c-myc Repression of TSC2 Contributes to Control of Translation Initiation and Myc-Induced Transformation (original) (raw)
. Author manuscript; available in PMC: 2011 Jan 18.
Abstract
The c-myc oncogene plays a key role in cellular growth control, and translation initiation factors are among the transcriptional targets of Myc. Here, we describe a defect in translation initiation control in myc-null cells due to alterations in the mammalian target of rapamycin (mTOR) pathway. Myc loss increased sensitivity to dominant inhibition of eukaryotic translation initiation factor 4E function. Polysomal profiles of myc−/− cells revealed decreased translation initiation rates, which were accompanied by decreased 40S/60S ribosomal subunit ratios. Because the 40S small ribosomal subunit contains the key regulatory ribosomal protein S6 (rpS6), we considered that myc loss might affect expression of components of the mTOR signaling pathway that regulate rpS6 function. Among mTOR signaling components, Myc directly affected transcription of tuberous sclerosis 2 (TSC2), as shown by quantitative mRNA analysis and by Myc binding to its promoter in chromatin immunoprecipitation assays. Importantly, Myc acted as a strong and direct repressor for TSC2 expression because its loss increased TSC2 mRNA in myc-null and in HL60 shRNA experiments, activation of a mycER construct in myc−/− cells suppressed TSC2 induction in a myc box II–dependent manner, and mycER activation recruited Myc to the TSC2 promoter. The biological significance of the effect of Myc on TSC2 expression was shown by markedly reduced TSC2 mRNA levels in myc-transformed cells, stimulation of S6 kinase activity in myc-null cells by TSC2 siRNA, and decreased Myc-induced soft agar colony formation following retroviral transduction of TSC2. Together, these findings show that regulation of TSC2 can contribute to the effects of Myc on cell proliferation and neoplastic growth.
Introduction
DNA synthesis is initiated and a cell divides only after it grows beyond a minimum size threshold (1). Dysregulation of the growth apparatus is therefore a key step in the development of malignancy (2). Growth and cell division are coordinated by the mRNA translation apparatus through translation initiation control, and the translation initiation step is a key rate-limiting step in cell proliferation (3). Initiation requires assembly of the eukaryotic initiation factor eukaryotic translation initiation factor 4E (eIF4E) and the mRNA cap binding apparatus at the 7-methyl-guanosine cap structure. The target of rapamycin (TOR) signaling pathway regulates translation initiation through S6 kinase (S6K) phosphorylation of ribosomal protein S6 (rpS6) and through eIF4E activation (4). We previously showed transcriptional control of eIF4E expression by c-myc (5). In this report, we further evaluate the effects of Myc loss on translation initiation control via the mammalian TOR (mTOR) pathway.
Myc is an immediate-early gene involved in the growth response to mitogenic stimulation that is crucial for both G0–G1 and G1-S cell cycle transitions (6); Myc is frequently overexpressed in human cancers (7). Known targets of growth regulation by myc include RNA polymerases I and III, ribosomal proteins, and eIF4E (8). Somatic knockout Myc-null cells suffer from a growth defect (9), although the contribution of translation initiation control to this defect has not been directly evaluated.
A broad range of signaling pathways regulate translation initiation (10). Of these pathways, TOR signaling has received increasing attention because rapamycin is an emerging cancer therapeutic (11). Rapamycin is an immunosuppressant with potent antiproliferative and antineoplastic effects, which inhibits mTOR/raptor/mLST8 (mTOR complex 1) activation (12). Rapamycin blocks cell growth and proliferation through inhibition of mTOR-mediated rpS6 phosphorylation (13). Its potential therapeutic use has been shown in myc-induced cancers in which eIF4E expression levels were critical to the effects of rapamycin (14). Tuberous sclerosis genes TSC1 and TSC2 are key components of the TOR signaling pathway (15). The TSC2 gene product (tuberin) is a GTPase-activating protein, which functions with TSC1 (hamartin) to negatively regulate cell growth via mTOR (16). Loss of TSC function specifically activates mTOR complex activation (15). mTOR activity then regulates the S6K that phosphorylates ribosomal protein S6 (rpS6; ref. 17).
Both the Drosophila myc (dMyc) and tuberous sclerosis genes control cell size (18). Interestingly, dMyc overexpression blocks Drosophila eye size reduction resulting from dTSC1/2 overexpression. In mammalian cells, c-Myc also restores normal proliferation rates to cells overexpressing tuberin (19). A relevant connection between Myc and TSC2 is further supported by evidence of TSC2 down-regulation in Burkitt’s lymphoma cells (20). Whereas these data suggest functional antagonism between c-Myc and TSC2, they identify no direct molecular regulatory connection between the two gene products. Interestingly, TSC2 also regulates the G1-S transition, and antisense inhibition blocks entry into a quiescent (G0) state (21). Thus, both c-Myc and TSC2 have emerged as key nodal points in the regulation of translation, protein synthesis, and growth control, and both have significant roles in differentiation and carcinogenesis.
In this report, we investigated the growth defect in myc-null cells, starting with the hypothesis that these cells might be more sensitive to blockade of the mTOR pathway than their wild-type counterparts. Because myc-null cells proved to be sensitive to a dominant inhibitor of translation initiation, we analyzed translation initiation control using polysomal analysis. We find that c-myc directly regulates the TSC2 gene and that loss of negative TSC2 regulation by c-myc contributes to defective rpS6 activation and translation initiation in myc-null cells.
Materials and Methods
Plasmids and retroviral production
Packaged retroviruses expressing HA-TSC2, c-myc, or empty pBABE-Puro controls were made with the EcoPac retroviral packaging vector; 293T cells were cotransfected with pBABE-HA-TSC2 (gift of Dr. James Brugarolas), pBABE-c-myc (gift of Dr. William Hahn), or empty pBABE vector using standard calcium phosphate techniques. Our vectors containing no insert (Vec), eIF4E (eIF4E), wild-type 4E binding protein (4EBP), or a 4EBP mutant whose serine phosphorylation targets of the mTOR pathway were all mutated to alanines (4EBPμ) were described previously (22). Mission retroviral constructs expressing shRNAs for c-myc and a scrambled control sequence in the plasmid pLKO were purchased from Sigma Aldrich (NM_002467) and retroviruses were generated according to the accompanying instructions.
Cell lines and transfections
Wild-type cells (myc+/+; TGR), myc-null cells (myc−/−; HO15), myc-null cells expressing mycERtmx (myc−/− mycERtmx), and myc-null cells expressing Δ106-143mycERtmx (myc−/− ΔMBII-mycERtmx) cells were obtained from Dr. John Sedivy (23). Rat1A cells that can be directly transformed by Myc were previously described (22).
Myc+/+ and myc−/− cells were transfected using Lipofectin reagents (Life Technologies, Inc.) with control vector (Vec), eIF4E, wild-type 4EBP, or a 4EBP mutant (4EBPμ), and pooled transfectants were selected with G418. Max transfections were accomplished by transfecting pREP4 or pREP4-max together with pCEP4 or pCEP-eIF4E. Colonies selected in the presence of 500 μg/mL Geneticin and 200 μg/mL hygromycin were stained and photographed to assess the combined effects of constructs on cell survival. Individual colonies were initially isolated with trypsin-impregnated cloning filters. Because all colonies expressed the transfected proteins due to the double selection, pooled transfectants were used. All cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS).
