptional co-activator β-catenin, a protein that can stimulate cell-cycle entry. The loss of E-cadherin from the cell surface might trigger epithelial–mesenchymal transition
EPITHELIAL–MESENCHYMAL TRANSITION
Conversion from an epithelial to a mesenchymal phenotype, which is a normal component of embryonic development. In carcinomas, this transformation results in altered cell morphology, the expression of mesenchymal proteins and increased invasiveness
CONNEXIN
Functions as a subunit of the gap junction hemichannel. Several members of the connexin family have been identified
INTERSTITIAL MATRIX
The extracellular matrix (ECM) contained within the stroma
INTERMEDIATE FILAMENT
A component of the eukaryotic cytoskeleton. Intermediate filaments form a dense network extending from the nucleus to the plasma membrane
TERATOCARCINOMA
A malignant germ-cell tumour arising from the ovary or testis that is composed of embryonal carcinoma cells
INTEGRINS
A family of more than 20 heterodimeric cell-surface extracellular matrix (ECM) receptors. They connect the structure of the ECM with the cytoskeleton and can transmit signalling information bidirectionally
HEPATIC STELLATE CELLS
The principal fibrogenic cell type of the liver. They are located in a perivascular orientation and contain long cytoplasmic processes that interact with neighbouring cells
HEMIDESMOSOME
An adhesion complex located at the interface of epithelial cells with the basement membranes. Responsible for linking keratin intermediate filaments to components of the extracellular matrix
Footnotes
References
- 1.Roskelley CD, Srebrow A, Bissell MJ. A hierarchy of ECM-mediated signalling regulates tissue-specific gene expression. Curr Opin Cell Biol. 1995;7:736–747. doi: 10.1016/0955-0674(95)80117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartz MA, Baron V. Interactions between mitogenic stimuli, or, a thousand and one connections. Curr Opin Cell Biol. 1999;11:197–202. doi: 10.1016/s0955-0674(99)80026-x. [DOI] [PubMed] [Google Scholar]
- 3.Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 4.Radisky D, Muschler J, Bissell MJ. Order and disorder: the role of extracellular matrix in epithelial cancer. Cancer Invest. doi: 10.1081/cnv-120000374. (in the press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar NM, Gilula NB. The gap junction communication channel. Cell. 1996;84:381–388. doi: 10.1016/s0092-8674(00)81282-9. [DOI] [PubMed] [Google Scholar]
- 6.Green KJ, Gaudry CA. Are desmosomes more than tethers for intermediate filaments? Nature Rev Mol Cell Biol. 2000;1:208–216. doi: 10.1038/35043032. [DOI] [PubMed] [Google Scholar]
- 7.Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nature Rev Mol Cell Biol. 2001;2:285–293. doi: 10.1038/35067088. [DOI] [PubMed] [Google Scholar]
- 8.Runswick SK, O’Hare MJ, Jones L, Streuli CH, Garrod DR. Desmosomal adhesion regulates epithelial morphogenesis and cell positioning. Nature Cell Biol. 2001;3:823–830. doi: 10.1038/ncb0901-823. [DOI] [PubMed] [Google Scholar]
- 9.Christofori G, Semb H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem Sci. 1999;24:73–76. doi: 10.1016/s0968-0004(98)01343-7. [DOI] [PubMed] [Google Scholar]
- 10.Raghavan S, Bauer C, Mundschau G, Li Q, Fuchs E. Conditional ablation of β1 integrin in skin. Severe defects in epidermal proliferation, basement membrane formation, and hair follicle invagination. J Cell Biol. 2000;150:1149–1160. doi: 10.1083/jcb.150.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lochter A, et al. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol. 1997;139:1861–1872. doi: 10.1083/jcb.139.7.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hordijk PL, et al. Inhibition of invasion of epithelial cells by Tiam1-Rac signaling. Science. 1997;278:1464–1466. doi: 10.1126/science.278.5342.1464. [DOI] [PubMed] [Google Scholar]
- 13.Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392:190–193. doi: 10.1038/32433. Using E-cadherin knockout mice and dominant-negative forms of this protein, the authors show that loss of E-cadherin is associated with pancreatic β-cell carcinogenesis. [DOI] [PubMed] [Google Scholar]
