Resolution of Inflammation in Murine Autoimmune Arthritis Is Disrupted by Cyclooxygenase-2 Inhibition and Restored by Prostaglandin E2-Mediated Lipoxin A4 Production (original) (raw)
. Author manuscript; available in PMC: 2011 Jun 1.
Published in final edited form as: J Immunol. 2010 Apr 30;184(11):6418–6426. doi: 10.4049/jimmunol.0903816
Abstract
Acute inflammation follows defined phases of induction, inflammation and resolution, and resolution occurs by an active process that requires cyclooxygenase-2 (COX-2) activity. This study aims to address whether this paradigm extends to recognized model of chronic inflammation. We demonstrated that murine collagen-induced arthritis follows a similar sequential course. Interestingly, COX-2 and its metabolite, the presumably proinflammatory PGE2, are present in the joints during resolution, and blocking COX-2 activity and PGE2 production within this period perpetuated, instead of attenuated, inflammation. Repletion with PGE2 analogs restored homeostasis, and this function is mediated by the proresolving lipoxygenase metabolite, lipoxin A4, a potent stop signal. Thus, the study provided in vivo evidence for a natural, endogenous link between the cyclooxygenase–lipoxygenase pathways and showed that PGE2 serves as a feedback inhibitor essential for limiting chronic inflammation in autoimmune arthritis. These findings may explain the enigma regarding why COX-2 inhibitors are palliative rather than curative in humans, because blocking resolution may mitigate the benefit of preventing induction.
In the field of inflammation, there have been two seminal discoveries that precipitated a paradigm shift in the past decade. One is the discovery that, contrary to early belief, inflammatory response does not dissipate but requires an active process that is mediated by products of cyclooxygenase (COX) for subsiding. For example, Gilroy et al. (1) observed that blocking COX activity, whether with the selective COX-2 inhibitor _N_-[2-(cyclohexyloxy)-4-nitrophenyl]-methanesulfonamide (NS-398) or the dual COX-1/COX-2 inhibitor indomethacin, inhibited carrageenin-induced acute pleurisy in the inflammation phase, but then it significantly perpetuated inflammation by interfering with resolution. Similarly, Fukunaga et al. (2) have shown that pharmacologic inhibition or gene disruption of COX-2 blocked resolution of acid-induced lung injury. COX-2 has also been shown to mediate resolution of acute inflammation in the brain (3), liver (4), small bowel, and colon (5) as well.
Another major impact came from the discovery that a class of arachidonic acid metabolites facilitates resolution of inflammation. These eicosanoids, named lipoxins, resolvins, and protectins, are generated from lipoxygenases or aspirin-acetylated COX and serve as stop signals in the evolution of acute inflammation responses. They act by stimulating the uptake of apoptotic polymorphonuclear leukocytes at sites of inflammation to promote a programmed return to homeostasis (6–8). Analogs and precursors (ω-3-fatty acids) of lipoxins have been shown to relieve inflammatory-related pathology responses, especially in asthma and various airway injures (9). Currently, it is known that lipoxins are formed in vivo in the course of inflammation and animals with a mutation in their synthetic enzyme, ALOX12/15, suffer exacerbated inflammation (10). However, our knowledge on the endogenous pathway that regulates their biogenic synthesis is still very limited.
Autoimmune diseases are cyclical in nature. It is generally assumed that tissue destruction occurs because the inflammation is chronic (11). The first objective of this study is to determine the relevance of the eicosanoid-directed, proresolving process in an autoimmune condition so as to evaluate whether it can be harnessed for therapeutic intervention; and then to determine whether the mechanism is vulnerable to COX-2 inhibitors for these are blockbuster drugs that are widely prescribed for curbing inflammation (12). We used the murine collagen-induced arthritis (CIA) model, for it has been generally used for evaluating immunological and pharmacological treatments, including COX-2 inhibitors, for rheumatoid arthritis (13). We discovered that PGE2, generally viewed as a culprit for exacerbating arthritis, functions as a critical endogenous signal sufficient for mediating resolution. It coordinates resolution of inflammation by mediating biogenesis of lipoxin A4 (LXA4) for curbing inflammation. This link between PG and lipoxin may explain why clinical treatments with COX inhibitors relieve symptoms but would not halt progression of the disease in humans.
Materials and Methods
Induction of disease
Experimental protocol used in this study was approved by the Temple University Institutional Animal Care and Use Committee. Arthritis was induced in 6- to 8-wk-old, male DBA/1 mice (The Jackson Laboratory, Bar Harbor, ME) with intradermal injection of chicken collagen II (Chondrex, Redmond, WA) in CFA according to the procedure of Terato et al. (14). Each mouse received a 0.05 ml volume 50 µg collagen emulsified in CFA intradermally in the tail. Immunity was boosted with another injection of 50 µg collagen II in IFA at day 21. Pathogenesis was assessed in a double-blinded manner by measuring the thickness of the hind footpads twice a week with a constant-tension caliper.
Defining resolution
Ability to resolve inflammation was determined by the percent of footpads that has decreased, increased, or remains unchanged in their thickness. A numerical index for each footpad was deduced from the slope of a secant line. In the vehicle control group, the line was drawn from the peak of inflammation to the point of sacrifice or total resolution (< 10% swollen) in the resolution phase. For therapeutically treated groups, the line was drawn from the beginning of NS-398 administration. A value of “0” represents no change in thickness. Negative values correspond to decrease in swelling and positive values indicate that swelling increased.
Radiology assessment
Mice were anesthetized with ketamine/xylazine then radiographs were taken using a Faxitron for small animals (model 43855, Faxitron X-Ray, Lincolnshire, IL).
Histological assessment
Knee joints were fixed in 10% neutral buffered formalin for 24 h, decalcified in EDTA, embedded in paraffin, and cut in serial sections for histopathological analysis. The sections were then stained with H&E, and safranin O to examine the integrity of the cartilage. Formalin-fixed tissue sections were stained with rabbit anti-murine COX-2 Ab (H-62, Santa Cruz Biotechnology, Santa Cruz, CA), then biotinylated anti-rabbit Ab, followed by streptavidin-HRP and DAB solution (all from Dako Scientific, Carpinteria, CA) to assess COX-2 expression. Rabbit normal Ig was used for isotype controls.
PG assays
Lipid was extracted before assaying for the PGs. Randomly selected footpads were harvested, snap frozen, and stored in liquid nitrogen until usage, and then weighed before lipid extraction. The frozen joint tissues were pulverized using a mortar and pestle to obtain a fine powder, which was homogenized (Polytron PRO200 homogenizer; PRO Scientific, Oxford, CT) and then sonicated in methanol with 0.01 M butylhydroxytoluene and 0.85% formic acid while surrounded by ice. After centrifugation, an aliquot of the supernatants was collected to perform Bradford assay for protein determination, then the homogenates were protein-precipitated by the addition of acetonitrite (pH 3.5). After centrifugation, the supernatants were loaded onto a C18 SPE column (3M, St. Paul, MN) to collect the lipid molecules. The cartridge was preconditioned by washing thoroughly with 10% methanol/0.1% formic acid and ethyl acetate. The lipid molecules were eluted with ethyl acetate, followed by methanol with 0.2% formic acid and 0.01 M butylhydroxytoluene, and then evaporated to dryness under nitrogen and reconstituted in ELISA buffer. The ELISA kits used for PGE2 and LXA4 were from Cayman Chemicals, Assay Design, and Neogen, respectively. Each sample was assessed in triplicate and at two to three dilutions to ascertain that the reactions occurred within the standard curve and did not reflect interference from cross-reactive substances. The amount of lipid produced was calculated as pg/mg of tissue. A minimum of three repeats were performed for each experiment and they were merged by normalization to the corresponding basal value within each experiment for statistical comparison.
