Role for Peroxynitrite in Sphingosine-1-Phosphate Induced Hyperalgesia in Rats (original) (raw)
. Author manuscript; available in PMC: 2012 Mar 1.
Abstract
Sphingosine-1-phosphate (S1P) is an important mediator of inflammation recently shown in in vitro studies to increase the excitability of small diameter sensory neurons at least in part via activation of the S1P1 receptor subtype. Activation of S1PR1 has been reported to increase the formation of NADPH oxidase-derived superoxide (O2•−) and nitric oxide synthase (NOS)-derived nitric oxide (NO). This process favors the formation of peroxynitrite (ONOO−, PN), a potent mediator of hyperalgesia associated with peripheral and central sensitization. The aims of our study were to determine whether S1P causes peripheral sensitization and thermal hyperalgesia via S1PR1 activation and PN formation. Intraplantar injection of S1P in rats led to a time-dependent development of thermal hyperalgesia that was blocked by the S1PR1 antagonist, W146 but not its inactive enantiomer, W140. The hyperalgesic effects of S1P were mimicked by intraplantar injection of the well characterized S1PR1 agonist, SEW2871. The development of S1P-induced hyperalgesia was blocked by apocynin, a NADPH oxidase inhibitor, L-NAME, a non-selective NOS inhibitor and by the potent PN decomposition catalysts (FeTM-4-PyP5+ and MnTE-2-PyP5+). Our findings provide mechanistic insight into the signaling pathways engaged by S1P in the development of hyperalgesia and highlight the contribution of the S1P1 receptor-to-PN signaling in this process.
Keywords: sphingosine-1-phosphate, peroxynitrite, sphingosine kinase, hyperalgesia, superoxide, nitric oxide
Introduction
The bioactive sphingosine-1-phosphate (S1P) is synthesized by phosphorylation of sphingosine by sphingosine kinases (SphK1 and 2) in a wide variety of cell types and in response to many extracellular stimuli including nerve growth factors and cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) [59; 64]. Once generated and released extracellularly, S1P binds its cognate receptors (S1PRs) with high affinity and initiates typical G-protein coupled receptor (GPCR) signaling pathways [63; 26; 64]. Five S1PRs have been identified to date (S1PR1–5) [63; 26; 64] and these are ubiquitously but differentially expressed on all cells [15; 64] and couple to different G proteins upon binding to S1P. Whereas S1PR1, S1PR4 and S1PR5 subtypes mainly couple to Gi/o, S1PR2 and S1PR3 subtype couple to Gi/o, Gq and G12/13 [40–41]. Over the last decade, S1P has emerged as a potent mediator of inflammation [58; 64] with important roles documented in several studies showing that inhibition of SphK (and in particular SphK1) and S1P1 receptor is anti-inflammatory in several animal models [58; 57]. Besides its role in inflammation, a potential role of S1P in peripheral sensitization and hyperalgesia is also documented by the observations that S1P directly increases the excitability of rat sensory neurons in vitro [69; 68] at least in part via activation of S1PR1 [11] and that S1P derived following bioconversion of ceramide, contributes to NGF-induced excitation of rat sensory neurons [70; 36].
Interestingly, S1P1 receptor stimulation activates the superoxide (O2•−)-generating enzyme NADPH (nicotinamide adenine dinucleotide phosphate) oxidase [37; 9; 61] and the nitric oxide (NO)-producing enzyme nitric oxide synthase (NOS) [20; 19; 42]. The interaction between O2•− and NO leads to the production of peroxynitrite (ONOO−, PN) [6] that acts as a potent proinflammatory nitroxidative species [44; 50; 49; 62] and a critical signaling molecule in the development of peripheral and central sensitization associated with pain of several etiologies [48; 23; 47]. In addition, we have recently reported that ceramide contributes to the development of morphine-induced hyperalgesia and antinociceptive tolerance [35] following its bioconversion to S1P in spinal cord which in turn signals via PN [32]. These observations prompt us to consider the potential contribution of PN in S1P-induced peripheral sensitization and hyperalgesia. Our results reveal that the S1P-to-S1P1 receptor pathway contributes to the development of S1P-induced thermal hyperalgesia in a peroxynitrite-dependent manner. These findings, when analyzed collectively with the emerging roles of sphingolipids such as ceramides and nitroxidative species, such as PN in pain, suggest that targeting the ceramide/S1P and nitroxidative pathways may offer novel approaches in pain management.
Materials and Methods
Materials
S1P, SEW2871, W146 and W140 were purchased from Cayman Chemical, Ann Arbor MI. Unless otherwise noted, all other chemicals and reagents were from Sigma-Aldrich (St. Louis MO). MnTE-2-PyP5+ was synthesized as described previously [4]. Charges on FeTM-4-PyP5+ and MnTE-2-PyP5+ are omitted for clarity on all Figures.
Experimental animals
Male Sprague Dawley rats (200–220 g) were purchased from Harlan (Indianapolis IN), housed 3–4 per cage, and maintained in a controlled environment (12 h light/dark cycles) with food and water available ad libitum. All experiments were performed in accordance with the International Association for the Study of Pain and the National Institutes of Health guidelines on laboratory animal welfare and the recommendations by Saint Louis University Institutional Animal Care and Use Committee.