- 14.Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol. 2000;148:779–790. doi: 10.1083/jcb.148.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Auersperg N, et al. E-cadherin induces mesenchymal-to-epithelial transition in human ovarian surface epithelium. Proc Natl Acad Sci USA. 1999;96:6249–6254. doi: 10.1073/pnas.96.11.6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicholson BJ, Weber PA, Chang HC, Lampe P, Goldberg G. The molecular basis of selective permeability of connexins is complex and includes both size and charge. Br J Med Biol Res. 2000;33:369–378. doi: 10.1590/s0100-879x2000000400002. [DOI] [PubMed] [Google Scholar]
- 17.Hanna EA, et al. Gap juncitonal intercellular communication and connexin43 expression in human ovarian surface epithelial cells and ovarian carcinomas in vivo and in vitro. Carcinogenesis. 1999;20:1369–1373. doi: 10.1093/carcin/20.7.1369. [DOI] [PubMed] [Google Scholar]
- 18.Locke D. Gap junctions in normal and neoplastic mammary gland. J Pathol. 1999;186:343–349. doi: 10.1002/(SICI)1096-9896(199812)186:4<343::AID-PATH189>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 19.Saunders MM, et al. Breast cancer metastatic potential correlates with a breakdown in homospecific and heterospecific gap junctional intercellular communication. Cancer Res. 2001;61:1765–1767. [PubMed] [Google Scholar]
- 20.Temme A, et al. High incidence of spontaneous and chemically induced liver tumors in mice deficient for connexin 32. Curr Biol. 1995;7:713–716. doi: 10.1016/s0960-9822(06)00302-2. [DOI] [PubMed] [Google Scholar]
- 21.Moennikes O, Buchmann A, Willecke K, Traub O, Schwarz M. Hepatocarcinogenesis in female mice with mosaic expression of connexin32. Hepatology. 2000;32:501–506. doi: 10.1053/jhep.2000.16598. [DOI] [PubMed] [Google Scholar]
- 22.Zhang ZQ, et al. Suppression of tumorigenicity of human lung carcinoma cells after transfection with connexin43. Carcinogenesis. 1998;19:1889–1894. doi: 10.1093/carcin/19.11.1889. [DOI] [PubMed] [Google Scholar]
- 23.King TJ, et al. Reduced levels of connexin43 in cervical dysplasia: inducible expression in a cervical carcinoma cell line decreases neoplastic potential with implications for tumor progression. Carcinogenesis. 2000;21:1097–1109. [PubMed] [Google Scholar]
- 24.Krutovskikh VA, et al. Differential effect of subcellular communication impairing gap junction protein connexin43 on tumor cell grown in vivo. Oncogene. 2000;19:505–513. doi: 10.1038/sj.onc.1203340. [DOI] [PubMed] [Google Scholar]
- 25.Mesnil M, et al. Negative growth control of HeLa cells by connexin genes: connexin species specificity. Cancer Res. 1995;55:629–639. [PubMed] [Google Scholar]
- 26.Ito A, et al. A role for heterologous gap junctions between melanoma and endothelial cells in metastasis. J Clin Invest. 2000;105:1189–1197. doi: 10.1172/JCI8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ronnov-Jessen L, Petersen OW, Bissell MJ. Cellular changes involved in conversion of normal to malignant breast: importance of the stromal reaction. Physiol Rev. 1996;76:69–125. doi: 10.1152/physrev.1996.76.1.69. [DOI] [PubMed] [Google Scholar]
- 28.Sager R. Expression genetics in cancer: shifting the focus from DNA to RNA. Proc Natl Acad Sci USA. 1997;94:952–955. doi: 10.1073/pnas.94.3.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sternlicht MD, Kedeshian P, Shoa ZM, Safarians S, Barsky SH. The human myoepithelial cell is a natural tumor suppressor. Clin Cancer Res. 1997;3:1949–1958. [PubMed] [Google Scholar]
- 30.Stoler DL, et al. The onset and extent of genomic instability in sporadic colorectal tumor progression. Proc Natl Acad Sci USA. 1999;96:15121–15126. doi: 10.1073/pnas.96.26.15121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frame S, Balmain A. Integration of positive and negative growth signals during Ras pathway activation in vivo. Curr Opin Genet Dev. 2000;10:106–113. doi: 10.1016/s0959-437x(99)00052-0. [DOI] [PubMed] [Google Scholar]
- 32.Deng G, Lu Y, Zlotnikov G, Thor AD, Smith HS. Loss of heterozygosity in normal tissue adjacent to breast carcinomas. Science. 1996;274:2057–2059. doi: 10.1126/science.274.5295.2057. [DOI] [PubMed] [Google Scholar]
- 33.Washington C, Dalbègue F, Abreo F, Taubenberger JK, Lichy JH. Loss of heterozygosity in fibrocystic change of the breast. Am J Pathol. 2000;157:323–329. doi: 10.1016/S0002-9440(10)64543-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bissell MJ, et al. Tissue structure, nuclear organization, and gene expression in normal and malignant breast. Cancer Res. 1999;59:1757–1763. [PubMed] [Google Scholar]