RT-PCR and PCR
Limbs harvested from euthanized mice were weighed, snap frozen, and stored at −80°C for assessing gene expression. The joints were crushed in liquid nitrogen and homogenized with a micro ultrasonic cell disrupter for RNA extraction in Trizol reagent (Invitrogen, San Diego, CA). Extracts from limbs of randomly selected mice from each group were pooled for reverse transcription and real-time PCR analysis as described in Adapala and Chan (15). Commercial primers and SYBR Green I PCR master mix were used (Superarray). The housekeeping gene 18S RNA was reverse transcribed with random hexamers, and its cDNA solution was diluted to attain an amplification efficiency that was comparable to that of the experimental gene, which cycling threshold values were normalized to those encoding 18S rRNA by the Ct method. A minimum of three repeats were performed for each experiment and, in each, relative units were deduced so the repeats can be normalized and merged for statistical comparison.
Data analyses
For statistical comparison, the data on gene expression and footpad thickness were tested for normality using the Kolmogorov-Smirnov test. Then, the normally distributed populations were compared using unpaired, one-way ANOVA, followed by Bonferroni test. Otherwise, the nonparametric Kruskal-Wallis test was used instead. A p value of 0.05 was chosen as the threshold for statistical significance throughout.
Results
Kinetic studies revealed a resolution phase in the pathogenesis of chronic inflammatory autoimmune arthritis
The study began with examining whether the pathogenesis of autoimmune arthritis in the CIA model would resemble acute injury in having discrete phases. Hind foot thickness showed that, similar to acute inflammation, swelling followed three phases: induction, inflammation, and resolution (Fig. 1_A_). From days 0–30, arthritis had not developed; the mice were asymptomatic and their footpads were not swollen. Footpad swelling became increasingly prevalent beginning at day 30 and continued to do so until around day 45 when >95% the hind feet were swollen by 20% or more in thickness, and the incidence of arthritis in the group of mice was 98%. Subsequently, the footpad swelling began to progressively subside with 40% resolved at day 55 and 90% at days 69–70. The progression from induction through inflammation to resolution is illustrated in footpad swelling by a representative footpad (Fig. 1_B_).
FIGURE 1.

Three phases of pathogenesis in arthritis. In A, open bars indicate the percent of footpads that have swollen by 20%. Filled bars indicate the percent of swollen limbs that have decreased by > 20% in thickness, n = 50–200. The number of footpads decreases as mice were sacrificed for cytokine analyses. Mean and SDs are plotted. The degree of swelling for each footpad was calculated as 100 × (thickness – thickness on day 0)/(thickness on day 0, range from 2.0–2.3 mm). B, The progression of inflammation in a typical arthritic limb is shown. In C, IL-17 and TNF-α mRNA were normalized against 18S RNA and then the relative level of expression was deduced. Three independent experimental repeats were performed. Within each experiment, 4–6 limbs from randomly selected mice were pooled, each data point is averaged from the three repeats, so total n = 12–18. The data were tested for normality, and unpaired one-way ANOVA, followed by Bonferroni test was used for statistical comparison. Their levels at peak of inflammation (day 0) were significantly different from when the joints were normal (day −40) or resolved (day +30), p < 0.05, but the levels in normal and resolved joints were not different, _p_ > 0.05. D, H&E shows a highly cellular synovium in the inflamed (70% swollen at day 40) but not in the resolved (subsided from 91% to 12% swelling at day 70) knee joints (original magnification ×40). E, Sequential x-ray of representative metatarsal joints of a footpad is shown; note the subsiding of soft tissue swelling and the return of joint integrity and bone density in the fifth phalange.
IL-17 and TNF-α are two of the several cytokines that drive the progression of inflammation (16, 17). Therefore, groups of mice were sacrificed at weekly intervals and the joints were collected for reverse transcriptase real-time PCR analysis of these transcriptionally regulated cytokines to verify the state of inflammation by their levels. Their mRNA expression increased by 4- to 6-fold within the inflammatory phase (days 25–55), then it subsided as inflammation resolved at day 70 (Fig. 1_C_). The state of inflammation was further documented by histology and radiography (Fig. 1_D_, 1_E_). The H&E sections show that the synovium of a knee joint at the peak of inflammation is fully infiltrated with neutrophils, whereas that of a resolved one, which footpad thickness had subsided to 18% from 91%, is cleared of infiltrates. The radiographs show that the metatarsophalangeal joints of the footpad regained alignment as inflammation resolved.
COX-2 was expressed in the resolution phase
Whereas the gene expression of IL-17 and TNF-α subsided as resolution occurred, COX-2 mRNA remained escalated (Fig. 2_A_). Kinetics studies demonstrated that COX-2 was upregulated by 2- to 3.5-fold during the inflammatory phase between days 35 and 45, declined slightly around day 55, then resurged during resolution through day 70. Comparison by unpaired, one-way ANOVA showed that the level of expression during resolution (day 70) and at the peak of inflammation, day 35 or 45, were both significantly higher than baseline (p < 0.005) but they did not differ between each other (_p_ > 0.05). This bimodal pattern was observed in more than three experimental repeats and was analogous to the nature of COX-2 expression in acute inflammation: Gilroy et al. (3) have shown that when carrageenin is used to induce acute pleurisy, COX-2 expression in the rat model shows a bimodal pattern and the enzyme is expressed 3.5-fold higher in the resolution phase (48 h after induction) than in the inflammation phase (2 h after induction). Blaho et al. (18) have also found a similar pattern in an infectious disease model, Lyme disease. Furthermore, Kapoor et al. (19) have shown that COX-2 expression is sustained for repair in the process of wound healing. The presence of COX-2 was confirmed at the protein level by immunohistochemical staining of the knee joints (Fig. 2_B_). The enzyme was detected in the synovium as well as the infiltrating cells of the resolving knees.
FIGURE 2.

COX-2 expression during resolution. In A, the gray, shaded area, duplicated from Fig. 1_A_,provides a background to show the progression from induction to inflammation to resolution. Kinetics of COX-2 expression (black bold line) was derived by real-time PCR. Three independent experiments were performed. In each, 4–6 limbs from randomly selected mice within a group were used to derive the data points, totaling 12–15 per point. The data were tested for normality, and statistical analysis was performed using unpaired, one-way ANOVA, followed by Bonferroni test. The levels at the peak of inflammation and during resolution (day 70) were not significantly different from each other, p > 0.08, but they were significantly different from when the joints were normal (day 0), p ≤ 0.006. B, Immunohistochemical staining of joints (representative of eight) is shown in the inflammation (left) and resolution phases (right) for the presence of COX-2 in the pannus and synovium. Positive staining for COX-2 appears as brown. Specificity of binding was verified by isotype control. The hematoxylin counterstain appears as blue, original magnification ×40 and ×600.