Drug administration and induction of thermal hyperalgesia
Apocynin, L-NAME, FeTMPyP, MnTE-2-PyP5+ (or their respective vehicle, saline) W146, W140 (or their respective vehicle, 20% DMSO in saline) were given by intraplantar injection into the right hindpaw of rats 15 minutes before intraplantar injections of S1P or SEW2871 (or their respective vehicle, 6% ethanol in saline). All drugs were injected in a 5 μl injection volume using a Hamilton gauge needle (31/2″) in lightly anesthetized rats [CO2 (80%)/O2 (20%)]. Hyperalgesic responses to heat were determined by the Hargreaves’ Method using a Basile Plantar Test (Ugo Basile; Comeria, Italy) [16] with a cut-off latency of 20 s employed to prevent tissue damage. Rats were individually confined to Plexiglas chambers. A mobile infrared generator was positioned to deliver a thermal stimulus directly to an individual hindpaw from beneath the chamber. The withdrawal latency period of injected paws was determined with an electronic clock circuit and thermocouple. Results are expressed as Paw-Withdrawal Latency (s). Experiments were conducted with the experimenters blinded to treatment conditions
Immunoprecipitation and Western Blot analysis of protein nitration
Total paw lysates were obtained from flash-frozen plantar soft tissues harvested at the 5h time point. Frozen tissues were pulverized in liquid nitrogen-chilled mortar and pestles; then homogenized in 0.3 – 1 ml (4 volumes) of homogenization buffer [20 mM Tris–Cl (pH 7.4), 150 mM NaCl, 16.3 mM CHAPS, 0.5% Triton X-100, 0.1% SDS, 2 mM EGTA, 5% glycerol, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM sodium molybdinate, 2 mM PMSF, 1X phosphatase inhibitor cocktail (Sigma, St. Louis, MO, USA) (containing: cantharidin, bromotetramisole, and microcystin LR), 1X protease inhibitor cocktail (Sigma, St. Louis, MO, USA) (final concentration: 1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), 15 μM pepstatin A, 14 μM E-64, 40 μM bestatin, 20 μM leupeptin, and 850 nM aprotinin)]. Each lysate was sonicated for 10 min at 4°C; incubated for 1 hr, rotating at 4°C; and clarified by centrifugation at 13,000g, 4°C for 10 min. Total protein concentrations were determined by bicinchoninic acid assay (Pierce, Rockford, IL, USA). Nitrotyrosine-containing proteins in whole paw lysates were immunoprecipitated as previously described [30]. Paw lysate (1 mg total protein) was treated with 12 μg of agarose-conjugated anti-3-nitrotyrosine antibody (Millipore, Billerica, MA, USA) and diluted to 500 μl total volume with PBS pH 7.4 supplemented with 2 mM PMSF and 1X protease inhibitor cocktail. Samples were incubated overnight at 4°C, washed four times in 1X PBS, and denatured with 40 μl of 2X Laemmli Buffer. Samples were boiled 5 min, resolved on 4–20% SDS-PAGE (Bio-Rad, Hercules, CA, USA), and immunoblotted to PVDF membrane in Towbin buffer. The membranes were blocked with 5% low-fat milk in PBS pH 7.4 and 0.05% Tween-20 (PBS-T) for 2 hr at RT before overnight incubation at 4°C with rabbit anti-3-nitrotyrosine affinity purified antibody [14; 17] (2 μg/ml) in 2.5% low-fat milk/PBS-T. Membranes were washed 1×10 min and 4×5 min in PBS-T and incubated for 1 hr with horseradish-conjugated goat anti-rabbit IgG (1:1000, Pierce, Rockford, IL, USA). The membranes were washed 1×10 min and 4×5 min in PBS-T and NT-positive bands were visualization by enhanced chemiluminescence detection using a Fujifilm LAS-3000 imaging system with Image Reader LAS-3000 v2.2 (Fujifilm, Japan) software.
Statistical Analysis
All data are expressed as a mean ± SEM. The differences in levels of thermal hyperalgesia were assessed by two-way analysis of variance (ANOVA) with Bonferroni post hoc comparisons to S1P-treated animals where significance is defined at P<0.05.
Results
Sphingosine-1-phosphate induces thermal hyperalgesia via the S1P1 receptor
When compared to the vehicle group (rats in this group received an intraplantar injection of the vehicle used for W146 or W140 followed by intraplantar injection of the vehicle used for S1P), intraplantar injection of S1P (0.03–0.3 μg, n=4) led to a dose and time-dependent development of thermal hyperalgesia, which peaked by 2h and remained sustained up to the 5h time-point (Fig. 1A). The development of S1P (0.3 μg, n=4) induced thermal-hyperalgesia was blocked in a dose-dependent manner by the well-characterized S1P1 receptor antagonist, W146 (0.1–0.6 μg, n=4, Fig. 1B) [51; 40] but not by W140 (1.2 μg, n=4 Fig. 1C) its inactive S-enantiomer with doses chosen from previous studies [51; 40]. When tested alone and compared to rats that received an intraplantar injection of the vehicle used for S1P, W146 (0.6 μg, n = 4) or W140 (1.2 μg, n=4) had no effect on baseline withdrawal latencies (Fig. 1B, C). The role for S1PR1 in the development of thermal hyperalgesia was strengthened by the use of SEW2871, a well-characterized S1P1 receptor agonist [40–41]. When compared to the vehicle group (rats in this group received an intraplantar injection of the vehicle used for W146 followed by intraplantar injection of the vehicle used for SEW2871), intraplantar injection of SEW-2871 (0.3 μg, n=4) elicited a time-dependent development of thermal hyperalgesia that was maximal by 2h and sustained up to the 5h time period (Fig. 2). This effect was blocked as expected by the S1PR1 receptor antagonist, W146 (0.6 μg, n=4, Fig. 2). When tested alone and compared to rats that received an intraplantar injection of the vehicle used for SEW-2871, W146 (0.6 μg, n=4) had no effect on baseline withdrawal latencies (Fig. 2).
Fig. 1. S1P induced thermal hyperalgesia is blocked by a S1P receptor antagonist.

