High Throughput Fingerprint Analysis of Large-Insert Clones (original) (raw)
Abstract
As part of the Human Genome Project, the Washington University Genome Sequencing Center has commenced systematic sequencing of human chromsome 7. To organize and supply the effort, we have undertaken the construction of sequence-ready physical maps for defined chromosomal intervals. Map construction is a serial process composed of three main activities. First, candidate STS-positive large-insert PAC and BAC clones are identified. Next, these candidate clones are subjected to fingerprint analysis. Finally, the fingerprint data are used to assemble sequence-ready maps. The fingerprinting method we have devised is key to the success of the overall approach. We present here the details of the method and show that the fingerprints are of sufficient quality to permit the construction of megabase-size contigs in defined regions of the human genome. We anticipate that the high throughput and precision characteristic of our fingerprinting method will make it of general utility.
Recent advances in DNA sequencing technology have allowed high throughput sequencing centers to generate millions of bases of raw sequence data on a weekly basis. The development of new technologies is expected to increase further sequencing throughput and decrease associated costs. These improvements will result in additional high throughput projects focused on genome-level sequencing. As demonstrated by the Saccharomyces cerevisiae and Caenorhabditis elegans sequencing projects, a detailed map of clones suitable for sequencing provides an efficient way to organize the sequencing effort. In both yeast and the worm, highly detailed, redundant physical maps constructed from sequence-ready reagents (Coulson et al. 1986, 1991; Olson et al. 1986; Riles et al. 1993) provided uninterrupted sources of material for sequencing. The high degree of redundancy of the maps was essential, allowing efficient selection of overlapping clones, which in turn has resulted in the generation of megabase lengths of contiguous sequence for both genomes (Wilson et al. 1994; Goffeau et al. 1996; The C. elegans Genome Sequencing Consortium, in prep.).
With this enhanced sequencing capacity in hand, an international effort to obtain the complete sequence of the human genome has begun. However, in contrast to the situation in yeast and C. elegans, most of the human genome lacks detailed physical maps constructed from sequenceable clones. Instead, the human physical map consists of landmarks, called sequence-tagged sites (STSs), ordered either against yeast artificial chromosome (YAC) libraries or radiation hybrid panels. Only in the former case is there a clone map, and this is composed of YACs that, because of instability, the high frequency of chimeras, and difficulties in manipulation and purification, are not ideal sequencing reagents. Therefore, the challenge is to develop an efficient strategy to convert the mapped STSs into contigs of clones that can be sequenced.
One strategy for STS-based sequence-ready map construction would involve using STSs to screen highly redundant genomic libraries to obtain large-insert low-copy-number bacterial clones, namely bacterial artificial chromosomes (BACs) and P1-derived artificial chromosomes (PACs). These clones are easily manipulated and, in our experience, more stable than cosmids. Clones identified by STS screening can be characterized by fingerprinting and the fingerprints used to build contigs. Using these contigs, appropriate clones can then be selected for sequencing and to develop probes for chromosome walking. Clones recovered in walking experiments can be fingerprinted and incorporated into contigs. This process, after a sufficient number of iterations, will result in closure of intercontig gaps.
The key to the success of the above approach is a robust method for high throughput fingerprint characterization of BAC and PAC clones. The polyacrylamide-based fingerprinting method used in the construction of the C. elegans physical map (Coulson et al. 1986), although effective (see Siden-Kiamos et al. 1990; Stallings et al. 1990; Taylor et al. 1996), involves radioactivity and in our hands has proven difficult to replicate. Furthermore, no information on clone size is recovered, and the absence of predictable signal intensity from band to band presents significant challenges for fully automated band calling. Another method under development is the multiple-complete-digest (MCD) mapping (Wong et al. 1997) in which three separate restriction digestions of a cosmid clone are analyzed by agarose gel electrophoresis and the data are used to construct a detailed restriction map. Here, the developers have not relied on the universally available BAC and PAC libraries, instead constructing custom cosmid libraries from redundant YACs.
We have developed a high throughput fingerprinting approach that borrows elements from the pioneering work that led to the construction of the yeast and C. elegans physical maps. Similar to studies by Olson et al. (1986) and Wong et al. (1997), data from restriction digests are collected on agarose gels. Then, using a strategy similar to that used by Coulson et al. (1986), we measure the relative mobilities of restriction fragments and use these to identify other clones that share a large proportion of fragments with the same relative mobilities, plus or minus a constant “tolerance”. In this way we infer the overlap of clones, and construct a contig where the relative positions of the clones reflect the extent to which they overlap. To our knowledge, ours is the first method to generate nonradioactive fingerprints for low-copy-number BAC, PAC, and fosmid clones in a high throughput fashion. Advantages offered by this approach include data that are comparatively free of artifacts, compatibility with pre-exisiting software developed at the Sanger Centre, and the high throughput necessary to fuel our sequencing goals. We report the details of our fingerprinting process, demonstrating the key features, and show that the data are of sufficient quality for purposes of contig construction.
RESULTS
We sought to develop and implement a high throughput fingerprinting scheme to allow the rapid selection of clones for DNA sequencing. Our general approach is diagrammed in Figure 1A with a representative fingerprinting gel shown in Figure 1B. A description of contig construction as currently performed at our Center is given in the Methods section in “Computer analysis and contig construction.” An overview of the application of the procedure is given in Figure 2.
Figure 1.