Contradictory duality of COX-2: resolution was COX-2 dependent
Because COX-2 is expressed, it is important to determine the function of COX-2 in the resolution phase. In this experiment, collagen-induced mice were divided into groups of positive control, which received vehicle only; negative control, which were age-matched normal mice; and the experimental, which received 10 µg NS-398, a COX-2–specific inhibitor, dissolved in saline with 10% ethanol. The dose of NS-398 given was 0.5 mg/kg, and it was administered by gavage every other day in two manners: 1) treatment began at the induction phase to imitate prophylactic use of COX inhibitor (Fig. 3_A_), or 2) after inflammation has established to mimic patients seeking treatment after symptoms have appeared (Fig. 3_B_).
FIGURE 3.

NS-398 increased the incidence and severity of swelling in the resolution phase. A, Depiction of the relative increase in thickness of footpad (mm) due to arthritis induced by chicken collagen. The maximum thickness obtained within days 30–55, the inflamed phase, was plotted for each foot. The data are compiled from four independent trials. The black diamonds (n = 112) represent footpads of mice that were induced for arthritis but fed vehicle as control. The gray triangles (n = 58) represent footpads of experimental mice that were fed NS-398 prior to manifestation of symptoms, and they are congregated at the bottom. B, The incidence and severity of swelling in the resolution phase. Each black diamond (n = 38) and gray triangle (n = 48) represents a footpad. The data were compiled from three independent experiments. The open squares, in A and B, both n = 20, represent footpads of normal mice that were not injected. The horizontal bars indicate the average value in each group, and the groups were compared by Kruskal-Wallis test, and p < 0.05 was considered significant. C and D, The difference in levels of mRNA in a subgroup of limbs randomly selected and harvested from the group in A or the group in B, is shown, respectively. The levels of mRNA expression were normalized against 18S RNA and the results shown are the mean ± SEM from 4–6 mice and representative of three experimental repeats. The data were tested for normality and then statistical analysis by ANOVA. The differences between NS-398–fed groups and the vehicle control group was significant (p < 0.05). E, Kinetically plots a representative limb from the vehicle control group (dashed line–open triangles) and a group where NS-398 treatment was initiated after inflammation has established (solid line–filled triangles). E is also used to delineate the three phases (lines) and illustrate treatment regimen. Closed arrow points to when therapeutic NS-398 treatment started.
The effect of NS-398 in the inflammation and the resolution phases is opposite. Blocking COX-2 prior to manifestation of symptoms (initiating NS-398 feeding at day 14) attenuated footpad swelling during the nominal inflammation phase. This was apparent when the prevalence of arthritis was enumerated by percent or by the degree of swelling in the hind footpads. Among mice in the vehicle control group, > 75% of the hind feet had swollen, whereas NS-398 feeding reduced the incidence to ~40%. The thickness of their footpads had increased by 40.4% on average, whereas those of the NS-398–fed group had swollen by only 21.4% (Fig. 3_A_). Correspondingly, the mRNA levels of proinflammatory cytokines, TNF-α and IL-17, in the footpads that were harvested between days 40 and 45 were also suppressed by NS-398 (Fig. 3_C_). Henceforth, considering that both groups of mice are comparable in weight, averaging ~20 g throughout the experiment, our results demonstrate what would be expected; NS-398 reduces inflammation when given in the induction phase.
In contrast, blocking COX-2 during the resolution phase perpetuates inflammation (Fig. 3_B_). This phenomenon is best illustrated by the percent of mice that have qualitatively been defined by a zero, a positive, or a negative slope. Among the footpads that have developed arthritis (82%), 87% of those in the vehicle control group had experienced resolution, as indicated by the rate of change in footpad thickness (negative slope, downward), and only 14% of them remained unchanged or continued to swell. In contrast, inflammation persisted or progressed (zero or positive slope) in 54% of the footpads in the group that was fed NS-398 after inflammation has established (therapeutically). When secant lines were drawn to deduce numbers, the slopes of the footpads in the vehicle control group average to −2.35, indicating inflammation was resolving naturally, whereas those of NS-398 groups that were fed therapeutically was +0.38, showing that resolution has failed to occur. The degree of inflammation corresponded to the levels of proinflammatory cytokines in the harvested limbs. Real-time PCR analysis (Fig. 3_D_) confirmed that the levels of TNF-α and IL-17 mRNA were significantly higher in the NS-398–treated groups, and the differences were statistically significant. Fig. 3_E_ illustrates how NS-398 affected the course of resolution kinetically using a representative limb from each group. The footpads in the vehicle control swelled and then subsided; however, resolution was impaired in the group that was fed NS-398. Even in mice that were fed NS-398 preventively (initiated in the induction phase), the suppression of inflammation was temporary in 67% of the footpads; the onset of swelling eventually occurred.
The medical consequence of loss of resolution was increased destruction of the joints (Fig. 4). Radiography showed that the degree of soft tissue swelling, digital misalignment, ankylosis, and loss of bone density were more severe in the limbs that had delayed onset (preventive treatment) or perpetuated inflammation (therapeutic treatment) than in those that were able to resolve (vehicle control). The degree of deterioration was further delineated by histological staining. The pannus in the knee joints of the NS-398–fed mice were more proliferative and the cartilage and bone were more severely damaged than those of the vehicle controls.
FIGURE 4.
