The oncogene HER2; Its signaling and transforming functions and its role in human cancer pathogenesis (original) (raw)
. Author manuscript; available in PMC: 2011 Jan 14.
Published in final edited form as: Oncogene. 2007 Apr 30;26(45):6469–6487. doi: 10.1038/sj.onc.1210477
Abstract
The year 2007 marks exactly two decades since HER2 was functionally implicated in the pathogenesis of human breast cancer (Slamon et al. 1987). This finding established the HER2 oncogene hypothesis for the development of some human cancers. The subsequent two decades have brought about an explosion of information about the biology of HER2 and the Human Epidermal Growth Factor Receptor (HER) family. An abundance of experimental evidence now solidly supports the HER2 oncogene hypothesis and etiologically links amplification of the HER2 gene locus with human cancer pathogenesis. The molecular mechanisms underlying HER2 tumorigenesis appear to be complex and a unified mechanistic model of HER2 induced transformation has not emerged. Numerous hypotheses implicating diverse transforming pathways have been proposed and are individually supported by experimental models and HER2 may indeed induce cell transformation through multiple mechanisms. Here I review the evidence supporting the oncogenic function of HER2, the mechanisms that are felt to mediate its oncogenic functions, and the evidence that links the experimental evidence with human cancer pathogenesis.
INTRODUCTION
HER2 and neu are the human and rodent homologues of an oncogenic growth factor receptor that were identified and named independently in the early 1980s from rodent and human models, but soon found to be homologues of each other. The neu oncogene was initially described as a transforming oncogene discovered in a carcinogen induced rat brain tumor model (Shih et al. 1981). This gene was found to be homologous to the v-erbB (avian erythroblastosis virus) viral oncogene and the cellular epidermal growth factor receptor (EGFR) gene (Schechter et al. 1985; Schechter et al. 1984). In independent studies, an EGFR-related gene was found to be amplified in a human breast cancer cell line and named Human Epidermal Growth Factor Receptor-2 (HER2)(King et al. 1985). The HER2 protein product was related to and had tyrosine kinase activity similar to EGFR (Akiyama et al. 1986). Subsequent cloning of two other related human genes and the post-genome characterization of the human kinome completed the description of this family of four members (Kraus et al. 1989; Plowman et al. 1993; Manning et al. 2002). These four members are commonly referred to as EGFR (HER1, erbB1), HER2 (erbB2, HER2/neu), HER3 (erbB3), and HER4 (erbB4).
The analysis of tumors from mouse models and from human cancer cells has led to the description of numerous variants and mutants of HER2 and neu and adherence to appropriate nomenclature is essential in order to avoid confusion. While ErbB2 is used to refer to the gene across both human and rodent species, HER2 is used in reference to the human gene and gene product and neu is used in reference to its rodent counterparts. Table 1 lists the various HER2 and neu species that have been described in the literature and the nomenclature and aliases that are often used to refer to them. Each of these species are discussed in the sections below and Table 1 is provided for reference and to clarify the distinctions between these variants.
TABLE 1.
Common nomenclature of HER2 rodent and human variants
| Name, aliases | Description | Site mutated(figure 1) | Where found |
|---|---|---|---|
| neu, c-neu, wtneu,neu proto-oncogene | rodent cellular homolog of HER2 | - | in rat and mouse cells |
| neuT, neuNT, neu oncogene,neuV664E | rat neu containing a transmembrane domain mutation,has potent transforming activity | C | disovered in a rat carcinogenesis model, used as apotent experimental oncogene |
| neu8142, neu8342 | rat neu containing deletion mutations in theextracellular domain | A | found in tumors from MMTV-c-neu transgenic mice |
| HER2, ErbB2, c-ErbB2 | human cellular HER2 | - | in human cells |
| HER2V659E | engineered transforming mutant of human HER2,analogous to neuT | C | never found in nature, engineered for experiments |
| ΔHER2 | isoform of human HER2 missing one exon | B | normally occuring minor isoform found whereverHER2 is expressed |
| HER2YVMA | HER2 with a kinase domain mutation and increasedkinase activity | D | found in some lung cancers |
THE SIGNALING FUNCTIONS OF HER PROTEINS
The HER family proteins are type I transmembrane growth factor receptors that function to activate intracellular signaling pathways in response to extracellular signals. Their structure consists of an extracellular ligand binding domain, a transmembrane domain, and an intracellular tyrosine kinase domain (Figure 1). The function of this family is simplest in C. Elegans where signaling is mediated by a single ligand and a single receptor and slightly more complex in Drosophila where four ligands signal through a single receptor (Lacenere and Sternberg 2000; Moghal and Sternberg 2003). The system is far more complicated in mammalians where the functions of this family are performed by at least twelve ligands and four receptors. Readers are referred to several recent excellent reviews of HER family signaling and functions (Yarden and Sliwkowski 2001; Barnes and Kumar 2004; Mendelsohn and Baselga 2000; Prenzel et al. 2001; Olayioye et al. 2000). While the reasons behind such multiplicity in this system are not well understood, much is now known regarding the molecular basis underlying their signaling activities. Upon ligand binding to their extracellular domains, HER proteins undergo dimerization and transphosphorylation of their intracellular domains. These phosphorylated tyrosine residues dock numerous intracellular signaling molecules leading to activation of a plethora of downstream second messenger pathways and crosstalk with other transmembrane signaling pathways leading to diverse biological effects (reviewed in (Barnes and Kumar 2004; Prenzel et al. 2001; Bazley and Gullick 2005; Yarden and Sliwkowski 2001)). The structural bases for receptor dimerization have been coming to light in the past few years by crystollagraphic data. The extracellular domain of HER proteins can exist in a closed inhibited or an open active conformation. Ligand binding induces a conformational change in their extracellular domain that induces the active conformation and promotes their dimerization and consequent transphosphorylation (Burgess et al. 2003). Partner selection appears to be a key determinant of signaling activity among HER proteins and their signaling functions follow a distinct hierarchical order favoring heterodimers over homodimers. HER2 has the strongest catalytic kinase activity and HER2-containing heterodimers have the strongest signaling functions (Tzahar et al. 1996; Graus-Porta et al. 1997). The expansion of the HER family in mammalian systems has been associated with functional differentiation necessitating interdependence rather than promoting independent or redundant functions. This is exemplified by HER2 and HER3 which are functionally incomplete receptor molecules. Unlike the other members of the family, the extracellular domain of HER2 does not pivot between active and inactive conformations and constitutively exists in an activated conformation (Garrett et al. 2003; Cho et al. 2003). Consistent with its constitutively active conformation, HER2 lacks ligand binding activity and its signaling function is engaged by its ligand-bound heterodimeric partners (Sliwkowski 2003). On the other hand HER3, unlike the other members, lacks ATP binding within its catalytic domain and is catalytically inactive (Sierke et al. 1997). Consistent with this, the signaling functions of HER3 are mediated entirely through the kinase activity of its heterodimeric partners (Kim et al. 1998). Even chimeric kinase-active HER3 constructs fail to signal without hetero-partners suggesting that HER3 even lacks the ability to homodimerize and is an obligate heterodimerization partner (Berger et al. 2004). Although individually they are incomplete signaling molecules, a large body of evidence not only establishes HER2 and HER3 as obligate partners but their complex forms the most active signaling heterodimer of the family and essential for many biologic and developmental processes (Sliwkowski et al. 1994; Tzahar et al. 1996; Britsch et al. 1998; Vijapurkar et al. 2003; Goodearl et al. 2001; Vartanian et al. 2000; Keely and Barrett 1999; Horan et al. 1995; Wallasch et al. 1995b).
Figure 1.

Structure of the HER2 and Neu proteins. The domain structure is shown on the left consisting of two ligand binding regions (LD1 & LD2), two cysteine-rich regions (CR1 & CR2), a short transmembrane domain (TM), a catalytic tyrosine kinase domain (TK), and a carboxy terminal tail (CT). Numerous sites of tyrosine phosphorylation wiithin the TK and CT domains are indicated by circled P.The letters on the right point to specific areas that are altered or mutated in certain naturally occuring or experimentally induced cancers discussed in the text. A) site of somatic mutations found in tumors arising in MMTV-neu mice. B) site of the 48bp deletion in the naturally occuring human ΔHER2 isoform. C) site of the mutation in the neuT oncogene initially discovered in a rat carcinogen induced tumor model and subsequently used in numerous in vitro and transgenic experimental models. D) site of mutations found in rare cases of human lung cancers.
The HER proteins are widely expressed and functonally important in non-hematopoeitic tissues (Press et al. 1990; Real et al. 1986). They are each essential in mammalian development and gene disruption models in mice reveal that they are critically involved in the development of multiple organ systems including the brain, skin, lung, and gastrointestinal tract (Miettinen et al. 1995; Sibilia and Wagner 1995; Morris et al. 1999; Sibilia et al. 1998; Threadgill et al. 1995; Lee et al. 1995; Gassmann et al. 1995; Riethmacher et al. 1997; Britsch et al. 1998; Erickson et al. 1997). Their functional importance in adult mammary gland development escapes anlaysis in these germline deletion models due to embryonic lethality but is nicely demonstrated by alternative models that inactivate EGFR or HER2 in pubertal mammary tissue (Andrechek et al. 2005; Xie et al. 1997).
THE TRANSFORMING POTENTIAL OF HER2 AND NEU
Transformation by neu
The data supporting the transforming potential of human HER2 and rodent neu is irrefutable. The rodent neu oncogene was initially identified in a screen for oncogenes using a rat carcinogen-induced tumor model and shown to transform NIH3T3 cells (Shih et al. 1981; Schechter et al. 1984). The neu oncogene also transforms mammary epithelial cells in vitro (Brandt et al. 2001). After cloning of the normal c-neu allele, it was determined that transforming function in the neu oncogene was conferred by a point mutation within the transmembrane domain resulting in a V664E mutated protein named neuT (Bargmann et al. 1986)(figure 1, region C). The V664E mutation promotes receptor dimerization and enhanced tyrosine kinase activity (Weiner et al. 1989b). Numerous mouse transgenic models have confirmed the role of this oncogene in tumorigenesis. The activated neu oncogene (_neu_T) expressed in mice mammary tissue (MMTV-_neu_T mice) induces adenocarcinomas. This appears as a single step whole gland transformation or in a more typical stochastic tumor formation in different backgrounds (Muller et al. 1988; Bouchard et al. 1989). The c-neu protooncogene overexpressed in mammary tissue of mice (MMTV-neu mice) also induces tumor formation, although in the majority of tumors this occurs after acquisition of deletion mutations within the extracellular juxtamembrane region which promote dimerization and enhanced kinase activity (figure 1, region A)(Guy et al. 1992; Siegel and Muller 1996; Siegel et al. 1994). The development of tumors in MMTV-neu transgenic mice likely involves the acquisition of additional genetic abnormalities since these tumors frequently have LOH at specific genomic loci (Cool and Jolicoeur 1999; Ritland et al. 1997). In particular, the inactivation of p53 is descriptively and etiologically linked with neu induced mouse mammary tumors (Li et al. 1997). Additional transgenic models demonstrate that overexpression or activation of neu is tumorigenic in other tissues as well, including skin, biliary tract, and prostate (Table 2). The tumorigenic function of activated neu has also been confirmed in two rat models by luminal retroviral infection (Wang et al. 1991). The transforming functions of neu require its tyrosine kinase activity as demonstrated by kinase-inactivating mutational studies (Weiner et al. 1989a). The rodent data amounts to a body of highly consistent and indisputable evidence that activation of neu is tumorigenic. The MMTV-neu mice have become the workhorse model of HER2 tumorigenesis in vivo.
TABLE 2.
Reported mouse transgenic models of HER2/neu
| Reference | Promoter | Gene | Phenotype |
|---|---|---|---|
| (Muller et al. 1988) | MMTV | neuT transgene | early, multiple mammary tumors by 3 months, short survival |
| (Bouchard et al. 1989) | MMTV | neuT transgene | mammary tumors stochastically, 5-10 months |
| (Guy et al. 1992) | MMTV | c-neu transgene | mammary tumors stochastically, 4-12 months, frequent lung metastases |
| (Andrechek et al. 2000) | endogenous | neuT knock-in | late mammary tumors, 12-17 months, with transgene amplification |
| (Weinstein et al. 2000) | murine neu | c-neu transgene | abnormal involution, focal mammary abnormalities in multiparous females, 1-2 years |
| (Weinstein et al. 2000) | murine neu | neuT transgene | abnormal involution, some late mammary tumors in multiparous females, 14 months |
| (Finkle et al. 2004) | MMTV | human HER2 transgene | multiple mammary tumors, 7 months, some lung mets |
| (Moody et al. 2002) | MMTV-rtTA / TetO | neuT transgene | rapid mammary tumors within 6 weeks and frequent lung mets, regress without dox |
| (Xie et al. 1998) | human Keratin 14 | neuT transgene | severe hyperplastic lesions of skin, hair follicles, esophagus, perinatal lethal |
| (Bol et al. 1998) | bovine keratin 5 | neuT transgene | severe hyperplastic lesions of skin, esophagus, papillomas, early death |
| (Kiguchi et al. 2000; Kiguchi et al. 2001) | bovine keratin 5 | c-neu transgene | hyperplasia, allopecia, papillomas, skin carcinomas, biliary carcinomas, survive 6-12 months |
| (Xie et al. 1999) | K14-rtTA-TetRE | neuT transgene | rapid dox induction of hyperplastic lesions in skin, hair follicels, esophagus, regress without dox |
| (Li et al. 2006) | probasin | neuT transgene | prostatic intraepithelial neoplasia and invasive prostate cancer, 1-2 years |
Transformation by HER2
The data with regards to human HER2 is also compelling however there appears to be significant differences between the rodent and human genes. While rodent neu appears to require mutational activation for tumorigenicity, human HER2 appears to hold tumorigenic potential through overexpression alone. An engineered mutation within the transmembrane region of HER2 (figure 1, region C) does increase its kinase and transforming activities similar to neu, however this mutation does not appear to occur spontaneously (Segatto et al. 1988). Overexpression of HER2 in NIH3T3 or NR6 mouse fibroblasts induces cell transformation and tumorigenic growth (Di Fiore et al. 1987; Hudziak et al. 1987; Chazin et al. 1992). Overexpression of HER2 in MCF-7 breast cancer cells increases their invasiveness and tumorigenicity (Benz et al. 1992). Overexpression of HER2 in human mammary epithelial cells induces proliferative advantage, transformed characteristics, tumorigenic growth, and in 3D models induces proliferative and anti-apoptotic changes that mimick early stages of epithelial cell transformation (Muthuswamy et al. 2001; Woods Ignatoski et al. 2003). Transgenic expression of the human HER2 cDNA in mouse mammary breast tissue induces metastatic mammary tumors similar to neu (Finkle et al. 2004). However in this cross-species MMTV-HER2 transgenic model, the resulting mouse tumors have frequent deletion mutations within the extracellular domain of the HER2 transgene similar to that seen with MMTV-neu mice. However neither mutations within the transmembrane domain (analogous to neuT, see figure 1 region C) or the extracellular domain (analogous to neu8142/neu8342, figure 1 region A) of HER2 have ever been reported in naturally occuring human cancers and despite these experimentally acquired mutations, human breast cancers appear to be always characterized by overexpression of wildtype HER2. Interesting theories regarding this discrepancy have been proposed and are reviewed below in the section concerning ΔHER2.