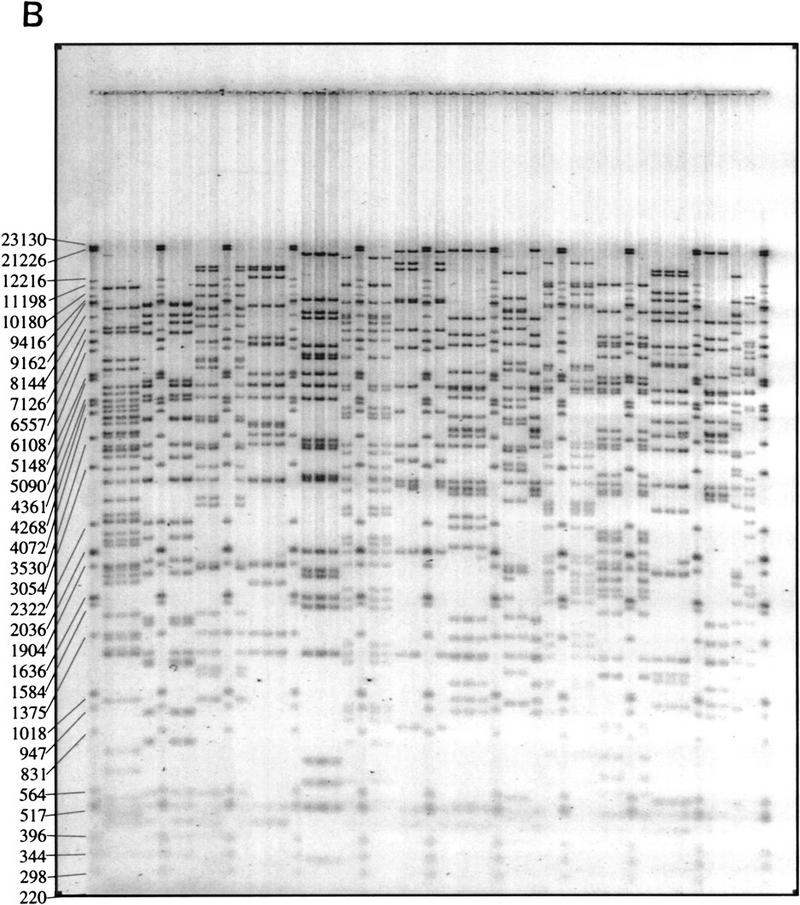
Schematic illustration of the role played by fingerprinting in the construction of sequence-ready contigs. (A) For construction of “local” sequence-ready maps, STSs, and probes specific for a small region, typically 1–2 Mb, are used to identify clones, either by hybridization or PCR methods. The fingerprinting produces higher resolution clone-specific information and facilitates contig construction and selection of clones for sequencing. Intercontig gaps are closed by chromosomal walks using probes developed from end sequences. Clones identified during walks are fingerprinted to determine their relationship to the contigs, permitting recognition of clones spanning intercontig gaps. (B) A typical agarose-mapping gel showing human PACs digested with _Hin_dIII. Clones are present in triplicate to verify stability during propagation and to control for the possibility of cross-contaminated glycerol stocks in the intial 384-well format. DNA size standards, which are a mixture of three commercially available markers (see Methods), are present every fifth lane. The sizes, in base pairs, of the marker fragments are indicated.
Figure 2.


A specific example of contig construction, on human chromosome 7, in FPC. Contigs are constructed as described in Methods (computer analysis and contig construction). Windows A, B, and C show the results of searching individual clones against FPC. The PAC clones 0897M19a, 1136G02b, and 0975M14a, used here to query the FPC database, correspond to the theoretical “clone 1,” “clone 2,” and “clone 3” described in Methods (computer analysis and contig construction). Window D shows the FPC fingerprint viewing tool displaying the fingerprints of these three clones as a representation of the original agarose gel fingerprint data output from the program Image. Note, however, that the lanes displayed are substantially altered versions of the original agarose gel image collected on the Molecular Dynamics FluorImager. The band positions have been normalized to the band positions of the marker DNA and only a one pixel-wide “slice” of the original 17 pixel-wide agarose gel image can be displayed. Manually verified band positions are indicated by the hashmarks flanking the lanes. Only these bands are used by FPC in database comparisons. Other band-like entities present in the lanes correspond to computer artifacts generated during the original gel image modification for display in FPC, to fluorescent foreign particles embedded in the agarose detected by the FluorImager during data acquisition, and rarely to legitimate bands that were not identified during the manual band calling routine conducted in Image. Examination of the original agarose gel images, retained in hard copy and electronic format, allows resolution of these alternate possibilities. Window E shows the restriction fragment sizes (in base pairs) for the clone 0897M19a. The + symbols to the right of the fragment sizes indicate fragments that have been user-selected by clicking with a mouse on the hashmarks in the FPC fingerprint viewer (D). Clicking a second time on the hashmark deselects the fragment. The sum of the sizes of the selected fragments is given at the bottom of window E, as is the total estimated size of the clone. The total size is calculated by summing the sizes of all the restriction fragments. This latter feature is a non-FPC function added at Washington University. Window F is an FPC display of a manually generated diagram of the contig after analysis is complete. The asterisks beside some clones indicate that redundant clones are contained entirely within these clones. Redundant clones are referred to as “buried” clones and are not visible in this view. A total of 40 clones have been incorporated into this contig. The clones considered specifically in this example are indicated with a box. Not shown are the additional search results used to construct this contig. G provides an example of the raw data used by FPC in performing the calculation of the Sulston score. Shown are the normalized relative mobilities (in tenths of a millimeter) for all of the detected restriction fragments for the clones 0897M19a and 1136G02b. The shaded values are considered common between these two clones. The values contained within open boxes indicate fragments unique to 1136G02b, all of which (except two fragments that are presumed to be the anomalous vector-insert junction fragments found in PACs digested with _Hin_dIII) are “confirmed” (Methods) by their prescence in the clone 0975M14a. Only the confirming fragments for 0975M14a are shown. These are indicated with a Y (for Yes). Details of FPC search results: The result of searching a PAC clone 0897M19a against the FPC database is given in Figure 2A. Shown in the window labeled xterm are the Sulston scores (see Methods) for the PAC clones 0741B13a and 1136G02b. The smaller score, indicating relatively more extensive overlap with 0897M19a (Methods), is exhibited by 0741B13a. Manual comparison of the fingerprints (not shown) of these two clones revealed that 0741B13a (24 restriction fragments) contains only two novel restriction fragments, which may correspond to the PAC vector-insert junction fragments. Manual comparison (Figure 2D) of the 1136G02b fingerprint (43 restriction fragments) with that of 0897M19a (31 restriction fragments) revealed that 20 of these fragments were unique to 1136G02b. 1136G02b was then used to query FPC. The results of this search are shown in Figure 2B. Strong matches to the previously mentioned clones are indicated, as are matches to the clones 0975M14a (40 restriction fragments) and 0659J06a (41 restriction fragments). Manual comparison of the 0975M14a fingerprint to that of 1136G02b (Figure 2D) reveals that the restriction fragments identified as unique to 1136G02b in the 1136G02b–0897M19a comparison are present in the fingerprint of 0975M14a (see Fig. 2D,G). These fragments are thus said to have been “confirmed” (Methods) and 0975M14a can be incorporated into the nascent contig.