- 35.Sieweke MH, Bissell MJ. The tumor-promoting effect of wounding: a possible role for TGF-β-induced stromal alterations. Crit Rev Oncog. 1994;5:297–311. doi: 10.1615/critrevoncog.v5.i2-3.90. [DOI] [PubMed] [Google Scholar]
- 36.Dolberg DS, Hollingsworth R, Hertle M, Bissell MJ. Wounding and its role in RSV-mediated tumor formation. Science. 1985;230:676–678. doi: 10.1126/science.2996144. [DOI] [PubMed] [Google Scholar]
- 37.Sieweke MH, Thompson NL, Sporn MB, Bissell MJ. Mediation of wound-related Rous sarcoma virus tumorigenesis by TGF-β. Science. 1990;248:1656–1660. doi: 10.1126/science.2163544. [DOI] [PubMed] [Google Scholar]
- 38.Mintz B, Silvers WK. Transgenic mouse model of malignant skin melanoma. Proc Natl Acad Sci USA. 1993;90:8817–8821. doi: 10.1073/pnas.90.19.8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barcellos-Hoff MH, Ravani SA. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res. 2000;60:1254–1260. Radiation-induced mutation of the stromal microenvironment can contribute to neoplastic progression of epithelial cells in vivo, indicating that disruption of solid tissue interactions is a method by which ionizing radiation acts as a carcinogen. [PubMed] [Google Scholar]
- 40.Ehrhart EJ, Segarini P, Tsang ML, Carroll AG, Barcellos-Hoff MH. Latent transforming growth factor β1 activation in situ: quantitative and functional evidence after low-dose γ-irradiation. FASEB J. 1997;11:991–1002. doi: 10.1096/fasebj.11.12.9337152. [DOI] [PubMed] [Google Scholar]
- 41.Moinfar F, et al. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis. Cancer Res. 2000;60:2562–2566. An examination of loss of heterozygosity (LOH) in microdissected mammary stromal and epithelial tissue samples. LOH at several loci was observed exclusively in stromal cells, indicating that genetic instability in the stroma can be a contributing factor to tumour progression. [PubMed] [Google Scholar]
- 42.Jacoby RF, et al. A juvenile polyposis tumor suppressor locus at 10p11 is deleted from nonepithelial cells in the lamina propria. Gastroenterology. 1997;112:1398–1403. doi: 10.1016/s0016-5085(97)70156-2. [DOI] [PubMed] [Google Scholar]
- 43.Howe JR, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086–1088. doi: 10.1126/science.280.5366.1086. Alterations in stromal function predispose colonic epithelial cells to carcinoma. [DOI] [PubMed] [Google Scholar]
- 44.Thomasset N, et al. Expression of autoactivated stromelysin-1 in mammary glands of transgenic mice leads to a reactive stroma during early development. Am J Pathol. 1998;153:457–467. doi: 10.1016/S0002-9440(10)65589-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tlsty TD. Stromal cells can contribute oncogenic signals. Semin Cancer Biol. 2001;11:97–104. doi: 10.1006/scbi.2000.0361. [DOI] [PubMed] [Google Scholar]
- 46.Vu TH, Werb Z. Matrix metalloproteinases: effectors of development and normal physiology. Genes Dev. 2000;14:2123–2133. doi: 10.1101/gad.815400. [DOI] [PubMed] [Google Scholar]
- 47.Sethi T, et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nature Med. 1999;5:662–668. doi: 10.1038/9511. [DOI] [PubMed] [Google Scholar]
- 48.Mitsiades N, Yu W, Poulaki V, Tsokos M, Stamenkovic I. Matrix metalloproteinase-7-mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res. 2001;61:577–581. [PubMed] [Google Scholar]
- 49.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 50.Sternlicht MD, et al. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 1999;98:137–146. doi: 10.1016/s0092-8674(00)81009-0. Shows that sustained expression of stromelysin-1, a stromal enzyme that destroys the basement membrane, can lead to epithelial tumorigenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wright J, et al. A switch from stromal to tumor cell expression of stromelysin-1 mRNA is associated with the conversion of squamous to spindle cell carcinomas during mouse skin tumor progression. Mol Carcinogen. 1994;10:207–215. doi: 10.1002/mc.2940100405. [DOI] [PubMed] [Google Scholar]