HER2 overexpression in human cancer
The relevance of the experimental data to human disease is supported by a substantial body of clinical data. Overexpression of the HER2 protein, either through gene amplification or through transcriptional deregulation is seen in approximately 25-30% of breast and ovarian cancers and confers worse biological behavior (Slamon et al. 1989). Initial conflicting reports regarding the prognostic relevance of HER2 were resolved with improved methodologies and the overwhelming data now confirms this initial landmark genetic-biologic finding (nicely reviewed in (Ross et al. 2003)). Breast cancers can have up to 25-50 copies of the HER2 gene and up to 40-100 fold increase in HER2 protein expression resulting in up to 2 million receptors expressed at the tumor cell surface (Kallioniemi et al. 1992; Lohrisch and Piccart 2001; Venter et al. 1987) (Figure 2). Evidence suggests that HER2 amplification is an early event in human breast tumorigenesis. HER2 amplification is seen in nearly half of all in situ ductal carcinomas without any evidence of invasive disease (Liu et al. 1992; Park et al. 2006). HER2 status is maintained during progression to invasive disease, nodal metastasis and distant metastasis (Park et al. 2006; Latta et al. 2002; Carlsson et al. 2004; Tsuda et al. 2001). Therefore although HER2 amplified breast cancers appear to have a higher propensity for progression and worse prognosis compared to non-amplified breast cancers, the amplification is an early event and defines a subtype of breast cancer not a later stage of it. This is further underscored by gene expression profiling studies that show that HER2 amplified breast cancers comprise a specific disease subset with a unique molecular portrait and this portrait is maintained during progression to metatatic disease (Perou et al. 2000; Weigelt et al. 2005). HER2 amplified breast cancers have biologic characteristics that distinguish them from other types of breast cancers. These include increased sensitivity to certain cytotoxic chemotherapeutic agents, resistance to certain hormonal agents, and increased propensity to metastasize to the brain (Ross et al. 2003; Gabos et al. 2006). HER2 overexpression and amplification is also seen in subsets of gastric, esophageal, and endometrial cancers, also associated with worse disease (Morrison et al. 2006; Yano et al. 2006; Mimura et al. 2005) and also seen rarely in cancers of the oropharynx, lung, and bladder (Khan et al. 2002; Hirsch et al. 2002; Latif et al. 2003). The etiologic role of HER2 overexpression in diseases other than breast cancer remain to be defined.
Figure 2.
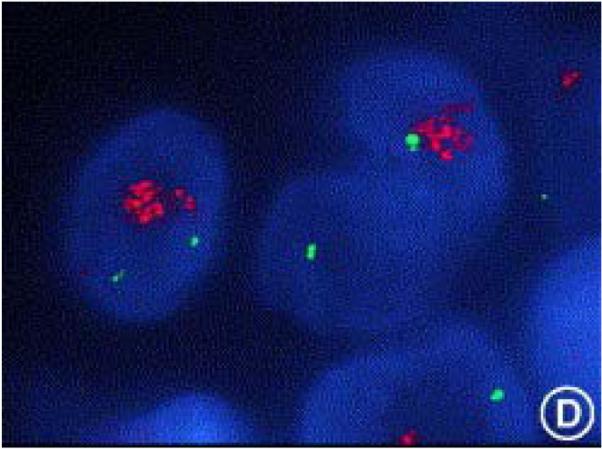
FISH analysis of HER2 amplification. A human breast cancer specimen hybridized with a HER2 gene probe (in green) and a chromosome 17 centromeric probe (in red) showing significantly increased HER2 gene copy number compared with the chromosome 17 control. (Hicks and Tubbs 2005) Reprinted from Human Pathology 36, p256, Copyright 2005, with permission from Elsevier.
HER2 kinase domain mutations in human tumors
Large scale resequencing efforts have only begun to screen human tumors for somatic mutations. Somatic mutations within the HER2 kinase domain have been described in a small subset of lung cancers, predominantly in Asian populations, and very rarely in gastric, colorectal, and breast cancers (figure 1 region D)(Stephens et al. 2004; Shigematsu et al. 2005; Lee et al. 2006). Much less is currently known about the biology of these rare HER2 mutations. At least one of these mutations has been shown to enhance HER2 kinase activity and increase its transforming efficiency (Wang et al. 2006). Although numerous mouse models have established the tumorigenic potential of neu through overexpression, HER2 mutation in lung cancers is not known to be associated with overexpression or gene amplification. As such, a tumorigenic function of this mutated HER2 cannot be presumed and awaits the appropriate transgenic mouse models for analysis. In breast tissue, activated neu is not by itself transforming and requires amplification and overexpression to induce tumorigenesis (Andrechek et al. 2000). Much of the data discussed in this review pertains to the overexpression model of HER2 transformation. Our understanding of the biology of kinase-domain mutated HER2 is still in its infancy and ongoing studies in this area will undoubtedly shed further light on the biological characteristics of this HER2 mutant, its role in tumorigenesis, and its similarities or differences with overexpressed HER2.
HER2 polymorphism in humans
Although somatic mutations in the transmembrane domain of HER2 analgous to the neu oncogene have not been seen in human tumors, polymorphism in the transmembrane domain of HER2 (figure1 region C) has been described. In particular, the I665V variant of HER2 has increased potential for dimerization and signaling (Fleishman et al. 2002). This has led some investigators to propose that the I665V allele of HER2 may confer increased susceptibility to breast cancer. Although an initial large study indeed found such an association with breast cancer risk, several subsequent studies have shown conflicting results with the majority of them showing no risk associated with the I665 genotype (Xie et al. 2000; Benusiglio et al. 2005; Montgomery et al. 2003). The preponderance of evidence at this time does not appear to support an association between HER2 polymorphism and breast cancer risk, although this continues to be debated.
MECHANISMS OF HER2-MEDIATED TUMORIGENESIS
The evidence that increased expression and activity of neu or HER2 induces cell transformation and tumorigenesis is overwhelming and was reviewed above. The data regarding the pathways that mediate, that are required for, and that cooperate with HER2 induced transformation are much more diverse and most likely HER2 induces transformation through a number of signaling pathways. The relative contribution of these pathways to tumorigenesis are difficult to know. The mechanisms and pathways for which there is significant evidence are reviewed below and are schematically depicted in Figure 3.
Figure 3.

Schematic of the signaling abnormalities resulting from HER2 overexpression that are felt to contribute to tumorigenesis. HER2 overexpression results in increased HER2 containing dimers of all kinds. Increased HER2-EGFR dimers drive proliferative and invasive functions. Increased HER2 homodimers disrupt cell polarity. Increased HER2-HER3 dimers drive proliferative, survival, invasive, and metabolic functions. Increased HER2 expression results in an increase in the rare ΔHER2 isoform with more potent signaling characteristics.Several transcription factors are induced in HER2 overexpressingcells resulting in a plethora of gene expression changes.
Overactivity vs overexpression
In the simplest mechanistic model, HER2 induces transformation through increased kinase activity and overphosphorylation of itself and cellular substrates. In this model, the sole consequence of overexpression is to increased cellular HER2 activity. But a limitation of this model has been that it fails to explain why in human cancers HER2 activity is always elevated through overexpression and mutational activation of HER2, analogous to rodent neuT, is never seen in human breast cancers. One explanation that has been offered is that neu can be activated through a single base pair mutation whereas the analogous activation of HER2 requires a two base pair mutation, making this much less likely to occur spontaneously in the human gene. However a more plausible explanation has been that increased expression of HER2 is an essential aspect of its transforming function. This presents a more complex model of transformation, but there is substantial evidence to support it. While high level expression of neu or neuT is tumorigenic in transgenic models, converting the endogenous neu to neuT by knock-in methodology leads to architectural distortion but is insufficient to induce tumorigenesis (Andrechek et al. 2000). These mice do eventually develop tumors with long latency, however these tumors are characterized by amplification and overexpression of the mutant neu allele supporting the hypothesis that overexpression is mechanistically required for transformation by HER2. A number of mechanistic models are able to propose how overexpression of HER2 can disrupt signaling and promote tumorigenic functions. Overexpression of HER2 can change the composition of HER family dimers, significantly increasing HER2-containing heterodimers and HER2 homodimers. Evidence discussed below suggests these increases can deregulate cell polarity and cell adhesion. In addition, evidence discussed below also shows that HER2-containing dimers have prolonged signaling activity and evade signal attenuation increasing signaling potency. Increased total expression of HER2 carries with it an increase in expression of subsets of HER2 including nuclear HER2 and the rare ΔHER2 isoform, and the increase in each of these may be functionally relevant to cell transformation. These are also discussed individually below.
The recent discovery that rare types of lung cancers have HER2 kinase domain mutations that confer increased kinase activity without overexpression may be inconsistent with the overexpression model. But it is too soon to challenge this model since very little is yet known about the biology of the kinase domain mutated HER2. Studies forthcoming in the coming years will determine whether this HER2 mutant is transforming without overexpression and establish its role in the evolution of lung cancer. A unified model of HER2 transformation that also explains why HER2 is deregulated through overexpression in breast and ovarian cancers but through mutation in lung cancers is impossible to propose at this time and must await a much better understanding of its biology in lung cancers.
Deregulation of HER family signaling dynamics
At the lateral level HER2 overexpression causes increased HER2 heterodimerization with EGFR and HER3 (Hendriks et al. 2003a; Karunagaran et al. 1996)(figure 3). This interferes with the endocytic regulation of EGFR. EGFR is unique among the HER family in that it undergoes endocytic degradation after ligand mediated activation and homodimerization, in contrast to the other members of the family which undergo endocytic recycling (Baulida et al. 1996). EGFR-HER2 heterodimers similarly evade endocytic degradation in favor of the recycling pathway and have increased signaling duration and potency (Lenferink et al. 1998; Waterman et al. 1998). Therefore HER2 overexpression results in increased EGFR membrane expression and activity (Huang et al. 1999; Wang et al. 1999; Hendriks et al. 2003b). But it seems unlikely that HER2 requires EGFR for its transforming function since HER2 also transforms NR6 fibroblasts which lack EGFR expression (Chazin et al. 1992). The analogous dispensibility of EGFR in epithelial models of HER2 transformation has not been specifically demonstrated. Overall, compared with other members of the family, HER2 is least subject to inactivating mechanisms and its recruitment into heterodimeric signaling complexes leads to prolonged signaling. As such, HER2 overexpressing cells have significantly prolonged activation of downstream MAPK and c-jun following stimulation with EGFR or HER3 ligands compared with low HER2 cells (Karunagaran et al. 1996).
Signaling through HER3 and downstream PI3K/Akt
The functional role of HER3 is much better established. HER3 is widely expressed in breast cancers (Lemoine et al. 1992). Heregulin regulates HER2 through coexpressed HER3 (Wallasch et al. 1995b). HER2 and HER3 cooperatively induce transformation and in fact HER3 is an obligate partner in HER2 induced transformation (Holbro et al. 2003; Alimandi et al. 1995). Tumors from MMTV-neu mice have increased HER3 expression and phosphorylation and elevated HER3 expression is similarly seen in HER2 overexpressing human tumors (Siegel et al. 1999). The most important oncogenic signaling function of the HER2-HER3 complex appears to be activation of the PI3K/Akt pathway (figure 3). HER2 lacks binding sites for the p85 subunit of PI3K whereas HER3 has seven p85 binding phosphotyrosine containing motifs (Prigent and Gullick 1994; Soltoff et al. 1994; Schulze et al. 2005). Growth factor mediated activation of PI3K and Akt by HER2 is mediated specifically through phosphorylation of HER3 (Soltoff et al. 1994; Hellyer et al. 2001; Fedi et al. 1994). Cell transformation by overexpressed HER2 is also associated with increased HER3 phosphorylation and activation of PI3K/Akt signaling (Holbro et al. 2003; Alimandi et al. 1995; Ram and Ethier 1996; Tokunaga et al. 2006; Wallasch et al. 1995a). Tumors from MMTV-neu mice also have activation of PI3K signaling (Amundadottir and Leder 1998). This is corroborated by clinical studies which show the frequent activation of Akt in HER2 overexpressing tumors (Tokunaga et al. 2006; Zhou et al. 2004). The existing data strongly suggests that the transactivation of HER3 and downstream PI3K/Akt seems to be a critical tumorigenic function of overexpressed HER2. This is consistent with numerous other lines of evidence that highlight a central role for Akt in tumorigenesis. Akt functions in the crossroads of multiple signal transduction pathways that regulate numerous cellular functions critically important for cancer cells including cell proliferation and survival, cell size and response to nutrient availability, glucose metabolism, epithelial-mesenchymal transition and cell invasiveness, genome stability, and angiogenesis. The critical function of Akt in cancer is now widely recognized but will not be discussed in this review. Readers are referred to a number of excellent recent reviews of this topic ((Vivanco and Sawyers 2002; Luo et al. 2003; Paez and Sellers 2003; Testa and Bellacosa 2001) and the entire Oncogene vol 24; issue 50 (11/14/2005)). The abundance of data reviewed above suggests that HER2 induced tumors activate Akt through HER3 and PI3K.
