Core and Panmetabolism in Escherichia coli (original) (raw)
Abstract
Escherichia coli exhibits a wide range of lifestyles encompassing commensalism and various pathogenic behaviors which its highly dynamic genome contributes to develop. How environmental and host factors shape the genetic structure of E. coli strains remains, however, largely unknown. Following a previous study of E. coli genomic diversity, we investigated its diversity at the metabolic level by building and analyzing the genome-scale metabolic networks of 29 E. coli strains (8 commensal and 21 pathogenic strains, including 6 Shigella strains). Using a tailor-made reconstruction strategy, we significantly improved the completeness and accuracy of the metabolic networks over default automatic reconstruction processes. Among the 1,545 reactions forming E. coli panmetabolism, 885 reactions were common to all strains. This high proportion of core reactions (57%) was found to be in sharp contrast to the low proportion (13%) of core genes in the E. coli pangenome, suggesting less diversity of metabolic functions compared to that of all gene functions. Core reactions were significantly overrepresented among biosynthetic reactions compared to the more variable degradation processes. Differences between metabolic networks were found to follow E. coli phylogeny rather than pathogenic phenotypes, except for Shigella networks, which were significantly more distant from the others. This suggests that most metabolic changes in non-Shigella strains were not driven by their pathogenic phenotypes. Using a supervised method, we were yet able to identify small sets of reactions related to pathogenicity or commensalism. The quality of our reconstructed networks also makes them reliable bases for building metabolic models.
Escherichia coli is a versatile species encompassing commensal organisms, as well as intraintestinal E. coli (InPEc) and extraintestinal E. coli (ExPEc) pathogens (27, 49). This variety of lifestyles has been seen as a consequence of the huge E. coli genome plasticity (51). However, linking genomic elements to phenotypic behaviors is not trivial because several layers of biological processes separate genes from their phenotypic effects, and in extreme cases, the evolutionary path can lead either to the functional convergence of distinct sets of genes or to the functional divergence of an initially common set of genes. Consequently, in order to establish links between genomes and phenotypes, one needs an integrative layer. A recent study on a set of 20 E. coli strains (51) has shown that a large fraction of the shared genomic elements with known function is related to metabolism. Because it is now feasible to reconstruct metabolic networks at the genome scale (7, 13, 16, 26), these metabolic networks can, in principle, be used as functional bridges between genomic diversity and phenotypic differences. Currently, such reconstructions are performed automatically from the annotation of input genomes, using algorithms that match these annotations with the contents of reference metabolic databases (13, 16).
In this work, we studied the metabolic diversity of the E. coli species from an evolutionary point of view, with a focus on (i) the extent of metabolic diversity compared to that of genomic diversity, (ii) the correlation between metabolic diversity and phylogeny, and (iii) the metabolic functions associated with pathogenicity.
To these ends, we reconstructed and compared the metabolic networks of 29 strains of E. coli, for which genome sequences and annotations were available (51). This set of strains comprises 23 E. coli strains covering all main phylogenetic groups (A, B1, B2, D, E, and F) (11) and various pathogenic or nonpathogenic behaviors (commensal, ExPEc, InPEc), as well as 6 Shigella strains, which are human obligate intraintestinal pathogens belonging to the E. coli species (15, 44). To obtain metabolic networks suitable for comparative analyses, we first developed a high-quality automated reconstruction process which builds homogenized genome annotations and combines metabolic evidence from the EcoCyc and MetaCyc databases (7, 28a). This reconstruction process is also able to infer enzyme complexes by similarity with K-12 MG1655 complexes. In a second step, we defined the core and variable parts of E. coli metabolic networks and analyzed their metabolic roles. We then confronted differences in metabolic networks with E. coli phylogeny and phenotypes to assess which factors influenced most changes in E. coli metabolism. As most differences were found to be uncorrelated with phenotypes, we finally performed a supervised search for metabolic differences specific to E. coli pathogenic phenotypes.
MATERIALS AND METHODS
Reannotation of E. coli genomes.
Building upon a previous annotation work performed for 20 E. coli/Shigella strains in the context of the ColiScope project (51) with the MicroScope platform (52), we added nine newly published E. coli genomes (strains ATCC 8739, E24377A [45], SE11 [38], LF82 [35], O127:H6 E2348/69 [23], O157:H7 EC4115, HS [45], 042 [9], and SMS-3-5 [17]). All of these publicly available genomes were reannotated using the following process. First, all genomes were integrated in the ColiScope database using MICheck, a method which enables rapid verification of sets of annotated genes and frameshifts in previously published bacterial genomes (10). Second, functional annotations of our previously annotated E. coli genes were automatically transferred in the new strains to genes showing very strong sequence similarity (85% identity on at least 80% of the length of the smallest protein). The remaining genes, i.e., those without any ortholog in any ColiScope genome, were left with their original functional annotations. All genome annotations are available through the MicroScope web platform (http://www.genoscope.cns.fr/agc/microscope/coliscope).
Metabolic network reconstruction.
Our metabolic network reconstruction process is mostly based on Pathway Tools (version 14.0), which is the BioCyc reconstruction software (28), and its associated metabolic database, MetaCyc (7). We used as input all genome annotations coming from our reannotation process, including genes, pseudogenes, partial genes, and insertion sequence-like and prophage-like elements.
By default, Pathway Tools associates genes with metabolic reactions from MetaCyc by examining gene ontology terms, gene product names, and EC number terms found in the genome annotation. Those reactions will be denoted matched reactions. Due to wrongly formatted or unspecific EC numbers or insufficiently explicit textual annotations, Pathway Tools may in some cases either overpredict or miss enzymatic reactions. To improve the accuracy of this gene-reaction association step, we exploited the expert curation done in the EcoCyc metabolic database for E. coli K-12 MG1655 (28a) by transferring gene-reaction associations found in EcoCyc to orthologous genes in the other strains. For this, we mapped genes from K-12 MG1655 to genes of each E. coli strain using the best bidirectional hit (BBH), computed by BLAST (2), with similarity rates above 70% and overlap above 80% of the shorter gene length. Direct associations between each gene having an ortholog in K-12 MG1655 and the corresponding EcoCyc reactions were then specified in a dictionary file given as an additional input to Pathway Tools. Pathway Tools was finally executed using this file and the homogenized genome annotations. All reconstructed networks are available from the Metacoli project website (http://www.genoscope.cns.fr/agc/metacoli) and are included in the MicroCyc repository (http://www.genoscope.cns.fr/agc/microcyc).
Since Pathway Tools infers full metabolic pathways (28), some reactions lacking an associated gene were retrieved on the basis of their presence in an inferred pathway. These purely inferred reactions were left in the MicroCyc databases to allow users to examine complete metabolic pathways but were removed for all comparative analyses done in this work.
Similarly, reactions associated only with pseudogenes were kept in the MicroCyc databases but were removed from our comparative analyses.