In the development of our fingerprinting method there were several important considerations. The method had to yield precise data, yet be sufficiently simple and robust to allow routine application on a large scale. Radioisotopic detection of DNA was considered undesirable because of the complications arising from the manipulation, storage, and disposal of large quantities of radioactive material. The method had to be amenable to high throughput generation of sequence-ready clone maps generated in different vector types, including BACs and PAC. The necessity for high throughput made preparation of DNA in 96-well format a prerequisite. In turn, the small quantities of DNA achievable with low copy number vectors in this format required high sensitivity detection. Furthermore, we considered it desirable that the method yield an approximation of the sizes of the restriction fragments. The size information has several potential uses, including the estimation of the number of reactions required during shotgun sequencing of the clone (Wilson and Mardis 1997) and provides the ability to assay for gross rearrangements that might occur during propagation of the clone. Furthermore, restriction fragment size data can be used to estimate the magnitude of the overlap between clones in a contig. We present here a detailed description of the fingerprinting method along with an analysis of our fingerprint data, focusing on its precision, accuracy, and utility in contig construction.
DNA Yield
The yield for 40 randomly selected fosmids and 40 randomly selected BAC DNA preparations was measured (see Methods). Our preparation method resulted in the isolation of, on average, 1.2 μg of DNA for BAC clones and 1.6 μg of DNA for fosmid clones. Values are not adjusted for the unknown amounts of Escherichia coli genomic DNA contaminating each preparation, and therefore, yields should be considered maximal. For both fosmids and BACs the clone-to-clone yields appeared to approximate a normal distribution with a narrow standard deviation (not shown). The cause of the difference in mean yield between fosmids and BACs is unknown. Both vector types have an F-factor origin of replication, and therefore should maintain a low number of clone copies per cell. One possible explanation is that the different clone types were cultured in different strains of E. coli; fosmid clones were propagated in XL1-Blue MR cells (Stratagene), whereas the BAC clones were propagated in DH10B cells (Life Technologies). Whatever the cause, the slightly reduced amount of DNA recovered from the BACs is of little consequence to our fingerprinting procedure as only approximately one-thirtieth of the total BAC DNA yield is required per lane on an agarose gel (see Methods).
Assay for Precision
Requisite in a fingerprinting methodology is the generation of data that permits the recognition of clone overlap although the clone fingerprints were not present on the same gel or generated in the same time period. To achieve this the data must be precise. We assayed our method for precision by attempting to identify automatically all of the ribosomal DNA (rDNA) clones present in a large database of C. briggsae fosmid fingerprints constructed from data generated over a 3.5-month period. rDNA clones were chosen for this analysis because they occur frequently (∼2.5% of all fingerprinted clones) in our fosmid library, and because double digestion of C. briggsae rDNA clones with _Hin_dIII and _Pst_I (see Methods) yields an easily recognized characteristic pattern of restriction fragments (not shown). We reasoned that if our data were precise, we should be able to recognize the clones that were derived from rDNA and group only these clones into a single cluster (or “contig”).
We surveyed manually agarose gel images for fosmid clones exhibiting the C. briggsae rDNA restriction pattern, commencing with a recent gel image (Gel 225, dated October 28, 1996) and working backward (to Gel 97, dated July 10, 1996) until we had identified 65 gels containing 107 clones that matched the rDNA digestion pattern. The “bands” files (ASCII files that contain the relative mobilities of each restriction fragment on a gel) and gel files (that contain a representation of the original FluorImager scan of each gel) corresponding to the 65 gels were then used to create an FPC database (see Methods) containing all of the data from these gels. This database contained the relative mobilities of 34,172 restriction fragments derived from 2600 clones, 107 of which we had scored manually as matching the rDNA restriction pattern. We then conducted automated contig assembly in FPC with various tolerances (see Methods). The results of this analysis are summarized in Table 1.
Table 1.
Assay for Precision
| Tolerancea | Clones identifiedb | Commentc |
|---|---|---|
| 3 | 108 | false negative |
| 5 | 109 | |
| 7 | 109 | |
| 9 | 110 | false positive |
| 11 | 110 | false positive |
When contig assembly was conducted using a tolerance of 3 (see Methods: Computer Analysis and Contig Construction) all 107 rDNA clones that had been identified manually were placed into a single contig by FPC. One additional clone (G31K19) was also identified and placed in this contig by FPC. Comparison of this clone’s restriction fragment pattern to the restriction fragment patterns of manually identified rDNA clones revealed that G31K19 contained restriction fragments common to other rDNA clones. Therefore, G31K19 appears to be a bona fide member of the rDNA contig that was overlooked during manual inspection of the gel images.
Contig assembly was repeated using identical parameters except that the tolerance parameter was increased to 5. This experiment and the subsequent one, conducted with a tolerance of 7, yielded identical results. In both experiments 109 clones were grouped into a single contig by FPC analysis. These 109 clones included all of the 107 manually identified clones, the clone G31K19 identified using a tolerance of 3, and an additional clone, G41G20. The restriction pattern of G41G20 was inspected in FPC and compared to the restriction patterns of manually identified rDNA clones. This comparison showed that two of the smaller restriction fragments were slightly shifted with respect to those of other rDNA clones. However, the restriction fragment pattern demonstrated clearly that this clone belonged in the rDNA contig, and should have been so identified during manual inspection of the agarose gel images.
Contig assemblies were conducted with tolerances of 9 and 11. Both assemblies consisted of 110 clones, which included the 107 manually identified clones as well as G31K19 and G41G20. In addition to these, one new clone, G34B11, was identified. This latter clone was found to have been placed incorrectly in the rDNA contig because of the relaxed tolerance parameters. Manual re-examination of agarose gel images confirmed that the number of identifiable rDNA clones in the data set used to conduct these experiments was 109.
Assay for Accuracy
Accuracy, or the ability to determine the sizes of restriction fragments correctly, is less important than precision as far as the detection of overlaps during contig construction is concerned. However, a method that yielded data that was both precise and accurate would be of added value. For example, accurate restriction fragment sizes could be used as a tool for verification of the correct assembly of sequencing projects conducted using the “shotgun” sequencing approach (Wilson and Mardis 1997). The final sequence of the clone can be used to generate a list of “restriction fragments” and their sizes, which can then be compared to the sizes of the restriction fragments obtained by electrophoretic analysis. If an error in assembly of the shotgun sequences has occurred, discrepancies would become evident provided that the fragment sizes obtained experimentally were accurate enough to allow correlation to the sizes predicted from DNA sequence.