When compared to rats administered intraplantar W146/W140 vehicle and S1P vehicle (Veh, △), an intraplantar injection of S1P given at 0.03 μg (▲), 0.1 μg (●), or 0.3 μg (■) led to a time and dose-dependent development of thermal hyperalgesia (A). The development of S1P-mediated (0.3 μg, ■) thermal hyperalgesia was attenuated in a dose-dependent manner by the active S1PR1 antagonist, W146, given at 0.1 μg (▼), 0.3 μg (◆), or 0.6 μg (●) (B) but not of its inactive enantiomer W140 (1.2 μg, ▽, C). When tested alone W146 (0.6 μg, ○, B) or W140 ( 1.2 μg, ◇, C) and compared to animals receiving S1P vehicle W146 or W140 (△), had no effect on baseline withdrawal latency. Results are expressed as mean ± SEM for 4 rats. *P < 0.001 versus Veh, and † † P < 0.01 or ††P < 0.001 versus S1P by ANOVA with Bonferroni post hoc test.
Fig. 2. S1PR1 agonist, SEW2871, induces thermal hyperalgesia.
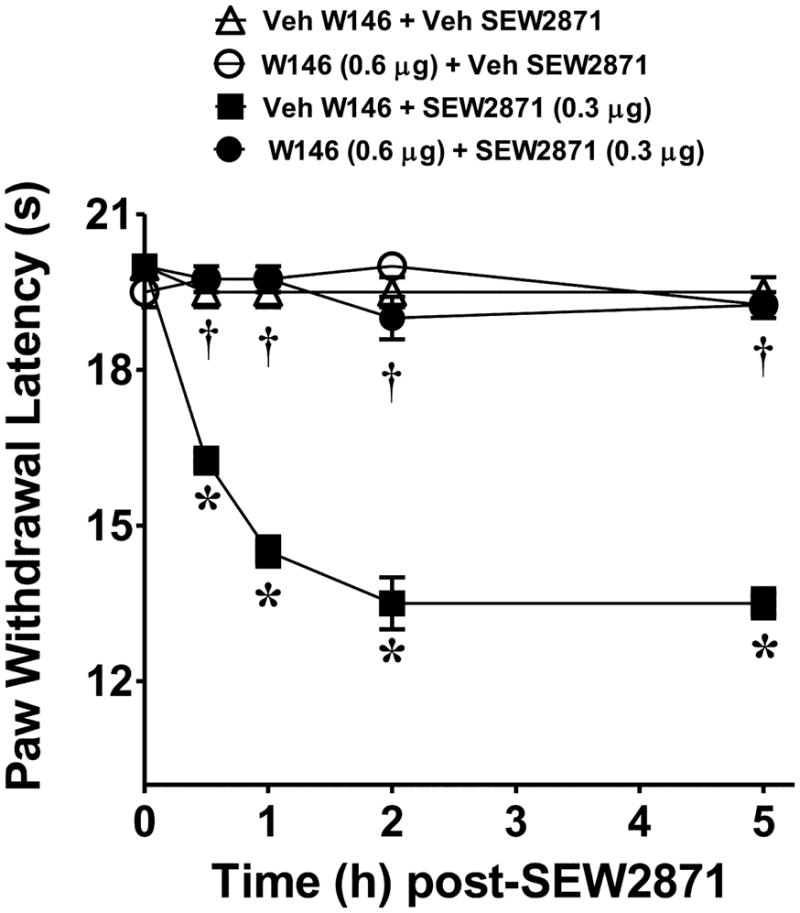
When compared to rats administered intraplantar W146/W140 vehicle and SEW2871 vehicle (Veh, △), an intraplantar injection of the S1PR1-specific agonist, SEW2871, given at 0.3 μg (■) induced time-dependant thermal hyperalgesia that was attenuated by intraplantar W146 (1.2 μg, ●). When tested alone and compared to animals receiving SEW2871 vehicle (△), intraplantar injection of W146 (1.2 μg, ○) had no effect on baseline withdrawal latency. Results are expressed as mean ± SEM for 4 rats. *P < 0.001 versus Veh, and †P < 0.001 versus SEW2871 by ANOVA with Bonferroni post hoc test.
Role for superoxide and nitric oxide in S1P induced thermal hyperalgesia
At doses known to block NADPH oxidase activity, intraplantar injection of apocynin (1 μg, n=4), a well characterized inhibitor of superoxide derived from activation of the NADPH oxidase [56; 60], attenuated the development of S1P (0.3 μg, n=4)-induced hyperalgesia (Fig. 3A). Likewise, the well characterized and non-selective inhibitor of nitric oxide synthase, NG-nitro-L-arginine methyl ester (L-NAME, 1.5 μg, n=4) [29], at doses known to block NOS activity mitigated the development of hyperalgesia in response to S1P (Fig. 3B). When tested alone and compared to rats that received an intraplantar injection of the vehicle used for S1P, apocynin or L-NAME had no effect on baseline withdrawal latencies (Fig. 3A, B).
Fig. 3. Role of superoxide and nitric oxide in S1P induced thermal hyperalgesia.

When compared to rats administered intraplantar apocynin/L-NAME vehicle and S1P vehicle (Veh, △), an intraplantar injection of S1P (0.3 μg, ▲, A and B) led to a time-dependent development of thermal hyperalgesia that was attenuated by intraplantar administration of apocynin (1 μg, ◆, A) or L-NAME (1.5 μg, ●, B). When tested alone and compared to animals receiving S1P vehicle (Veh, △), intraplantar injection of apocynin (1 μg, ◇, A) or L-NAME (1.5 μg, ○, B) had no effect on baseline withdrawal latency. Results are expressed as mean ± SEM for 4 rats. *P < 0.001 versus Veh, and †P < 0.001 versus S1P by ANOVA with Bonferroni post hoc test.