- 52.Boudreau N, Sympson CJ, Werb Z, Bissell MJ. Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science. 1995;267:891–893. doi: 10.1126/science.7531366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sympson CJ, et al. Targeted expression of stromelysin-1 in mammary gland provides evidence for proteinases in branching morphogenesis and the requirement for an intact basement membrane for tissue-specific gene expression. J Cell Biol. 1994;125:681–693. doi: 10.1083/jcb.125.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Witty JP, Wright JH, Matrisian LM. Matrix metalloproteinases are expressed during ductal and alveolar mammary morphogenesis, and misregulation of stromelysin-1 in transgenic mice induces unscheduled alveolar development. Mol Biol Cell. 1995;6:1287–1303. doi: 10.1091/mbc.6.10.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rudolph-Owen LA, Chan R, Muller WJ, Matrisian LM. The matrix metalloproteinase matrilysin influences early-stage mammary tumorigenesis. Cancer Res. 1998;58:5500–5506. [PubMed] [Google Scholar]
- 56.Boulay A, et al. High cancer cell death in syngeneic tumors developed in host mice deficient for the stromelysin-3 matrix metalloproteinase. Cancer Res. 2001;61:2189–2193. [PubMed] [Google Scholar]
- 57.Ha HY, et al. Overexpression of membrane-type matrix metalloproteinase-1 induces mammary gland abnormalities and adenocarcinoma in transgenic mice. Cancer Res. 2001;61:984–990. [PubMed] [Google Scholar]
- 58.Kaplan DH, et al. Demonstration of an interferon-γdependent tumor surveillance system in immunocompromised mice. Proc Natl Acad Sci USA. 1998;95:7556–7561. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakajima C, et al. A role of interferon-γ (IFN-γ) in tumor immunity: T cells with the capacity to reject tumor cells are generated but fail to migrate to tumor sites in IFN-γdeficient mice. Cancer Res. 2001;61:3399–3405. [PubMed] [Google Scholar]
- 60.Shankaran V, et al. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 61.Beatty GL, Paterson Y. IFN-γ-dependent inhibition of tumor angiogenesis by tumor angiogenesis of tumor-infiltrating CD4+ T cells requires tumor responsiveness to IFN-γ. J Immunol. 2001;166:2276–2282. doi: 10.4049/jimmunol.166.4.2276. [DOI] [PubMed] [Google Scholar]
- 62.Hanson HL, et al. Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity. 2000;13:265–276. doi: 10.1016/s1074-7613(00)00026-1. [DOI] [PubMed] [Google Scholar]
- 63.Griffith TS, et al. Monocyte-mediated tumoricidal activity via the tumor necrosis factor-related cytokine, TRAIL. J Exp Med. 1999;189:1343–1353. doi: 10.1084/jem.189.8.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smyth MJ, et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon-γ-dependent natural killer cell protection from tumor metastasis. J Exp Med. 2001;193:661–670. doi: 10.1084/jem.193.6.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maria DA, et al. Resistance to melanoma metastases in mice selected for high acute inflammatory response. Carcinogenesis. 2001;22:337–342. doi: 10.1093/carcin/22.2.337. [DOI] [PubMed] [Google Scholar]
- 66.Beatty GL, Paterson Y. IFN-γ can promote tumor evasion of the immune system in vivo by down-regulating cellular levels of an endogenous tumor antigen. J Immunol. 2000;165:5502–5508. doi: 10.4049/jimmunol.165.10.5502. [DOI] [PubMed] [Google Scholar]
- 67.Gati A, et al. Tumor cells regulate the lytic activity of tumor-specific cytotoxic T lymphocytes by modulating the inhibitory natural killer receptor function. Cancer Res. 2001;61:3240–3244. [PubMed] [Google Scholar]
- 68.Ganss R, Hanahan D. Tumor microenvironment can restrict the effectiveness of activated antitumor lymphocytes. Cancer Res. 1998;58:4673–4681. [PubMed] [Google Scholar]
- 69.Wei WZ, Fulton A, Winkelhake J, Heppner G. Correlation of natural killer activity with tumorigenesis of a preneoplastic mouse mammary lesion. Cancer Res. 1989;49:2709–2715. [PubMed] [Google Scholar]
- 70.Coussens LM, et al. Inflammatory mast cells upregulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999;13:1382–1397. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell. 2000;103:481–490. doi: 10.1016/s0092-8674(00)00139-2. References 70 and 71 showed that mast cells secrete MMP-9 following infiltration of developing squamous epithelial tumours, and that this action stimulates both development of malignancy and subsequent angiogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bergers B, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–739. doi: 10.1084/jem.193.6.727. Showed that macrophages potentiate neoplastic progression through paracrine factors, indicating that these factors are important to tumorigenesis as genetic mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fang KC, et al. Mast cell expression of gelatinases A and B is regulated by kit ligand and TGF-β. J Immunol. 1999;162:5528–5535. [PubMed] [Google Scholar]