We have compared _Hin_dIII restriction fragment sizes determined by agarose gel analysis to those predicted from the completed sequences of seven human BACs (Fig. 3). In general, correlation of the sequence-based fragment size to the appropriate restriction fragment was unambiguous. We also noted that the distribution of size deviations exhibited little variation between 500 and 12,000 bp, with 95% (102 of 107) of the data points in this size range falling between +1.5% and −0.75% deviation from true size. Furthermore, the data show that 96% (107 of 111) of the restriction fragments obtained by _Hin_dIII digestion and subsequent agarose gel analysis fall into a size range for which accurate sizes are obtained using our electrophoresis apparatus and conditions. There appears to be a bias toward a positive deviation from true size; that is, the restriction fragments tended to be sized slightly larger than predicted by sequence analysis. This positive trend, which fluctuated between +0.5% and +0.75% was found to be more or less constant for fragments 3000 to 12,000 bp in length. The four fragments larger than 12,000 bp exhibited greater deviation, indicating a decrease in accuracy in determining the sizes of these larger fragments. The magnitude of the error remained <5% of the true fragment size. A possible contributing factor to the increased error for the largest fragments is that our mixture of commercially available marker DNAs (see Methods) has no fragments in the size range 12,000–21,000 bp.
Figure 3.

Graph showing accuracy of restriction fragment sizes. On the y axis “% deviation from true size” is the size, in base pairs, predicted from agarose gel analysis divided by the size of the restriction fragment as determined from sequence analysis of the entire clone, converted to a percent value. The line indicates a moving average calculated at data-point intervals of 40. The box encases 95% (102 of 107) of the data points for fragments of <12 kilobases.
Utility in Contig Construction
For contig construction on human chromosomes 7 and X, clones were first identified using STSs that had been localized previously to a small region. The STS-positive clones were then subjected to fingerprint analysis, and the fingerprint data were used to assemble contigs from which clones were selected for either full shotgun sequencing or end sequencing (Marra et al. 1996), which was performed to develop additional probe reagents. Clones identified using these newly developed probes were incorporated into contigs based on their fingerprint (see Fig. 1).
We selected 17 STSs from the interval sWXD 1833–sWXD 1888 (Nagaraja et al. 1997), which spans ∼1 Mb of the X chromosome. Thus, the average marker density across this interval was one marker per 58 kb. The oligonucleotides corresponding to these STSs were labeled radioactively and hybridized (J. McPherson, unpubl.) to high density PAC and BAC filters. Positive clones were identified, and three single colonies corresponding to each clone were fingerprinted. The contigs resulting from our fingerprint analysis of these clones is shown in Figure 4.
Figure 4.

The FPC contig view of two X chromosome contigs juxtaposed, showing the relative positions of the various clones. Contig A is on the left and Contig B is on the right. Overlap between the PAC 0545D18a and the BAC R038K21a has yet to be verified.
After analysis, the fingerprinted clones cleanly resolved into two contigs, contig A and contig B. Including “buried” clones (clones that are contained entirely within other clones; Coulson et al. 1986), contig A consists of 22 clones, which span ∼440 kb. Contig B consists of 29 clones spanning ∼740 kb. Coverage of the region is deep; the recovery of clones from three libraries resulted in a high degree of redundancy, which was helpful during contig construction and facilitated the interclone verification of restriction fragments in the clones ultimately selected for sequencing.
Contig A’s right-most clone is the PAC 0545D18a. Contig B’s left-most clone is the BAC R038K21a. These two contigs are oriented according to the order of the STS markers (Nagaraja et al. 1997). Thus, 0545D18a and R038K21a might overlap. Indeed, close examination of the fingerprints for these clones reveals five common restriction fragments. This is an insufficient number of common fragments to declare overlap in our paradigm (see Methods: Computer Analysis and Contig Construction); thus, we have obtained end sequences from both clones and have initiated a walk to verify or refute the overlap between these two clones, and to provide additional depth of coverage for the contig ends.
As an additional example of the utility of our fingerprinting method in contig construction we analyzed clones from 7q22. Here, a slightly different paradigm was followed. First, clones were identified by PCR screening (E.D. Green, unpubl.) of commercially available BAC DNA pools (Research Genetics). PCR-positive clones were fingerprinted and contigs constructed. STS oligonucleotides were next used as hybridization probes (J. McPherson, unpubl.) against PAC and BAC high density filters, and the resulting positive clones fingerprinted and incorporated into the contigs. Fingerprint analysis of these clones produced numerous small contigs, with several of the contigs populated by a single clone. From these contigs, clones were selected for end sequencing. End sequences were used to either develop new STSs, or to design additional probes, which were pooled in batch hybridizations (J. McPherson, unpubl.) to the high-density filters. Positive clones identified in the PCR and hybridization experiments were then fingerprinted and the fingerprints used to assemble the clones into contigs. Incorporation of the end-walk clone fingerprints resulted in the production of a single contig of ∼2 Mb (Fig. 5).
Figure 5.

The FPC contig view of a chromosome 7q22 contig. The contig consists of 163 BAC and PAC clones in total. The majority of the redundant clones are hidden in this view. Clones labeled with a “w” were identified by hybridization. Clones labeled with a “p” were identified by PCR. The contig spans ∼2 Mb.
DISCUSSION
Large-scale genome sequencing projects can be organized efficiently using maps constructed from sequencable clones. This has been demonstrated by the success of both the yeast and C. elegans sequencing efforts, both of which relied on maps constructed before the start of large-scale sequencing. The goal of this study was to develop a high throughput precise fingerprinting method to facilitate construction of sequence-ready maps for localized regions of the human genome.
Fingerprint data generated with our method were found to be precise in a gel- and time-independent fashion. Had the data been imprecise, we presumed we would be unable to correctly and automatically recognize and group some fraction of the clones identified manually as deriving from rDNA. These would be classified as false negatives. Alternatively, we might have incorrectly identified clones as rDNA and erroneously incorporated them into a contig assembly. These would be classified as false positives. By varying the tolerance parameter in FPC we identified tolerances of 5 and 7 that, upon contig assembly, yielded a contig consisting of all of the appropriate clones and only these clones. A tolerance of 3 failed to identify one clone, and tolerances of 9 and 11 falsely identified a clone as derived from rDNA. At present, we routinely construct contigs in FPC using a tolerance of 7.