The myc−/− (HO15) cells were infected with either pBABE-Puro or pBABE-Myc to evaluate the myc specificity of the myc−/− cell translation initiation defect. To evaluate potential interactions between TSC2 and myc in transformation, we also infected Rat1A cells with the following combinations of virus vectors: pBABE-TSC2/pBABE-Myc, pBABE-Myc/pBABE, pBABE-TSC2/pBABE, or pBABE-Puro/pBABE-Puro. In both sets of infections, subconfluent cells were infected sequentially for 2 days with polybrene (4 μg/mL) and 50% viral supernatants of pBABE, pBABE-Myc, pBABE-TSC2, or their combinations, followed by puromycin selection (8 μg/mL). Additional Rat1A Myc and max transfectants were previously reported (24).
HL60 cells were obtained from Dr. David Sweetser (MGH Cancer Center, Boston, MA) and grown in RPMI medium supplemented with 10% FBS. They were induced to differentiate with 20 nmol/L phorbol 12-myristate 13-acetate (PMA).
Polysomal profile analysis
One 100-mm-diameter plate containing the indicated cell types was harvested for each polysomal analysis. Confluent cells were harvested and lysed in 300 μL of RSB [10 mmol/L NaCl, 10 mmol/L Tris-HCl (pH 7.4), 15 mmol/L MgCl2] containing 100 μg/mL heparin, 1.2% Triton X-100, and 0.12% deoxycholate (25). Nuclei were pelleted for 3 min in a microcentrifuge at 4 °C. The 300-μL extract was layered over 11.5 mL of a 15% to 45% (wt/wt) sucrose gradient with a 0.5-mL cushion of 45% sucrose. The gradients were centrifuged at 37,000 rpm for 2.5 h in an SW 41 (Beckman) rotor at 4 °C. After centrifugation, the A 260 was continuously monitored and recorded across the gradient using a Foxy Jr. Density Gradient fractionation system and UA-6 UV/VIS Detector (ISCO, Inc.).
Protein and mRNA expression analysis
For Western blot analyses, whole-cell extracts of cells were prepared with NP40 lysis buffer [50 mmol/L Tris (pH 7.5), 150 mmol/L NaCl, 50 mmol/L NaF, 1 mmol/L EDTA, 0.5% NP40, 50 mmol/L β-glycerophoshate, 1 mmol/L sodium orthovanadate, 1.25 μg/mL pepstatin] supplemented with 1 tablet of complete mini-protease inhibitor cocktail per 10 mL of buffer. Eighty micrograms of protein sample were subjected to SDS-PAGE in 7% to 12% gels depending on the molecular weight of the protein studied. After blotting, nitrocellulose membranes were hybridized with the indicated antibodies and detected by enhanced chemiluminescence (Amersham).
Steady-state mRNA levels were determined by quantitative real-time PCR with primers that spanned introns to avoid possible detection of contaminating genomic sequences. For serum induction experiments, mRNA was harvested 9 h after serum stimulation. Equal amounts (0.5–1.0 μg) of cellular RNA were subjected to reverse transcriptase reactions using the Superscript III First-Strand Synthesis system for real-time PCR and oligo-dT primers (Invitrogen, Inc.). Standardized quantities of gene-specific PCR products were generated from cDNAs derived from total rat cellular mRNA using Platinum Taq DNA polymerase (Invitrogen) and the same gene-specific primers as used in the quantitative real-time PCR. Molecular sizes of PCR products were verified. Equivalent amounts of the sample cDNAs or standardized quantities of the gene-specific PCR products were added to reactions containing iQ SYBR Green Supermix (Bio-Rad) and the respective gene-specific primers. The amount of starting material in each reaction was quantified in a Bio-Rad MYiQ Single Color Real-time PCR Detection System. Quantitative β-actin PCR reactions were done on additional aliquots as an internal standard. All mRNA quantifications were expressed as mRNA quantities normalized to β-actin levels. Primer sets are listed in Supplementary Table S1. Standard Northern blots were used to show decreased expression of c-myc in differentiated HL60 cells.
Promoter sequence analysis
Reference cDNA sequences for human, rat, and mouse mTOR pathway gene products were identified using the National Center for Biotechnology Information (NCBI) sequence database.3 Genomic promoter sequences and transcription initiation sites were identified using the Database of Human Transcription Start sites (26), the Advanced Biomedical Computing Center promoter analysis software,4 and the 5′ end of the reference sequences. Five thousand nucleotides of sequence upstream and downstream of the transcription initiation sites were downloaded and manipulated using Clone Manager Suite (Scientific and Educational Software). Myc target sites that were conserved between the three species were identified in the aligned regions using rVISTA (27). The conserved Myc sites are listed in Supplementary Table S2, together with their position in the rat promoter relative to the transcription start site.
Chromatin immunoprecipitation assays
We evaluated Myc promoter binding using the wild-type and myc-null cells, together with myc−/− mycERtmx cells, during logarithmic growth before and after 3-h induction with 200 nmol/L 4-hydroxy-tamoxifen. Our chromatin immunoprecipitation approach was previously described (24) and is further described in Supplementary methods. Chromatin immunoprecipitation assay promoter primers were chosen to flank candidate CACGTG sites identified in the IRS1, TSC1, TSC2, GβL, and rpS6 promoters (Supplementary Table S1). eIF4E promoter binding was compared as a positive control (24). S6K promoter primers were chosen to flank its transcription initiation site because its promoter lacked myc target sequences.
TSC2 and c-myc siRNA experiments
For TSC2 siRNA experiments, proliferating myc−/− cells were transfected with 10 or 50 nmol/L small interfering RNA (siRNA) for rat TSC2 (siGENOME SMARTpool, Dharmacon). These transfectants were then compared with cells transfected with a nontargeting firefly luciferase siRNA duplex (Dharmacon; nontargeting siRNA #2 sense, 5′-UAAGGCUAUGAAGAGAUACUU-3′; nontargeting siRNA #2 antisense, 5′-PGUAUCUCUUCAUAGCCUUAUU-3′). Transfections were accomplished using Oligofectamine (Invitrogen) according to the manufacturer’s instructions. Whole-cell extracts of transfected myc−/− cells were prepared after 72 h and analyzed for tuberin, phospho-S6, and phospho-S6K.
For the c-myc shRNA experiment in HL60 cells, c-myc shRNAs and control retroviruses were used to transfect HL60 cells according to the infection protocol described above. They were grown in RPMI in the presence of 8 μg/mL puromycin to select for successful transfectants of the shRNAs. After 10 days of selection, 3 × 104 cells were spun, lysed in SDS-PAGE gel loading buffer, and analyzed in an immunoblot for tuberin, Myc, and actin proteins.
Soft agar transformation assays
Rat1A soft agar transformation was assessed as described previously (22). After expansion, trypsinized pBABE-TSC2/pBABE-Myc, pBABE-Myc/pBABE, pBABE-TSC2/pBABE, or pBABE-Puro Rat1A cells were seeded in soft agar at 20,000 per well in six-well plates. Colony numbers were scored after 14 days.