Alternative HER2 transcript
The fact that deletion mutations in the extracellular region of the receptor seen in neu overexpressing mouse mammary tumors are not seen in human tumors has produced a dilemma and an interesting hypothesis to resolve it. A naturally occuring but rare human HER2 RNA transcript has been cloned that is alternatively spliced and lacks a single 48bp coding exon resulting in the in-frame deletion of 16 amino acids from the juxtamembrane region overlapping with the area of deletion mutations seen in neu induced tumors. This alternative HER2 protein, named ΔHER2, has increased ligand-independent signaling activity and increased transforming potency similar to the mutated neu gene (Kwong and Hung 1998; Siegel et al. 1999). This deletion removes cysteine residues in neu and in HER2, disrupting the disulfide bond structure of the proteins and leaving unpaired cysteine residues available for intermolecular bonding. Experimental models have confirmed that ΔHER2 has increased transforming activity compared with wtHER2 in several models, including in vitro and transgenic models, and ΔHER2 has significantly higher dimerization promoted by intermolecular disulfide-bond stabilization, and significantly increased tyrosine phosphorylation compared with wtHER2 (Kwong and Hung 1998; Siegel et al. 1999; Castiglioni et al. 2006). Although the activated ΔHER2 transcript is a normal byproduct of HER2 transcription, its significant increase in HER2 amplified tumors has been proposed as a mechanism underlying HER2 tumorigenesis. ΔHER2 transcripts have been detected in a majority of breast tumors and normal breast tissue and cell lines and reported by two groups to comprise 4-9% of total HER2 transcripts (Siegel et al. 1999; Castiglioni et al. 2006). This hypothesis of HER2 induced tumorigenesis implies that the presence of the naturally occuring ΔHER2 obviates the need for human HER2 to undergo mutational activation. The human MMTV-human HER2 mouse transgenic model is seemingly consistent with this. In this model, mouse mammary tumors are induced by overexpression of the human HER2 cDNA, which does not have a ΔHER2 transcriptional by product associated with it (Finkle et al. 2004). Interestingly, the majority of tumors in this model have acquired deletion mutations of the juxtamembrane region of the human HER2 transgene similar to the neu transgenic model. This is consistent with the hypothesis that there is a selection pressure for this mechanism of activation, whether it is through gene mutation or whether it is through increased expression of ΔHER2. More definitive proof of the causative role of ΔHER2 awaits the development of experimental reagents that selectively target and inactivate the ΔHER2 isoform.
Role of Src kinases
Since HER family proteins signal predominantly through the recruitment of proteins to their tyrosine phosphorylated residues, their transforming functions could be in large part mediated through these interacting proteins, in particular SH2 domain containing proteins. Evidence suggests that src kinase are important second messengers of HER2. Src kinases are potently transforming and tumorigenic when constitutively activated, and their activation is seen in many human tumor types, including breast cancers with or without HER2 overexpression (Reissig et al. 2001; Ottenhoff-Kalff et al. 1992; Belsches-Jablonski et al. 2001). An association between src activation and HER2 overexpression has been reported in pre-invasive carcinomas of the breast (Wilson et al. 2006). Mammary tumors that arise in MMTV-neu transgenic mice have activation of c-src and c-yes (Muthuswamy et al. 1994; Muthuswamy and Muller 1995a). In vitro models corroborate a direct link between src and HER2. Transformation of mammary epithelial cells by HER2, but not by H-Ras, results in the activation of c-src (Sheffield 1998). Therefore it seems plausible that src functions downstream of oncogenic HER2. But a clear mechanistic model of how HER2 activates src and proof that this activation is essential for tumorigenesis has not yet emerged. Src directly interacts with HER2 within the HER2 catalytic domain (Muthuswamy and Muller 1995b; Belsches-Jablonski et al. 2001; Kim et al. 2005). It has been suggested that HER2 activates src through increasing its expression and stability, or by directly phosphorylating src on tyr215 in its SH2 domain (Tan et al. 2005a; Vadlamudi et al. 2003). Inhibition of src kinases in HER2 overexpressing tumors cells using src-selective tyrosine kinase inhibitors has produced different phenotypes including the selective inhibition of invasive and prometastatic characteristics in one report and total growth inhibition and apoptosis in another report (Tan et al. 2005b; Belsches-Jablonski et al. 2001). A specific function in promoting migration, invasion, and metastasis would be highly consistent with the known functions of src in regulating focal adhesions and integrin signaling, and regulation of the actin cytoskeleton. Consistent with this, HER2 mediated activation of c-src in epithelial cells results in loss of polarity, disruption of cell-cell adhesions, and anchorage independent growth (Kim et al. 2005; Sheffield 1998). In contrast to a downstream function of c-src, recent reports show c-src can also function upstream of HER2. C-src enhances HER2-HER3 dimerization and increases their auto- and transphosphorylations and signaling activities (Ishizawar et al. 2006). C-src also phosphorylates HER2 at tyr877 within the activation loop of the kinase domain and increases the kinase activity of HER2 (Xu et al. 2007). These data imply that, in addition to functioning downstream, src kinases may also function upstream or midstream of the transforming functions of HER2 (figure 3). A multi-level involvement of src kinases would make them critical enhancers of HER2 driven tumorigenesis. Genetic crosses to prove an essential role of src in neu induced mammary tumorigenesis have been difficult to do due to the poor health and survival of src null mice.
Disruption of cell adhesion and cell polarity
A hallmark of many epithelial cancers is the loss of cell polarity and cell adhesion. Recent evidence demonstrates that deregulated HER2 signaling can disrupt cell polarity and cell adhesion. HER2, by virtue of its interaction with ERBIN (ErbB2 interacting protein), normally localizes to the basolateral surface of epithelial cells where it likely mediates crosstalk with ligand secreting stromal cells (Borg et al. 2000; Shelly et al. 2003; De Potter et al. 1989). The experimental homodimerization and activation of HER2 leads to disruption of tight junctions, loss of cell polarity, and proliferative disarray in breast epithelial cell acinar structures, although the basal membrane is preserved and invasive features are not induced (Muthuswamy et al. 2001). Activated HER2 disrupts apical-basal polarity through its interaction with components of the Par polarity complex including PAR6 (partition protein 6) and aPKC (atypical protein kinase C) (Aranda et al. 2006). On the other hand, the experimental heterodimerization of HER2 with EGFR promotes the invasive phenotype mediated through pathways including PI3K, Ras, and PLCγ (phospholipase Cγ) (Zhan et al. 2006). These studies were done using engineered synthetic ligand-activated constructs that can discriminate between the functions of homodimers and heterodimers of HER2 in cells grown in 3D models (Muthuswamy et al. 1999). It is inferred from these studies that overexpressed HER2 in cancers, through increased HER2 homodimers and HER2-EGFR heterodimers mediates the loss of polarity and deregulated cell adhesion typical of epithelial cancers (figure 3).
Promoting the invasive phenotype
Multiple downstream signals may be mediating the invasive phenotype associated with HER2 overexpression in tumor cells. Downstream signals that have been implicated in the invasive phenotype include activation of PI3K (Ignatoski et al. 2000), PKC-α and src (830}, FAK (Benlimame et al. 2005; Ignatoski et al. 2000), downregulation of α4 integrin (Woods Ignatoski et al. 2003), induction of β4 integrin (Gambaletta et al. 2000), and TGF-β (Seton-Rogers et al. 2004). The functional significance of β4 integrin in particular has recently been established in genetic models. HER2 physically interacts with β4 integrin (Falcioni et al. 1997) and tumors arising in MMTV-neu mice have delayed onset and reduced invasion and metastases if β4 integrin signaling is disrupted genetically (Guo et al. 2006). Furthermore, ex-vivo studies demonstrated that the proliferative phenotype of neu is mediated through c-jun and the disruption of cell adhesions is mediated through STAT3.
Cell cycle deregulation
Deregulation of cell cycle control, in particular G1/S checkpoint control, leads to uncontrolled proliferation and is a hallmark of many cancers. The two cell cycle regulators that have emerged as downstream targets of oncogenic HER2 are cyclin D1 and p27 (figure 3).
HER2 overexpression in breast epithelial cells deregulates G1/S control through upregulation of cyclin D1, E and cdk6, and degradation of p27 (Timms et al. 2002). Human tumor studies have only compared cyclin D1 expression among different breast cancers and have found no significant increase in cyclin D1 expression in HER2 overexpressing tumors compared to their non-overexpressing counterparts (Ahnstrom et al. 2005; Yang et al. 2004; Loden et al. 2003). It remains possible that cyclin D1 is widely overexpressed in breast cancers and it occurs through other pathways in tumors without HER2 overexpression. Consistent with this, a necessary but not rate-limiting role of cyclin D1 for HER2-induced tumorigenesis has been shown in transgenic models. MMTV-neu mice have almost complete protection from tumor formation in a cyclin D1-null background (Lee et al. 2000; Yu et al. 2001). With prolonged intervals hyperplastic lesions eventually develop due to cyclin E compensation (Bowe et al. 2002). Consistent with the requisite role of cyclin D1, its catalytic partner cdk4 is also required for tumorigenesis in MMTV-neu mice (Reddy et al. 2005). Other lines of evidence also corroborate the hypothesis that HER2 induces transformation through increased cyclin D1/cdk4 activity. Expression of the cyclin D1/cdk4 inhibitor p16 in breast tissue also protects against tumors in MMTV-neu mice (Yang et al. 2004).
HER2 regulates p27 through multiple mechanisms that regulate its localization and its proteolysis. Akt is activated in HER2 amplified breast cancers (reviewed above) and activated Akt phosphorylates p27 inhibiting its function by excluding it from the nucleus (Viglietto et al. 2002; Liang et al. 2002; Shin et al. 2002). Several clinical studies have also established that HER2 overexpressing breast cancers have reduced p27 expression compared to other types of breast cancers (Loden et al. 2003; Spataro et al. 2003; Newman et al. 2001). HER2 mediates p27 degradation likely through mechanisms that involve MAPK (Lenferink et al. 2001; Yang et al. 2000; Donovan et al. 2001). Consistent with a functional role for p27, HER2 induced cell transformation can be inhibited by forced expression of p27 (Yang et al. 2001). The cell cycle function of oncogenic HER2 is likely mediated specifically through its regulation of cyclin D1 and p27 since other G1/S defects such as loss of p16 or Rb are not seen in HER2 amplified breast cancers, whereas they are common in other types of breast cancer (Yang et al. 2004).
The role of transmembrane mucins
While in the simplest scenario, HER family receptor interactions are driven by their affinities for eachother, there is evidence that other transmembrane proteins can facilitate their interactions. HER2 stably interacts with the membrane mucin Muc4. The interaction is mediated through one of two EGF-like domains in ASPG-1, the membrane associated subunit of Muc4 and may play a role in regulating the polar localization of HER2 (Carraway, III et al. 1999; Ramsauer et al. 2003). The experimental induction of Muc4 expression in cells lacking Muc4 leads to increase cell surface retention of HER2 and HER3 and increased HER2-HER3 signaling activity (Funes et al. 2006)(figure 3). The evidence that Muc4 potentiates HER2 signaling has led to the hypothesis that it may promote HER2 tumorigenic signaling even in tumors that do not overexpress HER2. Consistent with this hypothesis, increased expression of Muc4 is associated with worse prognosis in several types of cancer (Tsutsumida et al. 2006; Tamada et al. 2006; Saitou et al. 2005; Shibahara et al. 2004). Further analysis of this hypothesis awaits models that more definitively demonstrate the role of Muc4 in promoting tumorigenic HER2 function.
The role of HER2 neighbors
While all the experimental preclinical models are generated by specific overexpression of the singular HER2 gene, the overexpression of HER2 in naturally occuring breast cancers is almost always due to amplification of a segment of chromosome 17 at 17q12-q21 that contains many genes in addition to HER2. Therefore the human disease involves a more complex genetic basis than the experimental models provide. The effort to identify the genes co-amplified with HER2 in human breast cancer have been greatly facilitated by modern techniques and access to the human genome database. The structure of the HER2 amplicon has now been characterized using a variety of techniques including southern blotting, FISH, and CGH, as well as looking for highly expressed neighboring genes by RT-PCR and microarray expression analyses ((Luoh 2002; Kauraniemi et al. 2001) and reviewed in (Kauraniemi and Kallioniemi 2006)). The HER2 amplicon is relatively small and fairly constant among many breast tumors and spans a minimal region of approximately 280kb centered around the HER2 locus (Kauraniemi et al. 2003)(figure 4). This region contains 10 transcribed genes, of which six are overexpresssed in tumors as a result of amplification of the region, suggesting that they may be the biologically relevant genes. These include HER2, GRB7, MLN64, PNMT, MGC9753, and MGC14832. GRB7 is an SH2-containing adaptor protein and is functionaly linked with HER2 as it is known to bind phosphorylated HER2 and mediate aspects of cell migration (Janes et al. 1997). MLN64 is a late endosomal membrane protein that binds sterols and is felt to be involved in cholesterol transport (Alpy and Tomasetto 2006). PNMT (phenylethanolamine N-methyltransferase) is a catecholamine biosynthetic enzyme and is unlikely to be important for HER2 tumorigenesis. The functions of the hypothetical proteins MGC9753 and MGC14832 are currently unknown. In the mouse knock-in model of HER2 tumorigenesis which occurs through spontaneous gene amplification, the mouse tumor HER2 amplicon is highly similar to the human breast cancer amplicon and contains the same genes, consistent with the hypothesis that genes in addition to HER2 are selected for in the amplification event (Hodgson et al. 2005). Studies to determine the functional relevance of these genes in HER2 driven tumorigenesis are ongoing.