Peroxynitrite formation from nitric oxide and superoxide is a key mediator in S1P induced thermal hyperalgesia
Intraplantar injection of FeTMPyP (1–6 μg, n=4, Fig. 4A) or MnTE-2-PyP5+ (1–6 μg, n=4, Fig. 4B) blocked the development of S1P (0.3 μg, n=4)-induced hyperalgesia in a dose and time-dependent manner. When tested alone and compared to rats that received an intraplantar injection of the vehicle used for S1P, FeTMPyP5+ (6 μg, n=4) or MnTE-2-PyP5+ (6 μg, n=4) had no effect on baseline withdrawal latencies (Fig. 4A, B). We have attempted to measure increased formation of 3-nitrotyrosine (NT), a well characterized and widely used marker of PN [31; 62] by immunoprecipitation in paw tissues following intraplantar injection of S1P. Our results yielded inadequate signal to detect changes in NT formation between the groups. These results may have resulted from insufficient sensitivity of the IP method for NT in these tissues as indicated by low signal or may have resulted from a highly localized production of NT in the paw that is capable of participating in the development of hyperalgesia, but whose signal is undetectable in a total paw preparation. Regardless, pharmacological targeting with well characterized PN decomposition catalysts such as FeTMPyP5+ and MnTE-2-PyP5+ clearly support the contribution of PN in S1P- mediated thermal hyperalgesia.
Fig. 4. Role of peroxynitrite in S1P induced thermal hyperalgesia.

When compared to rats administered intraplantar FeTMPyP or MnTE-2-PyP5+ vehicle and S1P vehicle (Veh, △), an intraplantar injection of S1P given at 0.3 μg (▲, A and B) led to a time-dependent development of thermal hyperalgesia that was attenuated in a dose-dependent manner by intraplantar FeTMPyP (A) or MnTE-2-PyP5+ (B) given at 1 μg (●), 3 μg (■) or 6 μg (◆). When tested alone and compared to animals receiving S1P vehicle (Veh, △), intraplantar administration (6 μg) of FeTMPyP (○, A) or MnTE-2-PyP5+ (○, B) had no effect on baseline withdrawal latency. Results are expressed as mean ± SEM for 4 rats. *P < 0.001 versus Veh, and †P < 0.001 versus S1P by ANOVA with Bonferroni correction
Discussion
In this study, we demonstrate that S1P is an important mediator of peripheral sensitization and thermal hyperalgesia acting via the S1PR1 –to- PN signaling pathway. To this end, S1P-mediated thermal hyperalgesia was attenuated by the well characterized S1PR1 antagonist W146 but not by its inactive enantiomer W140 [51; 40]. In addition, the hyperalgesic effects of S1P were mimicked by the well known S1PR1 agonist SEW2871 [51; 40] and SEW-2871 induced hyperalgesia was attenuated by W146. A literature survey of S1PR1 activation in research unrelated to pain mechanisms revealed that activation of S1PR1 subtype by S1P can activate in some cell types, the NADPH oxidase and to activate and/or induce the NO-generating enzyme, NOS [37; 39; 65–66; 9; 61]. The multicomponent membrane-associated NADPH oxidase system has been shown over the years to be a central source for O2•− generation during several pathological conditions [34]. This O2•− generating enzyme is dormant in resting cells and produces O2•− only upon activation. The principal regulation of NADPH oxidase is post-translational and depends on the assembly of several membrane-bound and cytosolic components to form an active enzyme complex [2]. In resting cells, the enzyme consists of two membrane-bound components, gp91_phox_ and p22_phox_, and several cytosolic components, including p47_phox_, p40_phox_, p67_phox_, and Rac1/2 [2]. Gp91_phox_ is a flavocytochrome and the catalytic core of the enzyme. Upon activation, the cytosolic components translocate to the membrane and associate with membrane components to form an assembled, activated, and O2•− producing enzyme complex [2]. Importantly, O2•− auto-augments its formation by up-regulating the expression of the Rac1 and gp91_phox_ subunits of the holoenzyme creating a self-perpetuating cascade [38; 33]. The mechanisms whereby S1P activates this enzyme are not fully understood but include activation of Rac small GTPase [66] and PKCζ [39; 65]. In our studies, the critical role of the NADPH oxidase as a source of O2•− in PN formation was highlighted by the findings that local administration of apocynin blocked the development of S1P induced hyperalgesia. Apocynin prevents serine phosphorylation of p47_phox_ and blocks its association with gp91phox, thus blunting NADPH oxidase activation [56; 60]. The important role of NOS as a source of NO-derived PN was highlighted in our studies by the findings that local administration of L-NAME, a well characterized non-selective inhibitor of NOS [29], blocked the development of S1P-induced hyperalgesia. Because NO is known to react with O2•− at a diffusion-limited rate to form PN [6] results obtained with apocynin and L-NAME indirectly suggest that PN from these reactive oxygen and nitrogen species is the common denominator in the molecular and biochemical pathways leading to S1P-induced thermal hyperalgesia. The role of PN was confirmed by the use two well characterized PN decomposition catalysts, namely, FeTMPyP5+ and MnTE-2-PyP5+ [27; 44; 50; 3]. These results establish that PN formed in response to S1P activation of S1PR1 is a key signaling molecule in the development of S1P-induced thermal hyperalgesia. It is also likely that S1P via activation of S1PR1 directly sensitizes peripheral sensory afferents as shown in vitro studies [11] thus contributing to peripheral sensitization and hyperalgesia.