- 75.Coussens LM, Werb Z. Inflammatory cells and cancer: think different! J Exp Med. 2001;193:F23–F26. doi: 10.1084/jem.193.6.f23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Frank BT, Rossall JC, Caughey GH, Fang KC. Mast cell tissue inhibitor of metalloproteinase-1 is cleaved and inactivated extracellularly by α-chymase. J Immunol. 2001;166:2783–2792. doi: 10.4049/jimmunol.166.4.2783. [DOI] [PubMed] [Google Scholar]
- 77.Mitchell RA, Bucala R. Tumor growth-promoting properties of macrophage migration inhibitory factor (MIF) Semin Cancer Biol. 2000;10:359–366. doi: 10.1006/scbi.2000.0328. [DOI] [PubMed] [Google Scholar]
- 78.Abe R, Peng T, Sailors J, Bucala R, Metz CN. Regulation of the CTL response by macrophage migration inhibitory factor. J Immunol. 2001;166:747–753. doi: 10.4049/jimmunol.166.2.747. [DOI] [PubMed] [Google Scholar]
- 79.Hudson JD, et al. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med. 1999;190:1375–1382. doi: 10.1084/jem.190.10.1375. Macrophage inhibitory factor (MIF) suppress p53-dependent transcriptional activity and blocks senescence of primary mouse embryonic fibroblasts. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shao C, et al. Chromosome instability contributes to loss of heterozygosity in mice lacking p53. Proc Natl Acad Sci USA. 2000;97:7405–7410. doi: 10.1073/pnas.97.13.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cordon-Cardo C, Prives C. At the crossroads of inflammation and tumorigenesis. J Exp Med. 1999;190:1367–1370. doi: 10.1084/jem.190.10.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maniotis AJ, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155:739–752. doi: 10.1016/S0002-9440(10)65173-5. Microscopic sectioning of uveal (ocular) tumours provided the first evidence that, in addition to directing the behaviour of stromal endothelium, tumours might also develop into functional channels capable of connencting to host vasculature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bissell MJ. Tumor plasticity allows vasculogenic mimicry, a novel form of angiogenic switch. Am J Pathol. 1999;155:675–679. doi: 10.1016/S0002-9440(10)65164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chang YS, et al. Mosaic blood vessels in tumors: frequency of cancer cells in contact with flowing blood. Proc Natl Acad Sci USA. 2000;97:14608–14613. doi: 10.1073/pnas.97.26.14608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shirikawa K, et al. Absence of endothelial cells, central necrosis, and fibrosis are associated with aggressive inflammatory breast cancer. Cancer Res. 2001;61:445–451. [PubMed] [Google Scholar]
- 86.Sood AK, et al. Molecular determinants of ovarian cancer plasticity. Am J Pathol. 2001;158:1279–1288. doi: 10.1016/S0002-9440(10)64079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hendrix MJC, et al. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: role in vasculogenic mimicry. Proc Natl Acad Sci USA. 2001;98:8018–8023. doi: 10.1073/pnas.131209798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Forberg R, Hendrix MJC, Maniotis AJ. Vasculogenic mimicry and tumor angiogenesis. Am J Pathol. 2000;156:361–381. doi: 10.1016/S0002-9440(10)64739-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Akashi K, Reya T, Dalma-Weiszhausz D, Weissman IL. Lympoid precursors. Curr Opin Immunol. 2000;12:144–150. doi: 10.1016/s0952-7915(99)00064-3. [DOI] [PubMed] [Google Scholar]
- 90.Khan AA, Bose C, Yam LS, Soloski MJ, Rupp F. Physiological regulation of the immunological synapse by agrin. Science. 2001;292:1681–1686. doi: 10.1126/science.1056594. [DOI] [PubMed] [Google Scholar]
- 91.Yang FC, et al. Rac and Cdc42 GTPases control hematopoietic stem cell shape, adhesion, migration, and mobilization. Proc Natl Acad Sci USA. 2001;98:5614–5618. doi: 10.1073/pnas.101546898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dror Y, Freedman MH. Schwachman–Diamond syndrome: an inherited preleukemic bone marrow failure disorder with aberrant hematopoietic progenitors and faulty marrow microenvironment. Blood. 1999;94:3048–3054. [PubMed] [Google Scholar]