Figure 4.

Structure of the HER2 amplicon in human breast cancer. Schematic and table listing of the genes surrounding the HER2/ERBB2 locus at 17q12-q21 that are frequently co-amplified with HER2/ERBB2. Specific cancers often have larger amplicons including genes outside of this region. But amplicon mapping studies identify the above minimal common region of amplification. Reprint from Endocrine-Related Cancer 13, p39-49, copyright 2006 with permission from Society for Endocrinology and A. Kallioniemi.
HER2 linked genomic abnormalities
In addition to the genes directly located within the 17q12 HER2 amplicon, HER2 amplified breast cancers often have numerous other genomic alterations that are characteristic of this tumor type and much less common in breast tumors without HER2 amplification. These include amplifications in 17q22-24, just distal to the HER2 amplicon as well as amplifications in 20q and deletions in 18q (Isola et al. 1999). These areas may also contain genes that through gain or loss of function could cooperate to establish the biology of HER2 amplified tumors. The topoisomerase IIa (TOP2A) gene on 17q21 is frequently co-amplified with HER2 and may explain why HER2 overrexpressing breast cancers are particularly sensitive to treatment with topoisomerase inhibitors (Mano et al. 2006).
Of particular interest, the PTPN1 gene encoding protein tyrosine phosphatase 1B (PTP1B) on 20q13 is often amplified in HER2 amplified breast cancers (Tanner et al. 1996). Although tyrosine phosphatases frequently play negative feedback roles in growth factor receptor signaling, the frequent amplification and overexpression of PTPN1 in HER2 amplified tumors has led to the hypothesis that it may have a tumor promoting function in these tumors. Consistent with this hypothesis, tumor formation in MMTV-neu mice is significantly delayed or prevented in the PTPN1-null background (tires-Alj and Neel 2007). The molecular mechanisms mediating the supportive tumorigenic role of PTP1B remain to be worked out.
TRANSCRIPTIONAL TARGETS OF HER2
While the function of HER proteins as transmembrane growth factor receptors is well understood and innumerable lines of evidence are consistent with its growth factor signaling function, a nuclear function as a transcription factor has also been proposed. In early two-hybrid screens for Neu interacting proteins, the cytoplasmic domain of Neu used as a bait was unexpectedly found to have transcriptional transactivating activity (Xie and Hung 1994). Consistent with a transcriptional function, a small percentage of cellular HER2 is seen in nuclei and DNA binding and transcriptional activation of at least one promoter has been reported (Wang et al. 2004). A transcriptional function inherent in HER2 significantly widens the realm of mechanisms by which overexpressed HER2 can mediate tumorigenic functions. The transcriptional function of HER2 is clearly not the principal mechanism underlying its transforming functions, since kinase function is essential for neu induced transformation (Weiner et al. 1989a). But transcriptional targets may be enhancing its tranforming functions. So far the COX-2 gene has been reported as a direct target of HER2. However the upregulation (and downregulation) of numerous other genes has been described in HER2 amplified tumors and in fact these tumors have a gene expression profile unique to HER2 amplified breast cancers (Perou et al. 2000). Many of these genes are likely consequences of HER2 overexpression, although some may be directly driven by HER2 transcriptional function. The pathways which current evidence suggests are functionally relevant are reviewed below. The role of many other genes remains to be defined.
Induction of COX-2
The inducible prostaglandin synthase cyclooxygenase-2 (COX-2) is overexpressed in HER2 amplified breast cancers (Subbaramaiah et al. 2002). HER2 directly regulates COX-2 expression through transcriptional induction (Vadlamudi et al. 1999; Wang et al. 2004). COX-2 knockout mice are impaired in their ability to support MMTV-neu induced mammary tumorigenesis (Howe et al. 2005). Interestingly, this phenotype seems to be related to impaired angiogenic signaling. Whether this is due to defective tumor cell angiogenic signaling or host angiogenic signaling is not currently defined. As such, the role of tumor COX-2 in HER2-induced transformation remains to be defined.
Induction of CXCR4
HER2 induces the expression of the chemokine receptor CXCR4 in transfection models and indeed increased expression of CXCR4 is seen in HER2 overexpressing breast cancers (Li et al. 2004). Chemokine receptors have been functionally linked with the metastatic properties of breast cancers (Muller et al. 2001) leading to the hypothesis that the prometastatic properties of HER2 overexpressing tumors may be mediated through the increased expression of relevant chemokine receptors. Consistent with this hypothesis, the increased migratory activity induced by the experimental overexpression of HER2 can be suppressed by anti-CXCR4 antibodies (Li et al. 2004).
Induction of ETS
ETS (E26 transformation specific) transcription factors are almost universally overexpressed in HER2 amplified breast cancers and they are also overexpressed and are essential in MMTV-neu induced mammary tumorigenesis (Shepherd et al. 2001; Neve et al. 2002). HER2 overexpression specifically leads to the bimodal activation of the ETS transcription factor ER81 (Goel and Janknecht 2003) which mediates the induction of expression of the catalytic subunit of telomerase (Goueli and Janknecht 2004).
Other downstream or cooperating pathways
HER2 overexpression leads to increased expression of hypoxia inducible factor 1α (HIF-1α) through Akt leading to increased expression of VEGF and increased surface expression of fibronectin receptors (Laughner et al. 2001; Li et al. 2005; Spangenberg et al. 2006). VEGF expression is also induced by HER2 overexpression through a HIF-1α independent mechanism (Loureiro et al. 2005).
Numerous other pathways have been implicated in HER2-mediated transformation, although they are much less well characterized. HER2 overexpressing breast cancers are characterized by increased expression of MMP-2 (matrix metalloproteinase -2) and MMP-9 (matrix metalloproteinase 9) (Pellikainen et al. 2004). HER2 overactivity also promotes the activation of the nuclear factor kappa-B (NF-kB) anti-apoptotic pathway (Makino et al. 2004). Involvement of myc downstream of HER2 has been proposed although it remains to be defined and may be complex (Hynes and Lane 2001).
TUMOR DEPENDENCE ON HER2
While the tumorigenic potential of HER2 overexpression has been clearly demonstrated in numerous model systems (see above), its suitability as a drug target depends on whether clinically advanced tumors continue to dependent on HER2 for survival and progression. This dependency, recently described as oncogene-addiction, implies that such cancers can be effectively treated and possibly cured with drugs that inactivate the oncogene product (Weinstein 2002). Alternatively, genomic instability can lead to the mutational activation of additional pathways that could compensate for the pharmacologic inactivation of the tumor-initiating oncogene making such an oncogene less effective as a drug target.
HER2 knockdown models
Since the identification of HER2 as an oncogene in human tumors, numerous approaches have been undertaken to demonstrate the dependence of HER2 overexpressing tumors on HER2. A number of studies using antisense, ribozyme, or siRNA methodologies to suppress HER2 expression in human cancer cell lines consistently show that HER2 overexpressing tumor cells are dependent on HER2 and undergo growth inhibition and apoptosis in cell culture, or tumor regression in vivo, in the absence of HER2 expression, while tumor types that do not overexpress HER2 are not sensitive to HER2 knockdown (Colomer et al. 1994; Juhl et al. 1997; Roh et al. 2000; Faltus et al. 2004; Choudhury et al. 2004).A kinase-dead mutant of activated HER2 competes with and reverses the transformed phenotype induced by activated HER2 (Messerle et al. 1994). Intracellularly expressed single chain antibodies that target and inactivate HER2 revert HER2 induced transformation or induce apoptotic cell death in HER2 overexpressing tumor cells (Beerli et al. 1994; Deshane et al. 1996).
HER2 withdrawal models
Tetracycline inducible systems offer even more elegant models for analysis of oncogene addiction. NIH3T3 cells transformed by tetracycline regulated overexpression of HER2 revert from the transformed phenotype and their mouse implanted tumors regress when HER2 expression is withdrawn (Baasner et al. 1996; Schiffer et al. 2003). Tetracycline induced expression of activated HER2 in squamous epithelia of mice results in severe hyperplastic abnormalities of squamous epithelial tissues, which reverse upon withdrawal of the HER2 transgene expression (Xie et al. 1999). Tumors in MMTV-_neu_T mice are also dependent on continued oncogene expression. In the MMTV-rtTA/TetO-NeuNT bitransgenic variant of this model regulated by doxycycline, when expression of the _neu_T oncogene is induced in the mammary tissue of adult mice, this leads to the formation of multiple mammary tumors and lung metastases, and the entire primary tumor and metastatic disease fully regresses when _neu_T expression is withdrawn (Moody et al. 2002).
Each of these models is subject to specific criticisms. For example the antisense or siRNA approaches have non-specific effects, the NIH3T3 fibroblast models are not representative of what is principally an epithelial oncogene in humans, and the transgenic models may be too simplistic and understate the genetic complexity of the human disease. But taken in aggregate, the existing data from all the different models and approaches is highly consistent and collectively makes a highly compelling case that HER2 induced tumors are addicted to HER2. This has made HER2 one of the most sought after targets in cancer drug development.
CONCLUSION
Numerous experimental models now solidly support the hypothesis that HER2 overexpression promotes tumorigenesis and the finding of HER2 amplification and overexpression in many human breast cancers and its link with the biology of this disease now clearly implicate this oncogene in the pathogenesis of this type of human cancer. Numerous well supported experimental models implicate diverse mechanisms involved in HER2 mediated tumorigenesis and a unified mechanistic model cannot currently be proposed. It is possible that the position of HER2 in the signaling web places it in control of numerous pathways that can contribute to malignant progression such that the amplification of HER2 as a single event can substitute for several events in the multi-step model of carcinogenesis. Indeed HER2 amplified breast cancers occur at an earlier age than other types of breast cancers consistent with a fast-track route to oncogenesis in these cancers (Crowe et al. 2006; Swede et al. 2001). The highly oncogene-addicted nature of these cancers also attests to a HER2 function that is not easily supplanted, possibly due to its multiple oncogenic roles. The critical driving role of HER2 in human cancers, and the large number of patients affected by this type of cancer has made HER2 an ideal target for the development of rationally designed anti-cancer drugs.