Although our results have identified potential enzymatic sources in the formation of PN (NADPH oxidase and NOS), our results have not identified the cellular sources of PN. To this end and since NADPH oxidase and NOS enzymes are expressed not only in resident cells of the paw (i.e. endothelial cell and resident tissue neutrophils/macrophages and mast cells) [1; 25; 13; 21] but also in peripheral nociceptors [7–8], it is possible and likely that activation of S1P1 receptors in both contribute to PN formation. The mechanisms whereby PN contribute to S1P-induced hyperalgesia must be multiple since PN contributes to peripheral and central sensitization by acting at multiple sites and through several biomolecular signaling pathways [see [48; 23; 47] for updated review articles]. For example, and in the context of the results presented in this paper, it is possible that PN contributes to the development of S1P-induced peripheral sensitization and hyperalgesia by favoring the in situ production of mediators such as PGE2 or TNF-α and IL-1β that, in addition to their pro-inflammatory effects, are also known to sensitize peripheral nociceptors [52; 10; 53]. These possibilities are supported by the fact that PN activates/induces COX-1 and COX-2 to increase a variety of prostaglandins including PGE2 [see [45; 28] for review articles] and that PN can also activate redox-sensitive transcription factors and MAPK kinases to increase the production of various cytokines [see [48; 47] for review articles]. Additionally, PN may contribute to the development of S1P-induced hyperalgesia by altering the sensitivity of TRPV1 on peripheral nociceptors highlighted by evidence that NADPH oxidase augments capsaicin-induced TRPV1 activity in DRG neurons through PKC activation and translocation [18]; nitro-oleic acid, a fatty acid nitrated by NO or PN [5], evokes TRPV1-mediated Ca2+ currents in DRG neurons [54]; and S-nitrosylation of cysteine residues of TRPV1 elicit Ca2+ influx into transfected HEK cells [67]. In light of its potent chemotactic activity [44; 50; 46], PN also may contribute to the development of S1P-induced hyperalgesia by favoring neutrophil infiltration that in turn perpetuates the cycle by providing mediators able to sensitize peripheral nociceptors [22; 24] or directly activate them [43]. These possibilities highlighted in the schematic shown in Fig. 5 are currently being investigated in our laboratories. It should be recognized that S1P has also been reported to exert antinociceptive effects [12; 55]. However, our studies reveal a potential multifaceted role of S1P in nociceptive processing that could depend on the site of S1P biosynthesis and the relative distribution of the S1P receptors involved in signaling. Although in our study we focused on the involvement of S1P1 receptors, we are not excluding the contribution of other S1P receptors such as S1PR2that will be investigated in future studies. In summary, our results have identified S1P acting via the S1P1 receptor as a potent inducer of peripheral sensitization and hyperalgesia signaling via peroxynitrite.
Fig. 5. Illustration of proposed S1PR1-to-PN signaling in S1P-induced hyperalgesia.

Activation of S1PR1 with its S1P ligand induces the apocynin-sensitive translocation of cytosolic subunits of NADPH oxidase to membrane-bound gp91_phox_ and p22_phox_ forming the active superoxide producing NADPH oxidase holoenzyme. NADPH oxidase-derived superoxide, together with nitric oxide produced from NOS activation, form PN leading to the development of peripheral sensitization and hyperalgesia. Thus, peroxynitrite decomposition with PN decomposition catalysts (i.e. FeTMPyP or MnTE-2-PyP5+) inhibit hyperalgesia. Possible mechanisms by which PN facilitates peripheral sensitization and hyperalgesia include increased TRPV1 sensitization and activation, leukocyte infiltration, and increased formation of prostaglandins (i.e. PGE2 synthesis) or cytokines (i.e. TNF-α and IL-1β).
Acknowledgments
Supported by R01 DA024074 and R21 DA023056 (DS). The authors declare no conflicts of interest. We thank Dr Ines Batinic-Haberle (Duke University) for providing MnTE-2-PyP5+ and Dr Harold Ischiropolous (Children’s Hospital of Philadelphia Research Institute) for providing the anti-3-nitrotyrosine antibody.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Babior BM, Kipnes RS, Curnutte JT. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J Clin Invest. 1973;52(3):741–744. doi: 10.1172/JCI107236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys. 2002;397(2):342–344. doi: 10.1006/abbi.2001.2642. [DOI] [PubMed] [Google Scholar]
- 3.Batinic-Haberle I, Spasojevic I, Tse HM, Tovmasyan A, Rajic Z, St Clair DK, Vujaskovic Z, Dewhirst MW, Piganelli JD. Design of Mn porphyrins for treating oxidative stress injuries and their redox-based regulation of cellular transcriptional activities. Amino Acids. 2010 doi: 10.1007/s00726-010-0603-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batinic-Haberle ISI, Stevens RD, Hambright P, Fridovich I. Manganese(III) Meso Tetrakis Ortho N-alkylpyridylporphyrins. Synthesis, Characterization and Catalysis of O2•− Dismutation. J Chem Soc, Dalton Trans. 2002;(30):2689–2696. [Google Scholar]
- 5.Batthyany C, Schopfer FJ, Baker PR, Duran R, Baker LM, Huang Y, Cervenansky C, Branchaud BP, Freeman BA. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J Biol Chem. 2006;281(29):20450–20463. doi: 10.1074/jbc.M602814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87(4):1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bredt DS, Hwang PM, Snyder SH. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347(6295):768–770. doi: 10.1038/347768a0. [DOI] [PubMed] [Google Scholar]