Our extensive application of the fingerprinting method has revealed variable band detection for fragments <300 bp in length (Fig. 3). Because we strive to avoid overloading of samples on the gels, the amount of DNA contained within these small bands is very near the limit of detection sensitivity for the MD FluorImager and SYBR green I. In addition, in two instances in different sequenced BAC clones we were unable to predict accurately band multiplicity, which had been inferred by manual examination of bands with increased relative intensities. Both of these shortcomings are of little practical consequence for the detection of overlapping clones by fingerprint analysis, but they prevent an absolute correlation of a small number of experimentally determined fragments to ones predicted from the sequence. We note that the data exhibit a predictable decrease in signal intensity with decreasing band size. Currently, we are attempting to exploit this feature of the data to provide accurate automatic assignments of band multiplicity and robust automatic band calling.
The availability of accurate restriction fragment sizes provides not only a means of checking assembly of finished shotgun sequencing projects, but also a method for estimating the number of sequencing reactions that are required for a BAC or PAC clone in the shotgun phase. This is advantageous because of the wide variation in the sizes of individual PAC and BAC clones. Restriction fragment sizes are also used during selection of clones for sequencing to determine the amount of DNA shared by overlapping clones with the goal of selecting clones that exhibit minimal overlap.
The ability to collect highly reproducible data has allowed us to impose a stringent criterion on clones selected for sequencing. We now attempt to account for all restriction fragments, confirming the presence of each fragment in an overlapping clone. In practice this is possible if the restriction enzyme used to generate the fingerprint is the one used to construct the genomic library from which the clone was recovered. If so, digestion cleanly liberates the vector from the cloned DNA, and no anomalous vector-insert junction fragments are produced. If not, a maximum of two anomalous unaccounted-for fragments are allowed per fingerprint. These fragments do not confound contig construction if clone coverage is redundant, nor do they prohibit integration of clones contained in different vector types into the contig. In our opinion, the principle of accounting for each fragment in a clone provides some assurance that the clone selected for sequencing is a faithful representation of the genome. Additional assurance is provided by the analysis of multiple, independently prepared bacterial colonies representing each clone.
In a 9-month period, we fingerprinted and entered into FPC 28,582 large insert bacterial clones. These clones correspond to a variety of mapping projects, including an estimated sevenfold redundant sampling of the C. briggsae genome cloned in fosmids, and PAC and BAC clones spanning >50 Mb of human chromosome 7. Throughput has averaged 893 lanes of data per week since the inception of the effort, including the time for development and implementation of the current protocols. Recent throughput routinely exceeds 1300 lanes per week. The current fingerprinting team consists of five technicians: one full-time employee involved in preparation and digestion of DNA and recordkeeping, two full-time employees performing interactive band calling using the program Image, one full-time employee inoculating 96-well cultures, constructing, and curating glycerol stocks, one half-time contig constructor, and one half-time supervisor. Gel pouring, loading, and postelectrophoresis staining duties are shared by these team members.
The high throughput and high quality of the data have provided clones in excess of that required to fuel our current sequencing goals. These features of our method, coupled with inexpensive reagents and stable large-insert clones make fingerprint analysis of large genomic segments eminently feasible.
METHODS
Preparation of DNA
Culture volumes of 1200 μl of 2X YT (Sambrook et al. 1989) containing 12.5 μg/ml of chloramphenicol (Sigma; fosmids and BACs) or kanamycin (Sigma; P1 and PAC clones) or the appropriate quantity of antibiotic for the cosmids under study were inoculated with a single colony from a freshly streaked plate. Cultures were grown in 2-ml 96-well blocks (Beckman; part 140504) for 24 hr at 37°C with agitation at 300 rpm in a Labline incubator shaker. After growth glycerol stocks in 96-well format were prepared by combining 50 μl of 80% glycerol with 100 μl of culture and mixing with a 12-channel pipettor. The microplates were sealed with Scotch brand heavy duty aluminum foil tape and stored at −80°C.
Bacterial cell cultures (96-well) were pelleted by centrifugation at 2700 rpm for 15 min in a Jouan model GR-422 floor centrifuge fitted with microplate carriers. The supernatant was decanted away from the pellet, and the 96-well block left inverted on paper towel for 5 min to drain excess culture media. The inverted block was rapped vigorously on fresh paper towel until excess culture media was removed and then placed immediately on ice. Alternatively, after removal of the culture media, blocks were sealed with foil tape and stored at −80°C until DNA preparation.
DNA preparation was performed using a modified alkaline lysis procedure (Sambrook et al. 1989). The cell pellet was resuspended by addition of 50 μl of chilled GET/RNase buffer [50 mm glucose, 25 mm Tris-HCl (pH 8.0), 10 mm EDTA (pH 8.0), 0.12 mg/ml RNase (Sigma R6513)] and vigorous vortexing. After the pellet was thoroughly resuspended an additional 150 μl of GET/RNase was added followed by gentle vortexing to mix. Cell lysis was achieved by addition of 200 μl of a mixture containing 0.2 n NaOH/1% SDS (freshly prepared), rotation of the block 90° along its long axis 20 times, followed by incubation on the bench for 5 min. Ice cold 3 m potassium acetate (200 μl) (KAc; pH 5.5) was then added to each well, the block tightly sealed with foil tape, and rapidly inverted three times before a 10-min incubation in ice water. For fosmids, we have found that cleaner DNA preparations, as assayed by examination of digested DNA run on agarose gels, are achieved using 3 m KAc (pH 4.9). However, use of this reagent in BAC DNA preparations invariably results in reduced yield compared to KAc at higher pH. The taped block was inverted rapidly once after the 10-min incubation. Cell debris was then pelleted by centrifugation of the block for 15–20 min at 4000 rpm in a Jouan GR-422 centrifuge maintained at a temperature of 4°C. After centrifugation, blocks were immediately placed on ice. During the last few minutes of the centrifugation, 600 μl of isopropanol were added to each well of a fresh 96-well block (Beckman). This isopropanol-filled block was then inserted into a vacuum manifold (Qiavac 96; Qiagen) and a Qiafilter 96 filter (Qiagen, part 19663) was placed on top of the manifold in preparation for filtration of the supernatant-containing DNA.