Results
Myc-null cells are defective in translation initiation
To better understand the molecular basis for growth regulation by c-myc, we evaluated translation initiation capacity in myc−/− cells (HO15) that were created by somatic inactivation in a diploid rat cell line (TGR; ref. 9). We evaluated the contribution of translation initiation control to cell survival using a 4EBP mutant lacking four TOR-regulated target sites. We previously developed a constitutively active form of the 4EBP by mutating its four serine phosphorylation target sites to alanines (4EBPμ; ref. 22) to render it resistant to inactivation by mTOR. We transfected wild-type and myc-null cells with 4EBPμ and compared its effects with those of a control vector, eIF4E, and a nonmutated 4EBP in clonal survival assays (Fig. 1_A_). No myc-null colonies appeared at any time after transfection with the 4EBPμ. Although myc wild-type colony formation seemed to be somewhat affected by 4EBPμ, proliferation rates in the resulting pooled 4EBP and 4EBPμ transfectants are actually somewhat decreased as previously reported (22). In addition, myc-null colonies readily appeared in transfections with the control, eIF4E, and 4EBP expression vectors, indicating that the 4EBPμ effect was specific for both its activating mutation and the myc-null phenotype.
Figure 1.

Myc-null cells are defective in translation initiation. A, myc−/− cells do not tolerate expression of an activated 4EBP mutant that constitutively blocks translation initiation. Myc+/+ and myc−/− cells were transfected with the indicated constructs expressing a control vector (Vec), eIF4E (eIF4E), wild-type 4EBP (4EBP), or a 4EBP mutant of which the serine phosphorylation targets of the mTOR pathway were all mutated to alanines (4EBPμ). All vectors expressed a colinear neomycin selection marker. Plates were each seeded with 10,000 cells and maintained in G418 selection. Shown are the crystal violet–stained dishes after 2 wk of colony growth. B, eIF4E enhanced colony formation when cotransfected with max. Rat1A cells were transfected with a control plasmid (Control) or a plasmid expressing max (Max) in the absence or presence of a vector expressing eIF4E (control or eIF4E). Transfections were done as in A except that dual hygromycin and G418 selection were applied. After 3 wk, colonies were stained with methylene blue. Western blots were done to evaluate expression of the transfected plasmids in pooled transfectants containing dual control vectors, max alone, eIF4E alone, or the combination of eIF4E and max. Shown are standards evaluated using the indicated antibodies. C, polysomal profile analysis of c-myc-null versus wild-type cells. Cells were grown in 100-mm plates and arrested by confluence followed by serum starvation for 48 h. Cells were >90% viable with this treatment as assessed by trypan blue exclusion. Cytoplasmic RNA was harvested and the lysates were centrifuged in sucrose gradients at 37,000 rpm to establish polysomal gradients. The gradients were harvested and A 260 was continuously monitored to evaluate the distribution of monosomal versus polysomal fractions. Shown are the profiles for arrested myc wild-type (black) and null (gray) cells showing a decreased polysomal fraction in myc-null cells. The region containing polysomes is indicated and the 40S, 60S, and 80S peaks are labeled. D, c-myc-null cells exhibit a diminished 40S/60S ratio in the monosomal fractions. Total absorbance in the 40S and 60S fractions were calculated by summing the area under each peak. Left, ratios of the 40S/60S peak fractions were determined for four separate experiments for each cell type (columns, mean; bars, SD). Myc-null cells also have less RNA in the polysomal fractions. Right, columns, mean total absorbance in the polysomal fractions calculated for four separate experiments for each cell type; bars, SD.
Max-transfected cells proliferate slowly because excess max promotes homodimerization that blocks myc:max heterodimer formation (28). To determine whether translation initiation regulation could be downstream of a myc:max regulated pathway, we cotransfected translation initiation factor eIF4E with max to determine whether enhancement of translation initiation could overcome the inhibitory effects of max transfection on clonal survival (Fig. 1_B_). As expected, max inhibited colony formation in max-transfected Rat1A cells (Fig. 1_B_). In contrast, colony numbers and morphology were not altered when max and eIF4E were cotransfected. We tested pooled transfectants for expression of the transfected proteins and found that the max construct is profoundly inhibitory for Rat1A cells because no max protein could be detected in the rare colonies that survived max transfection (Fig. 1_B_, lane 3). Importantly, excess max could be expressed when cotransfected with eIF4E (lane 4). eIF4E potentially corrects a growth defect caused by max because cells expressing both eIF4E and max survive.
Translation initiation steps include (a) eIF4 complex binding to the 7-methyl-guanosine cap; (b) unwinding of the 5′ end of mRNA; (c) positioning of the methionyl-initiator tRNA; and (d) assembly of the 40S and 60S ribosomal subunits. Once the initiation complex is assembled, ribosomes process along the mRNA at a fixed rate exceeding the initiation rate. Because more ribosomes are found on actively translating mRNAs, the accumulation of ribosomes into polysomal fractions is the best measure of translation initiation rates. We therefore compared polysomal profiles of myc+/+ and myc−/− cells to evaluate the effect of myc loss on translation initiation (Fig. 1_C_). Two changes are apparent in myc−/− polysomal profiles during growth arrest. First, the 40S peak is smaller than the 60S peak in myc-null cells, and second, less RNA is contained in polysomal regions larger than the 80S monosomes in myc−/− cells (Fig. 1_C_). This difference between the 40S and 60S peaks was confirmed by averaging four repetitions of the profiles (Fig. 1_D_, left). The difference in polysomal RNA content was also confirmed by four repetitions (Fig. 1_D_, right).
Serum stimulation restores the 40S peak in myc-null cells to higher levels than the 60S peak, but the polysomal region remains depressed in serum-stimulated myc-null cells (Fig. 2_A_). To determine whether these effects were specific to the loss of c-myc in the null cells, we reintroduced c-myc into myc-null cells using the retroviral pBABE-myc construct. We compared polysomal profiles between empty vector–infected cells (pBABE) and those transduced with pBABE-myc (Fig. 2_B_ and C). In growth-arrested cells, the 40S/60S ratio remained decreased in the myc-null cells transduced with pBABE, whereas the 40S peak became larger than the 60S peak in the myc-transduced cells (Fig. 2_B_). Reintroduction of myc also led to a net increase in the entire polysomal region of the transduced cells. Serum stimulation again reversed the 40S/60S ratio in the vector-transduced myc-null cells after serum stimulation (Fig. 2_C_).
Figure 2.

Restoration of Myc to myc-null cells alleviates their defect in translation initiation. A, myc-null cells are defective in translation initiation during serum induction. Cells were treated as in A, except that they were stimulated with 10% serum for 9 h before harvesting. Plots in gray are myc−/− and those in black are myc+/+. B and C, reintroduction of myc using retroviral transduction shows that the myc-null cell translation initiation defect is myc specific. B, myc-null cells transduced with an empty vector control virus (pBABE) or pBABE-myc (pMyc) were arrested and polysomal analysis was done as described in Fig. 1_C_. Plots in gray are myc−/− transfected with pBABE-Puro (pBABE) and plots in black are from myc−/− transfected with pBABE-myc. C, polysomal profiles were evaluated as in B, except that they were stimulated with 10% serum for 9 h before harvesting. Plots in gray are myc−/− transfected with pBABE-Puro and plots in black are from myc−/− transfected with pBABE-myc.