- 93.Damiano JS, Cress AE, Hazelhurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93:1658–1667. [PMC free article] [PubMed] [Google Scholar]
- 94.Aziz KA, Till KJ, Zuzel M, Cawley JC. Involvement of CD44–hyaluronan interaction in malignant cell homing and fibronectin synthesis in hairy cell leukemia. Blood. 2000;96:3161–3167. [PubMed] [Google Scholar]
- 95.Børset M, Hjertner Ø, Yaccoby S, Epstein J, Sanderson RD. Syndecan-1 is targeted to the uropods of polarized myeloma cells where it promotes adhesion and sequesters heparin-binding proteins. Blood. 2000;96:2528–2536. [PubMed] [Google Scholar]
- 96.Barillé S, et al. Metalloproteinases in multiple myeloma: production of matrix metalloproteinase-9 (MMP-9), activation of pro-MMP-2, and induction of MMP-1 by myeloma cells. Blood. 1997;90:1649–1655. [PubMed] [Google Scholar]
- 97.Kossakowska AE, et al. Interleukin-6 regulation of matrix-metalloproteinase (MMP-2 and MMP-9) and tissue inhibitor of metalloproteinase (TIMP-1) expression in malignant non-Hodgkin’s lymphomas. Blood. 1999;94:2080–2089. [PubMed] [Google Scholar]
- 98.Michigami T, et al. Cell–cell contact between marrow stromal cells and myeloma cells via VCAM-1 and α4β1-integrin enhances production of osteoclast-stimulating activity. Blood. 2000;96:1953–1960. [PubMed] [Google Scholar]
- 99.Sanz-Rodríguez F, Hidalgo A, Teixidó J. Chemokine stromal cell-derived factor-1α modulates VLA-4 integrin-mediated multiple myeloma cell adhesion to CS-1/fibronectin and VCAM-1. Blood. 2001;97:346–351. doi: 10.1182/blood.v97.2.346. [DOI] [PubMed] [Google Scholar]
- 100.Vacca A, et al. Bone marrow neovascularization, plasma cell angiogenic potential, and matrix metalloproteinase-2 secretion parallel progression of human multiple myeloma. Blood. 1999;93:3064–3073. [PubMed] [Google Scholar]
- 101.Padró T, et al. Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood. 2000;95:2637–2644. [PubMed] [Google Scholar]
- 102.Lagneaux L, Delforge A, Bron D, De Bruyn C, Styckmans P. Chronic lympocytic leukemic B cells but not normal B cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood. 1998;91:2387–2396. [PubMed] [Google Scholar]
- 103.Shain KH, Landowski TH, Dalton WS. The tumor microenvironment as a determinant of cancer cell survival: a possible mechanism for de novo drug resistance. Curr Opin Oncol. 2000;12:557–563. doi: 10.1097/00001622-200011000-00008. [DOI] [PubMed] [Google Scholar]
- 104.Mudry RE, Fortney JE, York T, Hall BM, Gibson LF. Stromal cells regulate survival of B-lineage leukemic cells during chemotherapy. Blood. 2000;96:1926–1932. [PubMed] [Google Scholar]
- 105.Cheung WC, Van Ness B. The bone marrow stromal microenvironment influences myeloma therapeutic response in vitro. Leukemia. 2001;15:264–271. doi: 10.1038/sj.leu.2402022. [DOI] [PubMed] [Google Scholar]
- 106.Moreno A, et al. Interleukin-6 dimers produced by endothelial cells inhibit apoptosis of B-chronic lymphocytic leukemia cells. Blood. 2001;97:242–249. doi: 10.1182/blood.v97.1.242. [DOI] [PubMed] [Google Scholar]
- 107.Illmensee K, Mintz B. Totipotency and normal differentiation of single teratocarcinoma cells cloned by injection into blastocysts. Proc Natl Acad Sci USA. 1976;73:549–553. doi: 10.1073/pnas.73.2.549. Malignant mouse teratocarcinoma cells, grown through 200 transplant generations over 8 years as in vivo ascites tumours, were microinjected into developing blastocysts. The resulting genetic mosaics were normal, and tumour cells were able to develop into normal tissues. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dolberg DS, Bissell MJ. Inability of Rous sarcoma virus to cause sarcomas in the avian embryo. Nature. 1984;309:552–556. doi: 10.1038/309552a0. [DOI] [PubMed] [Google Scholar]
- 109.Stoker AW, Hatier C, Bissell MJ. The embryonic environment strongly attenuates v-src oncogenesis in mesenchymal and epithelial tissues, but not in endothelia. J Cell Biol. 1990;111:217–228. doi: 10.1083/jcb.111.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Olumi AF, et al. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–5011. doi: 10.1186/bcr138. A direct demonstration of the interdependence of the tumour epithelium and the tumour-associated stromal cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Szabowski A, et al. c-Jun and JunB antagonistically control cytokine-regulated mesenchymal–epidermal interaction in skin. Cell. 2000;103:745–755. doi: 10.1016/s0092-8674(00)00178-1. [DOI] [PubMed] [Google Scholar]