After centrifugation, supernatant-containing DNA was separated from the cell debris by inserting a 12-channel pipettor into the block until the tips touched the bottom of the well. Moving the tips slightly created a channel in the cell debris, which facilitated removal of the supernatant while leaving the majority of the debris in the well. The supernatant was then transferred to a Qiafilter. When transfer of the supernatant was complete, a vacuum was applied to the Qiafilter manifold, which served to draw the supernatant through the Qiafilter into the isopropanol containing block positioned below. In this way residual SDS/cellular debris, which had not pelleted during centrifugation, was removed.
The block was tightly sealed with foil tape and inverted rapidly three times to mix the supernatant and isopropanol. Precipitation of the DNA was achieved by room temperature incubation for 15 min followed by a 30 min centrifugation at 4000 rpm. The foil tape was removed and the block inverted to remove the supernatant. The DNA pellet was then washed with 200 μl of 80% ethanol added to the side of the well, and then collected in the bottom of the well by a 10-min centrifugation at 4000 rpm after sealing the block with foil tape. The tape was removed and the block inverted on paper towels for 5 min to drain excess ethanol away from the pellet. The block was then placed in a Savant DNA 110 SpeedVac set at medium heat for 5 min to dry the DNA. The dried pellet was resuspended in 30 μl of TE [10 mm Tris-HCl (pH 8.0), 0.1 mm EDTA (pH 8.0)] in the case of fosmid, BAC, PAC, and P1 clones, or 150 μl of TE for cosmid clones. Resuspension of the DNA was achieved by incubating the sealed block for 30 min in a 37°C water bath followed by brief vortexing. The DNA was collected in the bottom of the wells by a brief centrifugation and transferred to a nontissue culture treated microplate which was sealed with foil tape for storage at −20°C.
Alternatively, DNA was prepared by serial addition of 150 μl each of GET/RNase, SDS/NaOH, and KAc pH 5.5 as described above. After addition of KAc, the sealed block was inverted gently three times and then placed in ice water for at least 10 min. The block was inverted twice vigorously before centrifugation, as described. While samples were undergoing centrifugation, 330 μl of 100% ethanol were aliquoted into each well of a 96-well polystyrene “Uni-Filter 800” receiver plate (Polyfiltronics). A 0.45 μm cellulose acetate 96-well filter plate (Polyfiltronics) was then mounted on top of the receiver plate and taped securely in place.
After centrifugation, a 12-channel pipette (Costar) was used to transfer 400 μl of supernatant-containing DNA to the 96-well filter plate mounted on top of the receiver plate. The assembly, consisting of filter plate and receiver plate, was then subjected to an additional centrifugation at 4000 rpm for 15 min. After centrifugation, the filter plate assembly was dismantled and the ethanol decanted. The DNA pellet was washed with 250 μl of 80% ethanol, dried, and resuspended in the appropriate volume of 10 mm Tris-HCl, 0.1 mm EDTA. This alternative procedure has the advantage of being somewhat more rapid and substantially less expensive due to the use of Polyfiltronics plasticware.
Restriction Enzyme Digestion
For PAC, P1, and BAC DNAs, individual restriction digests consisted of 3.75 μl of ddH20, 1 μl of 10× buffer “B” (Boehringer-Mannheim), 0.25 μl of _Hin_dIII (40 U/μl; Boehringer-Mannheim), and 5 μl of DNA. For fosmids, individual restriction digestions contained 2.75 μl of ddH20, 1 μl of 10× buffer “B” (Boehringer-Mannheim), 0.125 μl of _Hin_dIII (40 U/μl; Boehringer-Mannheim), 0.1 μl of _Pst_I (100 U/μl; NEB), and 6 μl of DNA. For cosmids, each digest contained 6.75 μl of ddH20, 1 μl 10× buffer “B”, 0.25 μl of _Hin_dIII (40 U/μl; Boehringer-Mannheim), and 2 μl of DNA. Components of the digestion cocktail were assembled in 96-well thin wall cycle plates (Robbins Scientific). Digestion was achieved by incubation of the cycle plates at 37°C for 4.5 hr in a 96-well thermocycler (MJ Research). After digestion a brief centrifugation collected the DNA in the bottom of the wells and 2 μl of 6× loading dye (0.25% bromophenol blue, 0.25% xylene cyanol FF, 15% Ficoll; Sambrook et al. 1989) was added to each well. Cycle plates were sealed with foil tape and stored at 4°C overnight before agarose gel electrophoresis.
Agarose Gel Electrophoresis and Data Acquisition
One percent agarose (SeaKem LE; FMC BioProducts) gels were prepared in 1× TAE (Sambrook et al. 1989). Molten agarose was cooled to 46°C in a water bath with occasional stirring and then poured into 20 by 25-cm UV transparent trays (Life Technologies) resting on a level surface. The comb was then inserted. For each gel 150 ml of molten agarose was used resulting in a gel thickness of approximately 3.5 mm. The comb formed 51 wells (D. Panussis, unpubl.) with the following dimensions: 2 mm wide by 1 mm thick by 3 mm deep, where thick is the dimension in the direction of DNA migration. After the gel solidified the comb was removed, the gel was wrapped in Saran Wrap and stored at 4°C until electrophoresis. This storage time period never exceeded 3 days. Gels were removed from 4°C storage and placed into electrophoresis units containing buffer at the desired electrophoresis temperature for at least 10 min before sample loading. The restriction enzyme digestion/loading dye mixture (1.75 μl) was loaded into each well. In the first well and every fifth well thereafter 1 μl of a standard “marker” DNA sample was loaded. Marker DNA was a mixture of 1 kb ladder (Life Technologies) and both Marker II and Marker III (Boehringer-Mannheim) in the following proportions: 0.83 μl (1 μg/μl) 1 kb ladder, 3.33 μl (250 ng/μl) Marker II, 3.33 μl (250 ng/μl) Marker III, 92.51 μl TE [10 mm Tris (pH 8.0), 0.1 mm EDTA (pH 8.0)], 25 μl 6× loading dye. Immediately before electrophoresis 20 μl of this mixture was removed to a separate tube, diluted by the addition of 17 μl of TE and 3 μl of 6× loading dye and incubated at 60°C for 5 min.