Myc regulates mTOR pathway signaling components through myc box II–dependent repression of TSC2 and regulation of rpS6 phosphorylation
We evaluated expression levels of the proteins involved in the canonical mTOR signaling pathway to test the possibility that myc regulation of rpS6 function might contribute to its control of translation initiation. We focused on proteins whose expression was related to myc status both during growth arrest and after serum stimulation because the translation initiation defect was observed in both circumstances. Immunoblot analysis was done on extracts of arrested cells and after stimulation for 20 h with 10% FBS. Western blots for the major proteins in the canonical mTOR pathway identified potential myc inputs into the pathway (Fig. 3_A_). First, TSC2 and IRS1 protein levels were higher in myc-null cells than in wild-types. These differences were seen in both arrested and stimulated cells for both gene products. Second, Myc expression in wild-type cells supported higher levels of GβL, S6K, and rpS6, compared with their lower levels in null cells. These effects were amplified downstream in the pathway as shown by large increases in rpS6 phosphorylation in wild-type cells. Serum stimulation increased phosphorylation of S6K and rpS6 to higher levels in serum-stimulated wild-type cells compared with myc-null cells, suggesting that effects upstream of TOR were likely to be particularly significant. We also observed up-regulation of S6K and rpS6 during serum stimulation, an effect that was greatly diminished in myc-null cells. Because this degree of inducibility of rpS6 and S6K was unexpected, we repeated the evaluations of the differentially regulated proteins using a shorter time course of serum induction (Fig. 3_B_). Serum-induced increases in S6K and rpS6 were reproduced at each of the shorter time points, and the differential expression of the protein targets between myc wild-type and null cells was also reproduced throughout the earlier time points. Tuberin remained elevated in myc-null cells.
Figure 3.

TSC2 expression is increased and other mTOR cascade components are altered in myc−/− cells. A, myc wild-type (lanes 1 and 2) and null (lanes 3 and 4) cells were grown as described in Materials and Methods. Immunoblot analysis was done on extracts of arrested cells (A) and after stimulation for 20 h with 10% (S). Eighty micrograms of whole-cell protein extracts from each sample were separated by SDS-PAGE and transferred electrophoretically onto nitrocellulose membranes. The membranes were probed with the indicated antibodies. Antibodies to phosphorylated forms of the proteins are labeled as α-P. The indicated molecular weights were identified with protein molecular weight size markers in each gel. TSC2 and IRS1 protein levels were higher in myc-null cells than in wild types in arrested and stimulated cells. Myc expression in wild-type cells supported higher levels of GβL, S6K, and rpS6, compared with their lower levels in null cells in arrested and stimulated cells. Rheb levels were somewhat different in arrested cells but not in serum-stimulated cells, so its expression patterns were not further evaluated in subsequent experiments. Importantly, lack of Myc in myc-null cells led to elevated tuberin levels. B, total levels of GβL, S6K, and rpS6 are induced by serum treatment of myc wild-type and null cells. Because the inducibility of S6K and rpS6 was unanticipated, we evaluated their levels at shorter time intervals after serum induction. Shown are protein levels at 1, 2, and 8 h after addition of serum for the indicated proteins using the indicated antibodies. C, genomic analysis of promoter regions of the mTOR pathway components for Myc binding sites. Reference cDNA sequences for human, rat, and mouse mTOR pathway gene products were identified using the NCBI sequence database. Genomic promoter sequences and transcription initiation sites were then identified as described in Materials and Methods (26). Five thousand nucleotides of sequence upstream and downstream of their transcription initiation sites were evaluated for Myc target sites that were conserved between human, rat, and mouse sequences in aligned regions using rVISTA (27). Nonconserved myc sites are designated as e boxes in the schematic diagram where the open rectangle identifies exon 1 of each of the candidate genes. Myc sites conserved in human, rat, and mouse promoters (top to bottom in each diagram) are identified as E boxes. The sequences of the conserved E boxes are provided in Supplementary Table S2, together with their position in the rat promoter relative to the transcription start site. D, chromatin immunoprecipitation (ChIP) analysis shows binding of tamoxifen-induced c-myc to the TSC2, GβL, and rpS6 promoters in mycER cells. Chromatin immunoprecipitation analysis with primers specific for rat TSC2, GβL, rpS6, and eIF4E promoters and anti–c-Myc antibodies was done on extracts from myc−/− mycER cells induced to express c-myc after 3 h of treatment with 200 nmol/L tamoxifen. No Myc binding was detected in the absence of tamoxifen treatment (not shown). Primers for the rat 5S RNA promoter were used in parallel as an internal loading control for input DNA and the data were normalized to the 5S signals. The difference between tamoxifen induced and noninduced was significant by t test for TSC2 (P = 0.0043) and for rpS6 (P = 0.0005). The GβL differences were not significant.
We then evaluated genomic sequences of the TOR pathway genes and found conserved canonical E box motifs in the IRS1, TSC1, TSC2, GβL, and rpS6 promoters (Fig. 3_C_; Supplementary Table S2). Myc protein bound to the TSC2 and rpS6 E box-containing promoters in chromatin immunoprecipitation experiments after induction of an inducible mycER construct (Fig. 3_D_). Although IRS1 and the TSC1 promoters contained myc sites, chromatin immunoprecipitation did not detect Myc at their promoters (not shown). Although the GβL promoter exhibited only weak and nonsignificant binding after mycER induction, Myc was readily detected at the GβL promoter in myc wild-type cells compared with the null cells (data not shown). Importantly, the S6K promoter neither contained myc sites nor bound Myc. Differential changes in IRS1 and S6K proteins were probably therefore an indirect consequence of the absence or presence of myc.
Because Drosophila and human Myc antagonized the effects of tuberin (18, 19), and TSC2 was decreased in microarrays in Burkitt’s lymphoma cells (20), we focused on myc repression of TSC2 expression as the central contributor to the effects of myc on cell proliferation and transformation. We first used quantitative real-time PCR assays to assess mRNA levels of TSC2 mRNA in wild-type and myc-null cells in growth-arrested and growth-stimulated conditions (Fig. 4_A_). The transferrin receptor (RTR), a known Myc-responsive gene, increased 6.3-fold between null and wild-type cells in arrested conditions. In contrast, the presence of myc decreased TSC2 mRNA 4.8- and 11-fold, comparing null to wild-type cells.
Figure 4.
