Everolimus (original) (raw)
. Author manuscript; available in PMC: 2011 Mar 1.
Abstract
Everolimus, an orally adminstered rapamycin analog, has recently been approved by the FDA for treatment of renal cell carcinoma (RCC) refractory to inhibitors of vascular endothelial growth factor (VEGF) receptor signaling. Everolimus significantly increased progression-free survival (median PFS for the everolimus treated group was 4.0 months versus 1.9 months for the placebo group), although tumor regressions were observed only infrequently. While the target for everolimus, (the serine/threonine kinase mTOR) is well established, the mechanism by which this agent retards tumor growth is not well defined. Further, biomarkers that predict tumor sensitivity are still elusive. The mechanism of action, preclinical antitumor activity, and clinical activity of everolimus against RCC are reviewed.
The Food and Drug Administration recently approved everolimus, a rapamycin analog (rapalog), for the treatment of patients with advanced renal cell carcinoma after failure of treatment with sorafenib or sunitinib. Rapalogs have been shown to suppress hypoxia-induced increases in hypoxia inducible factor-1α (HIF-1α), and inhibit the response of vascular endothelial cells to stimulation by vascular endothelial growth factor (VEGF). In clear cell renal cell carcinoma (RCC) mutation in the von Hippel-Lindau gene (VHL) is the causative genetic event leading to stabilization of the transcription factor HIF-1α, elevated expression of genes associated with an ‘hypoxia phenotype’ and increased secretion of VEGF. Another rapalog, temsirolimus, and kinase inhibitors sunitinib and sorafenib that target the VEGF receptors amongst other kinases, are approved for treatment of RCC, strengthening the association of dysregulated VEGF to the etiology of this cancer.
Preclinical Data
Everolimus (Afinitor®, RAD-001 (40-_O_-(2-hydroxyethyl)-rapamycin) is a rapamycin analog (rapalog) that is being developed as an antitumor agent. Like rapamycin, everolimus binds the cyclophilin FKBP-12, and this complex binds the serine/threonine kinase, mTOR (mammalian target of rapamycin) when it is associated with raptor and mLST8 to form a complex (mTORC1), and inhibits signaling downstream. Importantly, rapalogs are not direct mTOR kinase inhibitors at pharmacologically achievable drug concentrations. mTORC1 lies downstream of phosphotidylinositol 3′ kinase (PI3K), in a pathway that is very frequently activated in human cancers. Hence mTORC1 represents a pivotal target for cancer therapy (Figure 1). mTORC1 regulates cap-dependent translation, transcription, cell cycle progression, and survival. This complex coordinates cell growth and metabolism by acting as a restriction point in cells under stress conditions(1-5) such as low oxygen tension (hypoxia) (6-8). The best characterized pathways regulated by mTORC1 are phosphorylation and activation of ribosomal S6 kinase-1 (S6K1) and phosphorylation and inactivation of 4E-BP1, the suppressor of the mRNA cap-binding protein eIF4E. mTORC1 mediated phosphorylation of 4E-BP1 leads to its disassociation from eIF4E, and subsequent association of eIF4E with a scaffolding protein (eIF4G) and recruitment of initiation factors to form the pre-initiation translation complex (eIF4F) required for efficient translation of mRNA species that have highly structured 5′-untranslated regions (5′-UTRs) (9). These include cell cycle regulators Cyclin D1 and ornithine decarboxylase (ODC) required for transit from G1 phase to S phase, and transcription factors c-MYC and HIF-1α (10).
Figure 1.