The occurrences of all reactions (gene-associated, inferred, pseudogene-associated, and spontaneous reactions) can be found in Table S1 in the supplemental material.
Inference of complexes.
Even though BioCyc databases are able to represent protein complexes, the Pathway Tools reconstruction software does not automatically infer them. Benefiting from the protein complexes stored in EcoCyc for E. coli K-12 MG1655, we inferred by homology complexes for all strains using the following procedure.
First, for each protein complex experimentally identified in E. coli K-12 MG1655 and extracted from EcoCyc, we recursively analyzed its composition in terms of subunits. An equivalent subunit was inferred in the studied E. coli strain if and only if we could find in its genome an orthologous polypeptide using BBH computed by BLAST (2). Second, when an orthologous complex could be inferred, the functional annotations of the K-12 MG1655 complex were transferred to the reconstructed protein complex. Third, the functional annotations associated initially with each subunit of the complex were deleted if they were shared with the reconstructed complex. This final step ensures that the enzymatic function is held only by the complex, if appropriate. This procedure was implemented using the CyClone application programming interface (31), and all complexes are directly stored with the metabolic networks in the MicroCyc repository (http://www.genoscope.cns.fr/agc/microcyc). The list of inconsistencies raised during the complex reconstruction process (i.e., complexes with missing subunits) is available in Table S2 in the supplemental material.
Computation of pan- and core genome/metabolism.
To compute pan- and core genomes, we considered genes that were not pseudogene, partial gene, insertion sequence-like, or prophage-like elements. We clustered genes using the orthoMCL program (version 1.4) (32) for proteins with similarities above 70% and overlap above 70%. We obtained 14,986 clusters of genes that we called the pangenome and 1,957 clusters encompassing at least one gene from each strain that we called the core genome. To evaluate how core and pangenomes evolve when strains are added or removed, we computed them as a function of the number of strains for 5,000 random input orders of strains.
Similar analyses were conducted on metabolic networks. Core metabolism was defined as the set of reactions present in all strains, and panmetabolism was defined as the set of all reactions of all strains. Core metabolism was composed of 885 reactions, and panmetabolism contained 1,545 reactions. Evolution of the sizes of core and panmetabolism was studied by computing them for 10,000 random input orders of metabolic networks.
Computation of genetic distances and phylogenetic tree.
We computed the phylogenetic tree using a six-step procedure. (i) First, we built a modified core genome including pseudogenes and the genome of an outgroup reference organism, Escherichia fergusonii (29). Gene homologies were determined by nucleotide sequence comparisons of genes with similarities of ≥80% and coverage of ≥80%. This modified core genome gathered a set of 1,388 common genes. (ii) We performed multiple alignments on the sequences of these core genes using the MUSCLE program (version 3.6) (14). (iii) Sequence blocks of good alignment were then selected with the GBLOCKS program (version 0.91) (8). (iv) We concatenated those blocks to build one long sequence for each organism. (v) We reconstructed the phylogenetic tree on the basis of these long sequences with the PHYML program (version 3.1) (20), using maximum likelihood and a GTR+gamma model. The genetic distance was directly derived from the branch length of the generated tree. (vi) Finally, 100 bootstrap experiments were performed on the previous step to assess the robustness of the tree topology.
Computation of metabolic distances.
We defined the metabolic distance between two metabolic networks to be the number of distinct gene-associated reactions between them. We computed it using reaction occurrence vectors: each component of this vector corresponds to a reaction of panmetabolism and specifies whether the reaction is present (value = 1) or absent (value = 0) in the considered metabolic network. Metabolic distance is therefore directly computed as the Manhattan distance between reaction occurrence vectors, 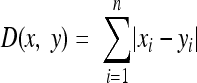 , for reaction i in reaction occurrence vectors x and y of length n. Using this distance, we created a metabolic tree by neighbor joining with R (46) and the R package ape (40).
, for reaction i in reaction occurrence vectors x and y of length n. Using this distance, we created a metabolic tree by neighbor joining with R (46) and the R package ape (40).
MCA.
Factorial multiple-correspondence analysis (MCA) is a projection technique that provides a low-dimensional graphical representation of a set of elements by capturing the maximal amount of variability from the variables describing those elements. We conducted an MCA on the reconstructed metabolic networks for the 23 E. coli non-Shigella strains using R (46) and the package FactoMineR (30). We took as active variables the occurrence of reactions from panmetabolism. Considering the first two eigenvalues was sufficient to explain 34% of the data set diversity. We extracted reactions which had a significant contribution effect on the first two dimensions using the dimdesc function with a multiple-test correction (Bonferroni correction) and a P value lower than 0.05.
Compactness and separation measures.
We computed two measures to assess the compactness and separation of phylogenetic and phenotypic groups according to the metabolic distance. We first defined a center for each group by taking the mean of the occurrence vectors of all groups' strains. Group compactness was then defined as the average metabolic distance between the group center and all groups' strains. Separation between two groups was defined as the metabolic distance between the group centers. Both measures were computed in R (46) using the package clv (http://CRAN.R-project.org/package=clv).
Classification tree analysis.
We used classification and regression tree analysis (CART) (6), a supervised method, to determine which combinations of reactions separate strains according to their pathogenicity. We used the R (46) package rpart (3) with the Gini index as the criterion of homogeneity to build the trees. We removed reactions from the core metabolism which carry no discriminating information and grouped together reactions with the same occurrence in the strains (called the occurrence profile). We obtained 155 different profiles. We computed three different groups of CARTs: commensal versus other phenotypes, ExPEc versus other phenotypes, and InPEc versus other phenotypes. We focused on groups of reactions belonging to the first nodes of the most homogeneous trees.
RESULTS
Reconstruction of metabolic networks.
In order to link phenotypic and genomic diversity through metabolism, one needs to accurately pinpoint similarities and differences in metabolic function in the set of strains under scrutiny. Although several tools are provided to automatically reconstruct metabolic networks from genome annotation only (7, 13, 16, 26), their level of accuracy and the completeness of the resulting networks are usually not sufficient to allow detailed downstream analyses, unless manual curation is carried out (13, 16). Here, we exploited the proximity of all strains to the well-studied E. coli K-12 MG1655 strain to develop a more efficient automatic reconstruction process. To improve the accuracy of the default BioCyc reconstruction process, our reconstruction strategy uses improved genome annotations: EcoCyc, the highly curated metabolic database for E. coli K-12 MG1655 (28a), and Pathway Tools, the BioCyc metabolic reconstruction software (28). This strategy was applied in two steps (Fig. 1).
FIG. 1.

Metabolic network reconstruction process. Genome annotations are homogenized using the MicroScope platform. Then, metabolic networks are reconstructed with BioCyc software tools using the reference metabolic database EcoCyc to benefit from expert curation on the K-12 MG1655 strain and infer enzymatic complexes and the panorganism metabolic database MetaCyc to retrieve non-K-12 MG1655 reactions.
First, annotations of all E. coli genomes were improved and homogenized. In the context of the ColiScope project, an important manual annotation work of the newly sequenced E. coli strains was performed on genes and regions not found in K-12 MG1655, thus allowing, at the end of the process, the reannotation of orthologs in the previously available E. coli and Shigella genomes (51). In the current study, nine new E. coli strains have been added to the ColiScope project within the MicroScope platform (52), and their genomes were reannotated in terms of both syntactic prediction and functional annotations on the basis of orthologs available in the ColiScope project (see Materials and Methods). This reannotation process revealed some inaccurate or missed gene annotations in these new strains and allowed us to standardize the definition and identification of pseudogenes. As a result, a set of consistent functional annotations for all 29 genomes was obtained and made available at the following URL: http://www.genoscope.cns.fr/agc/microscope/coliscope.
In the second step, we translated all genome annotations, encompassing genes, pseudogenes, and partial genes, into metabolic networks by first identifying metabolic reactions from EcoCyc for genes having orthologs in the K-12 MG1655 genome and then executing Pathway Tools with MetaCyc to translate the annotations of the remaining genes (see Materials and Methods for the detailed procedure). Using the highly curated EcoCyc database as the main pivot to reconstruct the metabolism of all E. coli species significantly improves the translation efficiency, as shown afterwards, since it prevents Pathway Tools from performing false predictions for genes orthologous to K-12 genes. Previous pivot-based reconstruction methodologies have already been applied to other organisms (37, 50) but were often unable to predict reactions absent from the pivot organisms. Here, our strategy also takes advantage of the panorganism MetaCyc database (7) to consider reactions beyond those present in K-12 MG1655. All of our reconstructed networks can be browsed, queried, and downloaded from the MicroCyc website (http://www.genoscope.cns.fr/agc/microcyc).
Pathway Tools infers full metabolic pathways (28); therefore, some reactions with no associated gene are retrieved on the basis of their sole occurrence in an inferred pathway. No direct evidence supports these inferred reactions, which often serve as candidates to fill missing biochemical activities (19). Since we kept our reconstruction process fully automatic and performed no further curation on the inferred pathways, we separated these inferred reactions from matched reactions (reactions associated with genes).
To evaluate the benefits of our optimized strategy, we reconstructed the networks using three increasing levels of improvements and compared their respective qualities. The three levels of reconstruction were done using (i) raw genome annotations directly extracted from the GenBank database and the default Pathway Tools process (strategy a), (ii) updated genome annotations from ColiScope and the default Pathway Tools process (strategy b), and (ii) updated genome annotations from ColiScope and the combined EcoCyc/Pathway Tools process (strategy c, our optimized reconstruction process). We estimated the quality of the reconstructed networks with the following criteria: number of matched reactions in the networks to assess their comprehensiveness, number of inferred reactions to estimate their levels of confidence, and number and completion of metabolic pathways.
Genome annotation quality directly impacted the number of matched reactions (Fig. 2 A, strategy b versus strategy a). On average, homogenization of genome annotations increased the number of matched reactions in each strain by an average of 31% and decreased the number of inferred reactions by an average of 10% (Fig. 2B). In the case of E. coli O157:H7 EDL933, the number of matched reactions increased more than 2-fold, jumping from 578 to 1,224 reactions. The use of EcoCyc as a pivot (Fig. 2, strategy c versus strategy b) resulted in networks with a small increase in size (2%, on average). The number of matched reactions slightly increased (5%, on average), while the number of inferred reactions considerably decreased (22%, on average). This shows that our process manages to transfer some of the curation performed in EcoCyc to the other reconstructed networks, mainly preventing the inference of wrong reactions.
FIG. 2.

Number of matched (A) and inferred (B) reactions for each network according to the reconstruction strategy. Strategy a, use of raw genome annotations directly extracted from GenBank database and the default Pathway Tools process; strategy b, use of updated genome annotations from ColiScope and the default Pathway Tools process; strategy c, use of updated genome annotations from ColiScope and the combined EcoCyc/Pathway Tools process.
As regards metabolic pathways, we observed that their total number decreased when improved genome annotations were used (451 versus 386 pathways, on average, for strategies a and b, respectively). This effect is mostly the consequence of the removal of falsely inferred reactions (on the basis of erroneous annotations and erroneous EC number-reaction associations), which triggered the inclusion of wrong pathways. Strategy c, however, slightly increased the number of pathways (2.5% increase with 396 pathways, on average), adding curated pathways from EcoCyc and also removing some false-positive pathways. The completion of pathways also improved when we optimized the reconstruction strategy. Starting from 45% of pathways with holes in strategy a, this proportion decreased to 42% in strategy b and reached 34% in strategy c. Furthermore, more than 44% of the pathways with holes in strategy c included only one hole. The improvement was most noticeable when we employed EcoCyc as a pivot, suggesting again that curation done on a reference metabolic network can be efficiently adapted to closely related organisms.
Table 1 shows the main characteristics of the final metabolic networks. On average, they include 1,491 reactions (1,274 matched, 217 inferred), with small variations occurring around that number: 1,300 to 1,564 (1,054 to 1,338 for matched reactions). The reaction count is slightly lower for Shigella strains (1,437 total, on average) than for non-Shigella strains (1,504 total, on average), a trend that is even stronger when inferred reactions and those associated with pseudogenes are removed (1,173 versus 1,301 gene-associated reactions for Shigella and non-Shigella strains, respectively). Shigella strains actually exhibit a significantly higher number of pseudogenes than non-Shigella strains, an observation that is consistent with their evolution to become obligate pathogens (44).
TABLE 1.
Main characteristics of the reconstructed metabolic networks
| Strain | Phylogenetic group | Phenotype | No. of genes | No. of reactions | No. of metabolites | No. of pathways | |||
|---|---|---|---|---|---|---|---|---|---|
| Total | With gene | With pseudogene | Without gene | ||||||
| Escherichia coli | |||||||||
| ATCC 8739 | A | Commensal | 4,411 | 1,499 | 1,301 | 11 | 187 | 1,454 | 347 |
| HS | A | Commensal | 4,541 | 1,510 | 1,300 | 9 | 201 | 1,443 | 349 |
| K-12 MG1655 | A | Commensal | 4,182 | 1,439 | 1,269 | 4 | 166 | 1,385 | 340 |
| K-12 W3110 | A | Commensal | 4,394 | 1,461 | 1,273 | 7 | 181 | 1,425 | 344 |
| 55989 | B1 | InPEc | 4,961 | 1,473 | 1,268 | 6 | 199 | 1,440 | 348 |
| E24377A | B1 | InPEc | 5,346 | 1,521 | 1,308 | 7 | 206 | 1,473 | 351 |
| IAI1 | B1 | Commensal | 4,412 | 1,486 | 1,271 | 3 | 212 | 1,450 | 351 |
| SE11 | B1 | Commensal | 5,071 | 1,504 | 1,318 | 4 | 182 | 1,451 | 345 |
| 536 | B2 | ExPEc | 4,654 | 1,499 | 1,290 | 18 | 191 | 1,452 | 344 |
| APEC O1 | B2 | ExPEc | 4,874 | 1,482 | 1,289 | 4 | 189 | 1,392 | 340 |
| CFT073 | B2 | ExPEc | 5,396 | 1,532 | 1,312 | 25 | 195 | 1,456 | 345 |
| ED1a | B2 | Commensal | 5,103 | 1,507 | 1,292 | 11 | 204 | 1,361 | 340 |
| LF82 | B2 | InPEc | 4,584 | 1,483 | 1,299 | 4 | 180 | 1,378 | 332 |
| O127:H6 E2348/69 | B2 | InPEc | 4,944 | 1,485 | 1,296 | 14 | 175 | 1,423 | 336 |
| S88 | B2 | ExPEc | 4,848 | 1,503 | 1,288 | 6 | 209 | 1,433 | 343 |
| UTI89 | B2 | ExPEc | 5,305 | 1,512 | 1,314 | 4 | 194 | 1,464 | 346 |
| 042 | D | InPEc | 5,031 | 1,509 | 1,311 | 6 | 192 | 1,463 | 343 |
| UMN026 | D | ExPEc | 5,046 | 1,564 | 1,338 | 3 | 223 | 1,452 | 352 |
| O157:H7 EC4115 | E | InPEc | 5,784 | 1,534 | 1,327 | 11 | 196 | 1,446 | 344 |
| O157:H7 EDL933 | E | InPEc | 5,267 | 1,531 | 1,313 | 8 | 210 | 1,445 | 346 |
| O157:H7 Sakai | E | InPEc | 5,431 | 1,524 | 1,307 | 13 | 204 | 1,459 | 344 |
| IAI39 | F | ExPEc | 4,740 | 1,531 | 1,307 | 10 | 214 | 1,484 | 352 |
| SMS-3-5 | F | Commensal | 5,128 | 1,514 | 1,323 | 3 | 188 | 1,457 | 347 |
| Shigella | |||||||||
| S. boydii Sb227 | S1 | Shigellosis | 4,717 | 1,461 | 1,188 | 52 | 221 | 1,413 | 332 |
| S. dysenteriae Sd197 | SD1 | Shigellosis | 4,867 | 1,300 | 1,054 | 75 | 171 | 1,238 | 304 |
| S. flexneri 2a 2457T | S3 | Shigellosis | 4,339 | 1,475 | 1,213 | 51 | 211 | 1,425 | 340 |
| S. flexneri 2a 301 | S3 | Shigellosis | 4,675 | 1,472 | 1,195 | 69 | 214 | 1,433 | 338 |
| S. flexneri 5 8401 | S3 | Shigellosis | 4,393 | 1,480 | 1,197 | 66 | 211 | 1,426 | 337 |
| S. sonnei Ss046 | SS | Shigellosis | 4,938 | 1,434 | 1,193 | 28 | 213 | 1,358 | 337 |
We included in the networks enzymatic complexes generated by similarity with strain K-12 MG1655 complexes described in EcoCyc (see Materials and Methods). Among the 712 homomeric complexes found in EcoCyc, 707 (99%) could be transferred to at least another strain (missing complexes were associated with pseudogenes in EcoCyc) and 458 (65%) were common to all networks. Among the 285 heteromeric complexes from EcoCyc, 278 (97%) were created for at least one strain and 107 (38%) were common to all strains. When Shigella strains were removed, the number of common heteromeric complexes reaches 157 (55%). We found in the networks an average of 237 complete heteromeric complexes and an average of 31 heteromeric complexes for which only part of the subunits could be identified. Since we could not automatically identify the reason for the subunit absence (possible reasons include missing gene, annotation error, or another gene with an equivalent product) and since we had evidence for at least a part of the complex, we decided to keep the reactions linked to these incomplete complexes. The names and compositions of all these complexes can be found in Table S2 in the supplemental material.
Using a unified source of genome annotations and a common reconstruction process for all metabolic networks limits the biases originating from the reconstruction process, thus making our networks reliably comparable. In order to focus on the most reliable reactions, we performed our comparative analyses using the set of gene-associated reactions (matched reactions) and discarded reactions associated only with pseudogenes or with no gene.
Core and variable parts of metabolism.
We separated metabolic reactions into three categories according to their occurrence in strains: panmetabolism, core metabolism, and variable metabolism (see Materials and Methods and Table S1 in the supplemental material). Panmetabolism is the set of all reactions of all strains, i.e., the global metabolic network of E. coli species. Core metabolism is the set of reactions common to all strains. Variable metabolism is the difference between pan- and core metabolism, i.e., the set of reactions that are missing from at least one strain.
Panmetabolism included 1,545 reactions. Among them, 885 reactions belonged to core metabolism (57% of the number for panmetabolism) and 660 reactions belonged to variable metabolism (43% of the number for panmetabolism). In each strain, these 885 core reactions represented the major part of the metabolic network (59%, on average), with only 416 reactions, on average, belonging to variable metabolism. The occurrence of variable reactions was not uniformly distributed among the strains, and its distribution exhibited a U-like shape (Fig. 3): variable reactions tended to be either common to all but a few strains or specific to one or a few strains. Relatively few reactions were shared by medium-size subsets of strains. A peak was yet visible at eight occurrences: these were mainly reactions specific to the eight strains of the B2 group.
FIG. 3.

Distribution of reaction occurrences in strains for reactions not in core metabolism.
When Shigella strains were removed, panmetabolism remained nearly identical (1,543 reactions), while core metabolism increased to 1,065 reactions (69% of the number for panmetabolism). This showed that E. coli reconstructed networks are well conserved and that Shigella has mostly lost reactions since its divergence (22). A set of 180 reactions was therefore absent from Shigella core metabolism. It may well include metabolic functions that were no longer required for Shigella strains to live in their current habitats (Shigella has a parasitic lifestyle) and were thereby lost in these strains. These lost reactions include, for instance, the d-allose degradation pathway (18) and about 10 pathways involved in aromatic compound (e.g., phenylethylamine and phenylacetate) degradation or in amino acid (e.g., histidine) degradation. Lost core reactions were also found among biosynthesis pathways linked to amino acid, nucleotide, and fatty acid anabolism.
Missing reactions from our networks reflected to some extent the auxotrophies found experimentally for Shigella strains (1). We observed, for instance, that the nicotinic acid biosynthesis pathway lacks the essential l-aspartate oxidase activity (genes ndaA and ndaB [42, 43]) in all Shigella strains except Shigella dysenteriae Sd197, a result that corroborates exactly the auxotrophies for NAD experimentally determined in a previous work (1). Similarly, the absence of homoserine _O_-transsuccinylase (metA gene [54]) in Shigella flexneri 2a strain 301 may explain the methionine auxotrophy reported for some S. flexneri strains in the same work. A few other reported auxotrophies could not, however, be interpreted by simply looking at reaction presence/absence. Turning these metabolic networks into mathematical models of metabolism may help with investigating these cases, as several modeling methods are available to study growth environments in a more systematic manner (13, 16).
The core metabolism/panmetabolism ratio was in sharp contrast to the core metabolism/panmetabolism ratio for the genome (see Materials and Methods for details on core and pangenome computation). For our set of strains, the core genome represented only 13% of the pangenome (1,957 common clusters over 14,986 clusters) (Fig. 4 A), a ratio much smaller than that for core metabolism. In addition, an assessment of the variation of the sizes of panmetabolism and pangenome as a function of the number of strains (Fig. 4B) showed that the size of panmetabolism approached a plateau at 29 strains, whereas the pangenome size was still steadily increasing. These results suggest that diversity is more limited within E. coli metabolic networks than it is within all gene functions. Two main interpretations can be hypothesized from this observation. First, this estimation of metabolic diversity is limited to the set of reactions already known. Consequently, panmetabolism may lack many unknown reactions, especially those specific to poorly studied organisms. In contrast, the pangenome is more confidently estimated since most genes, even those whose functions remain unknown, are detected on genomes. Because of this limitation, adding new strains to the study would not significantly expand panmetabolism if the strain-specific reactions are unknown, which is often the case for newly sequenced organisms. This observation has actually motivated several initiatives which focus on the search for novel enzymatic activities rather than on the mere sequencing of additional genomes (4, 5). Second, genes coding for enzymatic functions may vary less than those coding other functions. Diversity in metabolism could be traced back to a relatively small number of distinct enzymes; genomic diversity may involve nonenzymatic processes such as regulation, which contributes to another level of metabolic diversity via the control of metabolism (33).
FIG. 4.

Evolution of E. coli core metabolism (A, curve a), core genome (A, curve b), panmetabolism (B, curve a), and pangenome (B, curve b), according to the number of included strains. Boxes delimit the first and third quartiles of 10,000 different input orders of metabolic networks and 5,000 different input orders of genomes.
We next examined in more detail how core and variable reactions were distributed among metabolic categories (Table 2). Interestingly, the proportion of core reactions was significantly higher in biosynthetic processes (68%) than in other metabolic categories (the Fisher exact test, P < 10−15). This contrasts with degradation processes, which contain a significantly lower proportion of core reactions (29%) than other metabolic categories (the Fisher exact test, P < 10−15). Biosynthesis reactions actually constitute the majority of reactions from core metabolism (57%, 508 reactions). This result can be interpreted by the fact that, when environments are changing, metabolic functions closely related to metabolites from the environment (e.g., degradation pathways) are more likely to vary than biosynthetic reactions, which usually use ubiquitous basic metabolites as precursors. A similar effect has been observed in a previous study among the functions of horizontally transferred genes (i.e., variable genes), which were found to be involved more often in transport and peripheral degradation pathways than in central biosynthetic processes (39).
TABLE 2.
Distribution of reactions of core, variable, and panmetabolism across metabolic processes, as defined in BioCyc databasesa
| Process | No. of metabolic occurrences | ||
|---|---|---|---|
| Core | Variable | Pan | |
| Biosynthesis | 508 | 236 | 744 |
| Degradation | 200 | 224 | 424 |
| Detoxification | 9 | 5 | 14 |
| Energy metabolism | 68 | 29 | 97 |
| Transport pathways | 2 | 2 | 4 |
| Other | 262 | 231 | 493 |
| Total | 885 | 660 | 1,545 |
Reactions involved in sucrose degradation are a good illustration of variable metabolism. The ability to use sucrose as a sole carbon source is a highly variable phenotype in enterobacteria. Among commensal strains, E. coli K-12 MG1655, K-12 W3110, HS, ATCC 8739, and SMS-3-5 cannot utilize sucrose, whereas the IAI1 and SE11 strains can. This phenotype is also highly variable for E. coli pathogenic strains. Chromosomal genes associated with sucrose degradation are organized in a cluster of two operons coding for a non-phosphotransferase system permease (cscB gene) and a fructokinase (cscK gene) in the first operon and a sucrose hydrolase (cscA gene) in the second operon, with both being controlled by an adjacent repressor (cscR gene) (24). This cluster is integrated next to a tRNA-Arg gene, and the codon adaptation index (CAI) of the cluster genes is among the lowest of all E. coli genes (among the 8% of genes with the lowest CAI), suggesting acquisition of the csc genes by horizontal gene transfer.
Structure of E. coli metabolic diversity.
To study how metabolic diversity is distributed within the E. coli species, we analyzed the metabolic distances, defined by the number of distinct reactions between two strains (see Materials and Methods), between strains. We first grouped strains according to metabolic distance and obtained the tree shown in Fig. 5 A. Overall, strain groups matched phylogenetic groups relatively well. Group B2, D, E, and F strains clearly clustered according to their groups. The F group is a new group composed of strains previously included in the D one (25), a fact that was visible from the genomic point of view (Fig. 5B) but also from the metabolic one (Fig. 5A). Strains from the A and B1 groups are, however, mixed together. Group A and B1 strains are actually phylogenetically close (Fig. 5B), and the evolutionary distance between them may be too small to imply a significant difference in their metabolic networks.
FIG. 5.

Evolution tree of E. coli according to metabolic (A) and genetic (B) distances. *, nodes with a bootstrap value greater than 70% for the metabolic tree. All nodes of the genetic tree have bootstrap values greater than 70%. Phylogenetic groups are defined according to references 44 and 11 for E. coli and Shigella strains, respectively.
All Shigella strains were markedly more distant from the other strains (Fig. 5A). Shigella strains have evolved from multiple distinct phylogenetic groups (15, 44), and this effect is still visible from the strain phylogenetic tree, since they are spread among E. coli groups (Fig. 5B). However, the high metabolic distances that separate them from other strains have blurred this signal, suggesting that evolution of their metabolism has been rapid.
To further study the link between metabolism and genetic diversity, we directly compared metabolic and genetic distances for all pairs of strains (Fig. 6; see Materials and Methods). A Mantel test performed on this pair of distances showed that they are significantly correlated (P ≤ 0.01), yet they have a relatively large dispersion due to Shigella (linear regression, _r_2 = 0.15). When the focus is on non-Shigella strains, linear regression between the two distances significantly improved (linear regression, _r_2 = 0.54), showing that metabolic distance increases with genetic distance. Strains of the same phylogenetic groups (blue symbols in Fig. 6) were separated by sets of 50 to 150 reactions, and this number did not vary with genetic distance. Metabolic distances between non-Shigella strains from distinct phylogenetic groups were slightly higher but still in the range 100 to 250 reactions. Here again, group A and B1 strains behaved as if they formed a single phylogenetic group, and their genetic and metabolic distances were comparable to intragroup distances: sets of 75 to 125 reactions (set of leftmost black symbols in Fig. 6).
FIG. 6.

Plot of genetic distances (x axis) versus metabolic distances (y axis) for all pairs of strains, colored according to strain phylogenetic groups. Blue, both strains in each pair are non-Shigella strains from the same phylogenetic group; black, strains are from distinct groups but both are non-Shigella; green, both are Shigella strains; red, strains are from distinct groups, with one being Shigella and the other being non-Shigella.
As observed above, for similar genetic distances, Shigella metabolic networks were markedly more distant from other networks than were non-Shigella metabolic networks. Furthermore, metabolic distances between Shigella strains were comparable to metabolic distances between Shigella and non-Shigella strains, while the distance between Shigella strains from the same phylogenetic group (i.e., those of the S3 Shigella group) was equal to the intragroup E. coli metabolic distance. This suggests that their metabolic networks have quickly evolved by genetic drift (11) and that most metabolic differences were not common to all Shigella strains. Among the 176 pseudoreactions (linked only to pseudogenes) found in at least one Shigella strain, none were pseudoreactions in all 6 Shigella strains and 92 were pseudoreactions in only one Shigella strain. Nevertheless, convergent inactivation of a few metabolic characters has been reported, indicating adaptive evolution (1, 11, 34, 42). This could be a consequence of its parasitic lifestyle, which removes requirements for some degradation/biosynthesis pathways, as mentioned above.
In order to examine in more detail metabolic diversity within non-Shigella strains, we performed an MCA (see Materials and Methods) on reaction occurrences (Fig. 7). The first two factorial axes accounted for 34% of all variability. There were more than a hundred reactions with a significant contribution (see Materials and Methods) to the first axis. Half of the reactions with a high contribution were involved in biosynthetic processes, especially lipid biosynthesis (71% of them). Another 23% of high-contribution reactions were associated with degradation, in particular, aromatic compound degradation (37% of them). Most of the remaining reactions were not part of any pathway. Similarly, we observed on the second axis that 57% of high-contribution reactions were linked to biosynthetic processes (with 82% of them being lipid biosynthesis), and 25% were associated with degradation (with 42% of them being aromatic compound degradation).
FIG. 7.

Plot of the first two axes of MCA of reaction occurrences in E. coli non-Shigella strains, labeled according to phylogenetic groups (A) and phenotypes (B). MCA was performed on reactions associated with genes. The distance between strains can be interpreted as the most significant dissimilarities between their reaction absence/presence profiles.
The large number of reactions with high contributions on each of these axes made our MCA robust to addition or removal of reactions. Moreover, when the MCA was computed while discarding dozens of reactions with the best contributions, only minor changes to the distribution of strains were observed (data not shown).
In agreement with observations on metabolic distances, Fig. 7A shows that phylogenetic groups were relatively well separated by the first two axes of the MCA for all except strains of groups A and B1, which are mixed. Group F strains were separated from group D strains on both axes, confirming the existence of metabolic differences between them. Such a clear separation supports the separation of group F strains from group D strains (25).
When strains were grouped according to their phenotypes (commensal, ExPEc, or InPEc; Table 1 and Fig. 7B), no clear separation could be seen from the MCA. Indeed, reaction occurrence in strains seemed to be poorly correlated with strain phenotypes. In order to compare more robustly phenotypic and phylogenetic groups with metabolic distances, we computed compactness (mean distance between group centers and group members) and separation (distance between two group centers) measures for all groups and all pairs of groups using the metabolic distances (Table 3 ) (see Materials and Methods). These two measures globally evaluate the closeness of strains within a group and their separation between two groups, according to the chosen distance (21). Compactness measures confirmed that strains grouped by phylogeny were markedly closer to each other than strains grouped by phenotype (26 to 68 for phylogenetic groups versus 92 to 138 for phenotypic groups). Furthermore, when compactness measures are compared with separation measures, phylogenetic groups appeared to be globally distinct, except for the A and B1 groups, which here again showed overlap. Metabolic separation between phenotypic groups was, in contrast, not significantly higher than within-group distances. Strains from phenotypic groups were nearly as distant from each other than from strains of other phenotypic groups. Therefore, pathogenicity phenotypes did not appear to drive large changes in reaction occurrence in these strains.
TABLE 3.
Compactness and separation measures for phylogenetic and phenotypic groups, according to the metabolic distance
| Phylogenetic group or phenotype | Compactness | Separation | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B1 | B2 | D | E | F | S1 | S3 | SD1 | Commensal | ExPEc | InPEc |
| A | 62 | ||||||||||
| B1 | 64 | 50 | |||||||||
| B2 | 68 | 149 | 148 | ||||||||
| D | 30 | 128 | 118 | 134 | |||||||
| E | 22 | 153 | 142 | 162 | 93 | ||||||
| F | 32 | 117 | 110 | 103 | 97 | 131 | |||||
| S1 | NAa | 188 | 191 | 196 | 197 | 205 | 187 | ||||
| S3 | 26 | 181 | 169 | 201 | 169 | 193 | 182 | 150 | |||
| SD1 | NA | 282 | 280 | 303 | 308 | 290 | 297 | 238 | 258 | ||
| SS | NA | 161 | 156 | 221 | 189 | 199 | 188 | 163 | 181 | 207 | |
| Commensal | 92 | ||||||||||
| ExPEc | 88 | 119 | |||||||||
| InPEc | 102 | 92 | 114 | ||||||||
| Shigellosis | 138 | 159 | 199 | 177 |
As the presence of small sets of specific reactions can, however, induce notable changes in phenotypes, we looked in more detail for specific differences between networks grouped by pathogenicity. As no reaction was found to be completely specific to any pathogenicity phenotype, we used a supervised method able to slightly relax the specificity constraint and find such characteristic sets of reactions (CART; see Materials and Methods). We applied this method to each pathogenicity phenotype (commensal, InPEc, and ExPEc).
We observed that most commensal strains (except ED1a and SMS-3-5) possess reactions able to degrade phenylacetate and phenylethylamine (12) (paa transcription unit), which are absent from InPEc and ExPEc strains (except E24377A and 55989). E24377A and SMS-3-5 were further separated from the commensal strains by the presence of a plasmid-encoded toxin (PET) serine precursor (gene sat), which is known to be an important virulence factor (47) associated with both intestinal and extraintestinal infections.
ExPEc strains were mainly characterized by the absence of psicose and psicoselysine degradation pathways (frl transcription unit). They also specifically possess a putative transporter of capsular polysaccharide (gene kpsT), a virulent element used by the virulent strain E. coli K1 during neonatal septicemia and meningitis (41, 53).
Reactions characteristic of InPEc strains could be less clearly identified. Most of them are putative reactions, like a maleylacetoacetate isomerase (a locus similar to maiA in Salmonella), and another one has a high similarity with glutathione _S_-transferase and a cobalamine adenosyltransferase (gene glmL).
Results from this analysis can be found in Table S3 in the supplemental material.
DISCUSSION
Establishing a link between genomes and phenotypes is difficult because several layers of biological processes intervene between genes and their phenotypic effects. Metabolism is one of these layers, and thanks to automated metabolic reconstruction tools, it can be studied at the genome scale for sequenced organisms. However, identifying sound metabolic differences between distinct organisms and assessing diversity within a set of metabolic networks, as was done in this work, require sufficiently detailed metabolic networks that standard automated methods usually do not produce without curation (13). Here, we were able to improve an automated reconstruction strategy by leveraging the proximity of all strains with E. coli K-12 MG1655, whose genome and metabolism are incomparably well-known. As a result, we provide high-quality metabolic networks for 29 E. coli strains, including 6 Shigella strains, all of which are suitable for comparative analyses (available at http://www.genoscope.cns.fr/agc/metacoli/). Most noteworthy, a large improvement in network completeness was achieved by updating and homogenizing genome annotations for all E. coli strains, while using EcoCyc as a primary reconstruction pivot allowed the transfer of some of the manual curation done on K-12 MG1655 metabolism and thereby limit the proportion of falsely inferred reactions. We were also able to infer enzyme complexes similar to those known in K-12 MG1655.
The reconstructed networks were composed of a majority of E. coli core reactions and relatively few variable reactions. Moreover, examining the evolution of the size of panmetabolism as a function of the number of networks indicates that reconstructing the metabolism of new strains will add only little diversity to the current panmetabolism. The size of panmetabolism is yet likely to be underestimated, as many reactions remain unknown. Characterizing missing enzyme activities in the current strains will most probably contribute to expanding the knowledge of panmetabolism at least as much as sequencing and annotating new strains do.
We observed that biosynthetic reactions were mostly part of core metabolism and that degradation processes, on the other hand, were mainly found in variable metabolism. This can be interpreted by the fact that the selection pressure acting on biosynthetic processes is likely to be similar for all strains, as these processes, which take as inputs common central metabolic precursors, are only weakly influenced by the environment. Conversely, degradation processes are directly linked to compounds from the environment, and their selection therefore depends on the environment and strain lifestyles (39).
This evolutionary interpretation is supported by the large metabolic differences separating the six Shigella strains from the others. These strains, whose parasitic lifestyles make large parts of E. coli panmetabolism dispensable, have actually lost many reactions still present in all non-Shigella strains. These differences make their metabolic networks sufficiently distinct from the other E. coli networks to blur their phylogenetic origin (see metabolic tree in Fig. 5A).
When the Shigella strains were removed from the study, we observed that differences between metabolic networks were significantly correlated with the strains' phylogenies but not with their commensal/pathogenic phenotypes. This suggests that changes in metabolic networks occurred with strain divergence and were mostly not driven by strain phenotypes, as was yet the case for the Shigella phenotype.
The fact that E. coli commensal/pathogenic phenotypes do not globally influence their metabolic networks does not mean that no metabolic characteristic can be associated with them. First, the presence or absence of only a few enzymes may be related to these phenotypes. Using a supervised classification method, we were able to identify such cases, with some having already been described in literature. Second, diversity in metabolic behaviors does not originate from enzyme diversity only. Diversity in enzyme regulation and activity also influences metabolism and cannot be assessed by solely studying reconstructed metabolic networks. It involves, for instance, studying regulatory networks or experimentally measuring how metabolism actually operates in each strain. Our reconstructed networks represent a first step toward such investigations, as they form a solid basis on which to build the metabolic models needed to integrate and interpret such experimental data.
Supplementary Material
[Supplemental material]
Acknowledgments
This work is supported by a grant from the French National Research Agency (ANR) to the Metacoli project (contract number ANR-08-SYSC-011) and by MICROME, a collaborative project funded by the European Commission within its FP7 Program, contract number 222886-2. E.D. is partly supported by the Fondation pour le Recherche Médicale. V. Sabarly is partly supported by Délégation Géminale pour l'Armement.
Footnotes
▿
Published ahead of print on 14 January 2011.
REFERENCES
- 1.Ahmed, Z. U., M. R. Sarker, and D. A. Sack. 1988. Nutritional requirements of shigellae for growth in a minimal medium. Infect. Immun. 56**:**1007-1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215**:**403-410. [DOI] [PubMed] [Google Scholar]
- 3.Atkinson, E. J., and T. M. Therneau. 2000. An introduction to recursive partitioning. Technical report. Mayo Foundation, Rochester, MN.
- 4.Baran, R., W. Reindl, and T. R. Northen. 2009. Mass spectrometry based metabolomics and enzymatic assays for functional genomics. Curr. Opin. Microbiol. 12**:**547-552. [DOI] [PubMed] [Google Scholar]
- 5.Beloqui, A., et al. 2009. Reactome array: forging a link between metabolome and genome. Science 326**:**252-257. [DOI] [PubMed] [Google Scholar]
- 6.Breiman, L., J. Friedman, R. Olshen, and C. Stone. 1984. Classification and regression trees, new edition. Chapman & Hall/CRC, New York, NY.
- 7.Caspi, R., et al. 2010. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 38**:**D473-D479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castresana, J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17**:**540-552. [DOI] [PubMed] [Google Scholar]
- 9.Chaudhuri, R. R., et al. 2010. Complete genome sequence and comparative metabolic profiling of the prototypical enteroaggregative Escherichia coli strain 042. PLoS One 5**:**e8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cruveiller, S., et al. 2005. MICheck: a web tool for fast checking of syntactic annotations of bacterial genomes. Nucleic Acids Res. 33**:**W471-W479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denamur, E., B. Picard, and O. Tenaillon. 2010. Population genetics of pathogenic Escherichia coli, p. 269-286. In D. A. Robinson, D. Falush, and E. J. Feil (ed.), Bacterial population genetics in infectious disease. Wiley-Blackwell, West Sussex, United Kingdom.
- 12.Diaz, E., A. Ferrandez, M. A. Prieto, and J. L. Garcia. 2001. Biodegradation of aromatic compounds by Escherichia coli. Microbiol. Mol. Biol. Rev. 65**:**523-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Durot, M., P. Bourguignon, and V. Schachter. 2009. Genome-scale models of bacterial metabolism: reconstruction and applications. FEMS Microbiol. Rev. 33**:**164-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edgar, R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32**:**1792-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Escobar-Páramo, P., C. Giudicelli, C. Parsot, and E. Denamur. 2003. The evolutionary history of Shigella and enteroinvasive Escherichia coli revised. J. Mol. Evol. 57**:**140-148. [DOI] [PubMed] [Google Scholar]
- 16.Feist, A. M., et al. 2007. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 3**:**121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fricke, W. F., et al. 2008. Insights into the environmental resistance gene pool from the genome sequence of the multidrug-resistant environmental isolate Escherichia coli SMS-3-5. J. Bacteriol. 190**:**6779-6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukiya, S., H. Mizoguchi, T. Tobe, and H. Mori. 2004. Extensive genomic diversity in pathogenic Escherichia coli and Shigella strains revealed by comparative genomic hybridization microarray. J. Bacteriol. 186**:**3911-3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green, M. L., and P. D. Karp. 2004. A Bayesian method for identifying missing enzymes in predicted metabolic pathway databases. BMC Bioinform. 5**:**76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guindon, S., and O. Gascuel. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52**:**696-704. [DOI] [PubMed] [Google Scholar]
- 21.Handl, J., J. Knowles, and D. B. Kell. 2005. Computational cluster validation in post-genomic data analysis. Bioinformatics 21**:**3201-3212. [DOI] [PubMed] [Google Scholar]
- 22.Hershberg, R., H. Tang, and D. A. Petrov. 2007. Reduced selection leads to accelerated gene loss in Shigella. Genome Biol. 8**:**R164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iguchi, A., et al. 2009. Complete genome sequence and comparative genome analysis of enteropathogenic Escherichia coli O127:H6 strain E2348/69. J. Bacteriol. 191**:**347-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jahreis, K., et al. 2002. Adaptation of sucrose metabolism in the Escherichia coli wild-type strain EC3132. J. Bacteriol. 184**:**5307-5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaureguy, F., et al. 2008. Phylogenetic and genomic diversity of human bacteremic Escherichia coli strains. BMC Genomics 9**:**560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanehisa, M., et al. 2007. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36**:**D480-D484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaper, J. B., J. P. Nataro, and H. L. T. Mobley. 2004. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2**:**123-140. [DOI] [PubMed] [Google Scholar]
- 28.Karp, P. D., et al. 2010. Pathway Tools version 13.0: integrated software for pathway/genome informatics and systems biology. Brief. Bioinform. 11**:**40-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28a.Keseler, I. M., C. Bonavides-Martínez, J. Collado-Vides, S. Gama-Castro, R. P. Gunsalus, D. A. Johnson, M. Krummenacker, L. M. Nolan, S. Paley, I. T. Paulsen, M. Peralta-Gil, A. Santos-Zavaleta, A. G. Shearer, and P. D. Karp. 2009. EcoCyc: a comprehensive view of Escherichia coli biology. Nucleic Acids Res. 37**:**D464-D470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lawrence, J. G., H. Ochman, and D. L. Hartl. 1991. Molecular and evolutionary relationships among enteric bacteria. J. Gen. Microbiol. 137**:**1911-1921. [DOI] [PubMed] [Google Scholar]
- 30.Lê, S., J. Josse, and F. Husson. 2008. FactoMineR: an R package for multivariate analysis. J. Stat. Softw. 25**:**1-18. [Google Scholar]
- 31.Le Fèvre, F., S. Smidtas, and V. Schächter. 2007. Cyclone: Java-based querying and computing with Pathway/Genome databases. Bioinformatics 23**:**1299-1300. [DOI] [PubMed] [Google Scholar]
- 32.Li, L., C. J. Stoeckert, and D. S. Roos. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13**:**2178-2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maslov, S., S. Krishna, T. Y. Pang, and K. Sneppen. 2009. Toolbox model of evolution of prokaryotic metabolic networks and their regulation. Proc. Natl. Acad. Sci. U. S. A. 106**:**9743-9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maurelli, A. T., R. E. Fernández, C. A. Bloch, C. K. Rode, and A. Fasano. 1998. “Black holes” and bacterial pathogenicity: a large genomic deletion that enhances the virulence of Shigella spp. and enteroinvasive Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 95**:**3943-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miquel, S., et al. 2010. Complete genome sequence of Crohn's disease-associated adherent-invasive E. coli strain LF82. PLoS One 5**:**e12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reference deleted.
- 37.Notebaart, R. A., F. H. J. van Enckevort, C. Francke, R. J. Siezen, and B. Teusink. 2006. Accelerating the reconstruction of genome-scale metabolic networks. BMC Bioinform. 7**:**296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oshima, K., et al. 2008. Complete genome sequence and comparative analysis of the wild-type commensal Escherichia coli strain SE11 isolated from a healthy adult. DNA Res. 15**:**375-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pál, C., B. Papp, and M. J. Lercher. 2005. Adaptive evolution of bacterial metabolic networks by horizontal gene transfer. Nat. Genet. 37**:**1372-1375. [DOI] [PubMed] [Google Scholar]
- 40.Paradis, E., J. Claude, and K. Strimmer. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20**:**289-290. [DOI] [PubMed] [Google Scholar]
- 41.Pavelka, M. S., L. F. Wright, and R. P. Silver. 1991. Identification of two genes, kpsM and kpsT, in region 3 of the polysialic acid gene cluster of Escherichia coli K1. J. Bacteriol. 173**:**4603-4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prunier, A., et al. 2007. nadA and nadB of Shigella flexneri 5a are antivirulence loci responsible for the synthesis of quinolinate, a small molecule inhibitor of Shigella pathogenicity. Microbiology 153**:**2363-2372. [DOI] [PubMed] [Google Scholar]
- 43.Prunier, A., R. Schuch, R. E. Fernández, and A. T. Maurelli. 2007. Genetic structure of the nadA and nadB antivirulence loci in Shigella spp. J. Bacteriol. 189**:**6482-6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pupo, G. M., R. Lan, and P. R. Reeves. 2000. Multiple independent origins of Shigella clones of Escherichia coli and convergent evolution of many of their characteristics. Proc. Natl. Acad. Sci. U. S. A. 97**:**10567-10572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rasko, D. A., et al. 2008. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol. 190**:**6881-6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.R Development Core Team. 2009. R: a language and environment for statistical computing. R Development Core Team, Vienna, Austria.
- 47.Restieri, C., G. Garriss, M. Locas, and C. M. Dozois. 2007. Autotransporter-encoding sequences are phylogenetically distributed among Escherichia coli clinical isolates and reference strains. Appl. Environ. Microbiol. 73**:**1553-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reference deleted.
- 49.Tenaillon, O., D. Skurnik, B. Picard, and E. Denamur. 2010. The population genetics of commensal Escherichia coli. Nat. Rev. Microbiol. 8**:**207-217. [DOI] [PubMed] [Google Scholar]
- 50.Teusink, B., et al. 2005. In silico reconstruction of the metabolic pathways of Lactobacillus plantarum: comparing predictions of nutrient requirements with those from growth experiments. Appl. Environ. Microbiol. 71**:**7253-7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Touchon, M., et al. 2009. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 5**:**e1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vallenet, D., et al. 2009. MicroScope: a platform for microbial genome annotation and comparative genomics. Database 2009**:**bap021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Whitfield, C. 2006. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu. Rev. Biochem. 75**:**39-68. [DOI] [PubMed] [Google Scholar]
- 54.Zagaglia, C., et al. 1991. Virulence plasmids of enteroinvasive Escherichia coli and Shigella flexneri integrate into a specific site on the host chromosome: integration greatly reduces expression of plasmid-carried virulence genes. Infect. Immun. 59**:**792-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplemental material]