Samples were electrophoresed in Model H4 electrophoresis units (Life Technologies) at 90 V for 15 min after which time recirculation of the electrophoresis buffer (1× TAE; Sambrook et al. 1989) was initiated. Buffer was recirculated by pumping through 25 feet of small diameter tygon tubing (Tygon LFL 6429-17) immersed in a 16-liter tank containing water maintained at a constant temperature of 14°C. Temperature regulation of the water was achieved using a refrigerated recirculator (VWR Scientific, model 1170). A tank temperature of 14°C served to maintain a constant electrophoresis buffer temperature of 16°C. Total electrophoresis time was 8 hr.
After electrophoresis, gels were removed to custom designed plastic trays (D. Panussis, unpubl.) containing 400 ml of a 1:10,000 dilution of either SYBR Green (FMC BioProducts) or Vistra Green (Molecular Probes) in 1× TAE and agitated in the dark for 30–45 min. We routinely reused diluted SYBR Green I and Vistra Green solutions one more time. Diluted stains were stored at 4°C in a Rubbermaid (recycle number 5) container wrapped in foil. After staining gels were imaged using a Molecular Dynamics FluorImager SI with the following scan settings: pixel size, 200 μm; digital resolution, 16 bits; detection sensitivity, high; PMT voltage, 950 V; Filter, 530 nm. Gel images were first cropped and then converted from the proprietary 16-bit Molecular Dynamics format to 8-bit TIFF images, and transferred by ftp to Unix workstations for band calling and contig building. The Molecular Dynamics FluorImager was also used to measure the yield of DNA, prepared as described above, using protocols and Pico Green stain obtained from Molecular Dynamics.
Computer Analysis and Contig Construction
Computer-generated conceptual restriction digestions of sequences obtained from chromosome 7 BAC clones at Washington University Genome Sequencing Center were performed using an implementation of the program Nip (R. Staden, Version 7.1, July 1993).
Identification of restriction fragment bands was performed interactively using first an unmodified implementation of the program Image 2.0 (F. Wobus and R. Durbin, unpubl.) and subsequently Image 3.3 (D. Platt, F. Wobus, and R. Durbin, in prep.), suitably modified to accept gel images generated as described above. Band cell data were collected and used to perform contig assembly in the program FPC (C. Soderlund and I. Longden, Sanger Centre Technical Report SC-01-96, August 1996; Soderlund et al. 1997) using functions available in FPC and the program MAPSUB (Sulston et al. 1988). Image and FPC have been developed and are maintained at the Sanger Centre; documentation and user’s manuals are available on the Sanger Centre website (http://www.sanger.ac.uk).
All contig construction is performed using FPC as described here and illustrated in Figure 2. Our initial use of the software to build contigs of human clones has emphasized manual aspects of contig building. Automated features, used to assemble the C. briggsae rDNA contig, are provided by FPC and are described by Soderlund et al. (1997) and in the FPC user’s manual.
For human PAC and BAC we first select a clone (“clone 1”) and compared it to all clones in the FPC database at experimentally determined parameters of “tolerance”=7, “cutoff score”=10−8. The term tolerance refers to a window size; for example, if tolerance is set at 7, then two restriction fragments occurring in different fingerprints must have relative mobilities within seven-tenths of a millimeter to be considered equivalent fragments. A decrease in tolerance decreases the window size and therefore, increases the stringency of the comparison. It is important to note that all of the calculations we have performed in FPC have used the relative mobilities of the restriction fragments (for example, see Fig. 2G) and not the sizes of the restriction fragments.
The cutoff score is a threshold value representing the maximum allowable probability of a chance match between any two clones (the “Sulston score”). The smaller the Sulston score value, the lower the probability that the match has arisen by chance, and the more extensive the overlap between any two clones. Practical experience with our human fingerprint data has led us to apply a cutoff score of 10−8. Details describing the derivation of the scores and issues relating to the calculation of the Sulston score are presented by Sulston et al. (1988).
Matches between clone 1 and other clones are displayed (Fig. 2A). We select the clone (“clone 2”) exhibiting the best match (i.e., the matching clone exhibiting the smallest Sulston score) to clone 1 and manually compare, using a “fingerprint viewing tool” provided by FPC, its fingerprint to that of clone 1 to determine the number of shared fragments (Fig. 2D). The overlap between the clones can then be “drawn” manually in FPC (Fig. 2F). If the clone 2 fingerprint exhibits no unique restriction fragments, we “bury” (hide) clone 2 within clone 1. If unique fragments are observed in clone 2 we may then repeat the entire procedure using clone 2 for the next search against the FPC database (Fig. 2B). The best match (“clone 3”) is identified, and its fingerprint is compared manually against the fingerprints of clone 1 and clone 2 (Fig. 2D). To incorporate clone 3 into the nascent contig, we require that the unique restriction fragments exhibited by clone 2 be present in clone 3. These unique fragments are then considered “confirmed” (Fig. 2G). This constraint is imposed to ensure the internal consistency of the nascent contig and to provide additional assurance, through redundancy, that the clones represent faithfully the underlying genome. If this constraint cannot be met (a possibility that might arise because of, for example, a RFLP) the clone may still be incorporated into the contig and used as a mapping reagent, but will be labeled with a tag in FPC so that it will not be selected for other manipulations including DNA sequencing. For human PACs, which possess two variably sized vector-insert junction fragments, we allow two unconfirmed fragments per fingerprint.
The process of consecutive searches continues (e.g., Fig. 2C) until no matches better than the cutoff score can be identified and the contig cannot be extended further. An additional search, using the entire contig to query FPC, is performed to identify any remaining matching clones. If any are found they are incorporated into the contig as described above.
During contig assembly, only one of the three fingerprints from any given well address is incorporated into the nascent contig. The selection of the appropriate fingerprint, in the cases where differences are observed among the three fingerprints, is constrained to preserve the internal consistency of the contig. That is, all fragments (except for the two vector-insert junction fragments encountered in PAC clones) of a clone occupying an internal position in the contig are verified manually by direct comparison with the fragments of flanking clones. To declare overlap between any two clones ∼50% of the bands need be identified as common. In the context of a contig larger than two clones this parameter can, in practice, often be relaxed provided the constraint of internal consistency within the contig is met and new bands evident in a pairwise comparison between two clones are confirmed by the next clone entering the contig.