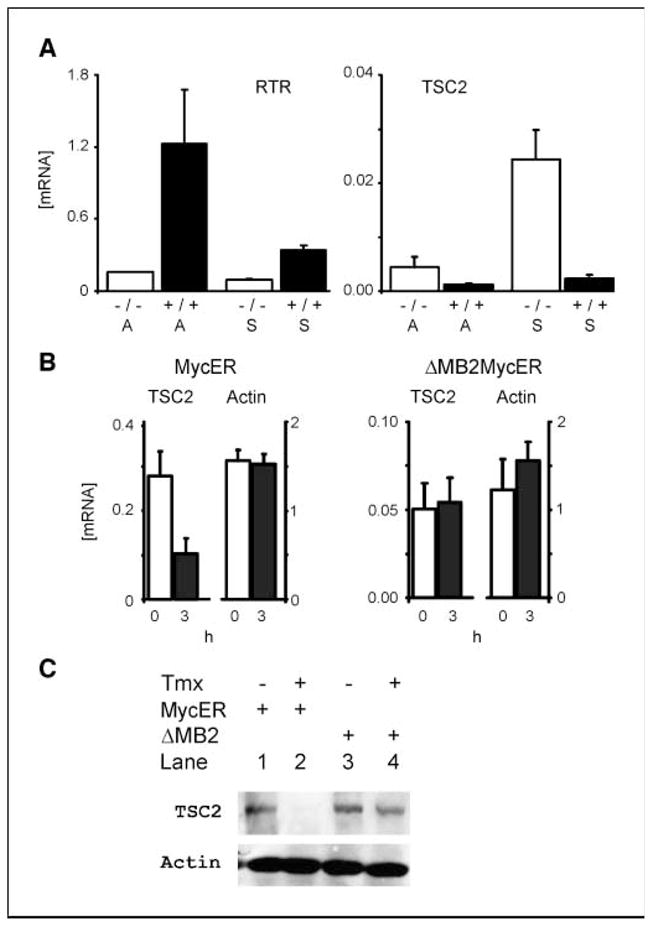
Myc directly represses TSC2 mRNA levels. A, steady-state levels of TSC2 mRNAs are increased in myc-null compared with wild-type cells. Cells were arrested by confluence followed by serum starvation for 48 h (A) or stimulated to reenter the cell cycle with 10% FBS (S). Treated cells were harvested for total RNA at 9 h. Total RNA (0.5 μg) was subjected to reverse transcriptase reactions using oligo-dT primers, and the resulting cDNAs were analyzed by quantitative real-time PCR (qrt-PCR) compared with standardized quantities of gene-specific PCR products generated with gene-specific primers. Equivalent amounts of cDNA from the same reverse transcription reactions were analyzed by quantitative PCR with standardized quantities of β-actin PCR product using primers specific for β-actin. The results are expressed as the absolute quantity of each mRNA species, normalized for their actin levels, which is then plotted along the y axis. The genotypes of the cells used are indicated as myc-null (−/−) or wild-type (+/+) along the x axis. The differences between wild-type and myc-null TSC2 levels were significant by t test in arrested (A; P = 0.038) and stimulated (S; P = 0.050) cells. B and C, Myc down-regulates TSC2 gene expression in a myc box II–dependent manner. B, induction of c-myc down-regulates TSC2 mRNA in myc−/− cells expressing a mycERtmx construct but not in ΔMBII-mycER cells. Total RNA was harvested from noninduced cells (0 h) or cells induced to express c-mycER or mutant mycER after 3-h treatment with 200 nmol/L tamoxifen (3 h), reverse transcribed, and analyzed by quantitative real-time PCR as in A. The difference between uninduced and induced TSC2 levels were significant in the mycERtmx cells (P = 0.0004). C, induction of conditional myc reduces tuberin protein levels. Tuberin protein levels were monitored in the mycER constructs treated as in B, except that protein lysates were harvested and immunoblots for tuberin and actin were done.
We further assessed TSC2 mRNA responses to changes in myc function using an inducible myc chimeric protein in myc−/− cells. Myc-null cells expressing a mycERtmx chimeric protein (23) were induced for 3 h with 4-hydroxy-tamoxifen to activate myc function, and TSC2 mRNA levels were quantified by quantitative real-time PCR (Fig. 4_B_). Induction of myc activity in these cells caused a 2-fold decrease of TSC2 message. By sharp contrast, induction of a similar chimeric construct containing a deleted myc box II (Δ106-143mycERtmx) failed to repress transcription of the TSC2 gene (Fig. 4_B_). An immunoblot for TSC2 confirmed the decrease of tuberin in the mycERtmx chimera compared with the Δ106-143mycERtmx (Fig. 4_C_).
Functional significance of myc repression of TSC2 evaluated with TSC2 siRNA and TSC2 antagonism of myc-mediated transformation
Our findings suggest that increased TSC2 protein expression may be a key cause for abnormal mTOR signaling in myc-null cells. To determine whether the known link between TSC2 and S6K regulation remains intact in myc−/− cells, we evaluated the effects of siRNA knockdown of TSC2 on myc−/− cells. Western blots showed successful TSC2 protein knockdowns in myc-null cells when they were transfected with TSC2 siRNAs but not with equivalent amounts of firefly luciferase siRNA (Fig. 5_A_). This down-regulation of TSC2 expression was paralleled by an increase in the amount of activated p70 S6K and phosphorylated rpS6 (Fig. 5_A_). This result is not unexpected because TSC2 knockdown is well known to increase p70 S6K activity (29) but does serve to show that the TSC2-mTOR connection is intact in myc-null cells and responsive to TSC2 protein levels.
Figure 5.
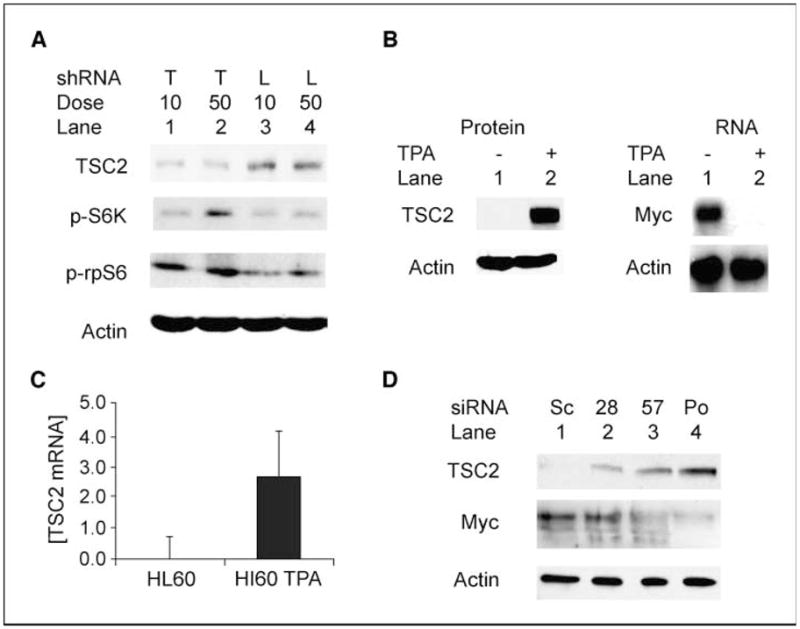
Inhibition of TSC2 expression restores S6K activation in myc-null cells, and TSC2 is regulated by c-myc in HL60 cells. A, TSC2 siRNA stimulates phosphorylation of S6K and rpS6 in myc−/− cells. Myc-null (myc−/−) cells were transiently transfected with 10 or 50 nmol/L siRNA for TSC2 (T) or with firefly luciferase siRNA (L) as a negative control. Levels of tuberin, phosphorylated S6K (p-S6K) protein, and phosphorylated rpS6 (p-rpS6) were analyzed by immunoblotting 72 h after transfection in confluent transfected cells. B, steady-state TSC2 protein is up-regulated in HL60 cells induced to differentiate with TPA. HL60 cells were given fresh medium and treated with TPA (20 nmol/L) or left untreated. After 48 h, untreated suspension cells were harvested by centrifugation, whereas TPA-treated differentiated cells were scraped from the bottom of the flask. Whole-cell protein extracts were separated by PAGE and probed for TSC2 and β-actin. As expected, a Northern blot shows that the PMA-induced loss of myc mRNA was complete in the differentiated HL60 cells. C, steady-state TSC2 mRNA is increased in HL60 cells induced to differentiate with TPA. HL60 cells were treated and harvested as in B, except that total RNA was extracted, subjected to reverse transcription, and analyzed by quantitative real-time PCR with primers specific to TSC2. Results are normalized to β-actin. D, Myc-specific shRNAs induce tuberin in HL60 cells. HL60 cells were infected with retroviral constructs expressing shRNAs for c-myc [NM_002467.2-1828slcl (28), NM_002467.2-1657slcl (57), and NM_002467-pooled (Po) shRNAs] and a negative control scrambled construct (Sc). Ten days after puromycin selection for the retroviral vectors was initiated immunoblots for tuberin, Myc and actin analyses were done using lysates from 3 × 104 cells per lane.