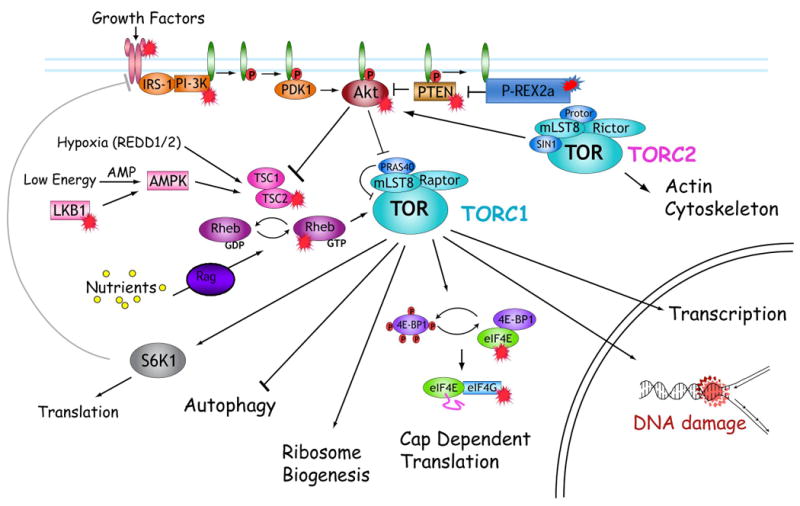
mTOR exists in two known complexes, mTORC1 inhibited by rapalogs controls translation, suppresses autophagy and regulates transcription and response to DNA damage. mTORC2 regulates actin cytoskeleton and activates Akt through phosphorylation of Ser473. mTORC complexes are activated by growth factors, and mTORC1 is regulated by the intracellular environment [O2, nutrients (amino acids, glucose) and energy charge]. The major (identified) substrates for mTORC1 are S6K1 and 4E-BP. S6K1 negatively regulates IGF-1 receptor signaling through phosphorylation of IRS-1 which leads to its proteasomal degradation. Phosphorylation of 4E-BP proteins facilitates formation of the translation initiation complex required for efficient translation of cell cycle regulators (Cyclin D1, ornithine decarboxylase) and transcription factors (c-MYC, HIF-1α). Signaling intermediates both upstream and downstream of mTORC1 are dysregulated in many human cancers or tumor prone syndromes are marked ( )
)
mTOR exists in a second complex (mTORC2) associated with rictor, sin1 and mLST8, that phosphorylates Akt at Ser-473, leading to full activation of Akt. Anecdotally, inhibition of mTORC1 by rapalogs leads to hyperphosphorylation of Akt(Ser473) in many cancer cell lines, probably through stabiization of IRS-1 subsequent to downregulation of the activity of the mTORC1 substrate S6K1 that normally negatively regulates signaling flux through the type-1 insulinlike growth factor receptor (IGF-1R) (11, 12). Importantly, activation of Akt may lead to survival when mTORC1 is inhibited or potentially increased VEGF production as PI3K/AKT signaling can induce tumor angiogenesis by regulating VEGF. This regulation occurs at both the mRNA and protein levels, and its regulation of VEGF mRNA appears to occur by both HIF-1α-dependent and -independent mechanisms (13).
Everolimus and other rapalogs potently inhibit growth of numerous human tumor cell lines, with 50% inhibition of growth in the sub-nanomolar concentration range, inhibit proliferation of human umbilical vein endothelial cells (HUVECs), and reduce expression of HIF-1α and VEGF in cultured tumor cells. The in vivo antitumor activity of rapamycin was identified almost thirty years ago in an NCI screen but for various reasons was not developed for cancer treatment. Everolimus, a hydroxyethyl ether derivative of rapamycin, has superior pharmaceutical characteristics to rapamycin, and was designed for oral administration. Unlike temsirolimus, everolimus is not converted to rapamycin in vivo.
Everolimus inhibits the growth of human tumor xenograft models in nude mice even where the cell line in vitro was insensitive to this agent. Similar results have been reported for rapamycin and temsirolimus, and suggest that tumor retardation is a consequence of the agent's antiangiogenic activity. Consistent with this is the reduced blood vessel density observed in several tumor models growing in mice treated with everolimus (14). However, many tumors appear intrinsically resistant to rapalogs, although the mechanism for resistance is as yet unknown. In some human sarcoma xenograft models rapamycin treatment significantly increased the level of tumor-associated VEGF (15). Of importance is that the spectrum of antitumor activity differs between everolimus and small molecule receptor tyrosine kinase inhibitors that target VEGF receptors (14). At doses in mice that demonstrated effective tumor control (0.5 to 5 mg/kg/day) everolimus was well tolerated, suggesting that it is a very promising chemotherapeutic agent.
Clinical Studies
The tolerability and efficacy of everolimus has been evaluated in several clinical settings. In the initial phase 1 trial, weekly oral administration at dosages up to 70 mg or daily oral dosing up to 10 mg were examined. Although dose-limiting toxicity was not determined in either arm of this trial, toxicity profiles and frequency were similar between weekly dosing at 70 mg and daily dosing at 10 mg. Most frequent drug-related adverse events were rash, stomatitis/mucositis, fatigue, nausea, and vomiting. Although NCI-CTC events ≥ grade 3 were rare, hyperglycemia, hyperglyceridemia and thrombocytopenia were common to both schedules, whereas the total number of grade 3 adverse events was slightly higher in the daily dosing arm (16). Notably, prolonged disease stabilization of RCC patients (≥ 6 months) was observed in both weekly schedules (n=3) and daily dosing schedules (n=2), as well as other tumors. Pharmacodynamic studies assessing mTORC1 inhibition in peripheral mononuclear cells showed sustained inhibition of S6K1 activity at ≥20 mg weekly and ≥5mg daily.