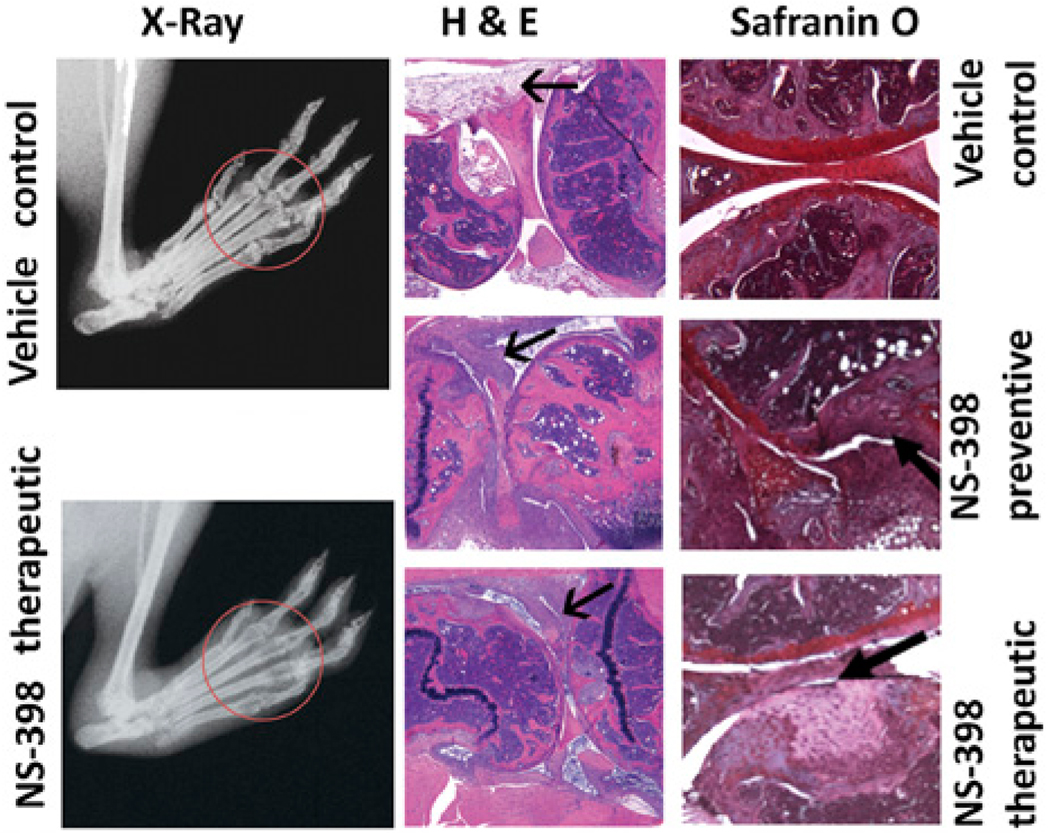
Joint damages incurred by NS-398. X-rays were taken before mice were sacrificed between days 65 and 70. Representative foot ads of mice that were fed vehicle and fed NS-398 after inflammation are shown. Note swelling (halo around ankle), misalignment and loss of space (circled: distinctive, round ending of the phalanges and metatarsal) and bone density (circled: loss of opacity) were more severe in the joints of mice that were treated with NS-398 than the ones from the vehicle. Paraffin sections of knee joints from a representative mouse that were induced for arthritis and fed vehicle, fed NS-398 from induction and have delayed onset of arthritis and fed NS-398 after inflammation has established were stained with H&E and safranin O. In the H&E sections, the bold black arrow points to the synovium, which is much more proliferative in those from mice that received NS-398 before or after their footpad swelled than that from the vehicle control. In the safranin O sections, the arrow points to the cartilage (crimson), which is intact in the vehicle control but chipped in those that became inflamed upon entering the purported resolution period but almost missing in those that failed to resolve (original magnification ×20).
PGE2 was produced in the resolution phase and COX-2 dependent
Among the COX-2 metabolites, PGE2 is usually considered as a culprit that causes inflammation, whereas 15-deoxy-Δ12,14-PGJ2 (15d-PGJ2) is a ligand for the immunosuppressive transcription factor peroxisome proliferator-activated receptor-γ and an inhibitor of NF-κB (20). A class switching from PGE2 to 15d-PGJ2 has been observed during transition from inflammation to resolution on acute models (19, 21). However, in murine CIA, the level of PGE2 is sustained.
In three independent experiments, similarly, the amount of PGE2 was elevated in the inflamed and the resolution phases. Compared with the normal control, the average increases in the two phases were 2.40 ± 0.43-fold and 2.57 ± 0.63-fold, respectively (Fig. 5_A_). They are statistically different from the normal control but not from each other. Intrigued by the synthesis of PGE2 in the phase of resolution, we proceeded to determine whether COX-2 is synthesizing the PG during the resolution phase. In the mice that were fed NS-398, the relative level of PGE2 decreased to 1.46 ± 0.13 from 2.22 ± 0.22, by ~60% when compared with the vehicle control (Fig. 5_B_, see legend for actual concentrations in pg/mg tissue).
FIGURE 5.

NS-398 inhibited production of PGE2 in resolution phase. The bars in A show the relative levels of PGE2 in the normal (day 0) footpads and footpads of mice sacrificed in the inflamed (day 35–40) and resolution phases (day 65–70). Three independent experimental repeats were performed and the total numbers of footpads per group were n = 12, n = 16, and n = 16, respectively. The values for the three experimental repeats were 36.1, 9.49, and 25.8 pg/mg tissue for normal, 103, 19.89, and 53.96 pg/mg tissue for inflamed, and 108.67, 17.96, and 78.6 pg/mg for resolved footpads. B shows PGE2 levels in footpads, harvested during resolution, from mice that were not induced for arthritis (n = 24), fed vehicle (n = 33), and treated with NS-398 (n = 46). For the individual experiments, the values for the normal control were 4.6, 24.9, and 5.86 pg/mg footpad, vehicle control were 9.46, 53.44, and 12.87, and NS-398–treated group 7.59, 35.86, and 7.52, respectively. In both A and B, relative units were derived by normalization to their respective normal control, and then compared by ANOVA, followed by Bonferroni procedure.
Reconstitution with PGE2 was associated with return of resolution
To determine whether PGE2 play a functional role in resolving arthritis, reconstitution studies were performed. Because PGE2 is unstable, we used stable analogs that resist degradation in vivo and would activate the PGE receptors, EP1–4, 16,16 dimethyl-PGE2 (dmPGE2), and misoprostol (22). The latter is a PGE1 analog used clinically for preventing nonsteriodal anti-inflammatory drug (NSAID)-induced stomach ulcers in humans (23). Arthritis was allowed to develop for 45 d and then the mice were divided into groups: 1) vehicle control that would resolve naturally, 2) NS-398 and PBS-treated that would fail to resolve, and 3) NS-398–treated ones reconstituted with PGE analogs, dmPGE2, and misoprostol. Subcutaneous injections were given in 50 µl volume along the thigh three times a week (Fig. 6). The analogs were effective in nanogram amounts, which were a significantly lower dose than what had been used in other studies, for example, daily injection of 60 mg per mouse i.p. (24).
FIGURE 6.

PGE reconstitution restores resolution of inflammation. A compares the rate of change in thickness in naturally resolving vehicle control (open diamond, n = 45), the NS-398–blocked ones that were reconstituted with 600 ng (open triangle, n = 28) or 1200 ng (gray triangle n = 15) of misoprostol, dmPGE2 (black triangle, n = 21), and not reconstituted (PBS control, black diamond, n = 42). Each marker represents a footpad. The groups were compared using the Kruskal-Wallis method. B illustrates the pathogenesis using a representative limb from the vehicle control group (dashed line with open diamonds), NS-398–fed group (solid line with filled diamonds), and the misoprostol- and dm PGE2- reconstituted groups (solid line with filled triangles). C shows H&E staining of knee joints that resolved naturally (no NS-398), inhibited from resolution by NS-398 (PBS) and resolved due to dmPGE2 reconstitution (original magnification ×40 and ×600). Neutrophils are identified by their multilooped nuclei (arrows).