We next sought to determine whether this functional linkage between myc and TSC2 might be relevant to the development of malignancy. HL60 leukemia cells overexpress c-myc, but their Myc levels can be reduced by PMA-induced differentiation leading to growth arrest (30). Immunoblots of differentiating HL60 cells showed that TSC2 increases from essentially absent levels in undifferentiated HL60 cells to high levels in PMA-differentiated cells (Fig. 5_B_). Quantitative real-time PCR revealed that steady-state TSC2 mRNA increased 850-fold in PMA-differentiated HL60 cells compared with untreated, undifferentiated cells (Fig. 5_C_). The increased TSC2 is directly linked to the decrease in c-Myc because retroviral constructs expressing shRNAs for c-myc also induced tuberin in HL60 cells (Fig. 5_D_).
Finally, we evaluated the potential contribution of TSC2 to single-step myc transformation of Rat1A cells (Fig. 6). Rat1A cells transfected with myc alone and combined myc/max showed marked myc-related decreases in TSC2 mRNA (Fig. 6_A_). These decreases accompanied large increases in soft agar colony formation (Fig. 6_B_). We then used retroviral transduction of myc, TSC2, and their combination to determine whether TSC2 loss contributes to myc transformation. Figure 6_C_ shows expression of Myc and tuberin in transfectants. Replicate wells of each cell type were then evaluated by soft agar assays. Whereas neither cells expressing TSC2 alone nor those transfected with empty pBABE vector produced colonies, the ectopic expression of c-myc in Rat1A cells generated numerous large colonies. Many large Myc-induced colonies showed large coronas of cells around the central mass (Fig. 6_D_). Coexpression of tuberin with Myc completely blocked the appearance of these larger, corona-bordered colonies (Fig. 6_D_). In addition to these striking qualitative results, 64% fewer 0.25- to 0.375-mm colonies appeared in pBABE/TSC2-pBABE/Myc transfections than in pBABE/Myc-pBABE transfectants (Supplementary Table S3).
Figure 6.
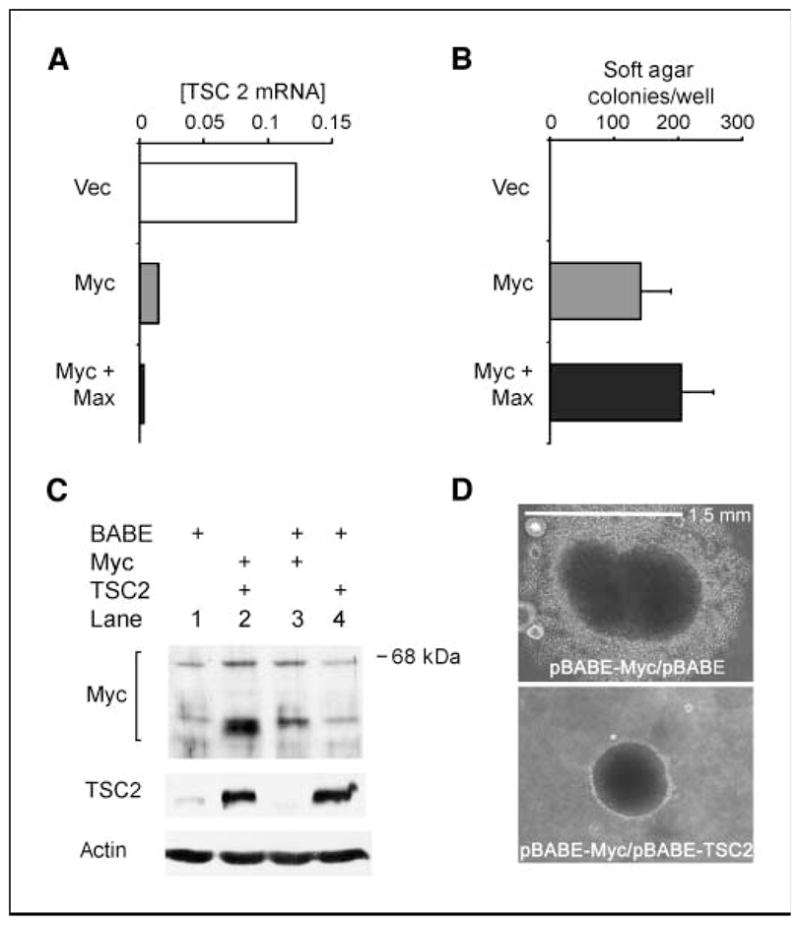
Exogenous Myc inhibits TSC2 mRNA expression and TSC2 blocks Myc-induced large colony formation of Rat1A cells in soft agar. A, transforming levels of c-myc inhibit TSC2 mRNA expression. Total RNA was harvested from confluent cultures of stable cell lines expressing a control vector, myc, and the combination of myc and max and subjected to reverse transcription followed by quantitative real-time PCR with primers specific for TSC2. Results are normalized to β-actin. B, myc transformation of Rat1A cells in soft agar assays. Plotted is the fold change in cells per well for six wells each in four separate repetitions of the experiment comparing transfected constructs to vector control cells for each repetition [columns, mean (n = 24 for each plot); bars, SE]. C, retroviral-mediated expression of myc and/or TSC2 in Rat1A cells. Rat1A cells were sequentially infected in pairs with retroviral constructs containing empty pBABE vector and pBABE-myc, or pBABE and pBABE-TSC2, or pBABE-TSC2 and pBABE-myc, or two rounds of retrovirus containing pBABE vector alone, and then stably selected in puromycin (8 μg/mL). Expression of c-myc and TSC2 proteins from cells in confluent cultures was assessed by immunoblotting. D, TSC2 blocks Myc-induced large colony formation of Rat1A cells grown in soft agar. Top, Rat1A cells transduced with pBABE-Myc were evaluated after growth in soft agar for 2 wk. Bottom, Rat1A cells transduced with pBABE-TSC2 in combination with pBABE-Myc were evaluated after growth in soft agar for 2 wk.
Discussion
Loss of rate-limiting proliferation controls contributes to malignant transformation, and uncontrolled proliferation can only occur if a malignant cell can add cell mass at a sufficient rate (31). Available evidence identifies translation initiation as the key rate-limiting component of translational control (3), which is especially shown by malignant transformation of cells overexpressing several translation initiation factors (32, 33). Increasing evidence that myc-driven oncogenesis is heavily dependent on its ability to drive cell growth processes (34) focused our attention on the effects of myc on translation initiation. Using genetic and polysomal profile analyses of cells lacking c-myc, we found that myc-null cells are sensitive to inhibition of translation initiation and exhibited a decreased capacity to initiate translation (Figs. 1 and 2). Whereas genetic evidence from other labs has shown that eIF4E can collaborate with c-myc in oncogenesis (14), taken together, our studies support the idea that c-myc may be regulating some component of the translational control apparatus in normal cells. Our studies parallel similar reports of myc effects on rapamycin sensitivity. One report found that rapamycin inhibits cap-dependent translation of c-myc, and Akt levels determine whether this change renders cells rapamycin-sensitive (35). Similarly, altered c-myc regulation correlated with rapamycin resistance in cells derived from childhood cancers (36). Our polysomal profiles revealed a myc-specific translation defect that seemed to especially involve the 40S ribosomal subunit containing the rpS6 target of the mTOR pathway.