- 8.Cao X, Demel SL, Quinn MT, Galligan JJ, Kreulen D. Localization of NADPH oxidase in sympathetic and sensory ganglion neurons and perivascular nerve fibers. Auton Neurosci. 2009;151(2):90–97. doi: 10.1016/j.autneu.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Catarzi S, Giannoni E, Favilli F, Meacci E, Iantomasi T, Vincenzini MT. Sphingosine 1-phosphate stimulation of NADPH oxidase activity: relationship with platelet-derived growth factor receptor and c-Src kinase. Biochim Biophys Acta. 2007;1770(6):872–883. doi: 10.1016/j.bbagen.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 10.Cheng JK, Ji RR. Intracellular signaling in primary sensory neurons and persistent pain. Neurochem Res. 2008;33(10):1970–1978. doi: 10.1007/s11064-008-9711-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chi XX, Nicol GD. The sphingosine 1-phosphate receptor, S1PR1, plays a prominent but not exclusive role in enhancing the excitability of sensory neurons. J Neurophysiol. 2010 doi: 10.1152/jn.00709.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coste O, Brenneis C, Linke B, Pierre S, Maeurer C, Becker W, Schmidt H, Gao W, Geisslinger G, Scholich K. Sphingosine 1-phosphate modulates spinal nociceptive processing. J Biol Chem. 2008;283(47):32442–32451. doi: 10.1074/jbc.M806410200. [DOI] [PubMed] [Google Scholar]
- 13.Forstermann U, Pollock JS, Schmidt HH, Heller M, Murad F. Calmodulin-dependent endothelium-derived relaxing factor/nitric oxide synthase activity is present in the particulate and cytosolic fractions of bovine aortic endothelial cells. Proc Natl Acad Sci U S A. 1991;88(5):1788–1792. doi: 10.1073/pnas.88.5.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fries DM, Paxinou E, Themistocleous M, Swanberg E, Griendling KK, Salvemini D, Slot JW, Heijnen HF, Hazen SL, Ischiropoulos H. Expression of inducible nitric-oxide synthase and intracellular protein tyrosine nitration in vascular smooth muscle cells: role of reactive oxygen species. J Biol Chem. 2003;278(25):22901–22907. doi: 10.1074/jbc.M210806200. [DOI] [PubMed] [Google Scholar]
- 15.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9(2):139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 16.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32(1):77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 17.Heijnen HF, van Donselaar E, Slot JW, Fries DM, Blachard-Fillion B, Hodara R, Lightfoot R, Polydoro M, Spielberg D, Thomson L, Regan EA, Crapo J, Ischiropoulos H. Subcellular localization of tyrosine-nitrated proteins is dictated by reactive oxygen species generating enzymes and by proximity to nitric oxide synthase. Free Radic Biol Med. 2006;40(11):1903–1913. doi: 10.1016/j.freeradbiomed.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Ibi M, Matsuno K, Shiba D, Katsuyama M, Iwata K, Kakehi T, Nakagawa T, Sango K, Shirai Y, Yokoyama T, Kaneko S, Saito N, Yabe-Nishimura C. Reactive oxygen species derived from NOX1/NADPH oxidase enhance inflammatory pain. J Neurosci. 2008;28(38):9486–9494. doi: 10.1523/JNEUROSCI.1857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Igarashi J, Bernier SG, Michel T. Sphingosine 1-phosphate and activation of endothelial nitric-oxide synthase. differential regulation of Akt and MAP kinase pathways by EDG and bradykinin receptors in vascular endothelial cells. J Biol Chem. 2001;276(15):12420–12426. doi: 10.1074/jbc.M008375200. [DOI] [PubMed] [Google Scholar]
- 20.Igarashi J, Michel T. Agonist-modulated targeting of the EDG-1 receptor to plasmalemmal caveolae. eNOS activation by sphingosine 1-phosphate and the role of caveolin-1 in sphingolipid signal transduction. J Biol Chem. 2000;275(41):32363–32370. doi: 10.1074/jbc.M003075200. [DOI] [PubMed] [Google Scholar]
- 21.Jones SA, O’Donnell VB, Wood JD, Broughton JP, Hughes EJ, Jones OT. Expression of phagocyte NADPH oxidase components in human endothelial cells. Am J Physiol. 1996;271(4 Pt 2):H1626–1634. doi: 10.1152/ajpheart.1996.271.4.H1626. [DOI] [PubMed] [Google Scholar]
- 22.Kochukov MY, McNearney TA, Yin H, Zhang L, Ma F, Ponomareva L, Abshire S, Westlund KN. Tumor necrosis factor-alpha (TNF-alpha) enhances functional thermal and chemical responses of TRP cation channels in human synoviocytes. Mol Pain. 2009;5:49. doi: 10.1186/1744-8069-5-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Little JW, Doyle T, Salvemini D. Reactive nitroxidative species and nociceptive processing: determining the roles for nitric oxide, superoxide, and peroxynitrite in pain. Amino Acids. 2010 doi: 10.1007/s00726-010-0633-0. [DOI] [PubMed] [Google Scholar]
- 24.Ma F, Zhang L, Westlund KN. Reactive oxygen species mediate TNFR1 increase after TRPV1 activation in mouse DRG neurons. Mol Pain. 2009;5:31. doi: 10.1186/1744-8069-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marletta MA, Yoon PS, Iyengar R, Leaf CD, Wishnok JS. Macrophage oxidation of L-arginine to nitrite and nitrate: nitric oxide is an intermediate. Biochemistry. 1988;27(24):8706–8711. doi: 10.1021/bi00424a003. [DOI] [PubMed] [Google Scholar]