In the vehicle control, >89% of the swollen hind footpads resolved naturally. In the NS-398–fed group in which PBS was administered, 53% of the footpads remained swollen and their thickness increased beyond the inflamed phase. Resolution was restored by PGE reconstitution. The swollen footpads in these groups resolved as in the vehicle control. The rate of change in thickness in the vehicle mice has an average slope of −2.21, whereas those fed NS-398 was −0.12, suggesting perpetuation of inflammation. The reconstituted groups have slopes of −2.49 (600 ng misoprostol), −2.55 (1200 ng misoprostol), and −2.96 (600 ng dmPGE2), indicating that resolution was restored. The footpads of normal mice did not show any change in thickness as they were never swollen (Fig. 6_A_). Statistical comparison by the Kruskal-Wallis method verified that the reconstituted group and vehicle control group were similar, but they were different from the group that received PBS instead of the PGE analogs. The resolution kinetics in the PGE-reconstituted footpads resembled that observed in footpads that resolved naturally (Fig. 6_B_). Histological sections (Fig. 6_C_) also revealed that they were not infiltrated with neutrophils, which were abundant in the PBS-treated joint of the NS-398–fed mice. Henceforth, the study supported the hypothesis that PGE2 restored resolution in the mice which COX-2 had been inhibited.
MPGES-1 did not synthesize the PGE2 during resolution
PGE2 is synthesized by the enzyme PG E synthase, which has several isoforms. The COX-2-associated inducible isoform, microsomal PGES-1 (mPGES-1), is currently regarded as a potential therapeutic target to replace COX-2, which inhibition has cardiovascular side effects (25, 26). Therefore, it is important to examine whether it is the source of PGE2 during resolution. Gene expression analysis showed that mPGES-1 was expressed only in the inflamed phase; its levels in the resolving footpads were the sameas basal (Fig. 7). Thus, mPGES-1 is not likely the downstream terminal synthase for PGE2, even though further investigation is needed to establish that it does not affect the resolution process.
FIGURE 7.

Kinetics of mPGES-1 expression. The bold black line shows the gene expression of mPGES-1. The number of footpads used in the analysis ranged from 12–16 per data point. Their RNA expression was normalized to 18S RNA, and then, the values in each experiment are normalized to the respective normal control to deduce the relative units. The results from three independent repeats, each showing a similar pattern, are averaged, tested for normality and compared by ANOVA, followed by Bonferroni procedure. MPGES-1 in normal footpads is only different from inflamed footpads (p < 0.001) but not when compared with the resolving footpads (_p_ > 0.99). The kinetics of COX-2 expression (gray line) has been included for comparison.
Return of resolution was associated with restoration of LXA4 production
Having established that PGE2 has a functional role, we were curious as to how it may promote the resolution of arthritis. Clearance of apoptotic neutrophils from the inflammatory tissue by professional phagocytes is a hallmark of LXA4, an anti-inflammatory eicosanoid from the LOX pathway; this prompted us to examine whether COX-2 affects its production (8). First, a kinetic study was conducted to determine whether LXA4 and ALOX12/15, the enzyme that controls production of LXA4, was upregulated during resolution. ALOX12/15 increased by 2- to 2.5-fold as inflammation from the primary injection subsided. After the booster, it returned to basal level during the inflamed phase (day 40) and then surged again on entering the resolution phase, returning to the peak level at day 70 (Fig. 8_A_). Correspondingly, the lipid was not produced during the peak of inflammation but increased as pathogenesis progressed into the resolution phase (Fig. 8_B_). Production was highest, 13.8 pg/mg, within the resolution phase, a 2-fold increase from the basal level of 6.1 pg/mg. Surprisingly, COX-2 inhibition reduced LXA4 in a pattern that paralleled the loss of the PGs. The level of LXA4 in the NS-398 group was decreased by 80%, 7.28 down from 13.8 pg/mg (Fig. 8_C_). Therefore, blocking COX-2 affected the synthesis of LXA4.
FIGURE 8.

Loss of LXA4 production with NS-398 treatment. A shows the kinetics of ALOX12/15 mRNA expression, measured by real-time PCR as described in Fig. 2. The results were averages derived by combining data from three independent repeats. The total numbers of footpads used were as follows: day 0 = 11, day 7 = 10, day 14 = 11, day 24 = 13, day 35 = 15, day 45 = 15, day 55 = 11, and day 70 = 14. COX-2 was included for comparison. The level at day 70 is different from those at day 45 and day 0 (p < 0.01), and the level at day 45 is similar to that at day 0 (_p_ > 0.5). In B and C, LXA4 were measured as pg/mg of tissue and then, the values in each experiment are normalized to the respective normal control to deduce the relative units. The results from three independent repeats, each showing a similar pattern, are averaged, tested for normality and compared by ANOVA, followed by Bonferroni procedure. B shows the levels of the lipid, LXA4. The number of limbs used in the analysis were normal control = 24, inflamed = 9, and resolved = 29. The levels were significantly different between normal and resolved (p = 0.001). C shows LXA4 production was reduced by NS-398, administered after inflammation had established. The relative levels in footpads of normal (n = 20), vehicle control (n = 23) and NS-398–treated (n = 42) mice are plotted. The levels were significantly different between vehicle and NS-398 treated (p = 0.001).
The subsequent experiment is to determine whether reconstituting PGE2 restores LXA4 production. For this, lipids were extracted from the limbs that were harvested in the reconstitution experiment (Fig. 6). As shown in Fig. 9, when PGE was replenished production of LXA4 returned (Fig. 9_A_). In the NS-398 group, the amount of LXA4 was reduced by 50%, from 3.5 pg/mg in the vehicle control group to 1.5 pg/mg. However, with administration of 600 and 1200 ng of misoprostol, in a dose-dependent manner, the levels of LXA4 returned to 2 and 2.7 pg/mg tissue, a reconstitution by 25% and 50%, respectively. Reconstitution with 600 ng of dmPGE2 also restored LXA4 production to 2.9 pg/mg tissue. Bonnans et al. (27) have shown that PGE2 induces the expression of ALX, the receptor of LXA4. DmPGE2 also reconstituted the expression of ALX, thus supporting the argument that PGE2 regulates the operation of a lipoxin-mediated resolution mechanism (Fig. 9_B_).
FIGURE 9.

Association of LXA4 reconstitution with restoration of resolution. A shows the levels of LXA4 and B shows the relative expression of ALX in the naturally resolving vehicle control (n = 15), normal nonarthritic mice, and NS-398–treated ones that injected with PBS control (n = 15) or were reconstituted with PGE1 misoprostol at 600 ng (n = 12) and 1200 ng (n = 5), and dmPGE2 (n = 6). (See Results for actual concentrations.) In B, the levels between vehicle control and the reconstituted sample were not significantly different (p < 0.08).
Discussion
Chronic inflammation has long been known as the cause of many tissue destructive diseases, such as arthritis, asthma, colitis, and periodontitis. In the past decade, investigations have uncovered that the impact of inflammation is much more extensive. It is the underlying culprit in many prevalent diseases that were previously not considered to be inflammatory in nature, such as coronary heart conditions, Alzheimer’s, and cancer. This revelation intensified interests in controlling inflammation for disease prevention and treatment. The momentum has led to a growing body of evidence within acute inflammation models, which shows that turning off the inflammatory response requires an active process mediated at least in part by products of the COX and lipoxygenase pathways. However, acute inflammation is self-limiting and without relapse; some of its models have been criticized as lacking the complexity of autoimmune diseases and incongruous with chronic inflammation, which are cyclic and laden with flares (28). For example, Seibert et al. (28) doubted whether COX-2 inhibitors will apply to rheumatoid arthritis when Gilroy et al. (1) first discovered the anti-inflammatory property of COX-2 in 1995. This study in a murine CIA, a model that, to a large extent, has been regarded as resembling the clinical disease, addresses the challenge. It critically expands the clinical significance of the COX-2/PGE-mediated resolution mechanism by revealing that it is an active and integral process, and hence, potentially can be harnessed for therapeutic intervention of the autoimmune conditions (Fig. 10).
FIGURE 10.

Proposed model for crosstalk between the COX and LOX pathways. PGE2 produced through COX activates the production of lipoxins from the LOX pathway to mediate resolution of inflammation in a feedback manner.
COX inhibitors are first-line therapies of arthritis and have been widely adopted for treatment of many chronic inflammatory disorders and autoimmune diseases (29). In the murine CIA, we found that a COX-2 inhibitor, NS-398, at a dose of 0.5 mg/kg, will interfere with resolution when given after the disease is symptomatic, and when given earlier will only delay onset. This may explain why NSAID and COX inhibitors are palliative rather than curative, and why they must be given relatively early to be effective and would not halt the progression of disease in humans (12).
There are examples that dual inhibitors of COX-1 and COX-2 induce clinical relapse in patients with osteoarthritis and inflammatory bowel diseases. Reijman et al. (30) observed that patients who received long-term treatment with diclofenac (180 d) had a 2.4-fold and a 3.2-fold higher risk of osteoarthritic damage in their hips and knee, respectively, than subjects who received the NSAID for a short term (1–30 d). In a small-scaled retrospective study, Matuk et al. (31) found that 40% of patients on coxib and 70% of those on celecoxib had a relapse within 6 wk. In a study that used 109 subjects, Takeuchi et al. (32) found that 17–28% of patients with quiescent Crohn’s disease or ulcerative colitis had a relapse after 4 wk on conventional NSAIDs (naproxen, diclofenac, or indomethacin), whereas no patients taking acetaminophen did. It has also been reported that indomethacin exacerbates psoriasis and rofecoxib prolongs the time needed to resolve inflammation caused by tooth extraction (33–35).
In several acute models, such as heart inflammation and skin wound healing, production switches from pro- to anti-inflammatory metabolites, PGE2 to 15d-PGJ2, as the inflammation evolves into resolution (19, 21). However, in chronic murine autoimmune arthritis, PGE2 production is sustained throughout both phases. Since the mid-1980s, there have been reports on the pro- as well as antiinflammatory effects of PGE2; however, the view that PGE2 promotes inflammation has dominated. More recently, the pendulum has begun to swing toward its anti-inflammatory nature. In cancer, for example, PGE2 was thought to promote tumor growth by activating cell proliferation, migration, apoptosis, and/or angiogenesis. However, it is now considered that COX-2 expression promotes carcinogenesis because PGE spares the transformed cells from immune elimination (36). The enzyme that degrades PGE, 15-PG dehydrogenase, is now classified as a tumor suppressor. PGE has been found to suppress many immune functions, for example, it activates regulatory T lymphocytes (37) and inhibits NF-κB activation to reduce the production of proinflammatory mediators (38). It has also been shown to activate the Th-2 subset of T cells, while suppressing the Th-1 subset (39). The PG signals cellular events by binding to its receptors, EP1, EP2, EP3 and EP4, which activate nonidentical functions. The EP1 receptor activates G protein, Gq, and enhances intracellular Ca2+ levels. The EP3 receptor inhibits adenylate cyclase via Gi to reduce cAMP levels, whereas, contrarily, the EP2 and EP4 receptors activate G proteins that stimulate cAMP production via adenylate cyclase activation (40). It has been reported that PGE2-signaling through EP3 suppresses skin inflammation in murine contact hypersensitivity (41). In contrast, it has also been shown that PGE2 can exert immunosuppressive effects by binding to EP2 and EP4 (42). To date, a consensus on which EP is pro- or anti-inflammatory has not been reached.
As early as the 1980s, fish oil has been shown to modulate arthritis in rodent models through products of COX and lipogenase (43, 44). In 1995, Clair and Serhan (45) showed that the in vivo anti-inflammatory action of the NSAID, aspirin, is actually conferred by inducing 15-epi-LXA4 synthesis. 15-epi-LXA4 and LXA4 are trihydroxytetraene-containing eicosanoids which promote resolution of inflammation by enhancing apoptotic removal of dying neutrophils. In this study, we showed that, in chronic inflammation, a sustained presence of PGE2 is essential for mediating production of LXA4 to maintain homeostasis’s return in vivo (Fig. 10). This protracted dependency is different from what happens in an acute condition or at single cell level. In vitro, with polymorphonuclear neutrophils extracted from air pouch and bronchial epithelial cells isolated from acid-injured lung, PGE2 is only transiently produced to provide a transitional signal for inducing LXA4 production and mediating evolution of inflammation to resolution (2, 46). Whether the need for LXA4 is particular to autoimmunre arthritis remains to be investigated. During resolution of murine _Borrelia burgdorferi_-induced arthritis, another lipid mediator 15-HETE is formed instead (47).
COX is the rate-limiting enzyme for the synthesis of PGE; however, downstream PGE2 is synthesized by one of three terminal enzymes, cytosolic PG E synthase, mPGES-1, and mPGES-2. The noninducible cytosolic PG E synthase colocalizes with COX-1. MPGES-1 is preferentially associated with COX-2 but mPGES-2 has no preference. Since the incidence of Vioxx, many drug developers have switched their strategy from targeting COX-2 to blocking mPGES-1 instead (48). We found that mPGES-1 is restricted to the inflamed phase, suggesting that this isozyme is not a likely source of the PG in the resolution phase. Thus, mPGES-1 inhibitors may possibly be able to limit the severity of inflammation without interfering with resolution. The source/s of the PGE2 molecule remains an enigma currently. Perhaps, mPGES-2 is a candidate for providing the PGE involved in tissue homeostasis.
In sum, this study provides a new mechanistic insight regarding the resolution of inflammation in autoimmune arthritis with specific emphasis on the function of COX-2 and PGE2 and LXA4. It demonstrates that preserving the PGE2-mediated signaling mechanisms is critical, because if lost, the benefit of preventing induction will be mitigated. Currently, COX-2 inhibitors are widely used as an anti-inflammatory therapy. This is a salient concern, in addition to the fact that they increase the risk of stroke, especially in view of the fact that most patients seek treatment after arthritis has been established. Apparently, a more in depth understanding on the intricacies of the endogenous lipid mediators from the COX–LOX pathways is urgently warranted.
Acknowledgments
We thank Dr. Dunne Fong and Dr. Samuel Spadone for valuable discussion and statistic analysis, Dr. Robert Christman for help in analyzing the x-ray, and Dr. Suresh Adapala, Dr. Xinmin Zhang, Rodger Brown, and Kyle Evans for assistance throughout the project.