Phase 2 testing of everolimus was in patients with predominantly clear cell RCC who had received 1 prior treatment or less, and had progressive measurable metastatic disease. Everolimus was administered orally on a continuous daily schedule, with dose modifications for toxicity. Patients were assessed every eight weeks (2 cycles) using Response Evaluation Criteria in Solid Tumors (RECIST). The population (n=37 evaluable patients) was predominantly male (78%), median age 60 years, and good performance status (Zubrod 0-1), and most had received prior therapy (93%). The median progression-free survival (PFS) was 11.2 months, and the median overall survival 22.1 months. Partial responses were observed in 14% patients (7% by independent review), stable disease ≥ 3 months occurred in 73%, and stable disease lasting ≥ 6months in 57% of patients. Frequent toxicities recapitulated those seen in the phase 1 trial with nausea (31%), anorexia (38%), diarrhea (31%), stomatitis (31%), pneumonitis (31%) and rash (26%) being reported. Severe toxicities (≥ grade 3) were pneumonitis (18%), elevated transaminases (10%), thrombocytopenia, hyperglycemia, alkaline phosphatase elevation (8% each) and hyperlipidemia (5%).
The phase 3 registration trial (NCT00410124) was a randomized, double-blind, placebo-controlled trial of everolimus in patients with metastatic RCC whose disease had progressed on sunitinib, sorafenib or both drugs (17). Patients were randomly assigned in a two to one ratio to receive everolimus 10 mg once daily (n=272) or placebo (n=138). The primary endpoint was PFS and was assessed by blinded, independent central review.
All 410 patients were included in efficacy analyses. Results from a second interim analysis indicated a significant difference in efficacy between arms. The trial was thus halted early after 191 progression events had been observed (101 [37%] events in the everolimus group, 90 [65%] in the placebo group; hazard ratio 0.30, 95% CI 0.22-0.40, p<0.0001. Median PFS for the everolimus treated group was 4.0 months [95% CI 3.7-5.5] vs 1.9 months [1.8-1.9]. The most commonly reported adverse events, stomatitis (everolimus: 40% vs placebo: 8%), rash (25% vs 4%) and fatigue (20% vs 16%) were mostly mild or moderate in severity. Pneumonitis was detected in 8% of patients receiving everolimus with eight having grade 3 severity. The conclusion from this phase 3 study was that everolimus prolongs PFS in patients with metastatic RCC that had progressed on agents that target vascular endothelial growth factor-targeted therapy. Thus, everolimus, like temsirolimus, has significant activity in the setting of RCC, being largely cytostatic and inducing RECIST-defined partial tumor regression only infrequently (1%). Whether the antitumor mechanism of action of everolimus is uniquely directed at the VEGF locus is less certain, although the study supports the non-clinical results indicating that everolimus has antiangiogenic properties distinct from VEGF receptor tyrosine kinase inhibitors (18).
Pharmacodynamic studies
Identifying robust pharmacodynamic markers that predict sensitivity to rapalogs has proven difficult. Phosphorylation of ribosomal protein S6, most frequently biomarker used, is equally inhibited in cell lines that are sensitive or insensitive to everolimus (18). Similarly, enhanced Akt Ser473 phosphorylation induced by mTORC1 inhibition does not correlate with cell sensitivity, or activation of Akt substrates. Other markers of rapalog sensitivity proposed have been treatment-induced downregulation of cyclin D1 and CDK4 transcripts, the basal expression of the antiapoptotic protein BCL-2 and the basal phosphorylation state of Akt. The most comprehensive clinical pharmacodynamic study reported (19) compared markers of mTORC1 inhibition in tumor and skin biopsies. Everolimus was administered either daily (5 or 10 mg) or weekly (20, 50 and 70 mg). There was good concordance between mTORC1 pathway inhibition in tumor and skin biopsies. Inhibition of mTORC1 signaling was observed at all doses and schedules with almost complete inhibition of S6 and eIF4G phosphorylation, although the magnitude of inhibition of 4E-BP phosphorylation was less in tumor than skin. At both dose levels on the daily schedule, proliferation was reduced in tumor and skin, and phospho-Akt increased in approximately half of tumor samples. These pharmacodynamic studies indicated that inhibition of mTORC1 was more robust and prolonged using daily dosing at 10 mg rather than weekly dosing schedules. However, of note was that while S6 phosphorylation was suppressed for 5 days after the last dose of everolimus (weekly schedule), hyperphosphorylation of Akt (Ser473) was not maintained. While these pharmacodynamic studies were valuable for selecting biologically effective dose levels for each schedule (10 mg daily and 50 mg/week), they did not identify a marker that predicts tumor sensitivity.