- 26.Melendez AJ. Sphingosine kinase signalling in immune cells: potential as novel therapeutic targets. Biochim Biophys Acta. 2008;1784(1):66–75. doi: 10.1016/j.bbapap.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 27.Misko TP, Highkin MK, Veenhuizen AW, Manning PT, Stern MK, Currie MG, Salvemini D. Characterization of the cytoprotective action of peroxynitrite decomposition catalysts. J Biol Chem. 1998;273(25):15646–15653. doi: 10.1074/jbc.273.25.15646. [DOI] [PubMed] [Google Scholar]
- 28.Mollace V, Muscoli C, Masini E, Cuzzocrea S, Salvemini D. Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacol Rev. 2005;57(2):217–252. doi: 10.1124/pr.57.2.1. [DOI] [PubMed] [Google Scholar]
- 29.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43(2):109–142. [PubMed] [Google Scholar]
- 30.Muscoli C, Cuzzocrea S, Ndengele MM, Mollace V, Porreca F, Fabrizi F, Esposito E, Masini E, Matuschak GM, Salvemini D. Therapeutic manipulation of peroxynitrite attenuates the development of opiate-induced antinociceptive tolerance in mice. J Clin Invest. 2007;117(11):3530–3539. doi: 10.1172/JCI32420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muscoli C, Cuzzocrea S, Riley DP, Zweier JL, Thiemermann C, Wang ZQ, Salvemini D. On the selectivity of superoxide dismutase mimetics and its importance in pharmacological studies. Br J Pharmacol. 2003;140(3):445–460. doi: 10.1038/sj.bjp.0705430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muscoli C, Doyle T, Dagostino C, Bryant L, Chen Z, Watkins LR, Ryerse J, Bieberich E, Neumman W, Salvemini D. Counter-regulation of opioid analgesia by glial-derived bioactive sphingolipids. J Neuroscience. 2010 doi: 10.1523/JNEUROSCI.2391-10.2010. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muzaffar S, Shukla N, Angelini GD, Jeremy JY. Superoxide auto-augments superoxide formation and upregulates gp91(phox) expression in porcine pulmonary artery endothelial cells: inhibition by iloprost. Eur J Pharmacol. 2006;538(1–3):108–114. doi: 10.1016/j.ejphar.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 34.Nauseef WM. The NADPH-dependent oxidase of phagocytes. Proc Assoc Am Physicians. 1999;111(5):373–382. doi: 10.1111/paa.1999.111.5.373. [DOI] [PubMed] [Google Scholar]
- 35.Ndengele MM, Cuzzocrea S, Masini E, Vinci MC, Esposito E, Muscoli C, Petrusca DN, Mollace V, Mazzon E, Li D, Petrache I, Matuschak GM, Salvemini D. Spinal ceramide modulates the development of morphine antinociceptive tolerance via peroxynitrite-mediated nitroxidative stress and neuroimmune activation. J Pharmacol Exp Ther. 2009;329(1):64–75. doi: 10.1124/jpet.108.146290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nicol GD. Nerve growth factor, sphingomyelins, and sensitization in sensory neurons. Sheng Li Xue Bao. 2008;60(5):603–604. [PubMed] [Google Scholar]
- 37.Paik JH, Chae S, Lee MJ, Thangada S, Hla T. Sphingosine 1-phosphate-induced endothelial cell migration requires the expression of EDG-1 and EDG-3 receptors and Rho-dependent activation of alpha vbeta3- and beta1-containing integrins. J Biol Chem. 2001;276(15):11830–11837. doi: 10.1074/jbc.M009422200. [DOI] [PubMed] [Google Scholar]
- 38.Puntambekar P, Mukherjea D, Jajoo S, Ramkumar V. Essential role of Rac1/NADPH oxidase in nerve growth factor induction of TRPV1 expression. J Neurochem. 2005;95(6):1689–1703. doi: 10.1111/j.1471-4159.2005.03518.x. [DOI] [PubMed] [Google Scholar]
- 39.Reinehr R, Becker S, Eberle A, Grether-Beck S, Haussinger D. Involvement of NADPH oxidase isoforms and Src family kinases in CD95-dependent hepatocyte apoptosis. J Biol Chem. 2005;280(29):27179–27194. doi: 10.1074/jbc.M414361200. [DOI] [PubMed] [Google Scholar]
- 40.Rosen H, Gonzalez-Cabrera P, Marsolais D, Cahalan S, Don AS, Sanna MG. Modulating tone: the overture of S1P receptor immunotherapeutics. Immunol Rev. 2008;223:221–235. doi: 10.1111/j.1600-065X.2008.00645.x. [DOI] [PubMed] [Google Scholar]
- 41.Rosen H, Gonzalez-Cabrera PJ, Sanna MG, Brown S. Sphingosine 1-phosphate receptor signaling. Annu Rev Biochem. 2009;78:743–768. doi: 10.1146/annurev.biochem.78.072407.103733. [DOI] [PubMed] [Google Scholar]
- 42.Roviezzo F, Bucci M, Delisle C, Brancaleone V, Di Lorenzo A, Mayo IP, Fiorucci S, Fontana A, Gratton JP, Cirino G. Essential requirement for sphingosine kinase activity in eNOS-dependent NO release and vasorelaxation. FASEB J. 2006;20(2):340–342. doi: 10.1096/fj.05-4647fje. [DOI] [PubMed] [Google Scholar]
- 43.Russell FA, Fernandes ES, Courade JP, Keeble JE, Brain SD. Tumour necrosis factor alpha mediates transient receptor potential vanilloid 1-dependent bilateral thermal hyperalgesia with distinct peripheral roles of interleukin-1beta, protein kinase C and cyclooxygenase-2 signalling. Pain. 2009;142(3):264–274. doi: 10.1016/j.pain.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 44.Salvemini D, Jensen MP, Riley DP, Misko TP. Therapeutic manipulations of peroxynitrite. Drug News Perspect. 1998;11(4):204–214. [PubMed] [Google Scholar]
- 45.Salvemini D, Masferrer JL. Interactions of nitric oxide with cyclooxygenase: in vitro, ex vivo, and in vivo studies. Methods Enzymol. 1996;269:12–25. doi: 10.1016/s0076-6879(96)69005-3. [DOI] [PubMed] [Google Scholar]
- 46.Salvemini D, Mazzon E, Dugo L, Riley DP, Serraino I, Caputi AP, Cuzzocrea S. Pharmacological manipulation of the inflammatory cascade by the superoxide dismutase mimetic, M40403. Br J Pharmacol. 2001;132(4):815–827. doi: 10.1038/sj.bjp.0703841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salvemini D, Neumann W. Targeting peroxynitrite driven nitroxidative stress with synzymes: A novel therapeutic approach in chronic pain management. Life Sci. 2010;86(15–16):604–614. doi: 10.1016/j.lfs.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 48.Salvemini D, Neumann WL. Peroxynitrite: a strategic linchpin of opioid analgesic tolerance. Trends Pharmacol Sci. 2009;30(4):194–202. doi: 10.1016/j.tips.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 49.Salvemini D, Riley DP, Cuzzocrea S. SOD mimetics are coming of age. Nat Rev Drug Discov. 2002;1(5):367–374. doi: 10.1038/nrd796. [DOI] [PubMed] [Google Scholar]
- 50.Salvemini D, Wang ZQ, Stern MK, Currie MG, Misko TP. Peroxynitrite decomposition catalysts: therapeutics for peroxynitrite-mediated pathology. Proc Natl Acad Sci U S A. 1998;95(5):2659–2663. doi: 10.1073/pnas.95.5.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanna MG, Wang SK, Gonzalez-Cabrera PJ, Don A, Marsolais D, Matheu MP, Wei SH, Parker I, Jo E, Cheng WC, Cahalan MD, Wong CH, Rosen H. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol. 2006;2(8):434–441. doi: 10.1038/nchembio804. [DOI] [PubMed] [Google Scholar]
- 52.Schafers M, Lee DH, Brors D, Yaksh TL, Sorkin LS. Increased sensitivity of injured and adjacent uninjured rat primary sensory neurons to exogenous tumor necrosis factor-alpha after spinal nerve ligation. J Neurosci. 2003;23(7):3028–3038. doi: 10.1523/JNEUROSCI.23-07-03028.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schafers M, Sommer C, Geis C, Hagenacker T, Vandenabeele P, Sorkin LS. Selective stimulation of either tumor necrosis factor receptor differentially induces pain behavior in vivo and ectopic activity in sensory neurons in vitro. Neuroscience. 2008;157(2):414–423. doi: 10.1016/j.neuroscience.2008.08.067. [DOI] [PubMed] [Google Scholar]
- 54.Sculptoreanu A, Kullmann FA, Artim DE, Bazley FA, Schopfer F, Woodcock S, Freeman BA, de Groat WC. Nitro-oleic acid inhibits firing and activates TRPV1- and TRPA1-mediated inward currents in dorsal root ganglion neurons from adult male rats. J Pharmacol Exp Ther. 2010;333(3):883–895. doi: 10.1124/jpet.109.163154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sim-Selley LJ, Goforth PB, Mba MU, Macdonald TL, Lynch KR, Milstien S, Spiegel S, Satin LS, Welch SP, Selley DE. Sphingosine-1-phosphate receptors mediate neuromodulatory functions in the CNS. J Neurochem. 2009;110(4):1191–1202. doi: 10.1111/j.1471-4159.2009.06202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simons JM, Hart BA, Ip Vai Ching TR, Van Dijk H, Labadie RP. Metabolic activation of natural phenols into selective oxidative burst agonists by activated human neutrophils. Free Radic Biol Med. 1990;8(3):251–258. doi: 10.1016/0891-5849(90)90070-y. [DOI] [PubMed] [Google Scholar]
- 57.Snider AJ, Orr Gandy KA, Obeid LM. Sphingosine kinase: Role in regulation of bioactive sphingolipid mediators in inflammation. Biochimie. 2010;92(6):707–715. doi: 10.1016/j.biochi.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4(5):397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 59.Spiegel S, Milstien S. Functions of the multifaceted family of sphingosine kinases and some close relatives. J Biol Chem. 2007;282(4):2125–2129. doi: 10.1074/jbc.R600028200. [DOI] [PubMed] [Google Scholar]
- 60.Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11(1):95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- 61.Straub AC, Klei LR, Stolz DB, Barchowsky A. Arsenic requires sphingosine-1-phosphate type 1 receptors to induce angiogenic genes and endothelial cell remodeling. Am J Pathol. 2009;174(5):1949–1958. doi: 10.2353/ajpath.2009.081016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6(8):662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 63.Taha TA, Argraves KM, Obeid LM. Sphingosine-1-phosphate receptors: receptor specificity versus functional redundancy. Biochim Biophys Acta. 2004;1682(1–3):48–55. doi: 10.1016/j.bbalip.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 64.Takabe K, Paugh SW, Milstien S, Spiegel S. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. 2008;60(2):181–195. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Won JS, Singh I. Sphingolipid signaling and redox regulation. Free Radic Biol Med. 2006;40(11):1875–1888. doi: 10.1016/j.freeradbiomed.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 66.Yi F, Zhang AY, Li N, Muh RW, Fillet M, Renert AF, Li PL. Inhibition of ceramide-redox signaling pathway blocks glomerular injury in hyperhomocysteinemic rats. Kidney Int. 2006;70(1):88–96. doi: 10.1038/sj.ki.5001517. [DOI] [PubMed] [Google Scholar]
- 67.Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, Tominaga M, Shimizu S, Sato Y, Mori Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol. 2006;2(11):596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
- 68.Zhang YH, Fehrenbacher JC, Vasko MR, Nicol GD. Sphingosine-1-phosphate via activation of a G-protein-coupled receptor(s) enhances the excitability of rat sensory neurons. J Neurophysiol. 2006;96(3):1042–1052. doi: 10.1152/jn.00120.2006. [DOI] [PubMed] [Google Scholar]
- 69.Zhang YH, Vasko MR, Nicol GD. Ceramide, a putative second messenger for nerve growth factor, modulates the TTX-resistant Na(+) current and delayed rectifier K(+) current in rat sensory neurons. J Physiol. 2002;544(Pt 2):385–402. doi: 10.1113/jphysiol.2002.024265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang YH, Vasko MR, Nicol GD. Intracellular sphingosine 1-phosphate mediates the increased excitability produced by nerve growth factor in rat sensory neurons. J Physiol. 2006;575(Pt 1):101–113. doi: 10.1113/jphysiol.2006.111575. [DOI] [PMC free article] [PubMed] [Google Scholar]