Using a candidate protein approach, we investigated proteins in the mTOR signaling pathway to identify proteins whose the levels differed between myc-null and wild-type cells (Fig. 3). We showed the potential for direct Myc transcriptional control of TSC2 by quantitative chromatin immunoprecipitation to evaluate Myc binding to their genomic promoters (Fig. 3). We focused on Myc inhibition of TSC2 expression, showing the failure of a mutant c-myc lacking myc box II to repress transcription of the TSC2 gene (Fig. 4_B_ and C). Because myc box II is critical for Myc-induced repression of Inr-dependent gene expression (37), we have identified TSC2 as an important new Myc-inhibited target.
Two of the protein expression differences we observed fit known feedback mechanisms in the TOR signaling pathway. Decreased IRS1 levels in myc wild-type cells fit with the known enhanced degradation of IRS1 that results from TOR pathway activation (38). The finding that Akt phosphorylation is relatively increased in myc-null cells is consistent with reports indicating feedback inhibition to Akt from mTOR (39). The changes in IRS1 and Akt are therefore likely to be consequences of lost feedback inhibition of TOR signaling in myc-null cells and are not likely to be a primary cause of their growth defect. In contrast, the TSC2-controlled step in the pathway is positioned to be a primary defective signaling component in myc-null cells, and our data show that it is a direct transcriptional target of c-myc (Figs. 3 and 4). TSC2 inhibition of Rheb-mTOR complexes that phosphorylate and activate S6K (15) is increased in myc-null cells, as shown by the marked loss of S6K activity and rpS6 phosphorylation (Fig. 3).
Several previous publications identified potential interactions between myc and the TSC genes, although they did not identify a direct regulatory interaction between the two genes. Antagonism between Myc and TSC2 was first shown for control of Drosophila eye size (18) and then confirmed for mammalian cell proliferation (19). Given these previous studies, the central position of TSC2 in the TOR pathway, and our demonstration that myc represses TSC2, we evaluated the functional significance of the control of TSC2 by myc. Our data indicate that myc regulation of TSC2 is important during differentiation related to myc and in myc-driven transformation (Figs. 5 and 6). siRNA knockdown of TSC2 increased phosphorylated S6K and rpS6, as expected if the mTOR pathway is direct and intact in myc-null cells (Fig. 5_A_). We found a marked increase of TSC2 protein and a major up-regulation of TSC2 mRNA during 12-_O_-tetradecanoylphorbol-13-acetate (TPA)–induced differentiation of HL60 cells (Fig. 5_B_ and C). shRNAs for c-Myc recapitulated that induction. Because Myc protein and myc/max complex formation are down-regulated during the differentiation of HL60 cells (30), our data indicate that c-myc represses TSC2 in the undifferentiated state, and TSC2 is derepressed as myc levels drop in differentiating cells. Myc overexpression in HL60 leukemia cells may lead to reduced TSC2 levels and thereby contribute to the neoplastic process by inhibiting normal differentiation of these cells.
Strikingly, Rat1A cells transduced with TSC2 and c-myc together grew significantly fewer medium-sized colonies and completely failed to form the largest colonies with large coronas or halos on their periphery, when compared with those transfected with myc alone (Fig. 6_D_). Hamartin and tuberin are known to suppress cell migration and metastasis, possibly through their regulation of the small GTPase Rho (40). Conversely, over-expression of c-myc is able to confer a metastatic phenotype, and metastasis-associated protein 1 is an essential downstream effector of the c-Myc oncoprotein (41). Intriguingly, the cells in the corona of the large myc-induced colonies extend out from, and are only loosely associated with, the central core of these large colonies. Thus, we suggest that inhibition of cell migration may underlie the ability of TSC2 to inhibit myc-induced large colony formation. One microarray screen for myc-regulated genes in Burkitt’s lymphoma cells further supports our findings (20). This screen identified TSC2 together with the p27 tumor suppressor in clusters of myc-down-regulated genes of which the expression was most changed in four leukemia-lymphoma cell lines expressing high myc levels.
Overall, the myc target genes we identify here add additional information about the global role of myc in cellular proliferation. The biggest dilemma facing myc studies is its relatively huge number of candidate targets (42). By focusing on a single signaling pathway, we identified TSC2 as the key target within the TOR signaling cascade that could be assisted by GβL S6K and rpS6 to amplify the relatively modest individual transcriptional effects of myc. This situation is similar to Myc effects on several genes within the glycolysis pathway (43). Taken together, our results suggest that functional antagonism between c-myc and TSC2 is a result of direct TSC2 regulation by c-myc that may be important in the pathogenesis of both Myc-induced cancers and tumors found in patients with tuberous sclerosis.
Supplementary Material
Supplementary materials
Acknowledgments
Grant support: Public Health Service grants RO1-CA63117 (M. Lynch, M. Ravitz, L. Chen, and E. Schmidt) and RO1-CA69069 (L. Chen and E. Schmidt) and NIH National Research Service Award training grant 5T32 CA009216-24 (M. Ravitz).
We thank Dr. John Sedivy (Molecular Biology, Brown University, Providence, RI) for the TGR and HO15 cells, Dr. James Brugarolas (Hematology-Oncology, UT Southwestern, Dallas, TX) for pBABE-HA-TSC2, Dr. William Hahn (Dana Farber Cancer Institute, Harvard Medical School, Boston, MA) for pBABE-c-myc, and Dr. David Sabatini (Whitehead Institute, Massachusetts Institute of Technology, Cambridge, MA) for antibodies to GβL.