This work was supported by National Institutes of Health Grant R21 AR051761 and funding from the American Institute for Cancer Research (to M.C.).
Abbreviations used in this paper
15d-PGJ2
15-deoxy-Δ12,14-PGJ2 dmPGE2, 16,16 dimethyl-PGE2
CIA
collagen-induced arthritis
COX-2
cyclooxygenase-2
LXA4
lipoxin A4
mPGES-1
microsomal PGES-1
NS-398
_N_-[2-(cyclohexyloxy)-4-nitrophenyl]-methanesulfonamide
NSAID
nonsteriodal anti-inflammatory drug
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat. Med. 1999;5:698–701. doi: 10.1038/9550. [DOI] [PubMed] [Google Scholar]
- 2.Fukunaga K, Kohli P, Bonnans C, Fredenburgh LE, Levy BD. Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J. Immunol. 2005;174:5033–5039. doi: 10.4049/jimmunol.174.8.5033. [DOI] [PubMed] [Google Scholar]
- 3.Blais V, Turrin NP, Rivest S. Cyclooxygenase 2 (COX-2) inhibition increases the inflammatory response in the brain during systemic immune stimuli. J. Neurochem. 2005;95:1563–1574. doi: 10.1111/j.1471-4159.2005.03480.x. [DOI] [PubMed] [Google Scholar]
- 4.Yin H, Cheng L, Langenbach R, Ju C. Prostaglandin I(2) and E(2) mediate the protective effects of cyclooxygenase-2 in a mouse model of immune-mediated liver injury. Hepatology. 2007;45:159–169. doi: 10.1002/hep.21493. [DOI] [PubMed] [Google Scholar]
- 5.Reuter BK, Asfaha S, Buret A, Sharkey KA, Wallace JL. Exacerbation of inflammation-associated colonic injury in rat through inhibition of cyclooxygenase-2. J. Clin. Invest. 1996;98:2076–2085. doi: 10.1172/JCI119013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nat. Rev. Drug Discov. 2004;3:401–416. doi: 10.1038/nrd1383. [DOI] [PubMed] [Google Scholar]
- 7.Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LA, Perretti M, Rossi AG, Wallace JL. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwab JM, Arita M, Serhan CN. Resolvin E1 and protectin D activate inflammation-resolution programmes. Nature. 2007;447:869–874. doi: 10.1038/nature05877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haworth O, Levy BD. Endogenous lipid mediators in the resolution of airway inflammation. Eur. Respir. J. 2007;30:980–992. doi: 10.1183/09031936.00005807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krönke G, Katzenbeisser J, Uderhardt S, Zaiss MM, Scholtysek C, Schabbauer G, Zarbock A, Koenders MI, Axmann R, Zwerina J, et al. 12/15-lipoxygenase counteracts inflammation and tissue damage in arthritis. J. Immunol. 2009;183:3383–3389. doi: 10.4049/jimmunol.0900327. [DOI] [PubMed] [Google Scholar]
- 11.Lawrence T, Gilroy DW. Chronic inflammation: a failure of resolution? Int. J. Exp. Pathol. 2007;88:85–94. doi: 10.1111/j.1365-2613.2006.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melnikova I. Future of COX2 inhibitors. Nat. Rev. Drug Discov. 2005;4:453–454. doi: 10.1038/nrd1755. [DOI] [PubMed] [Google Scholar]
- 13.Williams RO. Collagen-induced arthritis as a model for rheumatoid arthritis. In: Corti A, Ghezzi P, editors. Methods in Molecular Medicine Vol. 98 Tumor Necrosis Factor Methods and Protocols. Totowa, NJ: Humana Press Inc; 2004. pp. 207–216. [DOI] [PubMed] [Google Scholar]
- 14.Terato K, Hasty KA, Cremer MA, Stuart JM, Townes AS, Kang AH. Collagen-induced arthritis in mice. Localization of an arthritogenic determinant to a fragment of the type II collagen molecule. J. Exp. Med. 1985;162:637–646. doi: 10.1084/jem.162.2.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adapala N, Chan MM. Long-term use of an antiinflammatory, curcumin, suppressed type 1 immunity and exacerbated visceral leishmaniasis in a chronic experimental model. Lab. Invest. 2008;88:1329–1339. doi: 10.1038/labinvest.2008.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furuzawa-Carballeda J, Vargas-Rojas MI, Cabral AR. Autoimmune inflammation from the Th17 perspective. Autoimmun. Rev. 2007;6:169–175. doi: 10.1016/j.autrev.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Maini RN, Brennan FM, Williams R, Chu CQ, Cope AP, Gibbons D, Elliott M, Feldmann M. TNF-alpha in rheumatoid arthritis and prospects of anti-TNF therapy. Clin. Exp. Rheumatol. 1993;11 Suppl 8:S173–S175. [PubMed] [Google Scholar]
- 18.Blaho VA, Mitchell WJ, Brown CR. Arthritis develops but fails to resolve during inhibition of cyclooxygenase 2 in a murine model of Lyme disease. Arthritis Rheum. 2008;58:1485–1495. doi: 10.1002/art.23371. [DOI] [PubMed] [Google Scholar]
- 19.Kapoor M, Kojima F, Yang L, Crofford LJ. Sequential induction of pro- and anti-inflammatory prostaglandins and peroxisome proliferators-activated receptor-gamma during normal wound healing: a time course study. Prostaglandins Leukot. Essent. Fatty Acids. 2007;76:103–112. doi: 10.1016/j.plefa.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajakariar R, Hilliard M, Lawrence T, Trivedi S, Colville-Nash P, Bellingan G, Fitzgerald D, Yaqoob MM, Gilroy DW. Hematopoietic prostaglandin D2 synthase controls the onset and resolution of acute inflammation through PGD2 and 15-deoxyDelta12 14 PGJ2. Proc. Natl. Acad. Sci. USA. 2007;104:20979–20984. doi: 10.1073/pnas.0707394104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schuligoi R, Grill M, Heinemann A, Peskar BA, Amann R. Sequential induction of prostaglandin E and D synthases in inflammation. Biochem. Biophys. Res. Commun. 2005;335:684–689. doi: 10.1016/j.bbrc.2005.07.130. [DOI] [PubMed] [Google Scholar]
- 22.Kojima F, Kato S, Kawai S. Prostaglandin E synthase in the pathophysiology of arthritis. Fundam. Clin. Pharmacol. 2005;19:255–261. doi: 10.1111/j.1472-8206.2005.00316.x. [DOI] [PubMed] [Google Scholar]
- 23.Roth S, Agrawal N, Mahowald M, Montoya H, Robbins D, Miller S, Nutting E, Woods E, Crager M, Nissen C, Swabb E. Misoprostol heals gastroduodenal injury in patients with rheumatoid arthritis receiving aspirin. Arch. Intern. Med. 1989;149:775–779. [PubMed] [Google Scholar]
- 24.Sheibanie AF, Khayrullina T, Safadi FF, Ganea D. Prostaglandin E2 exacerbates collagen-induced arthritis in mice through the inflammatory interleukin-23/interleukin-17 axis. Arthritis Rheum. 2007;56:2608–2619. doi: 10.1002/art.22794. [DOI] [PubMed] [Google Scholar]