References
- Ahnstrom M, Nordenskjold B, Rutqvist LE, Skoog L, Stal O. Role of cyclin D1 in ErbB2-positive breast cancer and tamoxifen resistance. Breast Cancer Res Treat. 2005;91:145–151. doi: 10.1007/s10549-004-6457-4. [DOI] [PubMed] [Google Scholar]
- Akiyama T, Sudo C, Ogawara H, Toyoshima K, Yamamoto T. The product of the human c-erbB-2 gene: a 185-kilodalton glycoprotein with tyrosine kinase activity. Science. 1986;232:1644–1646. doi: 10.1126/science.3012781. [DOI] [PubMed] [Google Scholar]
- Alimandi M, Romano A, Curia MC, Muraro R, Fedi P, Aaronson SA, et al. Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene. 1995;10:1813–1821. [PubMed] [Google Scholar]
- Alpy F, Tomasetto C. MLN64 and MENTHO, two mediators of endosomal cholesterol transport. Biochem Soc Trans. 2006;34:343–345. doi: 10.1042/BST0340343. [DOI] [PubMed] [Google Scholar]
- Amundadottir LT, Leder P. Signal transduction pathways activated and required for mammary carcinogenesis in response to specific oncogenes. Oncogene. 1998;16:737–746. doi: 10.1038/sj.onc.1201829. [DOI] [PubMed] [Google Scholar]
- Andrechek ER, Hardy WR, Siegel PM, Rudnicki MA, Cardiff RD, Muller WJ. Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proc Natl Acad Sci U S A. 2000;97:3444–3449. doi: 10.1073/pnas.050408497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrechek ER, White D, Muller WJ. Targeted disruption of ErbB2/Neu in the mammary epithelium results in impaired ductal outgrowth. Oncogene. 2005;24:932–937. doi: 10.1038/sj.onc.1208230. [DOI] [PubMed] [Google Scholar]
- Aranda V, Haire T, Nolan ME, Calarco JP, Rosenberg AZ, Fawcett JP, et al. Par6-aPKC uncouples ErbB2 induced disruption of polarized epithelial organization from proliferation control. Nat Cell Biol. 2006;8:1235–1245. doi: 10.1038/ncb1485. [DOI] [PubMed] [Google Scholar]
- Baasner S, von MH, Klenner T, Hilgard P, Beckers T. Reversible tumorigenesis in mice by conditional expression of the HER2/c-erbB2 receptor tyrosine kinase. Oncogene. 1996;13:901–911. [PubMed] [Google Scholar]
- Bargmann CI, Hung MC, Weinberg RA. Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell. 1986;45:649–657. doi: 10.1016/0092-8674(86)90779-8. [DOI] [PubMed] [Google Scholar]
- Barnes CJ, Kumar R. Biology of the epidermal growth factor receptor family. Cancer Treat Res. 2004;119:1–13. doi: 10.1007/1-4020-7847-1_1. [DOI] [PubMed] [Google Scholar]
- Baulida J, Kraus MH, Alimandi M, Di Fiore PP, Carpenter G. All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J Biol Chem. 1996;271:5251–5257. doi: 10.1074/jbc.271.9.5251. [DOI] [PubMed] [Google Scholar]
- Bazley LA, Gullick WJ. The epidermal growth factor receptor family. Endocr Relat Cancer. 2005;12(Suppl 1):S17–S27. doi: 10.1677/erc.1.01032. [DOI] [PubMed] [Google Scholar]
- Beerli RR, Wels W, Hynes NE. Intracellular expression of single chain antibodies reverts ErbB-2 transformation. J Biol Chem. 1994;269:23931–23936. [PubMed] [Google Scholar]
- Belsches-Jablonski AP, Biscardi JS, Peavy DR, Tice DA, Romney DA, Parsons SJ. Src family kinases and HER2 interactions in human breast cancer cell growth and survival. Oncogene. 2001;20:1465–1475. doi: 10.1038/sj.onc.1204205. [DOI] [PubMed] [Google Scholar]
- Benlimame N, He Q, Jie S, Xiao D, Xu YJ, Loignon M, et al. FAK signaling is critical for ErbB-2/ErbB-3 receptor cooperation for oncogenic transformation and invasion. J Cell Biol. 2005;171:505–516. doi: 10.1083/jcb.200504124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benusiglio PR, Lesueur F, Luccarini C, Conroy DM, Shah M, Easton DF, et al. Common ERBB2 polymorphisms and risk of breast cancer in a white British population: a case-control study. Breast Cancer Res. 2005;7:R204–R209. doi: 10.1186/bcr982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz CC, Scott GK, Sarup JC, Johnson RM, Tripathy D, Coronado E, et al. Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF-7 cells transfected with HER2/neu. Breast Cancer Res Treat. 1992;24:85–95. doi: 10.1007/BF01961241. [DOI] [PubMed] [Google Scholar]
- Berger MB, Mendrola JM, Lemmon MA. ErbB3/HER3 does not homodimerize upon neuregulin binding at the cell surface. FEBS Lett. 2004;569:332–336. doi: 10.1016/j.febslet.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Bol D, Kiguchi K, Beltran L, Rupp T, Moats S, Gimenez-Conti I, et al. Severe follicular hyperplasia and spontaneous papilloma formation in transgenic mice expressing the neu oncogene under the control of the bovine keratin 5 promoter. Mol Carcinog. 1998;21:2–12. [PubMed] [Google Scholar]
- Borg JP, Marchetto S, Le BA, Ollendorff V, Jaulin-Bastard F, Saito H, et al. ERBIN: a basolateral PDZ protein that interacts with the mammalian ERBB2/HER2 receptor. Nat Cell Biol. 2000;2:407–414. doi: 10.1038/35017038. [DOI] [PubMed] [Google Scholar]
- Bouchard L, Lamarre L, Tremblay PJ, Jolicoeur P. Stochastic appearance of mammary tumors in transgenic mice carrying the MMTV/c-neu oncogene. Cell. 1989;57:931–936. doi: 10.1016/0092-8674(89)90331-0. [DOI] [PubMed] [Google Scholar]
- Bowe DB, Kenney NJ, Adereth Y, Maroulakou IG. Suppression of Neu-induced mammary tumor growth in cyclin D1 deficient mice is compensated for by cyclin E. Oncogene. 2002;21:291–298. doi: 10.1038/sj.onc.1205025. [DOI] [PubMed] [Google Scholar]
- Brandt R, Wong AM, Hynes NE. Mammary glands reconstituted with Neu/ErbB2 transformed HC11 cells provide a novel orthotopic tumor model for testing anti-cancer agents. Oncogene. 2001;20:5459–5465. doi: 10.1038/sj.onc.1204709. [DOI] [PubMed] [Google Scholar]
- Britsch S, Li L, Kirchhoff S, Theuring F, Brinkmann V, Birchmeier C, et al. The ErbB2 and ErbB3 receptors and their ligand, neuregulin-1, are essential for development of the sympathetic nervous system. Genes Dev. 1998;12:1825–1836. doi: 10.1101/gad.12.12.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP, Leahy DJ, et al. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12:541–552. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- Carlsson J, Nordgren H, Sjostrom J, Wester K, Villman K, Bengtsson NO, et al. HER2 expression in breast cancer primary tumours and corresponding metastases. Original data and literature review. Br J Cancer. 2004;90:2344–2348. doi: 10.1038/sj.bjc.6601881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraway KL, III, Rossi EA, Komatsu M, Price-Schiavi SA, Huang D, Guy PM, et al. An intramembrane modulator of the ErbB2 receptor tyrosine kinase that potentiates neuregulin signaling. J Biol Chem. 1999;274:5263–5266. doi: 10.1074/jbc.274.9.5263. [DOI] [PubMed] [Google Scholar]
- Castiglioni F, Tagliabue E, Campiglio M, Pupa SM, Balsari A, Menard S. Role of exon-16-deleted HER2 in breast carcinomas. Endocr Relat Cancer. 2006;13:221–232. doi: 10.1677/erc.1.01047. [DOI] [PubMed] [Google Scholar]
- Chazin VR, Kaleko M, Miller AD, Slamon DJ. Transformation mediated by the human HER-2 gene independent of the epidermal growth factor receptor. Oncogene. 1992;7:1859–1866. [PubMed] [Google Scholar]
- Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW, Jr., et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- Choudhury A, Charo J, Parapuram SK, Hunt RC, Hunt DM, Seliger B, et al. Small interfering RNA (siRNA) inhibits the expression of the Her2/neu gene, upregulates HLA class I and induces apoptosis of Her2/neu positive tumor cell lines. Int J Cancer. 2004;108:71–77. doi: 10.1002/ijc.11497. [DOI] [PubMed] [Google Scholar]
- Colomer R, Lupu R, Bacus SS, Gelmann EP. erbB-2 antisense oligonucleotides inhibit the proliferation of breast carcinoma cells with erbB-2 oncogene amplification. Br J Cancer. 1994;70:819–825. doi: 10.1038/bjc.1994.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cool M, Jolicoeur P. Elevated frequency of loss of heterozygosity in mammary tumors arising in mouse mammary tumor virus/neu transgenic mice. Cancer Res. 1999;59:2438–2444. [PubMed] [Google Scholar]
- Crowe JP, Patrick RJ, Rybicki LA, Escobar PF, Weng D, Thomas BG, et al. A data model to predict HER2 status in breast cancer based on the clinical and pathologic profiles of a large patient population at a single institution. Breast. 2006;15:728–735. doi: 10.1016/j.breast.2006.03.005. [DOI] [PubMed] [Google Scholar]
- De Potter CR, Quatacker J, Maertens G, Van DS, Pauwels C, Verhofstede C, et al. The subcellular localization of the neu protein in human normal and neoplastic cells. Int J Cancer. 1989;44:969–974. doi: 10.1002/ijc.2910440604. [DOI] [PubMed] [Google Scholar]
- Deshane J, Grim J, Loechel S, Siegal GP, Alvarez RD, Curiel DT. Intracellular antibody against erbB-2 mediates targeted tumor cell eradication by apoptosis. Cancer Gene Ther. 1996;3:89–98. [PubMed] [Google Scholar]
- Di Fiore PP, Pierce JH, Kraus MH, Segatto O, King CR, Aaronson SA. erbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science. 1987;237:178–182. doi: 10.1126/science.2885917. [DOI] [PubMed] [Google Scholar]
- Donovan JC, Milic A, Slingerland JM. Constitutive MEK/MAPK activation leads to p27(Kip1) deregulation and antiestrogen resistance in human breast cancer cells. J Biol Chem. 2001;276:40888–40895. doi: 10.1074/jbc.M106448200. [DOI] [PubMed] [Google Scholar]
- Erickson SL, O’Shea KS, Ghaboosi N, Loverro L, Frantz G, Bauer M, et al. ErbB3 is required for normal cerebellar and cardiac development: a comparison with ErbB2-and heregulin-deficient mice. Development. 1997;124:4999–5011. doi: 10.1242/dev.124.24.4999. [DOI] [PubMed] [Google Scholar]
- Falcioni R, Antonini A, Nistico P, Di SS, Crescenzi M, Natali PG, et al. Alpha 6 beta 4 and alpha 6 beta 1 integrins associate with ErbB-2 in human carcinoma cell lines. Exp Cell Res. 1997;236:76–85. doi: 10.1006/excr.1997.3695. [DOI] [PubMed] [Google Scholar]
- Faltus T, Yuan J, Zimmer B, Kramer A, Loibl S, Kaufmann M, et al. Silencing of the HER2/neu gene by siRNA inhibits proliferation and induces apoptosis in HER2/neu-overexpressing breast cancer cells. Neoplasia. 2004;6:786–795. doi: 10.1593/neo.04313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedi P, Pierce JH, Di Fiore PP, Kraus MH. Efficient coupling with phosphatidylinositol 3-kinase, but not phospholipase C gamma or GTPase-activating protein, distinguishes ErbB-3 signaling from that of other ErbB/EGFR family members. Mol Cell Biol. 1994;14:492–500. doi: 10.1128/mcb.14.1.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkle D, Quan ZR, Asghari V, Kloss J, Ghaboosi N, Mai E, et al. HER2-targeted therapy reduces incidence and progression of midlife mammary tumors in female murine mammary tumor virus huHER2-transgenic mice. Clin Cancer Res. 2004;10:2499–2511. doi: 10.1158/1078-0432.ccr-03-0448. [DOI] [PubMed] [Google Scholar]
- Fleishman SJ, Schlessinger J, Ben-Tal N. A putative molecular-activation switch in the transmembrane domain of erbB2. Proc Natl Acad Sci U S A. 2002;99:15937–15940. doi: 10.1073/pnas.252640799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funes M, Miller JK, Lai C, Carraway KL, III, Sweeney C. The mucin Muc4 potentiates neuregulin signaling by increasing the cell surface populations of ErbB2 and ErbB3. J Biol Chem. 2006 doi: 10.1074/jbc.M603225200. [DOI] [PubMed] [Google Scholar]
- Gabos Z, Sinha R, Hanson J, Chauhan N, Hugh J, Mackey JR, et al. Prognostic significance of human epidermal growth factor receptor positivity for the development of brain metastasis after newly diagnosed breast cancer. J Clin Oncol. 2006;24:5658–5663. doi: 10.1200/JCO.2006.07.0250. [DOI] [PubMed] [Google Scholar]
- Gambaletta D, Marchetti A, Benedetti L, Mercurio AM, Sacchi A, Falcioni R. Cooperative signaling between alpha(6)beta(4) integrin and ErbB-2 receptor is required to promote phosphatidylinositol 3-kinase-dependent invasion. J Biol Chem. 2000;275:10604–10610. doi: 10.1074/jbc.275.14.10604. [DOI] [PubMed] [Google Scholar]
- Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol Cell. 2003;11:495–505. doi: 10.1016/s1097-2765(03)00048-0. [DOI] [PubMed] [Google Scholar]
- Gassmann M, Casagranda F, Orioli D, Simon H, Lai C, Klein R, et al. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature. 1995;378:390–394. doi: 10.1038/378390a0. [see comment] [DOI] [PubMed] [Google Scholar]
- Goel A, Janknecht R. Acetylation-mediated transcriptional activation of the ETS protein ER81 by p300, P/CAF, and HER2/Neu. Mol Cell Biol. 2003;23:6243–6254. doi: 10.1128/MCB.23.17.6243-6254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodearl A, Viehover A, Vartanian T. Neuregulin-induced association of Sos Ras exchange protein with HER2(erbB2)/HER3(erbB3) receptor complexes in Schwann cells through a specific Grb2-HER2(erbB2) interaction. Dev Neurosci. 2001;23:25–30. doi: 10.1159/000048693. [DOI] [PubMed] [Google Scholar]
- Goueli BS, Janknecht R. Upregulation of the Catalytic Telomerase Subunit by the Transcription Factor ER81 and Oncogenic HER2/Neu, Ras, or Raf. Mol Cell Biol. 2004;24:25–35. doi: 10.1128/MCB.24.1.25-35.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graus-Porta D, Beerli RR, Daly JM, Hynes NE. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997;16:1647–1655. doi: 10.1093/emboj/16.7.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Pylayeva Y, Pepe A, Yoshioka T, Muller WJ, Inghirami G, et al. Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell. 2006;126:489–502. doi: 10.1016/j.cell.2006.05.047. [DOI] [PubMed] [Google Scholar]
- Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci USA. 1992;89:10578–10582. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellyer NJ, Kim MS, Koland JG. Heregulin-dependent activation of phosphoinositide 3-kinase and Akt via the ErbB2/ErbB3 co-receptor. J Biol Chem. 2001;276:42153–42161. doi: 10.1074/jbc.M102079200. [DOI] [PubMed] [Google Scholar]
- Hendriks BS, Opresko LK, Wiley HS, Lauffenburger D. Quantitative analysis of HER2-mediated effects on HER2 and epidermal growth factor receptor endocytosis: distribution of homo- and heterodimers depends on relative HER2 levels. J Biol Chem. 2003a;278:23343–23351. doi: 10.1074/jbc.M300477200. [DOI] [PubMed] [Google Scholar]
- Hendriks BS, Wiley HS, Lauffenburger D. HER2-mediated effects on EGFR endosomal sorting: analysis of biophysical mechanisms. Biophys J. 2003b;85:2732–2745. doi: 10.1016/s0006-3495(03)74696-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks DG, Tubbs RR. Assessment of the HER2 status in breast cancer by fluorescence in situ hybridization: a technical review with interpretive guidelines. Hum Pathol. 2005;36:250–261. doi: 10.1016/j.humpath.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Hirsch FR, Varella-Garcia M, Franklin WA, Veve R, Chen L, Helfrich B, et al. Evaluation of HER-2/neu gene amplification and protein expression in non-small cell lung carcinomas. Br J Cancer. 2002;86:1449–1456. doi: 10.1038/sj.bjc.6600286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson JG, Malek T, Bornstein S, Hariono S, Ginzinger DG, Muller WJ, et al. Copy number aberrations in mouse breast tumors reveal loci and genes important in tumorigenic receptor tyrosine kinase signaling. Cancer Res. 2005;65:9695–9704. doi: 10.1158/0008-5472.CAN-05-0755. [DOI] [PubMed] [Google Scholar]
- Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, III, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci USA. 2003;100:8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan T, Wen J, Arakawa T, Liu N, Brankow D, Hu S, et al. Binding of Neu differentiation factor with the extracellular domain of Her2 and Her3. J Biol Chem. 1995;270:24604–24608. doi: 10.1074/jbc.270.41.24604. [DOI] [PubMed] [Google Scholar]
- Howe LR, Chang SH, Tolle KC, Dillon R, Young LJ, Cardiff RD, et al. HER2/neu-induced mammary tumorigenesis and angiogenesis are reduced in cyclooxygenase-2 knockout mice. Cancer Res. 2005;65:10113–10119. doi: 10.1158/0008-5472.CAN-05-1524. [DOI] [PubMed] [Google Scholar]
- Huang G, Chantry A, Epstein RJ. Overexpression of ErbB2 impairs ligand-dependent downregulation of epidermal growth factor receptors via a post-transcriptional mechanism. J Cell Biochem. 1999;74:23–30. [PubMed] [Google Scholar]
- Hudziak RM, Schlessinger J, Ullrich A. Increased expression of the putative growth factor receptor p185HER2 causes transformation and tumorigenesis of NIH 3T3 cells. Proc Natl Acad Sci U S A. 1987;84:7159–7163. doi: 10.1073/pnas.84.20.7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes NE, Lane HA. Myc and mammary cancer: Myc is a downstream effector of the ErbB2 receptor tyrosine kinase. J Mammary Gland Biol Neoplasia. 2001;6:141–150. doi: 10.1023/a:1009528918064. [DOI] [PubMed] [Google Scholar]
- Ignatoski KM, Maehama T, Markwart SM, Dixon JE, Livant DL, Ethier SP. ERBB-2 overexpression confers PI 3′ kinase-dependent invasion capacity on human mammary epithelial cells. Br J Cancer. 2000;82:666–674. doi: 10.1054/bjoc.1999.0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizawar RC, Miyake T, Parsons SJ. c-Src modulates ErbB2 and ErbB3 heterocomplex formation and function. Oncogene. 2006 doi: 10.1038/sj.onc.1210138. Advance online publication; doi: 10.1038/sj.onc.1210138. [DOI] [PubMed] [Google Scholar]
- Isola J, Chu L, DeVries S, Matsumura K, Chew K, Ljung BM, et al. Genetic alterations in ERBB2-amplified breast carcinomas. Clin Cancer Res. 1999;5:4140–4145. [PubMed] [Google Scholar]
- Janes PW, Lackmann M, Church WB, Sanderson GM, Sutherland RL, Daly RJ. Structural determinants of the interaction between the erbB2 receptor and the Src homology 2 domain of Grb7. J Biol Chem. 1997;272:8490–8497. doi: 10.1074/jbc.272.13.8490. [DOI] [PubMed] [Google Scholar]
- Juhl H, Downing SG, Wellstein A, Czubayko F. HER-2/neu is rate-limiting for ovarian cancer growth. Conditional depletion of HER-2/neu by ribozyme targeting. J Biol Chem. 1997;272:29482–29486. doi: 10.1074/jbc.272.47.29482. [DOI] [PubMed] [Google Scholar]
- Kallioniemi OP, Kallioniemi A, Kurisu W, Thor A, Chen LC, Smith HS, et al. ERBB2 amplification in breast cancer analyzed by fluorescence in situ hybridization. Proc Natl Acad Sci U S A. 1992;89:5321–5325. doi: 10.1073/pnas.89.12.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunagaran D, Tzahar E, Beerli RR, Chen X, Graus-Porta D, Ratzkin BJ, et al. ErbB-2 is a common auxiliary subunit of NDF and EGF receptors: implications for breast cancer. EMBO J. 1996;15:254–264. [PMC free article] [PubMed] [Google Scholar]
- Kauraniemi P, Barlund M, Monni O, Kallioniemi A. New amplified and highly expressed genes discovered in the ERBB2 amplicon in breast cancer by cDNA microarrays. Cancer Res. 2001;61:8235–8240. [PubMed] [Google Scholar]
- Kauraniemi P, Kallioniemi A. Activation of multiple cancer-associated genes at the ERBB2 amplicon in breast cancer. Endocr Relat Cancer. 2006;13:39–49. doi: 10.1677/erc.1.01147. [DOI] [PubMed] [Google Scholar]
- Kauraniemi P, Kuukasjarvi T, Sauter G, Kallioniemi A. Amplification of a 280-kilobase core region at the ERBB2 locus leads to activation of two hypothetical proteins in breast cancer. Am J Pathol. 2003;163:1979–1984. doi: 10.1016/S0002-9440(10)63556-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keely SJ, Barrett KE. ErbB2 and ErbB3 receptors mediate inhibition of calcium-dependent chloride secretion in colonic epithelial cells. J Biol Chem. 1999;274:33449–33454. doi: 10.1074/jbc.274.47.33449. [DOI] [PubMed] [Google Scholar]
- Khan AJ, King BL, Smith BD, Smith GL, DiGiovanna MP, Carter D, et al. Characterization of the HER-2/neu oncogene by immunohistochemical and fluorescence in situ hybridization analysis in oral and oropharyngeal squamous cell carcinoma. Clin Cancer Res. 2002;8:540–548. [PubMed] [Google Scholar]
- Kiguchi K, Bol D, Carbajal S, Beltran L, Moats S, Chan K, et al. Constitutive expression of erbB2 in epidermis of transgenic mice results in epidermal hyperproliferation and spontaneous skin tumor development. Oncogene. 2000;19:4243–4254. doi: 10.1038/sj.onc.1203778. [DOI] [PubMed] [Google Scholar]
- Kiguchi K, Carbajal S, Chan K, Beltran L, Ruffino L, Shen J, et al. Constitutive expression of ErbB-2 in gallbladder epithelium results in development of adenocarcinoma. Cancer Res. 2001;61:6971–6976. [PubMed] [Google Scholar]
- Kim H, Chan R, Dankort DL, Zuo D, Najoukas M, Park M, et al. The c-Src tyrosine kinase associates with the catalytic domain of ErbB-2: implications for ErbB-2 mediated signaling and transformation. Oncogene. 2005;24:7599–7607. doi: 10.1038/sj.onc.1208898. [DOI] [PubMed] [Google Scholar]
- Kim HH, Vijapurkar U, Hellyer NJ, Bravo D, Koland JG. Signal transduction by epidermal growth factor and heregulin via the kinase-deficient ErbB3 protein. Biochem J. 1998;334(Pt 1):189–195. doi: 10.1042/bj3340189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CR, Kraus MH, Aaronson SA. Amplification of a novel v-erbB-related gene in a human mammary carcinoma. Science. 1985;229:974–976. doi: 10.1126/science.2992089. [DOI] [PubMed] [Google Scholar]
- Kraus MH, Issing W, Miki T, Popescu NC, Aaronson SA. Isolation and characterization of ERBB3, a third member of the ERBB/epidermal growth factor receptor family: evidence for overexpression in a subset of human mammary tumors. Proc Natl Acad Sci USA. 1989;86:9193–9197. doi: 10.1073/pnas.86.23.9193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong KY, Hung MC. A novel splice variant of HER2 with increased transformation activity. Mol Carcinog. 1998;23:62–68. doi: 10.1002/(sici)1098-2744(199810)23:2<62::aid-mc2>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Lacenere CJ, Sternberg PW. Regulation of EGF receptor signaling in the fruitfly D. melanogaster and the nematode C. elegans. Breast Dis. 2000;11:19–30. doi: 10.3233/bd-1999-11103. [DOI] [PubMed] [Google Scholar]
- Latif Z, Watters AD, Dunn I, Grigor KM, Underwood MA, Bartlett JM. HER2/neu overexpression in the development of muscle-invasive transitional cell carcinoma of the bladder. Br J Cancer. 2003;89:1305–1309. doi: 10.1038/sj.bjc.6601245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latta EK, Tjan S, Parkes RK, O’Malley FP. The role of HER2/neu overexpression/amplification in the progression of ductal carcinoma in situ to invasive carcinoma of the breast. Mod Pathol. 2002;15:1318–1325. doi: 10.1097/01.MP.0000038462.62634.B1. [DOI] [PubMed] [Google Scholar]
- Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001;21:3995–4004. doi: 10.1128/MCB.21.12.3995-4004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Soung YH, Seo SH, Kim SY, Park CH, Wang YP, et al. Somatic mutations of ERBB2 kinase domain in gastric, colorectal, and breast carcinomas. Clin Cancer Res. 2006;12:57–61. doi: 10.1158/1078-0432.CCR-05-0976. [DOI] [PubMed] [Google Scholar]
- Lee KF, Simon H, Chen H, Bates B, Hung MC, Hauser C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature. 1995;378:394–398. doi: 10.1038/378394a0. [see comment] [DOI] [PubMed] [Google Scholar]
- Lee RJ, Albanese C, Fu M, D’Amico M, Lin B, Watanabe G, et al. Cyclin D1 is required for transformation by activated Neu and is induced through an E2F-dependent signaling pathway. Mol Cell Biol. 2000;20:672–683. doi: 10.1128/mcb.20.2.672-683.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine NR, Barnes DM, Hollywood DP, Hughes CM, Smith P, Dublin E, et al. Expression of the ERBB3 gene product in breast cancer. British Journal of Cancer. 1992;66:1116–1121. doi: 10.1038/bjc.1992.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenferink AE, Busse D, Flanagan WM, Yakes FM, Arteaga CL. ErbB2/neu kinase modulates cellular p27(Kip1) and cyclin D1 through multiple signaling pathways. Cancer Res. 2001;61:6583–6591. [PubMed] [Google Scholar]
- Lenferink AE, Pinkas-Kramarski R, van de Poll ML, van Vugt MJ, Klapper LN, Tzahar E, et al. Differential endocytic routing of homo- and hetero-dimeric ErbB tyrosine kinases confers signaling superiority to receptor heterodimers. EMBO J. 1998;17:3385–3397. doi: 10.1093/emboj/17.12.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Rosen JM, Menamin-Balano J, Muller WJ, Perkins AS. neu/ERBB2 cooperates with p53-172H during mammary tumorigenesis in transgenic mice. Mol Cell Biol. 1997;17:3155–3163. doi: 10.1128/mcb.17.6.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YM, Pan Y, Wei Y, Cheng X, Zhou BP, Tan M, et al. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell. 2004;6:459–469. doi: 10.1016/j.ccr.2004.09.027. [DOI] [PubMed] [Google Scholar]
- Li YM, Zhou BP, Deng J, Pan Y, Hay N, Hung MC. A hypoxia-independent hypoxia-inducible factor-1 activation pathway induced by phosphatidylinositol-3 kinase/Akt in HER2 overexpressing cells. Cancer Res. 2005;65:3257–3263. doi: 10.1158/0008-5472.CAN-04-1284. [DOI] [PubMed] [Google Scholar]
- Li Z, Szabolcs M, Terwilliger JD, Efstratiadis A. Prostatic intraepithelial neoplasia and adenocarcinoma in mice expressing a probasin-Neu oncogenic transgene. Carcinogenesis. 2006;27:1054–1067. doi: 10.1093/carcin/bgi324. [DOI] [PubMed] [Google Scholar]
- Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–1160. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- Liu E, Thor A, He M, Barcos M, Ljung BM, Benz C. The HER2 (c-erbB-2) oncogene is frequently amplified in in situ carcinomas of the breast. Oncogene. 1992;7:1027–1032. [PubMed] [Google Scholar]
- Loden M, Perris F, Nielsen NH, Emdin SO, Landberg G. C-erbB2, p27 and G1/S aberrations in human primary breast cancer. Anticancer Res. 2003;23:2053–2061. [PubMed] [Google Scholar]
- Lohrisch C, Piccart M. An overview of HER2. Semin Oncol. 2001;28:3–11. [PubMed] [Google Scholar]
- Loureiro RM, Maharaj AS, Dankort D, Muller WJ, D’Amore PA. ErbB2 overexpression in mammary cells upregulates VEGF through the core promoter. Biochemical & Biophysical Research Communications. 2005;326:455–465. doi: 10.1016/j.bbrc.2004.11.053. [DOI] [PubMed] [Google Scholar]
- Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257–262. doi: 10.1016/s1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- Luoh SW. Amplification and expression of genes from the 17q11 approximately q12 amplicon in breast cancer cells. Cancer Genet Cytogenet. 2002;136:43–47. doi: 10.1016/s0165-4608(01)00657-4. [DOI] [PubMed] [Google Scholar]
- Makino K, Day CP, Wang SC, Li YM, Hung MC. Upregulation of IKKalpha/IKKbeta by integrin-linked kinase is required for HER2/neu-induced NF-kappaB antiapoptotic pathway. Oncogene. 2004;23:3883–3887. doi: 10.1038/sj.onc.1207485. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Mano MS, Rosa DD, De Azambuja E, Ismael GFV, Durbecq V. The 17q12-q21 amplicon: Her2 and topoisomerase-II[alpha] and their importance to the biology of solid tumours. Cancer Treatment Reviews. 2006;33:64–77. doi: 10.1016/j.ctrv.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19:6550–6565. doi: 10.1038/sj.onc.1204082. [DOI] [PubMed] [Google Scholar]
- Messerle K, Schlegel J, Hynes NE, Groner B. NIH/3T3 cells transformed with the activated erbB-2 oncogene can be phenotypically reverted by a kinase deficient, dominant negative erbB-2 variant. Mol Cell Endocrinol. 1994;105:1–10. doi: 10.1016/0303-7207(94)90029-9. [DOI] [PubMed] [Google Scholar]
- Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Werb Z, et al. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature. 1995;337:341. doi: 10.1038/376337a0. [DOI] [PubMed] [Google Scholar]
- Mimura K, Kono K, Hanawa M, Mitsui F, Sugai H, Miyagawa N, et al. Frequencies of HER-2/neu expression and gene amplification in patients with oesophageal squamous cell carcinoma. British Journal of Cancer. 2005;92:1253–1260. doi: 10.1038/sj.bjc.6602499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghal N, Sternberg PW. The epidermal growth factor system in Caenorhabditis elegans. Exp Cell Res. 2003;284:150–159. doi: 10.1016/s0014-4827(02)00097-6. [DOI] [PubMed] [Google Scholar]
- Montgomery KG, Gertig DM, Baxter SW, Milne RL, Dite GS, McCredie MR, et al. The HER2 I655V polymorphism and risk of breast cancer in women < age 40 years. Cancer Epidemiol Biomarkers Prev. 2003;12:1109–1111. [PubMed] [Google Scholar]
- Moody SE, Sarkisian CJ, Hahn KT, Gunther EJ, Pickup S, Dugan KD, et al. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell. 2002;2:451–461. doi: 10.1016/s1535-6108(02)00212-x. [DOI] [PubMed] [Google Scholar]
- Morris JK, Lin W, Hauser C, Marchuk Y, Getman D, Lee KF. Rescue of the cardiac defect in ErbB2 mutant mice reveals essential roles of ErbB2 in peripheral nervous system development. Neuron. 1999;23:273–283. doi: 10.1016/s0896-6273(00)80779-5. [DOI] [PubMed] [Google Scholar]
- Morrison C, Zanagnolo V, Ramirez N, Cohn DE, Kelbick N, Copeland L, et al. HER-2 is an independent prognostic factor in endometrial cancer: association with outcome in a large cohort of surgically staged patients. J Clin Onc. 2006;24:2376–2385. doi: 10.1200/JCO.2005.03.4827. [DOI] [PubMed] [Google Scholar]
- Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell. 1988;54:105–115. doi: 10.1016/0092-8674(88)90184-5. [DOI] [PubMed] [Google Scholar]
- Muthuswamy SK, Gilman M, Brugge JS. Controlled dimerization of ErbB receptors provides evidence for differential signaling by homo- and heterodimers. Mol Cell Biol. 1999;19:6845–6857. doi: 10.1128/mcb.19.10.6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001;3:785–792. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthuswamy SK, Muller WJ. Activation of Src family kinases in Neu-induced mammary tumors correlates with their association with distinct sets of tyrosine phosphorylated proteins in vivo. Oncogene. 1995a;11:1801–1810. [PubMed] [Google Scholar]
- Muthuswamy SK, Muller WJ. Direct and specific interaction of c-Src with Neu is involved in signaling by the epidermal growth factor receptor. Oncogene. 1995b;11:271–279. [PubMed] [Google Scholar]
- Muthuswamy SK, Siegel PM, Dankort DL, Webster MA, Muller WJ. Mammary tumors expressing the neu proto-oncogene possess elevated c-Src tyrosine kinase activity. Mol Cell Biol. 1994;14:735–743. doi: 10.1128/mcb.14.1.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RM, Ylstra B, Chang CH, Albertson DG, Benz CC. ErbB2 activation of ESX gene expression. Oncogene. 2002;21:3934–3938. doi: 10.1038/sj.onc.1205503. [DOI] [PubMed] [Google Scholar]
- Newman L, Xia W, Yang HY, Sahin A, Bondy M, Lukmanji F, et al. Correlation of p27 protein expression with HER-2/neu expression in breast cancer. Mol Carcinog. 2001;30:169–175. doi: 10.1002/mc.1025. [DOI] [PubMed] [Google Scholar]
- Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. [Review] [126 refs] EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottenhoff-Kalff AE, Rijksen G, van Beurden EA, Hennipman A, Michels AA, Staal GE. Characterization of protein tyrosine kinases from human breast cancer: involvement of the c-src oncogene product. Cancer Res. 1992;52:4773–4778. [PubMed] [Google Scholar]
- Paez J, Sellers WR. PI3K/PTEN/AKT pathway. A critical mediator of oncogenic signaling. Cancer Treat Res. 2003;115:145–167. [PubMed] [Google Scholar]
- Park K, Han S, Kim HJ, Kim J, Shin E. HER2 status in pure ductal carcinoma in situ and in the intraductal and invasive components of invasive ductal carcinoma determined by fluorescence in situ hybridization and immunohistochemistry. Histopathology. 2006;48:702–707. doi: 10.1111/j.1365-2559.2006.02403.x. [DOI] [PubMed] [Google Scholar]
- Pellikainen JM, Ropponen KM, Kataja VV, Kellokoski JK, Eskelinen MJ, Kosma VM. Expression of matrix metalloproteinase (MMP)-2 and MMP-9 in breast cancer with a special reference to activator protein-2, HER2, and prognosis. Clin Cancer Res. 2004;10:7621–7628. doi: 10.1158/1078-0432.CCR-04-1061. [DOI] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de RM, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Plowman GD, Culouscou JM, Whitney GS, Green JM, Carlton GW, Foy L, et al. Ligand-specific activation of HER4/p180erbB4, a fourth member of the epidermal growth factor receptor family. Proc Natl Acad Sci USA. 1993;90:1746–1750. doi: 10.1073/pnas.90.5.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prenzel N, Fischer OM, Streit S, Hart S, Ullrich A. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr Relat Cancer. 2001;8:11–31. doi: 10.1677/erc.0.0080011. [DOI] [PubMed] [Google Scholar]
- Press MF, Cordon-Cardo C, Slamon DJ. Expression of the HER-2/neu protooncogene in normal human adult and fetal tissues. Oncogene. 1990;5:953–962. [PubMed] [Google Scholar]
- Prigent SA, Gullick WJ. Identification of c-erbB-3 binding sites for phosphatidylinositol 3′-kinase and SHC using an EGF receptor/c-erbB-3 chimera. EMBO J. 1994;13:2831–2841. doi: 10.1002/j.1460-2075.1994.tb06577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram TG, Ethier SP. Phosphatidylinositol 3-kinase recruitment by p185erbB-2 and erbB-3 is potently induced by neu differentiation factor/heregulin during mitogenesis and is constitutively elevated in growth factor-independent breast carcinoma cells with c-erbB-2 gene amplification. Cell Growth Differ. 1996;7:551–561. [PubMed] [Google Scholar]
- Ramsauer VP, Carraway CA, Salas PJ, Carraway KL. Muc4/sialomucin complex, the intramembrane ErbB2 ligand, translocates ErbB2 to the apical surface in polarized epithelial cells. J Biol Chem. 2003;278:30142–30147. doi: 10.1074/jbc.M303220200. [DOI] [PubMed] [Google Scholar]
- Real FX, Rettig WJ, Chesa PG, Melamed MR, Old LJ, Mendelsohn J. Expression of epidermal growth factor receptor in human cultured cells and tissues: relationship to cell lineage and stage of differentiation. Cancer Res. 1986;46:4726–4731. [PubMed] [Google Scholar]
- Reddy HK, Mettus RV, Rane SG, Grana X, Litvin J, Reddy EP. Cyclin-dependent kinase 4 expression is essential for neu-induced breast tumorigenesis. Cancer Res. 2005;65:10174–10178. doi: 10.1158/0008-5472.CAN-05-2639. [DOI] [PubMed] [Google Scholar]
- Reissig D, Clement J, Sanger J, Berndt A, Kosmehl H, Bohmer FD. Elevated activity and expression of Src-family kinases in human breast carcinoma tissue versus matched non-tumor tissue. J Cancer Res Clin Oncol. 2001;127:226–230. doi: 10.1007/s004320000197. [DOI] [PubMed] [Google Scholar]
- Riethmacher D, Sonnenberg-Riethmacher E, Brinkmann V, Yamaai T, Lewin GR, Birchmeier C. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature. 1997;389:725–730. doi: 10.1038/39593. [DOI] [PubMed] [Google Scholar]
- Ritland SR, Rowse GJ, Chang Y, Gendler SJ. Loss of heterozygosity analysis in primary mammary tumors and lung metastases of MMTV-MTAg and MMTV-neu transgenic mice. Cancer Res. 1997;57:3520–3525. [PubMed] [Google Scholar]
- Roh H, Pippin J, Drebin JA. Down-regulation of HER2/neu expression induces apoptosis in human cancer cells that overexpress HER2/neu. Cancer Res. 2000;60:560–565. [PubMed] [Google Scholar]
- Ross JS, Fletcher JA, Linette GP, Stec J, Clark E, Ayers M, et al. The Her-2/neu gene and protein in breast cancer 2003: biomarker and target of therapy. Oncologist. 2003;8:307–325. doi: 10.1634/theoncologist.8-4-307. [DOI] [PubMed] [Google Scholar]
- Saitou M, Goto M, Horinouchi M, Tamada S, Nagata K, Hamada T, et al. MUC4 expression is a novel prognostic factor in patients with invasive ductal carcinoma of the pancreas. J Clin Pathol. 2005;58:845–852. doi: 10.1136/jcp.2004.023572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schechter AL, Hung MC, Vaidyanathan L, Weinberg RA, Yang-Feng TL, Francke U, et al. The neu gene: an erbB-homologous gene distinct from and unlinked to the gene encoding the EGF receptor. Science. 1985;229:976–978. doi: 10.1126/science.2992090. [DOI] [PubMed] [Google Scholar]
- Schechter AL, Stern DF, Vaidyanathan L, Decker SJ, Drebin JA, Greene MI, et al. The neu oncogene: an erb-B-related gene encoding a 185,000-Mr tumour antigen. Nature. 1984;312:513–516. doi: 10.1038/312513a0. [DOI] [PubMed] [Google Scholar]
- Schiffer IB, Gebhard S, Heimerdinger CK, Heling A, Hast J, Wollscheid U, et al. Switching off HER-2/neu in a tetracycline-controlled mouse tumor model leads to apoptosis and tumor-size-dependent remission. Cancer Res. 2003;63:7221–7231. [PubMed] [Google Scholar]
- Schulze WX, Deng L, Mann M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Molecular Systems Biology. 2005 doi: 10.1038/msb4100012. doi: 10.1038/msb4100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segatto O, King CR, Pierce JH, Di Fiore PP, Aaronson SA. Different structural alterations upregulate in vitro tyrosine kinase activity and transforming potency of the erbB-2 gene. Mol Cell Biol. 1988;8:5570–5574. doi: 10.1128/mcb.8.12.5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seton-Rogers SE, Lu Y, Hines LM, Koundinya M, LaBaer J, Muthuswamy SK, et al. Cooperation of the ErbB2 receptor and transforming growth factor beta in induction of migration and invasion in mammary epithelial cells. Proc Natl Acad Sci U S A. 2004;101:1257–1262. doi: 10.1073/pnas.0308090100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield LG. C-Src activation by ErbB2 leads to attachment-independent growth of human breast epithelial cells. Biochem Biophys Res Commun. 1998;250:27–31. doi: 10.1006/bbrc.1998.9214. [DOI] [PubMed] [Google Scholar]
- Shelly M, Mosesson Y, Citri A, Lavi S, Zwang Y, Melamed-Book N, et al. Polar expression of ErbB-2/HER2 in epithelia. Bimodal regulation by Lin-7. Dev Cell. 2003;5:475–486. doi: 10.1016/j.devcel.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Shepherd TG, Kockeritz L, Szrajber MR, Muller WJ, Hassell JA. The pea3 subfamily ets genes are required for HER2/Neu-mediated mammary oncogenesis. Curr Biol. 2001;11:1739–1748. doi: 10.1016/s0960-9822(01)00536-x. [DOI] [PubMed] [Google Scholar]
- Shibahara H, Tamada S, Higashi M, Goto M, Batra SK, Hollingsworth MA, et al. MUC4 is a novel prognostic factor of intrahepatic cholangiocarcinoma-mass forming type. Hepatology. 2004;39:220–229. doi: 10.1002/hep.20031. [DOI] [PubMed] [Google Scholar]
- Shigematsu H, Takahashi T, Nomura M, Majmudar K, Suzuki M, Lee H, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005;65:1642–1646. doi: 10.1158/0008-5472.CAN-04-4235. [DOI] [PubMed] [Google Scholar]
- Shih C, Padhy LC, Murray M, Weinberg RA. Transforming genes of carcinomas and neuroblastomas introduced into mouse fibroblasts. Nature. 1981;290:261–264. doi: 10.1038/290261a0. [DOI] [PubMed] [Google Scholar]
- Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8:1145–1152. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- Sibilia M, Steinbach JP, Stingl L, Aguzzi A, Wagner EF. A strain-independent postnatal neurodegeneration in mice lacking the EGF receptor. EMBO J. 1998;17:719–731. doi: 10.1093/emboj/17.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibilia M, Wagner EF. Strain-dependent epithelial defects in mice lacking the EGF receptor [published erratum appears in Science 1995 Aug 18;269(5226):909] Science. 1995;269:234–238. doi: 10.1126/science.7618085. [DOI] [PubMed] [Google Scholar]
- Siegel PM, Dankort DL, Hardy WR, Muller WJ. Novel activating mutations in the neu proto-oncogene involved in induction of mammary tumors. Mol Cell Biol. 1994;14:7068–7077. doi: 10.1128/mcb.14.11.7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel PM, Muller WJ. Mutations affecting conserved cysteine residues within the extracellular domain of Neu promote receptor dimerization and activation. Proc Natl Acad Sci U S A. 1996;93:8878–8883. doi: 10.1073/pnas.93.17.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel PM, Ryan ED, Cardiff RD, Muller WJ. Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. EMBO J. 1999;18:2149–2164. doi: 10.1093/emboj/18.8.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierke SL, Cheng K, Kim HH, Koland JG. Biochemical characterization of the protein tyrosine kinase homology domain of the ErbB3 (HER3) receptor protein. Biochemical Journal. 1997;322:757–763. doi: 10.1042/bj3220757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- Sliwkowski MX. Ready to partner. Nat Struct Biol. 2003;10:158–159. doi: 10.1038/nsb0303-158. [DOI] [PubMed] [Google Scholar]