To assay precision we used FPC to conduct five independent automated contig assemblies of C. briggsae rDNA clone fingerprints generated by double digestion with _Hin_dIII and _Pst_I. Treatment of C. briggsae rDNA clones with these enzymes produces nine restriction fragments. Two of these are specific to the pFOS1 (“fosmid”) vector and were not analyzed in Image or considered further. Of the seven remaining fragments one exhibited interclone variability. The minimum number of matching fragments required to identify correctly a clone as rDNA depends, for any specified tolerance value, on the cutoff score. An appropriate cutoff value was determined during the course of these experiments (below).
The contig assemblies were performed identically except that the specified tolerance was increased two-tenths of a millimeter and then repeated. For the contig assemblies the parameters set in FPC were cutoff=10−5, Diff=0.3, MinBands=3, Diffbury=0.10, MinEnd=8. Tolerances were varied from 3 (0.3 mm) to 11 (1.1 mm) in increments of 0.2 mm.
Acknowledgments
We acknowlege Dr. Alan Coulson for patient instruction in contig building, and Drs. Cari Soderlund, Darren Platt, Richard Durbin, Maynard Olson, Gane Wong, and Jun Yu for valuable discussion and assistance in many aspects of the work described here. Thanks to Simon Gregory and Gareth Howell of the Sanger Centre for much helpful advice on contig construction. We are grateful to Dr. Ung-Jin Kim for his generous gift of fosmid vector, Dr. Elaine Mardis and Lucinda Fulton for end sequence generation, Dr. Stephanie Chissoe, Dr. Jeff Woessner, and Sharon Gorski for comments on the manuscript and for support. Many thanks to Muno Sekhon, Kurt Hinds, Jacquie Fedele, Nancy Mudd, Proteon Pridgeon, Christine Cox, and Carrie Rhine for excellent technical support in all aspects of this work. Funding for this work has been provided by NHGRI grants 5P01HG00956 and 1P50HG01458.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
This paper is dedicated to the memory of Amerigo Marra, 1929–1997.
E-MAIL mmarra@watson.wustl.edu; FAX (314) 286-1810.
REFERENCES
- Coulson A, Sulston J, Brenner S, Karn J. Toward a physical map of the genome of the nematode Caenorhabditis elegans. Proc Natl Acad Sci. 1986;83:7821–7825. doi: 10.1073/pnas.83.20.7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson A, Kozono Y, Lutterbach B, Shownkeen R, Sulston J, Waterston R. YACS and the C. elegans genome. BioEssays. 1991;13:413–417. doi: 10.1002/bies.950130809. [DOI] [PubMed] [Google Scholar]
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin W, Oliver SG. Life with 6,000 genes. Science. 1996;274:546–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- Marra MA, Weinstock LA, Mardis ER. End sequence determination from large insert clones using energy transfer fluorescent primers. Genome Res. 1996;6:1118–1122. doi: 10.1101/gr.6.11.1118. [DOI] [PubMed] [Google Scholar]
- Nagaraja R, MacMillan S, Kere J, Jones C, Cox S, Schmatz M, Terrell J, Shomaker M, Jermak C, Hoff C, et al. X chromosome map at 75 kb STS resolution, revealing extremes of recombination and GC content. Genome Res. 1997;7:210–222. doi: 10.1101/gr.7.3.210. [DOI] [PubMed] [Google Scholar]
- Olson MV, Dutchik JE, Graham MY, Brodeur GM, Helms C, Frank M, MacCollin M, Scheinman R, Frank T. Random clone strategy for genomic restriction mapping in yeast. Proc Natl Acad Sci. 1986;83:7826–7830. doi: 10.1073/pnas.83.20.7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riles L, Dutchik JE, Baktha A, McCauley BK, Thayer EC, Leckie MP, Braden VV, Depke JE, Olson MV. Physical maps of the six smallest chromosomes of Saccharomyces cerevisiae at a resolution of 2.6 kilobase pairs. Genetics. 1993;134:81–150. doi: 10.1093/genetics/134.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Siden-Kiamos I, Saunders RDC, Spanos L, Majerus T, Treanear J, Savakis C, Louis C, Glover DM, Ashburner M, Kafatos FC. Towards a physical map of the Drosophila melanogaster genome: Mapping of cosmid clones within defined genomic divisions. Nucleic Acids Res. 1990;18:6261–6270. doi: 10.1093/nar/18.21.6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderlund, C., I. Longden, and R. Mott. 1997. FPC: A system for building contigs from restriction fingerprinted clones. CABIOS 13: (in press). [DOI] [PubMed]
- Stallings RL, Torney DC, Hildebrand CE, Longmire JL, Deaven LL, Jett JH, Doggett NA, Moyzis RK. Physical mapping of human chromosomes by repetitive sequence fingerprinting. Proc Natl Acad Sci. 1990;87:6218–6222. doi: 10.1073/pnas.87.16.6218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston J, Mallett F, Staden R, Durbin R, Horsnell T, Coulson A. Software for genome mapping by fingerprinting techniques. CABIOS. 1988;4:125–132. doi: 10.1093/bioinformatics/4.1.125. [DOI] [PubMed] [Google Scholar]
- Taylor K, Hornigold N, Conway D, Williams D, Ulinowski Z, Agochiya M, Fattorini P, de Jong P, Little PFR, Wolfe J. Mapping the human Y chromosome by fingerprinting cosmid clones. Genome Res. 1996;6:235–248. doi: 10.1101/gr.6.4.235. [DOI] [PubMed] [Google Scholar]
- Wilson R, Ainscough R, Anderson K, Baynes C, Berks M, Bonfield J, Burton J, Connell M, Copsey T, Cooper J, et al. 2.2 Mb of contiguous nucleotide sequence from chromosome III of C. elegans. Nature. 1994;368:32–38. doi: 10.1038/368032a0. [DOI] [PubMed] [Google Scholar]
- Wilson RK, Mardis ER. Fluorescence-based DNA sequencing and shotgun sequencing. In: Birren B, Green E, Heiter P, Myers R, editors. Genome analysis: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. . (In press). [Google Scholar]
- Wong GK, Yu J, Thayer EC, Olson MV. Multiple-complete-digest restriction fragment mapping: Generating sequence-ready maps for large-scale DNA sequencing. Proc Natl Acad Sci. 1997;94:5225–5230. doi: 10.1073/pnas.94.10.5225. [DOI] [PMC free article] [PubMed] [Google Scholar]