Footnotes
References
- 1.Baserga R. The biology of cell reproduction. Cambridge (MA): Harvard University Press; 1985. [Google Scholar]
- 2.Hahn WC, Dessain SK, Brooks MW, et al. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol. 2002;22:2111–23. doi: 10.1128/MCB.22.7.2111-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sonenberg N, Hershey JWB, Mathews M, editors. Translational control of gene expression. 2. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- 4.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 5.Jones RM, Branda J, Johnston KA, et al. An essential E box in the promoter of the gene encoding the mRNA cap-binding protein (eukaryotic initiation factor 4E) is a target for activation by c-myc. Mol Cell Biol. 1996;16:4754–64. doi: 10.1128/mcb.16.9.4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prathapam T, Tegen S, Oskarsson T, Trumpp A, Martin GS. Activated Src abrogates the Myc requirement for the G0/G1 transition but not for the G1/S transition. Proc Natl Acad Sci U S A. 2006;103:2695–700. doi: 10.1073/pnas.0511186103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prochownik EV. c-Myc as a therapeutic target in cancer. Expert Rev Anticancer Ther. 2004;4:289–302. doi: 10.1586/14737140.4.2.289. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt EV. The role of c-myc in regulation of translation initiation. Oncogene. 2004;23:3217–21. doi: 10.1038/sj.onc.1207548. [DOI] [PubMed] [Google Scholar]
- 9.Mateyak MK, Obaya AJ, Adachi S, Sedivy JM. Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ. 1997;8:1039–48. [PubMed] [Google Scholar]
- 10.Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–63. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- 11.Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5:671–88. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- 12.Hara K, Maruki Y, Long X, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–89. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 13.Shamji AF, Nghiem P, Schreiber SL. Integration of growth factor and nutrient signaling: implications for cancer biology. Mol Cell. 2003;12:271–80. doi: 10.1016/j.molcel.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 14.Wendel HG, De Stanchina E, Fridman JS, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–7. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 15.Kwiatkowski DJ, Manning BD. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet. 2005;14(Spec No 2):R251–8. doi: 10.1093/hmg/ddi260. [DOI] [PubMed] [Google Scholar]
- 16.Astrinidis A, Henske EP. Tuberous sclerosis complex: linking growth and energy signaling pathways with human disease. Oncogene. 2005;24:7475–81. doi: 10.1038/sj.onc.1209090. [DOI] [PubMed] [Google Scholar]
- 17.Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol. 2004;24:200–16. doi: 10.1128/MCB.24.1.200-216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tapon N, Ito N, Dickson BJ, Treisman JE, Hariharan IK. The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell. 2001;105:345–55. doi: 10.1016/s0092-8674(01)00332-4. [DOI] [PubMed] [Google Scholar]
- 19.Rosner M, Hofer K, Kubista M, Hengstschlager M. Cell size regulation by the human TSC tumor suppressor proteins depends on PI3K and FKBP38. Oncogene. 2003;22:4786–98. doi: 10.1038/sj.onc.1206776. [DOI] [PubMed] [Google Scholar]
- 20.Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc Natl Acad Sci U S A. 2003;100:8164–9. doi: 10.1073/pnas.1332764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soucek T, Pusch O, Wienecke R, DeClue JE, Hengstschlager M. Role of the tuberous sclerosis gene-2 product in cell cycle control. Loss of the tuberous sclerosis gene-2 induces quiescent cells to enter S phase. J Biol Chem. 1997;272:29301–8. doi: 10.1074/jbc.272.46.29301. [DOI] [PubMed] [Google Scholar]
- 22.Lynch M, Fitzgerald C, Johnston KA, Wang S, Schmidt EV. Activated eIF4E-binding protein slows G1 progression and blocks transformation by c-myc without inhibiting cell growth. J Biol Chem. 2004;279:3327–39. doi: 10.1074/jbc.M310872200. [DOI] [PubMed] [Google Scholar]
- 23.You Z, Madrid LV, Saims D, Sedivy J, Wang CY. c-Myc sensitizes cells to tumor necrosis factor-mediated apoptosis by inhibiting nuclear factor κB transactivation. J Biol Chem. 2002;277:36671–7. doi: 10.1074/jbc.M203213200. [DOI] [PubMed] [Google Scholar]
- 24.Lynch M, Chen L, Ravitz MJ, et al. hnRNP K binds a core polypyrimidine element in the eukaryotic translation initiation factor 4E (eIF4E) promoter, and its regulation of eIF4E contributes to neoplastic transformation. Mol Cell Biol. 2005;25:6436–53. doi: 10.1128/MCB.25.15.6436-6453.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyuhas O, Blierman Y, Pierandrei-Amaldi P, Amaldi F. Analysis of polysomal RNA. In: Krieg P, editor. A laboratory guide to RNA: isolation, analysis and synthesis. New York (NY): Wiley Liss; 1996. pp. 65–81. [Google Scholar]
- 26.Yamashita R, Suzuki Y, Wakaguri H, Tsuritani K, Nakai K, Sugano S. DBTSS: Data Base of Human Transcription Start Sites, progress report 2006. Nucleic Acids Res. 2006;34:D86–9. doi: 10.1093/nar/gkj129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loots GG, Ovcharenko I. rVISTA 2.0: evolutionary analysis of transcription factor binding sites. Nucleic Acids Res. 2004;32:W217–21. doi: 10.1093/nar/gkh383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato GJ, Lee WM, Chen LL, Dang CV. Max: functional domains and interaction with c-Myc. Genes Dev. 1992;6:81–92. doi: 10.1101/gad.6.1.81. [DOI] [PubMed] [Google Scholar]
- 29.Karbowniczek M, Cash T, Cheung M, Robertson GP, Astrinidis A, Henske EP. Regulation of B-Raf kinase activity by tuberin and Rheb is mammalian target of rapamycin (mTOR)-independent. J Biol Chem. 2004;279:29930–7. doi: 10.1074/jbc.M402591200. [DOI] [PubMed] [Google Scholar]
- 30.Xu D, Popov N, Hou M, et al. Switch from Myc/Max to Mad1/Max binding and decrease in histone acetylation at the telomerase reverse transcriptase promoter during differentiation of HL60 cells. Proc Natl Acad Sci U S A. 2001;98:3826–31. doi: 10.1073/pnas.071043198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Polymenis M, Schmidt EV. Coordination of cell growth with cell division. Curr Opin Genet Dev. 1999;9:76–80. doi: 10.1016/s0959-437x(99)80011-2. [DOI] [PubMed] [Google Scholar]
- 32.Lazaris-Karatzas A, Montine KS, Sonenberg N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature. 1990;345:544–7. doi: 10.1038/345544a0. [DOI] [PubMed] [Google Scholar]
- 33.Ruggero D, Montanaro L, Ma L, et al. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004;10:484–6. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- 34.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gera JF, Mellinghoff IK, Shi Y, et al. AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. J Biol Chem. 2004;279:2737–46. doi: 10.1074/jbc.M309999200. [DOI] [PubMed] [Google Scholar]
- 36.Hosoi H, Dilling MB, Liu LN, et al. Studies on the mechanism of resistance to rapamycin in human cancer cells. Mol Pharmacol. 1998;54:815–24. doi: 10.1124/mol.54.5.815. [DOI] [PubMed] [Google Scholar]
- 37.Li LH, Nerlov C, Prendergast G, MacGregor D, Ziff EB. c-Myc represses transcription in vivo by a novel mechanism dependent on the initiator element and Myc box Ii. EMBO J. 1994;13:4070–9. doi: 10.1002/j.1460-2075.1994.tb06724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takano A, Usui I, Haruta T, et al. Mammalian target of rapamycin pathway regulates insulin signaling via subcellular redistribution of insulin receptor substrate 1 and integrates nutritional signals and metabolic signals of insulin. Mol Cell Biol. 2001;21:5050–62. doi: 10.1128/MCB.21.15.5050-5062.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang H, Cicchetti G, Onda H, et al. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through down-regulation of PDGFR. J Clin Invest. 2003;112:1223–33. doi: 10.1172/JCI17222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henske EP. Metastasis of benign tumor cells in tuberous sclerosis complex. Genes Chromosomes Cancer. 2003;38:376–81. doi: 10.1002/gcc.10252. [DOI] [PubMed] [Google Scholar]
- 41.Zhang XY, DeSalle LM, Patel JH, et al. Metastasis-associated protein 1 (MTA1) is an essential downstream effector of the c-MYC oncoprotein. Proc Natl Acad Sci U S A. 2005;102:13968–73. doi: 10.1073/pnas.0502330102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guccione E, Martinato F, Finocchiaro G, et al. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat Cell Biol. 2006;8:764–70. doi: 10.1038/ncb1434. [DOI] [PubMed] [Google Scholar]
- 43.Kim JW, Zeller KI, Wang Y, et al. Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol Cell Biol. 2004;24:5923–36. doi: 10.1128/MCB.24.13.5923-5936.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials