SHPRH and HLTF Act in a Damage-specific Manner to Coordinate Different Forms of Post-replication Repair and Prevent Mutagenesis (original) (raw)
. Author manuscript; available in PMC: 2012 Apr 22.
SUMMARY
Post-replication repair (PRR) pathways play important roles in restarting stalled replication forks and regulating mutagenesis. In yeast, Rad5-mediated damage avoidance and Rad18-mediated translesion synthesis (TLS) are two forms of PRR. Two Rad5-related proteins, SHPRH and HLTF, have been identified in mammalian cells, but their specific roles in PRR are unclear. Here, we show that HLTF and SHPRH suppress mutagenesis in a damage-specific manner, preventing mutations induced by UV and MMS, respectively. Following UV, HLTF enhances PCNA monoubiquitination and recruitment of TLS polymerase eta, while also inhibiting SHPRH function. In contrast, MMS promotes the degradation of HLTF and the interactions of SHPRH with Rad18 and polymerase kappa. Our data not only suggest that cells differentially utilize HLTF and SHPRH for different forms of DNA damage, but also, surprisingly, that HLTF and SHPRH may coordinate the two main branches of PRR to choose the proper bypass mechanism for minimizing mutagenesis.
INTRODUCTION
The genome is particularly vulnerable during DNA replication since replication forks can stall at secondary DNA structures, repetitive sequences, certain protein-DNA complexes, and DNA damage. Although stalled forks are typically stabilized by the DNA damage response, they can also undergo collapse, a process associated with replisome dissociation, double-strand break formation, and the generation of gross chromosomal rearrangements (GCRs) (Branzei and Foiani, 2010). DNA damage tolerance pathways, also known as post-replication repair (PRR) pathways, help prevent fork collapse when DNA damage is present by allowing stalled forks to progress through lesions (Branzei and Foiani, 2010; Chang and Cimprich, 2009; Friedberg, 2005; Lee and Myung, 2008). Studies in both yeast and mammalian cells have suggested there are two major strategies for PRR. The first employs translesion synthesis (TLS) polymerases, a class of specialized, low-processivity polymerases able to replicate over distortions in DNA and directly bypass many lesions (Prakash et al., 2005; Waters et al., 2009). The second involves indirect bypass through a damage avoidance mechanism. Although the mechanism of damage avoidance is unclear, it is thought to involve template switching with the undamaged sister chromatid and/or homologous recombination (Broomfield et al., 2001; Unk et al., 2010). Thus, both the direct and indirect bypass pathways allow for resumption of DNA replication, leaving the repair of lesions for a later time.
Although PRR pathways prevent serious chromosomal abnormalities, lesion bypass itself can introduce mutations and represents an important mechanism by which mutations can result from DNA damage. Many fork-stalling lesions cause inaccurate base pairing (e.g. at abasic sites or alkylated bases). Thus, direct lesion bypass by TLS polymerases can result in either point or frameshift mutations, as these use damaged DNA as templates (Prakash et al., 2005; Waters et al., 2009). In contrast, when bypass is accomplished by damage avoidance, the chance of mutation may be significantly reduced, as undamaged sister chromatids are used as templates, thereby avoiding the lesion entirely (Broomfield et al., 2001; Friedberg, 2005). Moreover, certain TLS polymerases themselves are now known to bypass specific lesions with high efficiency and fidelity. For example, human Polη, loss of which leads to the UV-sensitive syndrome xeroderma pigmentosum variant XP-V, can bypass cyclobutane pyrimidine dimers in an error-free manner (Johnson et al., 1999; Masutani et al., 1999). Thus, while convention maintains that TLS is error-prone and that damage avoidance is error-free, we now know that TLS can be error-free, as well.
Although individual TLS polymerases have different activities and are likely tailored to specific types of fork-stalling lesions, the mechanism by which the cell promotes the use of one over another for a given lesion are not clear, nor is it clear what ultimately determines the choice between TLS and damage avoidance. A critical step in the regulation of PRR is the post-translational modification of PCNA (proliferating cell nuclear antigen), the replicative sliding clamp that plays an essential role in replication. Following many forms of DNA damage and replication stress, PCNA becomes modified on lysine 164 (K164) by the addition of either a mono- or polyubiquitin chain (Hoege et al., 2002; Lee and Myung, 2008; Stelter and Ulrich, 2003; Ulrich, 2009). Monoubiquitination of PCNA promotes some forms of direct lesion bypass by recruiting TLS polymerases to the replication fork through ubiquitin-interacting domains (Bienko et al., 2005; Kannouche et al., 2004; Waters et al., 2009), while polyubiquitination of PCNA promotes damage avoidance pathways through a process that is still poorly defined (Ulrich, 2009). The ubiquitination of PCNA is mediated by the Rad6 epistasis group in yeast, and two RING finger domain-containing proteins, Rad18 and Rad5, function as the ubiquitin ligases for this process (Hoege et al., 2002; Lee and Myung, 2008; Stelter and Ulrich, 2003; Ulrich, 2009). Rad18 mediates the monoubiquitination of PCNA, while Rad5 facilitates the further addition of K63-linked polyubiquitin chains. Despite the clear importance of PCNA ubiquitination in the regulation of PRR, it is insufficient, by itself, to account for the specificity of PRR pathway choice, as several TLS polymerases have ubiquitin-interacting motifs (Waters et al., 2009). This suggests additional specificity factors may be involved.
Genetic and biochemical studies in yeast suggest Rad5 may have additional roles in lesion bypass that are independent of its ubiquitin-conjugating activity. Rad5 contains a SWI/SNF-ATPase domain, which is important for its function in PRR (Gangavarapu et al., 2006). Biochemical studies have also shown that purified Rad5 can act as a helicase, mediating fork reversal in vitro, and it has been suggested that this activity might help to resolve stalled forks and promote template switching (Blastyak et al., 2007; Unk et al., 2010). Furthermore, Rad5 is required for at least one branch of TLS in yeast (Coulon et al., 2010; Pages et al., 2008). These findings suggest that Rad5 may have multiple roles in PRR, affecting both damage avoidance and TLS pathways.
In mammalian cells, there is a clear ortholog of Rad18 also involved in PCNA ubiquitination and PRR (Watanabe et al., 2004). However, two human Rad5-related proteins have been identified, SNF2 Histone-linker PHD-finger RING-finger Helicase (SHPRH) and Helicase-Like Transcription Factor (HLTF). Both HLTF and SHPRH are able to convert monoubiquitinated PCNA into the polyubiquitinated form in vitro, and they both appear to affect the polyubiquitination of PCNA in cells (Motegi et al., 2008; Motegi et al., 2006; Unk et al., 2008; Unk et al., 2006). Furthermore, loss of either HLTF or SHPRH increases the frequency of chromosome abnormalities observed following DNA damage (Motegi et al., 2008; Motegi et al., 2006). These findings suggest that both proteins are important for the maintenance of genomic stability in the presence of damage.
It is generally presumed that, like Rad5 in yeast, HLTF and SHPRH function to mediate polyubiquitination and damage avoidance. However, the specific functions of these two proteins in response to DNA damage have not been clearly addressed, and it is not known whether they may mediate different forms of damage avoidance or whether they might have other functions. Here, we investigated the roles of SHPRH and HLTF in PRR. Surprisingly, we found that SHPRH and HLTF act in non-redundant, damage-specific ways to suppress UV- or MMS-induced mutagenesis. The use of SHPRH versus HLTF appears to be regulated, at least in part, by a switch between Rad18-HLTF and Rad18-SHPRH complexes that is induced by DNA damage and the damage-specific degradation of HLTF. Furthermore, our data suggest SHPRH and HLTF promote error-free forms of PRR by helping to recruit the most appropriate TLS polymerase to a specific lesion. Thus, our data indicate that the functions of HLTF and SHPRH are not limited to the damage avoidance pathway and that they play a more central decision-making role in PRR to prevent mutagenesis.
RESULTS
Loss of HLTF or SHPRH increases mutagenesis caused by UV or MMS damage
The Rad5-mediated branch of PRR suppresses spontaneous and damage-induced mutagenesis in yeast (Broomfield et al., 2001; Johnson et al., 1992). To determine whether HLTF and SHPRH perform a similar function in mammalian cells, we assessed mutation frequency using a plasmid-based mutation assay that has been used for translesion synthesis (TLS) polymerases and other PRR proteins (Choi and Pfeifer, 2005; Huang et al., 2006; Parris and Seidman, 1992). In brief, a plasmid encoding a suppressor tRNA (SupF) was damaged with UV or MMS and then transfected into 293T cells together with diced siRNA pools targeted to either HLTF or SHPRH. The replicated plasmid was then isolated and transformed into an E. coli host containing a lacZ gene with a premature stop codon, and mutation frequency was assessed using blue-white selection. Diced siRNAs (d-siRNAs) targeting SHPRH and HLTF both efficiently reduced mRNA and protein levels (Fig 1A and data not shown) and had no significant effect on spontaneous mutagenesis. Importantly, as for yeast Rad5, loss of these Rad5-related proteins elevated the mutation frequency induced by DNA damaging agents (Fig 1B, C), and sequence analysis revealed the expected mutation spectrum for both UV and MMS (Fig S1F). Surprisingly, knockdown of HLTF greatly enhanced UV light-induced mutagenesis but had no significant effect on MMS-induced mutagenesis. Conversely, SHPRH knockdown promoted mutagenesis induced by MMS treatment, but had no significant effect on UV mutagenesis. Similar results were seen in HCT116 colon cancer cells, suggesting the effects are not limited to one cell type, and synthetic siRNAs targeted to HLTF and SHPRH had similar effects in 293T cells (Fig S1). Moreover, the mutagenic effects of the d-siRNAs could be rescued by ectopic expression of d-siRNA-resistant Xenopus HLTF and mouse SHPRH (Fig 1D & E, S1). These results indicate that HLTF and SHPRH play distinct roles in suppressing UV- and MMS-induced mutagenesis in human cells.
Figure 1. Loss of HLTF and SHPRH Leads to Increased Mutagenesis.

(A) Cells transfected with the indicated d-siRNAs were harvested after 48 hr, and protein levels were assessed by western blotting. (B&C) The mutation frequency was assessed 48 hr after cotransfection of cells with mock-treated, UV-irradiated or MMS-treated plasmid (pSP189) and the indicated d-siRNAs. (D&E) Rescue of HLTF and SHPRH knockdown. Mutation frequency was assessed as in B with the UV- (D) or MMS-treated plasmid (E) except that d-siRNA-resistant Xenopus HLTF, mouse SHPRH or empty vector was coexpressed as indicated. In all cases, data represent average +/− SEM (shown as error bar) from at least three independent experiments. See also Figure S1.
Damage-specific interactions of Rad18, HLTF and SHPRH
In addition to mediating PCNA ubiquitination, Rad18 also plays a structural role, recruiting Rad5 and other PRR proteins to stalled forks (Ulrich and Jentsch, 2000; Watanabe et al., 2004). Consistent with these findings, we and others have found that human Rad18 can interact with both SHPRH and HLTF. We also observed an interaction between SHPRH and HLTF, consistent with previous reports (Fig S2 & (Motegi et al., 2008; Motegi et al., 2006; Unk et al., 2008; Unk et al., 2006)). To test whether DNA damage affects these interactions, cells were transfected with FLAG-tagged Rad18 (F-Rad18), SHPRH and/or HLTF then either mock-treated or exposed to MMS or UV irradiation, and interactions with F-Rad18 were analyzed. In response to MMS treatment, but not UV irradiation, the interaction between SHPRH and F-Rad18 was significantly enhanced (Fig 2A). Similar results were observed across a range of MMS doses (Fig 2B) and with the endogenous proteins (Fig S2D). Interestingly, after UV treatment, the interaction between Rad18 and SHPRH decreased (Fig 2A, C), and the interaction between HLTF and SHPRH modestly increased (Fig S2). These observations indicate that UV and MMS alter the interactions between Rad18, SHPRH and HLTF in distinct and damage-specific ways, consistent with the damage-specific effects of HLTF and SHPRH loss in preventing mutagenesis.
Figure 2. Damage-Specific Interactions between SHPRH, HLTF and Rad18.
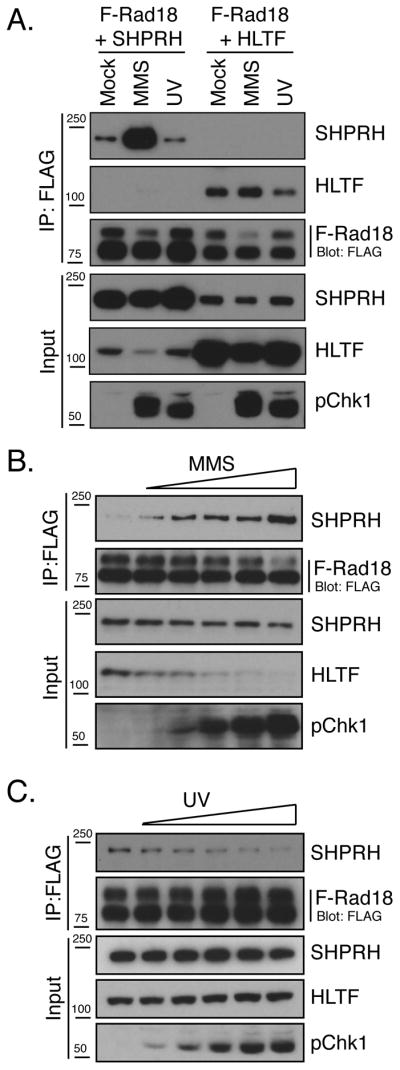
(A) MMS promotes the interaction of Rad18 and SHPRH. Cells expressing F-Rad18 and untagged HLTF or SHPRH were treated with MMS or UV. After 4hr, cells were lysed in a high salt/chromatin-extraction buffer (Condition A), then F-Rad18 and associated proteins were precipitated and analyzed by western blot. (B&C) UV and MMS dose effects on the Rad18-SHPRH interaction. Cells expressing F-Rad18 and SHPRH were treated with MMS (0, 0.1, 0.2, 0.3, 0.4 and 0.5 mM) or UV (0, 10, 20, 30, 40, 50 J/m2) and processed as in A. See also Figure S2.
MMS induces HLTF ubiquitination and degradation
In our coimmunoprecipitation assays, HLTF consistently decreased after MMS treatment (Fig 2B). Therefore, we asked if DNA damage affected the protein levels of endogenous HLTF and SHPRH. We found significant effects of MMS treatment, but not UV irradiation, on HLTF levels, but only minor effects of either treatment on SHPRH (Fig 3A and S3). The effects on HLTF were only observed following treatment with the alkylating agents MMS and ethyl methanesulfonate (EMS), and not following several other types of genotoxic stress (Fig S3).
Figure 3. MMS Induces HLTF Degradation through the Ubiquitin-Proteasome Pathway.

(A) HLTF protein levels are selectively reduced by MMS treatment. Cells were treated with UV (0, 25, 50, 100, 200, 400 J/m2) or MMS as in Fig 2. After 4 hr total cellular protein was isolated and samples were analyzed by western blotting. Protein levels were quantified and normalized to HSP90. (B) MMS enhances HLTF protein degradation. Cells mock-treated or treated with UV or MMS together with cycloheximide (CHX, 100 μM) were collected and analyzed as in (A) using tubulin as a control for protein quantification. (C) MMS-induced HLTF degradation is proteasome-dependent. Mock or MMS-damaged cells were treated with MG132 then analyzed as in A. (D) MMS increases HLTF ubiquitination. Cells transfected with F-SHPRH or F-HLTF with or without HA-Ub were mock-, UV- or MMS-treated. FLAG-tagged proteins were precipitated after 4 hr and blotted with α-HA antibodies. The same membranes were stained with coomassie blue. See also Figure S3.
To determine if the effects of MMS on HLTF levels are due to increased protein degradation, we used cycloheximide to block translation and assessed the stability of HLTF at various times after DNA damage. We found that HLTF levels decreased more rapidly following MMS treatment (t½= 1.9 hr) compared to mock treatment (t½= 6 hr) or UV irradiation (t½= 7.4 hr) indicating that the changes in HLTF are the result of accelerated protein degradation (Fig 3B & S3). This degradation was blocked by treatment with the proteosome inhibitors, MG132 or lactacystin (Fig 3C and S3). We also observed a lower mobility form of HLTF when lactacystin was present, suggesting that HLTF degradation may involve ubiquitination. To test this idea, we expressed FLAG-tagged SHPRH or HLTF together with HA-ubiquitin (HA-Ub) and examined the ubiquitination of F-SHPRH and F-HLTF after UV or MMS treatment. As shown in Figure 3D, HLTF ubiquitination was induced by MMS, while SHPRH ubiquitination was unaltered. These results indicate that MMS causes the degradation of HLTF, at least in part through the ubiquitin-proteasome pathway.
HLTF inhibits the interaction between SHPRH and Rad18
The effects of MMS on HLTF stability and the Rad18-SHPRH interaction could indicate that HLTF prevents the interaction of SHPRH with Rad18. Consistent with this idea, formation of the SHPRH-Rad18 complex coincided with the degradation of HLTF in a time and dose-dependent manner (Fig 4A & 2B). Additionally, increased expression of HLTF further suppressed the interaction between F-Rad18 and SHPRH in cells (Fig 4B), and competition for the binding of HLTF and SHPRH to Rad18 was also observed with purified proteins (Fig S4D). Interestingly, although increasing amounts of HLTF further disrupted the interaction of SHPRH with Rad18, they did not lead to the formation of additional HLTF-Rad18 complexes (Fig 4B & S4D). This may indicate that the effect of HLTF on the Rad18-SHPRH interaction depends, at least in part, on an interaction between HLTF and SHPRH that prevents the association of SHPRH with Rad18.
Figure 4. HLTF Inhibits the Rad18-SHPRH Interaction.

(A) Relationship between HLTF degradation and Rad18-SHPRH association following MMS treatment. Cells expressing SHPRH and F-Rad18 were treated with MMS, lysed using Hepes-Triton/100mM NaCl buffer (Condition B) and processed as in Fig 2A. (B) HLTF prevents the interaction of Rad18 and SHPRH. Cells expressing F-Rad18 and SHPRH were co-transfected with increasing amounts of the HLTF construct (0, 0.5, 1, 1.5, 2, 2.5 μg) and the interaction between F-Rad18 and SHPRH was assessed as in Fig 2B. (C) Proteosome inhibition suppresses the Rad18-SHPRH interaction. Cells expressing F-SHPRH and Rad18 were treated with MG132 (50 μM) and analyzed as in Fig 2B. (D) HLTF-CS mutant is resistant to MMS-induced degradation. F-HLTF or F-HLTF-CS was expressed and samples were analyzed as in Fig 3A. (E) HLTF-CS is not ubiquitinated after MMS treatment. Cells transfected with empty vector, F-HLTF or F-HLTF-CS and HA-Ub were analyzed as in Fig 3D. (F) Expression of HLTF-CS inhibits the interaction between SHPRH and Rad18 induced by MMS treatment. Cells were transfected with HLTF or HLTF-CS together with F-Rad18 and SHPRH then analyzed as in Fig 2A. See also Figure S4.
To test whether HLTF degradation is needed to promote the interaction of Rad18 with SHPRH, we tested the effect of proteasome inhibition on the SHPRH-Rad18 interaction induced by MMS. MG132 treatment blocked MMS-induced HLTF degradation as well as the Rad18-SHPRH interaction (Fig 4C), suggesting that HLTF degradation might be required to promote this interaction. To further test this hypothesis, we sought to identify a form of HLTF that does not degrade following MMS treatment. Surprisingly, we found that the MMS-induced degradation of HLTF was dramatically decreased when two conserved cysteines in the RING domain of HLTF were mutated to serine (HLTF-CS) (Fig 4D). This mutation is known to affect the ubiquitin ligase activity of RING-domain containing proteins (Creagh et al., 2004). Interestingly, the MMS-induced ubiquitination of HLTF-CS also decreased (Fig 4E), suggesting that the ubiquitin ligase activity of HLTF is important for its ubiquitination and degradation following MMS.
To determine whether HLTF-CS interfered with the ability of MMS to increase the association of SHPRH with Rad18, we coexpressed either wild-type HLTF or HLTF-CS with SHPRH and F-Rad18 and monitored the association of SHPRH with F-Rad18. The increased basal interaction between F-Rad18 and SHPRH observed in the presence of HLTF-CS is likely due to lower expression of HLTF-CS versus HLTF, consistent with the observations in Fig 4B. Importantly, expression of HLTF-CS prevented the increased association of SHPRH with Rad18 following MMS treatment while expression of wild-type HLTF did not (Fig 4F). These findings strongly suggest that the degradation of HLTF and its ubiquitin ligase activity is needed for the formation of the SHPRH-Rad18 complex following MMS treatment.
Because the interactions between HLTF, SHPRH and Rad18 are direct and competitive, we also asked if loss of HLTF is sufficient to promote formation of the Rad18-SHPRH complex in the absence of DNA damage. As shown in Fig S4E, knockdown of HLTF had no effect on the SHPRH-Rad18 interaction in mock-treated cells, indicating that loss of HLTF is not sufficient to promote the interaction between SHPRH and Rad18. Interestingly, however, loss of HLTF enhanced the interaction between Rad18 and SHPRH in UV-irradiated cells, although not to the extent induced by MMS. Taken together, these observations indicate that the presence of HLTF is sufficient to suppress the interaction of SHPRH with Rad18 under UV conditions. Following MMS damage, however, HLTF degradation is necessary, although not sufficient, to promote the Rad18-SHPRH interaction.
An imbalance of HLTF and SHPRH affects UV-induced mutagenesis
The competitive interactions between HLTF and SHPRH for Rad18 and the collective effects of MMS and UV on the formation of Rad18 complexes indicate that HLTF suppresses the interaction of Rad18 with SHPRH following UV. Furthermore, they raise the possibility that HLTF may be necessary to suppress a mutagenic function of SHPRH in UV-irradiated cells. To test this model, we performed the SupF mutation assay in cells expressing GFP-HLTF or GFP-SHPRH, reasoning that an increase in SHPRH levels might increase the rate of UV mutagenesis by preventing the action of HLTF and/or promoting the action of SHPRH. Consistent with this hypothesis, expression of SHPRH – but not HLTF – increased the mutation frequency following UV irradiation. There was no effect of expressing either HLTF or SHPRH on MMS-induced mutagenesis (Fig 5A, S5).
Figure 5. HLTF Suppresses SHPRH-mediated Mutagenic Bypass of UV-damaged DNA.

(A) Overexpression of SHPRH elevates UV-induced mutagenesis. Mutation frequency was assessed as in Fig 1B with cells expressing GFP, GFP-SHPRH or GFP-HLTF. (B) Knockdown of SHPRH rescues the UV-mutagenesis induced by knockdown of HLTF. UV (left) or MMS (right) induced mutation frequency was assessed as in Fig 1B using cells transfected with the indicated d-siRNAs. Data represent average +/− SEM (shown as error bar) from three independent experiments. See also Figure S5.
If the elevation in UV-induced mutagenesis in cells lacking HLTF is caused, at least in part, by the unregulated action of SHPRH, knockdown of SHPRH might suppress the effects of HLTF loss. Indeed, we found that the mutagenesis frequency was restored to the level of that observed in control cells when both HLTF and SHPRH were knocked down (Fig 5B & S5). In contrast, knockdown of both proteins had the same effect as SHPRH knockdown in MMS-treated cells. These observations indicate that the relative amounts of SHPRH and HLTF may determine the rate of UV-induced mutagenesis and suggest that HLTF suppresses the mutagenic function of SHPRH under UV conditions.
HLTF promotes PCNA monoubiquitination and UV-induced Polη recruitment
Previous studies have shown that HLTF and SHPRH, like Rad5, mediate the polyubiquitination of PCNA in vitro and in vivo (Motegi et al., 2008; Motegi et al., 2006; Unk et al., 2008; Unk et al., 2006). To ask if we could detect any differences in the effect of HLTF or SHPRH on PCNA ubiquitination, we expressed each protein together with HA-Ub in 293T cells and directly compared the effect of their expression on PCNA modifications. Although consistent effects on polyubiquitination were difficult to observe, ectopic expression of HLTF led to a modification in PCNA consistent with monoubiquitination (Fig 6A). This was also observed upon expression of Rad18, but not following expression of SHPRH or the HLTF-CS mutant. Because these observations suggest a previously unappreciated role for a Rad5-related protein in the control of monoubiquitination and, potentially, the recruitment of TLS polymerases, we sought to confirm that this was indeed PCNA monoubiquitination. To do so, we isolated ubiquitinated proteins via nickel-NTA-affinity purification from cells expressing His-Ub and wild-type HLTF, HLTF-CS or an ATPase mutant of HLTF. Expression of wild-type HLTF and the HLTF-ATPase mutant, but not HLTF-CS, increased the monoubiquitination of PCNA (Fig 6B). In addition, the levels of PCNA monoubiquitination were reduced by knockdown of HLTF or Rad18, but not SHPRH, and there was no greater decrease in monoubiquitination when both SHPRH and HLTF were knocked down (Fig 6C). These results strongly suggest that HLTF promotes PCNA monoubiquitination. To assess the role of HLTF in DNA damage-induced PCNA monoubiquitination, we also analyzed this modification in cells expressing no ectopic proteins, since expression of His-Ub increased basal PCNA ubiquitination and suppressed its damage inducibility (data not shown). Importantly, loss of HLTF reduced the monoubiquitination of PCNA specifically following treatment with UV or aphidicolin. After MMS treatment there was no effect of HLTF loss, consistent with the absence of HLTF protein after MMS treatment (Fig 6D). These observations suggest that HLTF, but not SHPRH, specifically promotes PCNA monoubiquitination. They also raise the possibility that the different effects of HLTF and SHPRH on PCNA ubiquitination might be related to their damage-specific effects on mutagenesis.
Figure 6. HLTF Regulates PCNA Monoubiquitination and UV-induced Polη Recruitment.

(A) Ectopic expression of HLTF promotes PCNA monoubiquitination. The indicated proteins were co-expressed with HA-Ub, and after 48 hr cells were lysed and PCNA ubiquitination was assessed by western blotting. (B) HLTF ligase activity is required to promote PCNA monoubiquitination. Cells were transfected with F-HLTF, F-HLTF-CS and an HLTF ATPase mutant (F-HLTF-mATPase) as well as His-Ub. Ubiquitinated proteins were isolated with Ni-NTA resin and PCNA ubiquitination was assessed by western blotting with α-PCNA antibody. (C) Loss of HLTF reduces PCNA monoubiquitination. The indicated d-siRNAs were transfected together with His-Ub and cells were processed as described in (B). (D) HLTF knockdown reduces PCNA monoubiquitination following DNA damage. d-siRNA-transfected cells were mock treated or treated with aphidicolin (10 μM), UV (100 J/m2) or MMS (0.5 mM). After 4 hr, lysates were analyzed by western blotting. (E–G) Effect of SHPRH and HLTF knockdown on Polη foci formation. XPV cells stably expressing GFP-tagged Polη were transfected with the indicated d-siRNAs, damaged with UV (40 J/m2) after 48 hr then fixed and stained 4h later with anti-Polη and anti-RPA32 antibodies as well as Hoechst (E). The nuclear intensity of GFP-Polη (F) and RPA32 (G) staining in cells was measured and the percentage of positive cells was determined. Data represent average +/− SEM (shown as error bar) from three independent experiments. See also Figure S6.
Because the TLS polymerase Polη can bypass UV lesions in an error-free manner and because its interaction with PCNA is ubiquitin-dependent (Johnson et al., 1999; Kannouche et al., 2004; Masutani et al., 1999), we next asked whether HLTF affects the recruitment of Polη. To do so, we transfected control, SHPRH or HLTF d-siRNAs into Polη-deficient XPV cells reconstituted with stably expressed GFP-Polη and monitored the localization of GFP-Polη following UV treatment. We found, as previously reported, that Polη formed triton-resistant nuclear foci following UV treatment, but that no Polη can be detected in undamaged cells after extraction (Fig 6E & (Kannouche et al., 2001)). To quantify the effect of HLTF knockdown, we determined the nuclear intensity of the GFP-Polη signal in individual cells after triton extraction and staining with antibodies to Polη. Cells were also stained with antibodies to replication protein A (RPA) as a marker for DNA damage and ssDNA formation. Following UV damage, the percentage of cells staining positive for both Polη and RPA increased (Fig 6E–G & S6). However, upon HLTF knockdown, the percentage of Polη positive cells was significantly reduced compared to that in the control or SHPRH knockdown, while the number of RPA-positive cells did not change. These observations indicate that HLTF promotes Polη chromatin binding following UV treatment.
Damage-specific functions of Polη and Polκ in mutagenesis and their interaction with SHPRH
The damage-specific effects of HLTF and SHPRH, as well as the effect of HLTF on Polη, could indicate that HLTF and SHPRH mediate the functions of specific branches of the TLS pathway. Polκ has been implicated in the bypass of alkylated bases and abasic sites and can bypass adducts that mimic damage induced by MMS in a 10-fold more accurate manner than Polη or Polι (Johnson et al., 2007; Ohashi et al., 2000; Plosky et al., 2008). Mouse cells deficient in Polκ are also hypersensitive to MMS (Takenaka et al., 2006). Therefore, we asked if there were damage-specific effects of Polη or Polκ loss in the mutagenesis assay. As previously observed, knockdown of Polη led to increased UV-induced mutagenesis (Fig 7A, B & (Choi and Pfeifer, 2005)). Furthermore, knockdown of Polκ but not Polη increased MMS-induced mutagenesis. These findings suggest that the TLS polymerases Polη and Polκ, like HLTF and SHPRH, suppress UV and MMS-induced mutagenesis, respectively.
Figure 7. Polκ and Polη Functions in Mutagenesis and Their Damage-specific Interactions with SHPRH.

(A) Effect of control, Polη or Polκ siRNAs on Polη or Polκ protein levels as assessed by western blotting. (B) Knockdown of Polη or Polκ enhances UV- or MMS-induced mutagenesis. Mutation frequency was assessed as in Fig 1B. (C) MMS effects on the interaction of Rad18 and SHPRH with Polη and Polκ. The indicated vectors were coexpressed with GFP-Polκ or YFP-Polη and coimmunoprecipitation was performed 4 hr after the indicated treatment as in Fig 2A. (D) Direct interaction between xPolκ and SHPRH. Beads bound to GST, GST-Rad18 or GST-xPolκ were incubated with purified F-Rad18 or F-SHPRH. Bound proteins were resolved by SDS-PAGE and analyzed by western blotting. (E) Polκ is required for SHPRH-mediated mutagenic bypass of UV-damaged DNA. Cells expressing GFP or GFP-SHPRH were co-transfected with the indicated siRNA. The mutation frequency of these cells was assessed as described in Fig 1B. In B and E, the data represent average +/− SEM (shown as error bar) from three independent experiments. (F) Model for the damage-specific roles of SHPRH and HLTF in promoting error-free bypass of UV and MMS-induced lesions. (i) In the absence of damage, both Rad18-HLTF and Rad18-SHPRH complexes are present. (ii) MMS induces the proteosome-dependent degradation of HLTF. This degradation, together with another unknown factor (represented as X) also induced by MMS, promotes the formation of SHPRH-Rad18 complexes and SHPRH-Polκ complexes to suppress mutagenesis. (iii) HLTF promotes error-free bypass following UV by promoting PCNA monoubiquitination and Polη recruitment as well as by suppressing SHPRH function and its interaction with Rad18. See also Figure S7.
Recently, yeast Rad5 was shown to physically interact with the TLS polymerase Rev1. Thus, we asked if either Polη or Polκ could interact with either HLTF or SHPRH. Cells were transfected with YFP-Polη or GFP-Polκ, and F-HLTF, F-SHPRH or F-Rad18, and then FLAG-tagged and associated proteins were precipitated from cells. Consistent with previous reports (Bi et al., 2006; Watanabe et al., 2004), we found Polη and Polκ in immunoprecipitates of F-Rad18. We also found that both Polη and Polκ interacted with SHPRH but not with HLTF (Fig S7). To test the preference of SHPRH for Polκ vs Polη, as well as the effect of DNA damage on these interactions, immunoprecipitations were performed in cells transfected with both YFP-Polη and GFP-Polκ, together with FLAG-tagged HLTF, SHPRH or Rad18. Importantly, we found that MMS, but not UV, promoted an interaction between SHPRH and both polymerases, while, again, no interaction of Polκ or Polη with HLTF was observed. Interestingly, we reproducibly found more Polκ than Polη in the SHPRH immunoprecipitates both in the presence and absence of DNA damage, suggesting that Polκ may interact more strongly with SHPRH. Increased interactions were also observed between these polymerases and Rad18 following MMS treatment (Fig 7C). These interactions appear to be direct, since GST-Polκ bound affinity-purified F-SHPRH and F-Rad18 in a pull-down experiment (Fig 7D).
The interaction between SHPRH and Polκ raises the possibility that the mutagenesis observed upon overexpression of SHPRH (see Fig 5A) results from the increased recruitment of Polκ to stalled forks. To test this idea, we knocked down Polκ in cells that overexpressed GFP-SHPRH, then carried out the mutagenesis assay with UV-damaged plasmid. Knockdown of Polκ prevented the mutagenesis observed upon GFP-SHPRH expression suggesting that SHPRH may be promoting aberrant Polκ function at stalled forks (Fig 7E). Taken together, these findings suggest that SHPRH directly interacts with the TLS polymerase Polκ to suppress MMS-induced mutagenesis.
DISCUSSION
Different types of DNA damage trigger different repair and signaling pathways, and the same type of DNA damage can even trigger different pathways, depending on the phase of the cell cycle in which it occurs (Branzei and Foiani, 2008). This type of specialization allows the cell to balance efficiency with accuracy in repair, but in most cases how such specialization is accomplished is not known. In this study, we provide evidence that two structurally similar Rad5-related proteins, SHPRH and HLTF, function in a dynamic equilibrium that is differentially affected by different types of DNA damage to prevent mutagenesis and genomic rearrangement.
Damage-Specific Responses of SHPRH and HLTF
We find that HLTF and SHPRH suppress mutagenesis in a damage-specific manner, with HLTF acting in response to DNA damage caused by UV treatment and SHPRH acting in response to damage caused by MMS treatment. It has been shown previously that SHPRH and HLTF interact both with Rad18 and with each other, and we show here that the interactions of HLTF and SHPRH with Rad18 are competitive. Our data suggest that different types of DNA damage promote or suppress the formation of specific Rad18 complexes to direct the damage-specific responses carried out by HLTF and SHPRH. They also suggest that the function of specific Rad18 complexes with either HLTF or SHPRH post-damage facilitates recruitment of the TLS polymerase best suited to bypass the lesion and suppress mutagenesis in a damage-specific manner. Thus, the damage-specific functions of HLTF and SHPRH are mediated, at least in part, by the TLS arm of the PRR pathway, as opposed to the damage avoidance arm.
SHPRH in the Suppression of MMS-Induced Mutagenesis
We find that SHPRH plays a critical role in preventing mutagenesis induced by MMS, and that its function at replication forks may be directed by its interaction with Rad18. Importantly, the MMS-induced interaction of SHPRH with Rad18 also seems to require the degradation of HLTF. Indeed, the concentrations of HLTF and Rad18 are higher than that of SHPRH in undamaged cells (Fig S4), so that degradation of HLTF may be needed to allow SHPRH to associate with Rad18. Consistent with this model, we show that inhibition of the proteosome or expression of a non-degradable mutant of HLTF suppresses the association of SHPRH with Rad18 induced by MMS. Nevertheless, HLTF degradation alone is not sufficient to cause formation of the Rad18-SHPRH complex which suggests that there might be other protein factors or post-translational modifications involved. Thus, we propose that MMS promotes SHPRH-mediated forms of PRR in at least two ways, by enhancing the interaction between SHPRH and Rad18 and by promoting the degradation of HLTF (Fig 7F). Intriguingly, we find that HLTF is ubiquitinated and degraded following MMS treatment and that these events are dependent upon its RING domain. These observations suggest that HLTF degradation involves autoubiquitination and that this process is triggered by a damage-specific event.
The dynamic interactions of SHPRH following DNA damage also provide insights into the mechanism by which SHPRH suppresses MMS-induced mutagenesis, as they link SHPRH to the recruitment of specific TLS polymerases. We find that MMS strongly induces an interaction of SHPRH with Polη and Polκ, with a preference for Polκ. Interactions of Rad18 with Polη and Polκ are also enhanced by MMS treatment, but in this case we observe a preference for Polη. The reasons for this difference are not clear, however, the net effect of these changes may be to alter the balance of TLS polymerases at the fork. Thus, the recruitment of SHPRH to Rad18 following MMS treatment may help to direct additional Polκ to the replication fork where it might suppress MMS-induced mutagenesis. Consistent with this model, we find that knockdown of Polκ increases mutagenesis following MMS (Fig 7B). Furthermore, previous findings suggest potential roles for Polκ in the response to MMS (Johnson et al., 2007; Ohashi et al., 2000; Plosky et al., 2008; Takenaka et al., 2006).
HLTF in the Suppression of UV-Induced Mutagenesis
We propose that HLTF suppresses UV-induced mutagenesis in at least two ways (Fig 7F). One effect of HLTF is to promote PCNA monoubiquitination after UV damage, thereby facilitating the error-free bypass of UV lesions. Consistent with this idea, knockdown of HLTF prevented the association of Polη with UV-irradiated chromatin. The second function for HLTF in suppressing UV-induced mutagenesis involves inhibition of SHPRH. Indeed, we find that overexpression of SHPRH elevates mutation frequency following UV irradiation. Furthermore, the UV-induced mutagenesis observed following HLTF loss is abrogated by knockdown of SHPRH. The observed interaction between SHPRH and Polκ may contribute to the mutagenic effects of SHPRH overexpression and HLTF loss, since the bypass of UV lesions by Polκ can be inaccurate (Ohashi et al., 2000; Prakash et al., 2005). Consistent with this model, loss of Polκ suppresses the mutagenesis observed upon expression of SHPRH. Interestingly, while we detect no significant effect of UV on HLTF-Rad18 interactions, we do find that UV suppresses the interaction between SHPRH and Rad18 via HLTF. Taken together, these findings suggest that negative regulation of Rad18-SHPRH complexes by HLTF may be important for the suppression of UV-induced mutagenesis.
Multiple Functions for HLTF and SHPRH
Altogether, our results suggest that SHPRH and HLTF regulate the interplay between different forms of PRR by directing the most appropriate TLS polymerase to the site of DNA damage. We suggest that these are unappreciated functions for HLTF and SHPRH that are likely to act together with other previously reported functions for these proteins, such as PCNA polyubiquitination. Indeed, a recent study in S. pombe showed that the enzymes needed for polyubiquitination of PCNA affect TLS (Coulon et al., 2010). Interestingly, Polη has only one ubiquitin binding domain while Polκ and some of the other TLS polymerases have two (Bienko et al., 2005). Thus, polyubiquitination of PCNA could allow for more efficient recruitment of Polκ and other polymerases with multiple ubiquitin binding domains to PCNA, whereas the monoubiquitination enforced by HLTF would favor the recruitment of Polη. These results, viewed in light of previous work suggesting roles for HLTF in fork reversal (Blastyak et al., 2010), indicate HLTF and SHPRH may have multiple functions in suppressing mutagenesis. Some of these functions may be overlapping (e.g. PCNA polyubiquitination and possibly fork reversal), whereas others, such as TLS recruitment and lesion bypass, may be non-redundant, damage-specific functions.
The Roles of HLTF and SHPRH in Maintaining Genome Stability
Loss of either SHPRH or HLTF under their damage-specific conditions leads to large genomic rearrangements and increased levels of point mutations (data not shown). Moreover, loss of either protein leads to increased GCRs in MEFs (Motegi et al., 2008; Motegi et al., 2006). Although these observations strongly suggest SHPRH and HLTF are needed to maintain genome stability, they also raise the question of how such complex mutations arise from defects in PRR pathways. In this context, we suggest it may be more appropriate to think of HLTF and SHPRH as playing critical roles in continued fork progression. The lack of HLTF- or SHPRH-mediated PRR could lead to slow progression of replication forks, causing prolonged stalling, fork collapse and GCRs. Indeed, by tracking replication forks on individual chromosomal fibers, it was shown that HLTF loss leads to increased numbers of stalled forks (Blastyak et al., 2010). Although loss of HLTF did not seem to affect MMS-induced mutagenesis in our assay, the effects of HLTF loss on replication forks and on GCRs were observed following MMS treatment (Blastyak et al., 2010; Motegi et al., 2008). This apparent discrepancy in damage specificity may result from a role for HLTF in preventing fork collapse by promoting efficient, though not necessarily accurate, bypass of MMS lesions. We hypothesize that both HLTF- and SHPRH-mediated pathways are initially used by the cell to promote efficient bypass. However, in order to achieve more accurate bypass in response to certain lesions, cells may have developed an additional regulatory mechanism to fine-tune this response. The degradation of HLTF following MMS treatment could help to channel MMS lesions through the more accurate SHPRH-mediated bypass. Thus, the presence of two Rad5-related genes in mammalian cells could ultimately allow the cell to optimize the accuracy and efficiency of its response to different types of lesions.
In summary, our results indicate that the balance between SHPRH and HLTF is critical to the outcome of PRR. They also suggest that deregulating the mechanisms governing the formation of Rad18-HLTF and Rad18-SHPRH complexes could drive mutagenesis and genome instability. Consistent with this idea, HLTF is frequently silenced in several types of cancer and changes in SHPRH have been observed as well (Unk et al., 2010). Further studies will be needed to better define the roles of HLTF and SHPRH and to determine how their different functions and domains specifically contribute to the maintenance of genome stability. Nevertheless, our work clearly shows these proteins are regulated in a damage-specific manner, functioning both in the TLS and damage avoidance arms of the PRR response to suppress mutagenesis and maintain genome stability.
EXPERIMENTAL PROCEDURES
Antibodies, Plasmids, DNA Damage and Cell Culture
293T cells were used in all experiments unless indicated. GFP-Polκ and YFP-Polη plasmids were provided by Dr. Vaziri (Bi et al., 2006), and the XPV-complemented cells were provided by Dr. Lehmann (Kannouche et al., 2001). Cells were damaged for 4 hr with 0.5 mM MMS (0.005%) or 50 J/m2 UV-C unless indicated. Sources for antibodies, reagents and cell lines and detailed descriptions of other cDNAs used are provided in the Supplemental Methods.
siRNA and Transfection
Diced siRNAs for human SHPRH and HLTF were generated as described using the primers provided in the Supplemental Data (Myers et al., 2003). Synthetic siRNA pools against SHPRH (MU-006167), HLTF (MU-006448), Polκ (MU-021038), Polη (MU-006454) and Rad18 (MU-004591) were purchased from Thermo Scientific. siRNA transfection was performed using Lipofectamine 2000 (Invitrogen) and the vendor’s protocol. Calcium phosphate was used for plasmid transfection.
Mutagenesis Assay
The protocol and reagents used for the SupF assay have been described (Parris and Seidman, 1992). Briefly, the reporter plasmid was damaged and purified then co-transfected with d-siRNAs or DNA as indicated. Replicated plasmid was retrieved and analyzed 48h later. Details are provided in the Supplemental Methods.
GST Pull-Downs, Immunoprecipitations and Protein Purification
FLAG-tagged proteins were isolated with M2 agarose beads. Extraction condition A: Cells were lysed in 600 mM NaCl/Hepes-Triton buffer (50 mM Hepes pH 7.5, 1 mM EDTA, 0.75% Triton X-100, 8% Glycerol, 1 mM DTT, 1 mM PMSF), diluted to 150 mM NaCl, then sonicated and clarified by centrifugation. Condition B: Cells were lysed in Hepes-Triton buffer (100mM NaCl) and clarified by centrifugation. Detailed conditions for cell lysis, protein purification and coimmunoprecipitations are described in the Supplemental Methods.
Immunofluorescence and Image Analysis
XPV-GFP cells were stained with Polη and RPA32 antibodies after pre-extraction in 0.5% Triton-X100. Cells were analyzed on a Zeiss Axioskope and 10x captured images were analyzed by ImageJ (NIH Image). Details on staining and fixation as well as image analysis are available in the Supplemental Methods. Overall 100 cells per experiment were counted and three independent results are pooled to calculated mean and SEM.
Statistical Analysis
Statistical analyses represent the mean of at least three independent experiments. Error bar represents standard error of mean (SEM). P-values were calculated using a two-tailed Student’s t test.
Supplementary Material
01
Acknowledgments
We would like to acknowledge Debbie J. Chang for technical assistance. We are also grateful to members of the Cimprich lab for helpful discussions and reading this manuscript. This work was supported by a Department of Defense (Breast Cancer Research Program) predoctoral fellowship (W81XWH–09–1–0026) and NIH training grant (R90 DK071499) to JRL, and by a grant to KAC from the NIH (ES016486).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bi X, Barkley LR, Slater DM, Tateishi S, Yamaizumi M, Ohmori H, Vaziri C. Rad18 regulates DNA polymerase kappa and is required for recovery from S-phase checkpoint-mediated arrest. Mol Cell Biol. 2006;26:3527–3540. doi: 10.1128/MCB.26.9.3527-3540.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, Kannouche P, Wider G, Peter M, Lehmann AR, et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310:1821–1824. doi: 10.1126/science.1120615. [DOI] [PubMed] [Google Scholar]
- Blastyak A, Hajdu I, Unk I, Haracska L. Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol Cell Biol. 2010;30:684–693. doi: 10.1128/MCB.00863-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blastyak A, Pinter L, Unk I, Prakash L, Prakash S, Haracska L. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol Cell. 2007;28:167–175. doi: 10.1016/j.molcel.2007.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9:297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11:208–219. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- Broomfield S, Hryciw T, Xiao W. DNA postreplication repair and mutagenesis in Saccharomyces cerevisiae. Mutat Res. 2001;486:167–184. doi: 10.1016/s0921-8777(01)00091-x. [DOI] [PubMed] [Google Scholar]
- Chang DJ, Cimprich KA. DNA damage tolerance: when it’s OK to make mistakes. Nat Chem Biol. 2009;5:82–90. doi: 10.1038/nchembio.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Pfeifer GP. The role of DNA polymerase eta in UV mutational spectra. DNA Repair (Amst) 2005;4:211–220. doi: 10.1016/j.dnarep.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Coulon S, Ramasubramanyan S, Alies C, Philippin G, Lehmann A, Fuchs RP. Rad8Rad5/Mms2-Ubc13 ubiquitin ligase complex controls translesion synthesis in fission yeast. EMBO J. 2010;29:2048–2058. doi: 10.1038/emboj.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creagh EM, Murphy BM, Duriez PJ, Duckett CS, Martin SJ. Smac/Diablo antagonizes ubiquitin ligase activity of inhibitor of apoptosis proteins. J Biol Chem. 2004;279:26906–26914. doi: 10.1074/jbc.M313859200. [DOI] [PubMed] [Google Scholar]
- Friedberg EC. Suffering in silence: the tolerance of DNA damage. Nat Rev Mol Cell Biol. 2005;6:943–953. doi: 10.1038/nrm1781. [DOI] [PubMed] [Google Scholar]
- Gangavarapu V, Haracska L, Unk I, Johnson RE, Prakash S, Prakash L. Mms2-Ubc13-dependent and -independent roles of Rad5 ubiquitin ligase in postreplication repair and translesion DNA synthesis in Saccharomyces cerevisiae. Mol Cell Biol. 2006;26:7783–7790. doi: 10.1128/MCB.01260-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R, D’Andrea AD. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol. 2006;8:339–347. doi: 10.1038/ncb1378. [DOI] [PubMed] [Google Scholar]
- Johnson RE, Henderson ST, Petes TD, Prakash S, Bankmann M, Prakash L. Saccharomyces cerevisiae RAD5-encoded DNA repair protein contains DNA helicase and zinc-binding sequence motifs and affects the stability of simple repetitive sequences in the genome. Mol Cell Biol. 1992;12:3807–3818. doi: 10.1128/mcb.12.9.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RE, Prakash S, Prakash L. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Pol eta. Science. 1999;283:1001–1004. doi: 10.1126/science.283.5404.1001. [DOI] [PubMed] [Google Scholar]
- Johnson RE, Yu SL, Prakash S, Prakash L. A role for yeast and human translesion synthesis DNA polymerases in promoting replication through 3-methyl adenine. Mol Cell Biol. 2007;27:7198–7205. doi: 10.1128/MCB.01079-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannouche P, Broughton BC, Volker M, Hanaoka F, Mullenders LH, Lehmann AR. Domain structure, localization, and function of DNA polymerase eta, defective in xeroderma pigmentosum variant cells. Genes Dev. 2001;15:158–172. doi: 10.1101/gad.187501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannouche PL, Wing J, Lehmann AR. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell. 2004;14:491–500. doi: 10.1016/s1097-2765(04)00259-x. [DOI] [PubMed] [Google Scholar]
- Lee KY, Myung K. PCNA modifications for regulation of post-replication repair pathways. Mol Cells. 2008;26:5–11. [PMC free article] [PubMed] [Google Scholar]
- Masutani C, Araki M, Yamada A, Kusumoto R, Nogimori T, Maekawa T, Iwai S, Hanaoka F. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. EMBO J. 1999;18:3491–3501. doi: 10.1093/emboj/18.12.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motegi A, Liaw HJ, Lee KY, Roest HP, Maas A, Wu X, Moinova H, Markowitz SD, Ding H, Hoeijmakers JH, et al. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc Natl Acad Sci U S A. 2008;105:12411–12416. doi: 10.1073/pnas.0805685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motegi A, Sood R, Moinova H, Markowitz SD, Liu PP, Myung K. Human SHPRH suppresses genomic instability through proliferating cell nuclear antigen polyubiquitination. J Cell Biol. 2006;175:703–708. doi: 10.1083/jcb.200606145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers JW, Jones JT, Meyer T, Ferrell JE., Jr Recombinant Dicer efficiently converts large dsRNAs into siRNAs suitable for gene silencing. Nat Biotechnol. 2003;21:324–328. doi: 10.1038/nbt792. [DOI] [PubMed] [Google Scholar]
- Ohashi E, Ogi T, Kusumoto R, Iwai S, Masutani C, Hanaoka F, Ohmori H. Error-prone bypass of certain DNA lesions by the human DNA polymerase kappa. Genes Dev. 2000;14:1589–1594. [PMC free article] [PubMed] [Google Scholar]
- Pages V, Bresson A, Acharya N, Prakash S, Fuchs RP, Prakash L. Requirement of Rad5 for DNA polymerase zeta-dependent translesion synthesis in Saccharomyces cerevisiae. Genetics. 2008;180:73–82. doi: 10.1534/genetics.108.091066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parris CN, Seidman MM. A signature element distinguishes sibling and independent mutations in a shuttle vector plasmid. Gene. 1992;117:1–5. doi: 10.1016/0378-1119(92)90482-5. [DOI] [PubMed] [Google Scholar]
- Plosky BS, Frank EG, Berry DA, Vennall GP, McDonald JP, Woodgate R. Eukaryotic Y-family polymerases bypass a 3-methyl-2′-deoxyadenosine analog in vitro and methyl methanesulfonate-induced DNA damage in vivo. Nucleic Acids Res. 2008;36:2152–2162. doi: 10.1093/nar/gkn058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- Stelter P, Ulrich HD. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003;425:188–191. doi: 10.1038/nature01965. [DOI] [PubMed] [Google Scholar]
- Takenaka K, Ogi T, Okada T, Sonoda E, Guo C, Friedberg EC, Takeda S. Involvement of vertebrate Pol kappa in translesion DNA synthesis across DNA monoalkylation damage. J Biol Chem. 2006;281:2000–2004. doi: 10.1074/jbc.M506153200. [DOI] [PubMed] [Google Scholar]
- Ulrich HD. Regulating post-translational modifications of the eukaryotic replication clamp PCNA. DNA Repair (Amst) 2009;8:461–469. doi: 10.1016/j.dnarep.2009.01.006. [DOI] [PubMed] [Google Scholar]
- Ulrich HD, Jentsch S. Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. EMBO J. 2000;19:3388–3397. doi: 10.1093/emboj/19.13.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unk I, Hajdu I, Blastyak A, Haracska L. Role of yeast Rad5 and its human orthologs, HLTF and SHPRH in DNA damage tolerance. DNA Repair (Amst) 2010;9:257–267. doi: 10.1016/j.dnarep.2009.12.013. [DOI] [PubMed] [Google Scholar]
- Unk I, Hajdu I, Fatyol K, Hurwitz J, Yoon JH, Prakash L, Prakash S, Haracska L. Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proc Natl Acad Sci U S A. 2008;105:3768–3773. doi: 10.1073/pnas.0800563105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unk I, Hajdu I, Fatyol K, Szakal B, Blastyak A, Bermudez V, Hurwitz J, Prakash L, Prakash S, Haracska L. Human SHPRH is a ubiquitin ligase for Mms2-Ubc13-dependent polyubiquitylation of proliferating cell nuclear antigen. Proc Natl Acad Sci U S A. 2006;103:18107–18112. doi: 10.1073/pnas.0608595103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004;23:3886–3896. doi: 10.1038/sj.emboj.7600383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, Walker GC. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev. 2009;73:134–154. doi: 10.1128/MMBR.00034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01