- Sliwkowski MX, Schaefer G, Akita RW, Lofgren JA, Fitzpatrick VD, Nuijens A, et al. Coexpression of erbB2 and erbB3 proteins reconstitutes a high affinity receptor for heregulin. J Biol Chem. 1994;269:14661–14665. [PubMed] [Google Scholar]
- Soltoff SP, Carraway KL, III, Prigent SA, Gullick WG, Cantley LC. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol Cell Biol. 1994;14:3550–3558. doi: 10.1128/mcb.14.6.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg C, Lausch EU, Trost TM, Prawitt D, May A, Keppler R, et al. ERBB2-mediated transcriptional up-regulation of the alpha5beta1 integrin fibronectin receptor promotes tumor cell survival under adverse conditions. Cancer Res. 2006;66:3715–3725. doi: 10.1158/0008-5472.CAN-05-2823. [DOI] [PubMed] [Google Scholar]
- Spataro VJ, Litman H, Viale G, Maffini F, Masullo M, Golouh R, et al. Decreased immunoreactivity for p27 protein in patients with early-stage breast carcinoma is correlated with HER-2/neu overexpression and with benefit from one course of perioperative chemotherapy in patients with negative lymph node status: results from International Breast Cancer Study Group Trial V. Cancer. 2003;97:1591–1600. doi: 10.1002/cncr.11224. [DOI] [PubMed] [Google Scholar]
- Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431:525–526. doi: 10.1038/431525b. [DOI] [PubMed] [Google Scholar]
- Subbaramaiah K, Norton L, Gerald W, Dannenberg AJ. Cyclooxygenase-2 is overexpressed in HER-2/neu-positive breast cancer: evidence for involvement of AP-1 and PEA3. J Biol Chem. 2002;277:18649–18657. doi: 10.1074/jbc.M111415200. [DOI] [PubMed] [Google Scholar]
- Swede H, Moysich KB, Freudenheim JL, Quirk JT, Muti PC, Hurd TC, et al. Breast cancer risk factors and HER2 over-expression in tumors. Cancer Detect Prev. 2001;25:511–519. [PubMed] [Google Scholar]
- Tamada S, Shibahara H, Higashi M, Goto M, Batra SK, Imai K, et al. MUC4 is a novel prognostic factor of extrahepatic bile duct carcinoma. Clin Cancer Res. 2006;12:4257–4264. doi: 10.1158/1078-0432.CCR-05-2814. [DOI] [PubMed] [Google Scholar]
- Tan M, Li P, Klos KS, Lu J, Lan KH, Nagata Y, et al. ErbB2 promotes Src synthesis and stability: novel mechanisms of Src activation that confer breast cancer metastasis. Cancer Res. 2005a;65:1858–1867. doi: 10.1158/0008-5472.CAN-04-2353. [DOI] [PubMed] [Google Scholar]
- Tan M, Li P, Klos KS, Lu J, Lan KH, Nagata Y, et al. ErbB2 promotes Src synthesis and stability: novel mechanisms of Src activation that confer breast cancer metastasis. Cancer Research. 2005b;65:1858–1867. doi: 10.1158/0008-5472.CAN-04-2353. [DOI] [PubMed] [Google Scholar]
- Tanner MM, Tirkkonen M, Kallioniemi A, Isola J, Kuukasjarvi T, Collins C, et al. Independent amplification and frequent co-amplification of three nonsyntenic regions on the long arm of chromosome 20 in human breast cancer. Cancer Res. 1996;56:3441–3445. [PubMed] [Google Scholar]
- Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci USA. 2001;98:10983–10985. doi: 10.1073/pnas.211430998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, et al. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science. 1995;269:230–234. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- Timms JF, White SL, O’Hare MJ, Waterfield MD. Effects of ErbB-2 overexpression on mitogenic signalling and cell cycle progression in human breast luminal epithelial cells. Oncogene. 2002;21:6573–6586. doi: 10.1038/sj.onc.1205847. [DOI] [PubMed] [Google Scholar]
- tires-Alj M, Neel BG. Protein-Tyrosine Phosphatase 1B Is Required for HER2/Neu-Induced Breast Cancer. Cancer Research. 2007;67:2420–2424. doi: 10.1158/0008-5472.CAN-06-4610. [DOI] [PubMed] [Google Scholar]
- Tokunaga E, Kimura Y, Oki E, Ueda N, Futatsugi M, Mashino K, et al. Akt is frequently activated in HER2/neu-positive breast cancers and associated with poor prognosis among hormone-treated patients. Int J Cancer. 2006;118:284–289. doi: 10.1002/ijc.21358. [DOI] [PubMed] [Google Scholar]
- Tsuda H, Akiyama F, Terasaki H, Hasegawa T, Kurosumi M, Shimadzu M, et al. Detection of HER-2/neu (c-erb B-2) DNA amplification in primary breast carcinoma. Interobserver reproducibility and correlation with immunohistochemical HER-2 overexpression. Cancer. 2001;92:2965–2974. doi: 10.1002/1097-0142(20011215)92:12<2965::aid-cncr10156>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Tsutsumida H, Goto M, Kitajima S, Kubota I, Hirotsu Y, Wakimoto J, et al. MUC4 expression correlates with poor prognosis in small-sized lung adenocarcinoma. Lung Cancer. 2006;55:195–203. doi: 10.1016/j.lungcan.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Tzahar E, Waterman H, Chen X, Levkowitz G, Karunagaran D, Lavi S, et al. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol Cell Biol. 1996;16:5276–5287. doi: 10.1128/mcb.16.10.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadlamudi R, Mandal M, Adam L, Steinbach G, Mendelsohn J, Kumar R. Regulation of cyclooxygenase-2 pathway by HER2 receptor. Oncogene. 1999;18:305–314. doi: 10.1038/sj.onc.1202307. [DOI] [PubMed] [Google Scholar]
- Vadlamudi RK, Sahin AA, Adam L, Wang RA, Kumar R. Heregulin and HER2 signaling selectively activates c-Src phosphorylation at tyrosine 215. FEBS Lett. 2003;543:76–80. doi: 10.1016/s0014-5793(03)00404-6. [DOI] [PubMed] [Google Scholar]
- Vartanian T, Goodearl A, Lefebvre S, Park SK, Fischbach G. Neuregulin induces the rapid association of focal adhesion kinase with the erbB2-erbB3 receptor complex in schwann cells. Biochem Biophys Res Commun. 2000;271:414–417. doi: 10.1006/bbrc.2000.2624. [DOI] [PubMed] [Google Scholar]
- Venter DJ, Tuzi NL, Kumar S, Gullick WJ. Overexpression of the c-erbB-2 oncoprotein in human breast carcinomas: immunohistological assessment correlates with gene amplification. Lancet. 1987;2:69–72. doi: 10.1016/s0140-6736(87)92736-x. [DOI] [PubMed] [Google Scholar]
- Viglietto G, Motti ML, Bruni P, Melillo RM, D’Alessio A, Califano D, et al. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med. 2002;8:1136–1144. doi: 10.1038/nm762. [DOI] [PubMed] [Google Scholar]
- Vijapurkar U, Kim MS, Koland JG. Roles of mitogen-activated protein kinase and phosphoinositide 3′-kinase in ErbB2/ErbB3 coreceptor-mediated heregulin signaling. Exp Cell Res. 2003;284:291–302. doi: 10.1016/s0014-4827(02)00040-x. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. [Review] [156 refs] Nature Reviews, Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Wallasch C, Weiss FU, Niederfellner G, Jallal B, Issing W, Ullrich A. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J. 1995b;14:4267–4275. doi: 10.1002/j.1460-2075.1995.tb00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallasch C, Weiss FU, Niederfellner G, Jallal B, Issing W, Ullrich A. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J. 1995a;14:4267–4275. doi: 10.1002/j.1460-2075.1995.tb00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Kennan WS, Yasukawa-Barnes J, Lindstrom MJ, Gould MN. Frequent induction of mammary carcinomas following neu oncogene transfer into in situ mammary epithelial cells of susceptible and resistant rat strains. Cancer Res. 1991;51:5649–5654. [PubMed] [Google Scholar]
- Wang SC, Lien HC, Xia W, Chen IF, Lo HW, Wang Z, et al. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell. 2004;6:251–261. doi: 10.1016/j.ccr.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Wang SE, Narasanna A, Perez-Torres M, Xiang B, Wu FY, Yang S, et al. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell. 2006;10:25–38. doi: 10.1016/j.ccr.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhang L, Yeung TK, Chen X. Endocytosis deficiency of epidermal growth factor (EGF) receptor-ErbB2 heterodimers in response to EGF stimulation. Mol Biol Cell. 1999;10:1621–1636. doi: 10.1091/mbc.10.5.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman H, Sabanai I, Geiger B, Yarden Y. Alternative intracellular routing of ErbB receptors may determine signaling potency. J Biol Chem. 1998;273:13819–13827. doi: 10.1074/jbc.273.22.13819. [DOI] [PubMed] [Google Scholar]
- Weigelt B, Hu Z, He X, Livasy C, Carey LA, Ewend MG, et al. Molecular portraits and 70-gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer Res. 2005;65:9155–9158. doi: 10.1158/0008-5472.CAN-05-2553. [DOI] [PubMed] [Google Scholar]
- Weiner DB, Kokai Y, Wada T, Cohen JA, Williams WV, Greene MI. Linkage of tyrosine kinase activity with transforming ability of the p185neu oncoprotein. Oncogene. 1989a;4:1175–1183. [PubMed] [Google Scholar]
- Weiner DB, Liu J, Cohen JA, Williams WV, Greene MI. A point mutation in the neu oncogene mimics ligand induction of receptor aggregation. Nature. 1989b;339:230–231. doi: 10.1038/339230a0. [DOI] [PubMed] [Google Scholar]
- Weinstein EJ, Kitsberg DI, Leder P. A mouse model for breast cancer induced by amplification and overexpression of the neu promoter and transgene. Mol Med. 2000;6:4–16. [PMC free article] [PubMed] [Google Scholar]
- Weinstein IB. CANCER: Enhanced: Addiction to Oncogenes--the Achilles Heal of Cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- Wilson GR, Cramer A, Welman A, Knox F, Swindell R, Kawakatsu H, et al. Activated c-SRC in ductal carcinoma in situ correlates with high tumour grade, high proliferation and HER2 positivity. Br J Cancer. 2006;95:1410–1414. doi: 10.1038/sj.bjc.6603444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods Ignatoski KM, Grewal NK, Markwart S, Livant DL, Ethier SP. p38MAPK induces cell surface alpha4 integrin downregulation to facilitate erbB-2-mediated invasion. Neoplasia. 2003;5:128–134. doi: 10.1016/s1476-5586(03)80004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie D, Shu XO, Deng Z, Wen WQ, Creek KE, Dai Q, et al. Population-based, case-control study of HER2 genetic polymorphism and breast cancer risk. J Natl Cancer Inst. 2000;92:412–417. doi: 10.1093/jnci/92.5.412. [DOI] [PubMed] [Google Scholar]
- Xie W, Chow LT, Paterson AJ, Chin E, Kudlow JE. Conditional expression of the ErbB2 oncogene elicits reversible hyperplasia in stratified epithelia and up-regulation of TGFalpha expression in transgenic mice. Oncogene. 1999;18:3593–3607. doi: 10.1038/sj.onc.1202673. [DOI] [PubMed] [Google Scholar]
- Xie W, Paterson AJ, Chin E, Nabell LM, Kudlow JE. Targeted expression of a dominant negative epidermal growth factor receptor in the mammary gland of transgenic mice inhibits pubertal mammary duct development. Mol Endocrinol. 1997;11:1766–1781. doi: 10.1210/mend.11.12.0019. [DOI] [PubMed] [Google Scholar]
- Xie W, Wu X, Chow LT, Chin E, Paterson AJ, Kudlow JE. Targeted expression of activated erbB-2 to the epidermis of transgenic mice elicits striking developmental abnormalities in the epidermis and hair follicles. Cell Growth Differ. 1998;9:313–325. [PubMed] [Google Scholar]
- Xie Y, Hung MC. Nuclear localization of p185neu tyrosine kinase and its association with transcriptional transactivation. Biochemical & Biophysical Research Communications. 1994;203:1589–1598. doi: 10.1006/bbrc.1994.2368. [DOI] [PubMed] [Google Scholar]
- Xu W, Yuan X, Beebe K, Xiang Z, Neckers L. Loss of Hsp90 association up-regulates Src-dependent ErbB2 activity. Mol Cell Biol. 2007;27:220–228. doi: 10.1128/MCB.00899-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Ionescu-Tiba V, Burns K, Gadd M, Zukerberg L, Louis DN, et al. The role of the cyclin D1-dependent kinases in ErbB2-mediated breast cancer. Am J Pathol. 2004;164:1031–1038. doi: 10.1016/S0002-9440(10)63190-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HY, Shao R, Hung MC, Lee MH. p27 Kip1 inhibits HER2/neu-mediated cell growth and tumorigenesis. Oncogene. 2001;20:3695–3702. doi: 10.1038/sj.onc.1204472. [DOI] [PubMed] [Google Scholar]
- Yang HY, Zhou BP, Hung MC, Lee MH. Oncogenic signals of HER-2/neu in regulating the stability of the cyclin-dependent kinase inhibitor p27. J Biol Chem. 2000;275:24735–24739. doi: 10.1074/jbc.C000147200. [DOI] [PubMed] [Google Scholar]
- Yano T, Doi T, Ohtsu A, Boku N, Hashizume K, Nakanishi M, et al. Comparison of HER2 gene amplification assessed by fluorescence in situ hybridization and HER2 protein expression assessed by immunohistochemistry in gastric cancer. Oncology Reports. 2006;15:65–71. [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–1021. doi: 10.1038/35082500. [DOI] [PubMed] [Google Scholar]
- Zhan L, Xiang B, Muthuswamy SK. Controlled activation of ErbB1/ErbB2 heterodimers promote invasion of three-dimensional organized epithelia in an ErbB1-dependent manner: implications for progression of ErbB2-overexpressing tumors. Cancer Res. 2006;66:5201–5208. doi: 10.1158/0008-5472.CAN-05-4081. [DOI] [PubMed] [Google Scholar]
- Zhou X, Tan M, Stone H,V, Klos KS, Lan KH, Yang Y, et al. Activation of the Akt/mammalian target of rapamycin/4E-BP1 pathway by ErbB2 overexpression predicts tumor progression in breast cancers. Clin Cancer Res. 2004;10:6779–6788. doi: 10.1158/1078-0432.CCR-04-0112. [DOI] [PubMed] [Google Scholar]