- 25.Kojima F, Kapoor M, Yang L, Fleishaker EL, Ward MR, Monrad SU, Kottangada PC, Pace CQ, Clark JA, Woodward JG, Crofford LJ. Defective generation of a humoral immune response is associated with a reduced incidence and severity of collagen-induced arthritis in microsomal prostaglandin E synthase-1 null mice. J. Immunol. 2008;180:8361–8368. doi: 10.4049/jimmunol.180.12.8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, Pandher K, Lapointe JM, Saha S, Roach ML, et al. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc. Natl. Acad. Sci. USA. 2003;100:9044–9049. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonnans C, Fukunaga K, Levy MA, Levy BD. Lipoxin A(4) regulates bronchial epithelial cell responses to acid injury. Am. J. Pathol. 2006;168:1064–1072. doi: 10.2353/ajpath.2006.051056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seibert K, Lefkowith J, Tripp C, Isakson P, Needleman P. COX-2 inhibitors—is there cause for concern? Nat. Med. 1999;5:621–622. doi: 10.1038/9465. [DOI] [PubMed] [Google Scholar]
- 29.Taylor PC. Rheumatoid Arthritis in Practice. London: Royal Society of Medicine Press; 2006. [Google Scholar]
- 30.Reijman M, Bierma-Zeinstra SM, Pols HA, Koes BW, Stricker BH, Hazes JM. Is there an association between the use of different types of nonsteroidal antiinflammatory drugs and radiologic progression of osteoarthritis? The Rotterdam Study. Arthritis Rheum. 2005;52:3137–3142. doi: 10.1002/art.21357. [DOI] [PubMed] [Google Scholar]
- 31.Matuk R, Crawford J, Abreu MT, Targan SR, Vasiliauskas EA, Papadakis KA. The spectrum of gastrointestinal toxicity and effect on disease activity of selective cyclooxygenase-2 inhibitors in patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2004;10:352–356. doi: 10.1097/00054725-200407000-00005. [DOI] [PubMed] [Google Scholar]
- 32.Takeuchi K, Smale S, Premchand P, Maiden L, Sherwood R, Thjodleifsson B, Bjornsson E, Bjarnason I. Prevalence and mechanism of nonsteroidal anti-inflammatory drug-induced clinical relapse in patients with inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 2006;4:196–202. doi: 10.1016/s1542-3565(05)00980-8. [DOI] [PubMed] [Google Scholar]
- 33.Cohen AD, Bonneh DY, Reuveni H, Vardy DA, Naggan L, Halevy S. Drug exposure and psoriasis vulgaris: case-control and case-crossover studies. Acta Derm. Venereol. 2005;85:299–303. doi: 10.1080/00015550510032823. [DOI] [PubMed] [Google Scholar]
- 34.Katayama H, Kawada A. Exacerbation of psoriasis induced by indomethacin. J. Dermatol. 1981;8:323–327. doi: 10.1111/j.1346-8138.1981.tb02551.x. [DOI] [PubMed] [Google Scholar]
- 35.Wang XM, Wu TX, Lee YS, Dionne RA. Rofecoxib regulates the expression of genes related to the matrix metalloproteinase pathway in humans: implication for the adverse effects of cyclooxygenase-2 inhibitors. Clin. Pharmacol. Ther. 2006;79:303–315. doi: 10.1016/j.clpt.2005.12.306. [DOI] [PubMed] [Google Scholar]
- 36.Ben-Baruch A. Inflammation-associated immune suppression in cancer: the roles played by cytokines, chemokines and additional mediators. Semin. Cancer Biol. 2006;16:38–52. doi: 10.1016/j.semcancer.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 37.Baratelli F, Lin Y, Zhu L, Yang SC, Heuzé-Vourc’h N, Zeng G, Reckamp K, Dohadwala M, Sharma S, Dubinett SM. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J. Immunol. 2005;175:1483–1490. doi: 10.4049/jimmunol.175.3.1483. [DOI] [PubMed] [Google Scholar]
- 38.Gomez PF, Pillinger MH, Attur M, Marjanovic N, Dave M, Park J, Bingham CO, 3rd, Al-Mussawir H, Abramson SB. Resolution of inflammation: prostaglandin E2 dissociates nuclear trafficking of individual NF-kappaB subunits (p65, p50) in stimulated rheumatoid synovial fibroblasts. J. Immunol. 2005;175:6924–6930. doi: 10.4049/jimmunol.175.10.6924. [DOI] [PubMed] [Google Scholar]
- 39.Shibata Y, Ohata H, Yamashita M, Tsuji S, Bradfield JF, Nishiyama A, Henriksen RA, Myrvik QN. Immunologic response enhances atherosclerosis-type 1 helper T cell (Th1)-to-type 2 helper T cell (Th2) shift and calcified atherosclerosis in Bacillus Calmette-Guerin (BCG)-treated apolipoprotein E-knockout (apo E−/−) mice. Transl. Res. 2007;149:62–69. doi: 10.1016/j.trsl.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 40.Sugimoto Y, Narumiya S. Prostaglandin E receptors. J. Biol. Chem. 2007;282:11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- 41.Honda T, Matsuoka T, Ueta M, Kabashima K, Miyachi Y, Narumiya S. Prostaglandin E(2)-EP(3) signaling suppresses skin inflammation in murine contact hypersensitivity. J. Allergy Clin. Immunol. 2009;124:809–818. doi: 10.1016/j.jaci.2009.04.029. e2. [DOI] [PubMed] [Google Scholar]
- 42.Ogawa M, Suzuki J, Kosuge H, Takayama K, Nagai R, Isobe M. The mechanism of anti-inflammatory effects of prostaglandin E2 receptor 4 activation in murine cardiac transplantation. Transplantation. 2009;87:1645–1653. doi: 10.1097/TP.0b013e3181a5c84c. [DOI] [PubMed] [Google Scholar]
- 43.Prickett JD, Trentham DE, Robinson DR. Dietary fish oil augments the induction of arthritis in rats immunized with type II collagen. J. Immunol. 1984;132:725–729. [PubMed] [Google Scholar]
- 44.Leslie CA, Gonnerman WA, Ullman MD, Hayes KC, Franzblau C, Cathcart ES. Dietary fish oil modulates macrophage fatty acids and decreases arthritis susceptibility in mice. J. Exp. Med. 1985;162:1336–1349. doi: 10.1084/jem.162.4.1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clària J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc. Natl. Acad. Sci. USA. 1995;92:9475–9479. doi: 10.1073/pnas.92.21.9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 47.Blaho VA, Buczynski MW, Brown CR, Dennis EA. Lipidomic analysis of dynamic eicosanoid responses during the induction and resolution of Lyme arthritis. J. Biol. Chem. 2009;284:21599–21612. doi: 10.1074/jbc.M109.003822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Couzin J. Drug safety. Withdrawal of Vioxx casts a shadow over COX-2 inhibitors. Science. 2004;306:384–385. doi: 10.1126/science.306.5695.384. [DOI] [PubMed] [Google Scholar]