Advantages over other agents
Three agents were approved for treatment of RCC prior to approval of everolimus. Sorafenib (Nexavar), a rather promiscuous kinase inhibitor with targets including Raf kinase, PDGF (platelet-derived growth factor), VEGF receptor 2 and 3 kinases and c Kit, the receptor for Stem cell factor, prolongs PFS in patients with advanced clear-cell RCC in whom previous therapy has failed; the median PFS was 5.5 months in the sorafenib group and 2.8 months in the placebo group (hazard ratio for disease progression in the sorafenib group, 0.44; 95% confidence interval [CI], 0.35 to 0.55; P<0.01).
Sunitinib (Sutent, SU011248), another relatively non-selective inhibitor targeting VEGF receptors, PDGFR and other tyrosine kinases including, KIT, FLT3, colony-stimulating factor 1 (CSF-1), and RET, was compared to interferon-α in naive RCC patients. Median PFS was 47.3 weeks (95% CI 42.6, 50.7) for sunitinib-treated patients and 22.0 weeks (95% CI 16.4, 24.0) for patients treated with interferon-α; the hazard ratio was 0.415 (95% CI .320, 0.539, p<0.000001). Objective response rate on the sunitinib arm was 27.5% (95% CI 23.0%, 32.3%) vs. 5.3% (95% CI 3.3%, 8.1%) on the interferon-α arm.
In contrast to sorafenib and sunitinib that have a relatively broad-spectrum of kinase targets, rapalogs are highly selective inhibitors of mTORC1 signaling. Temsirolimus (Torisel®, CCI779) a rapamycin pro-drug was approved for treatment of RCC in 2007. Temsirolimus demonstrated significant benefit over interferon-α for previously untreated patients with advanced RCC who had 3 or more of 6 poor prognostic factors and Karnofsky performance status of 60 or 70. Temsirolimus (25 mg weekly) was associated with a statistically significant improvement in overall survival when compared to interferon-α (hazard ratio 0.73 [95% CI: 0.58, 0.92]; p= 0.0078). The median overall survival was 10.9 months on the temsirolimus arm and 7.3 months on the interferon-α arm. Progression-free survival was 5.5 months on the temsirolimus arm and 3.1 months on the interferon-α arm [hazard ratio 0.66 (95% CI: 0.53, 0.81)]. The combination of 15 mg temsirolimus and interferon-α did not result in a significant increase in overall survival when compared with interferon-α alone and was associated with an increase in multiple adverse reactions. Everolimus was approved for RCC demonstrating activity in patients that progressed on tyrosine kinase inhibitors (sunitinib, sorafenib). Thus, because of significant differences in the population of patients selected for each trial it is difficult to understand if the rapalogs differ in any significant manner, or indeed whether rapamycin would have similar efficacy against this disease. Perhaps of importance is that everolimus demonstrated activity after failure on these kinase inhibitors, and thus potentially offers a survival benefit for patients with this dismal prognosis. Thus, in this setting everolimus compliments the limited options offered by available agents. Acquired resistance to rapamycin has been shown to be unstable in cell lines in vitro (20), hence there is a potential for patients who progress on rapalog therapy to become sensitive again following a period off treatment. However, such a strategy assumes alternative therapy can maintain at least stable disease until the rapalog treatment is continued.
Conclusions and Challenges
Everolimus has established activity for treatment of refractory RCC. Modest activity has been reported for non-small cell lung cancer patients failing chemotherapy or EGFR targeted treatment (21) or patients with recurrent or metastatic breast cancer (22). Activity has been observed also in hematologic malignancies (23) and everolimus is in phase 2 evaluation for treatment of childhood malignancies. The agent is largely cytostatic, inducing relatively few regressions, thus the challenge will be in combining everolimus with other targeted agents that together will be cytotoxic – a challenge not unique to this class of agent. Recent studies in childhood sarcoma xenografts suggests that combining rapamycin with an antibody that blocks ligand binding to the insulin-like growth factor receptor leads to synergistic antitumor activity (15). However, clinical trials combining rapalogs with other signaling inhibitors such as erlotinib have generally shown enhanced host toxicity, and combination with standard cytotoxic agents may have variable effects from synergy to antagonism. A second, and perhaps more important, challenge will be to identify biomarkers that predict response to everolimus. Preferably such biomarkers would be detected in pretreatment biopsies, rather than in response to therapy. The other, largely unanswered question is the biological relevance of Akt hyperphosphorylation following inhibition of mTORC1 signaling. If activation of Akt is important in signaling survival (supported by an extensive literature) one may anticipate superior activity of mTOR kinase inhibitors currently in phase 1 clinical trials, that inhibit both mTORC1 and mTORC2 signaling.
Acknowledgments
Original work discussed from this laboratory was supported by PHS awards CA23099 and CA77776 from the National Cancer Institute.
References
- 1.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 2.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204–9. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6:91–9. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 5.Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4:335–48. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- 6.Arsham AM, Howell JJ, Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem. 2003;278:29655–60. doi: 10.1074/jbc.M212770200. [DOI] [PubMed] [Google Scholar]
- 7.Brugarolas J, Lei K, Hurley RL, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deyoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2 mTOR signaling and tumor suppression through REDD1-mediated 14 3 3 shuttling. Genes Dev. 2008;22:239–51. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richter JD, Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. 2005;433:477–80. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- 10.Bjornsti MA, Houghton PJ. Lost in translation: dysregulation of cap-dependent translation and cancer. Cancer Cell. 2004;5:519–23. doi: 10.1016/j.ccr.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 11.O'Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Easton JB, Kurmasheva RT, Houghton PJ. IRS-1: auditing the effectiveness of mTOR inhibitors. Cancer Cell. 2006;9:153–5. doi: 10.1016/j.ccr.2006.02.027. [DOI] [PubMed] [Google Scholar]
- 13.Arsham AM, Plas DR, Thompson CB, Simon MC. Akt and hypoxia-inducible factor-1 independently enhance tumor growth and angiogenesis. Cancer Res. 2004;64:3500–7. doi: 10.1158/0008-5472.CAN-03-2239. [DOI] [PubMed] [Google Scholar]
- 14.Lane HA, Wood JM, McSheehy PM, et al. mTOR inhibitor RAD001 (everolimus) has antiangiogenic/vascular properties distinct from a VEGFR tyrosine kinase inhibitor. Clin Cancer Res. 2009;15:1612–22. doi: 10.1158/1078-0432.CCR-08-2057. [DOI] [PubMed] [Google Scholar]
- 15.Kurmasheva RT, Dudkin L, Billups C, Debelenko LV, Morton CL, Houghton PJ. The insulin-like growth factor-1 receptor-targeting antibody, CP-751,871, suppresses tumor-derived VEGF and synergizes with rapamycin in models of childhood sarcoma. Cancer Res. 2009;69:7662–71. doi: 10.1158/0008-5472.CAN-09-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Donnell A, Faivre S, Burris HA, 3rd, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol. 2008;26:1588–95. doi: 10.1200/JCO.2007.14.0988. [DOI] [PubMed] [Google Scholar]
- 17.Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–56. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 18.Breuleux M, Klopfenstein M, Stephan C, et al. Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Mol Cancer Ther. 2009;8:742–53. doi: 10.1158/1535-7163.MCT-08-0668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tabernero J, Rojo F, Calvo E, et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol. 2008;26:1603–10. doi: 10.1200/JCO.2007.14.5482. [DOI] [PubMed] [Google Scholar]
- 20.Dilling MB, Germain GS, Dudkin L, et al. 4E-binding proteins, the suppressors of eukaryotic initiation factor 4E, are down-regulated in cells with acquired or intrinsic resistance to rapamycin. J Biol Chem. 2002;277:13907–17. doi: 10.1074/jbc.M110782200. [DOI] [PubMed] [Google Scholar]
- 21.Soria JC, Shepherd FA, Douillard JY, et al. Efficacy of everolimus (RAD001) in patients with advanced NSCLC previously treated with chemotherapy alone or with chemotherapy and EGFR inhibitors. Ann Oncol. 2009;20:1674–81. doi: 10.1093/annonc/mdp060. [DOI] [PubMed] [Google Scholar]
- 22.Ellard SL, Clemons M, Gelmon KA, et al. Randomized phase II study comparing two schedules of everolimus in patients with recurrent/metastatic breast cancer: NCIC Clinical Trials Group IND.163. J Clin Oncol. 2009;27:4536–41. doi: 10.1200/JCO.2008.21.3033. [DOI] [PubMed] [Google Scholar]
- 23.Coiffier B, Ribrag V. Exploring mammalian target of rapamycin (mTOR) inhibition for treatment of mantle cell lymphoma and other hematologic malignancies. Leuk Lymphoma. 2009:1–15. doi: 10.3109/10428190903207548. [DOI] [PubMed] [Google Scholar]