- 112.Javaherian A, Vaccariello M, Fusenig NE, Garlick JA. Normal keratinocytes suppress early stages of neoplastic progression in stratified epithelium. Cancer Res. 1998;58:2200–2209. [PubMed] [Google Scholar]
- 113.Barcellos-Hoff MH, Aggeler J, Ram TG, Bissell MJ. Functional differentiation and alveolar morphogenesis of primary mammary cultures on reconstituted basement membrane. Development. 1989;105:223–235. doi: 10.1242/dev.105.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Petersen OW, Ronnov-Jessen L, Howlett AR, Bissell MJ. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc Natl Acad Sci USA. 1992;89:9064–9068. doi: 10.1073/pnas.89.19.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Weaver VM, et al. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137:231–245. doi: 10.1083/jcb.137.1.231. Showed that progression to tumorigenicity in a mammary epithelial tumour-progression cell-culture model is accompanied by upregulation of β1-integrins, and that β1-blocking antibodies can restore normal phenotype and suppress tumorigenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang F, et al. Reciprocal interactions between β1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc Natl Acad Sci USA. 1998;95:14821–14826. doi: 10.1073/pnas.95.25.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang F, Yoneda T, Barcellos-Hoff MH, Bissell MJ. Combinatorial modifications of multiple pathways reverts the malignant phenotype of mammary carcinoma cells MDA–MB231. Mol Biol Cell. 1999;10:2024. [Google Scholar]
- 118.Slamon DJ, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 119.Joensuu H, et al. Effect of the tyrosine kinase inhibitor STI-571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001;322:1052–1056. doi: 10.1056/NEJM200104053441404. [DOI] [PubMed] [Google Scholar]
- 120.Friedman SL, Maher JJ, Bissell DM. Mechanisms and therapy of hepatic fibrosis. Hepatology. 2000;32:1403–1408. doi: 10.1053/jhep.2000.20243. [DOI] [PubMed] [Google Scholar]
- 121.Bilimora MM, et al. Underlying liver disease, not tumor factors, predicts long-term survival after resection of hepatocellular carcinoma. Arch Surg. 2001;136:528–535. doi: 10.1001/archsurg.136.5.528. [DOI] [PubMed] [Google Scholar]
- 122.Jacobs TW, Byrne C, Colditz G, Connolly JL, Schnitt SJ. Radial scars in benign breast-biopsy specimens and the risk of breast cancer. N Engl J Med. 1999;340:430–436. doi: 10.1056/NEJM199902113400604. [DOI] [PubMed] [Google Scholar]
- 123.Mossman BT, Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Am J Respir Crit Care Med. 2000;157:1666–1680. doi: 10.1164/ajrccm.157.5.9707141. [DOI] [PubMed] [Google Scholar]
- 124.Sporn MB. The war on cancer. Lancet. 1996;347:1377–1381. doi: 10.1016/s0140-6736(96)91015-6. [DOI] [PubMed] [Google Scholar]
- 125.Sporn MB, Suh N. Chemoprevention of cancer. Carcinogenesis. 2000;21:525–530. doi: 10.1093/carcin/21.3.525. [DOI] [PubMed] [Google Scholar]
- 126.Bange J, Zwick E, Ullrich A. Molecular targets for breast cancer therapy and prevention. Nature Med. 2001;7:548–552. doi: 10.1038/87872. [DOI] [PubMed] [Google Scholar]
- 127.Oshima M, et al. Suppression of intestinal polyposis in ApcΔ716 knockout mice by inhibition of prostaglandin endoperoxide synthase-2 (COX2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 128.Tsujii M, et al. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93:705–716. doi: 10.1016/s0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- 129.Liu CH, et al. Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. J Biol Chem. 2001;276:18563–18569. doi: 10.1074/jbc.M010787200. [DOI] [PubMed] [Google Scholar]
- 130.Williams CS, Tsujii M, Reese J, Dey SK, DuBois RN. Host cyclooxygenase-2 modulates carcinoma growth. J Clin Invest. 2000;105:1589–1594. doi: 10.1172/JCI9621. Showed that the induction of angiogenesis depends upon COX2 expression within tumour-associated fibroblasts, rather than within the tumour itself. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Herbst RS, Lee AT, Tran HT, Abbruzzese JL. Clinical studies of angiogenesis inhibitors: the University of Texas MD Anderson Center trial of human endostatin. Curr Oncol Rep. 2001;3:131–140. doi: 10.1007/s11912-001-0013-8. [DOI] [PubMed] [Google Scholar]
- 132.Zucker S, Cao J, Chen WT. Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene. 2000;19:6642–6650. doi: 10.1038/sj.onc.1204097. [DOI] [PubMed] [Google Scholar]
- 133.Hidalgo H, Eckhart SG. Development of matrix metalloproteinase inhibitors in cancer therapy. J Natl Cancer Inst. 2001;94:178–193. doi: 10.1093/jnci/93.3.178. [DOI] [PubMed] [Google Scholar]
- 134.Reichmann E. Oncogenes and epithelial cell transformation. Semin Cancer Biol. 1994;5:157–165. [PubMed] [Google Scholar]
- 135.Bilder D, Li M, Perrimon N. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science. 2000;289:113–116. doi: 10.1126/science.289.5476.113. A genetic screen to identify proteins that mediate epithelial polarity in Drosophila identified a network of two known tumour suppressors and a new gene that are involved in the assembly and maintenance of gap junctions. [DOI] [PubMed] [Google Scholar]
- 136.Bilder D, Perrimon N. Localization of apical epithelial determinants by the basolateral PDZ protein Scribble. Nature. 2000;403:676–680. doi: 10.1038/35001108. [DOI] [PubMed] [Google Scholar]
- 137.Borg JP, et al. ERBIN: a basolateral PDZ protein that interacts with the mammalian ERBB2/HER2 receptor. Nature Cell Biol. 2000;2:407–414. doi: 10.1038/35017038. [DOI] [PubMed] [Google Scholar]
- 138.Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341:738–746. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- 139.Dvorak HF. Tumors: wounds that do not heal. N Engl J Med. 1984;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 140.Skobe M, Fusenig NE. Tumorigenic conversion of immortal human keratinocytes through stromal cell activation. Proc Natl Acad Sci USA. 1998;95:1050–1055. doi: 10.1073/pnas.95.3.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor β in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 142.Ronnov-Jessen L, Petersen OW, Koteliansky VE, Bissell MJ. The origin of the myofibroblasts in breast cancer. Recapitulation of tumor environment in culture unravels diversity and implicates converted fibroblasts and recruited smooth muscle cells. J Clin Invest. 1995;95:859–873. doi: 10.1172/JCI117736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Okamoto-Inoue M, Kamada S, Kimura G, Taniguchi S. The induction of smooth muscle α–actin in a transformed rat cell line suppresses malignant properties in vitro and in vivo. Cancer Lett. 1999;142:173–178. doi: 10.1016/s0304-3835(99)00150-0. [DOI] [PubMed] [Google Scholar]
- 144.Zajchowski DA, et al. Suppression of tumor-forming ability and related traits in MCF-7 human breast cancer cells by fusion with immortal mammary epithelial cells. Proc Natl Acad Sci USA. 1990;87:2314–2318. doi: 10.1073/pnas.87.6.2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sager R, Anisoxicz A, Neveu M, Liang P, Sotiropoulou G. Identification by differential display of α6-integrin as a candidate tumor suppressor gene. FASEB J. 1993;7:964–970. doi: 10.1096/fasebj.7.10.8344495. [DOI] [PubMed] [Google Scholar]
- 146.Lee SW, Reimer CL, Oh P, Campbell DB, Schnitzer JE. Tumor cell growth inhibition by caveolin re-expression in human breast cancer cells. Oncogene. 1998;16:1391–1397. doi: 10.1038/sj.onc.1201661. [DOI] [PubMed] [Google Scholar]
- 147.Hirshi KK, Xu C, Tsukamoto T, Sager R. Gap junction genes Cx26 and Cx43 individually suppress the cancer phenotype of human mammary carcinoma cells and restore differentiation potential. Cell Growth Differ. 1996;7:861–870. [PubMed] [Google Scholar]
- 148.Zou Z, et al. Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science. 1994;263:526–529. doi: 10.1126/science.8290962. [DOI] [PubMed] [Google Scholar]
- 149.Bani D, Riva A, Bigazzi M, Sacchi BT. Differentiation of breast cancer cells in vitro is promoted by the concurrent influence of myoepithelial cells and relaxin. Br J Cancer. 1994;70:900–904. doi: 10.1038/bjc.1994.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Liu QY, et al. Inhibitory effects of activin on the growth and morphogenesis of primary and transformed mammary epithelial cells. Cancer Res. 1996;56:1155–1163. [PubMed] [Google Scholar]