A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium (original) (raw)
. Author manuscript; available in PMC: 2011 Jun 6.
Published in final edited form as: Nat Genet. 2009 Oct 11;41(11):1182–1190. doi: 10.1038/ng.467
Abstract
The number and volume of cells in the blood affect a wide range of disorders including cancer and cardiovascular, metabolic, infectious and immune conditions. We consider here the genetic variation in eight clinically relevant hematological parameters, including hemoglobin levels, red and white blood cell counts and platelet counts and volume. We describe common variants within 22 genetic loci reproducibly associated with these hematological parameters in 13,943 samples from six European population-based studies, including 6 associated with red blood cell parameters, 15 associated with platelet parameters and 1 associated with total white blood cell count. We further identified a long-range haplotype at 12q24 associated with coronary artery disease in 9,479 cases and 10,527 controls. We show that this haplotype demonstrates extensive disease pleiotropy, as it contains known risk loci for type 1 diabetes, hypertension and celiac disease and has been spread by a selective sweep specific to European and geographically nearby populations.
The hematopoietic system is one of the best-studied cellular differentiation processes in mammals. The differentiation of the hematopoietic stem cell into its progeny is a tightly orchestrated process of fate determination and cell proliferation which results in a repertoire of different types of mature cells in the peripheral blood that supervise a range of functions including the transport of oxygen, innate and adaptive immunity, vessel wall surveillance, homeostasis and wound repair. The count and volume of the cellular elements in circulating blood are highly heritable and tightly regulated1,2 and vary widely between individuals. Such hematological traits, which include the concentration of hemoglobin (Hb), the numbers of white blood cells (WBC), red blood cells (RBC) and platelets (PLT), and the volumes of red blood cells and platelets (MCV and MPV, respectively), are commonly used parameters in the clinic. Deviations outside normal ranges for these parameters are indicative of many different disorders including cancer and infectious and immune diseases. Multiple reports confirm that high white cell counts are an independent risk factor for coronary artery disease (CAD) and myocardial infarction (MI)3–5. Increased platelet volume has also been variably associated with MI risk6.
We established the HaemGen Consortium in order to search for genetic loci contributing to variation in hematological parameters and to assess the potential correlation of these loci with disease phenotypes. In an initial cross-replication analysis of two independent genome-wide association (GWA) studies, we described four loci associated with MPV in Europeans. The four loci map in or near WDR66 (rs7961894), ARHGEF3 (rs12485738), TAOK1 (rs2138852) and PIK3CG (rs342293) and account for approximately 5.5% of the genetic variance7,8 in MPV. Here, we describe the findings of the first systematic genome-wide meta-analysis with independent replication of a broader range of eight clinically relevant hematological traits. We report 22 loci associated with these traits, one of which is also associated with increased risk of CAD.
RESULTS
GWA analysis of hematological parameters
The study design is shown in Figure 1. We analyzed a total of eight hematological parameters. Six of these parameters are measured directly: Hb, RBC and MCV for red cells, PLT and MPV for platelets and WBC for white cells. In addition, we tested the two derived red cell measures of mean corpuscular hemoglobin content (MCH) and mean corpuscular hemoglobin concentration (MCHC). Although they are derived from, and thus correlated to, the three measured red cell traits, we included MCH and MCHC because they are commonly used in the differential diagnosis of anemia.
Figure 1.

Summary of the study design.
We implemented a two-stage design involving a discovery set of 4,627 individuals sampled from three population-based samples and a replication set of 9,316 individuals from three additional studies (Fig. 1). All participants were of European ancestry. The characteristics of each sample collection are described in Supplementary Table 1a. After we applied stringent quality control criteria as described in the Supplementary Note, 2.11 million genotyped and imputed autosomal SNPs were available for analysis in all the three stage 1 samples. A uniform analysis plan was applied to each cohort, and individual summary statistics were combined using an inverse variance meta-analysis. There was no evidence of inflation of the summary statistics across the eight traits in the three discovery cohorts (Supplementary Note).
Following meta-analysis, we applied additional filtering criteria as described in the Online Methods to prioritize genomic regions for replication. A total of 88 independent regions met these criteria across the eight traits, including 11 for Hb, 10 for MCH, 3 for MCHC, 12 for MCV, 12 for RBC, 25 for MPV, 8 for PLT and 7 for WBC (Fig. 2). In each region we selected the SNP with the lowest P value for follow-up in the replication samples (‘leading SNP’). For one locus on chromosome 12q24, we selected two SNPs for follow-up (rs11065987 and rs11066301) that were in high linkage disequilibrium (LD) with each other (_r_2 = 0.82) but were located >500 kb apart (specifically, the two SNPs are 799 kb apart). The replication set included 9,316 individuals from three European population-based studies (Supplementary Table 1a). We applied the same uniform analysis plan and meta-analytical approach described in the Online Methods for analysis of the replication datasets and for combining summary statistics.
Figure 2.

Manhattan plots describing the association of 2.11 M SNPs with eight hematological traits in the three discovery samples (UKBS-CC1, TwinsUK and KORA F3 500K). SNPs with _P_≤ 10 5 are highlighted in green; SNPs exceeding the genome-wide significance threshold of 5 × 10 8 are shown in purple.
Characterization of 22 loci associated with hematological parameters
Of the 89 SNPs with replication data, 23 SNPs from 22 regions (including both SNPs in the 12q24 region) had nominally significant P values in the replication sample and reached genome-wide significance at the threshold of 5 × 10−8 in the combined sample of 13,943 individuals (Table 1; the summary statistics for loci that did not reach this threshold are given in Supplementary Table 2). Of the 22 loci, 7 are known loci for hematological parameters and the remaining 15 identify new association signals. We searched published literature, databases of mendelian human disease (Online Mendelian Inheritance in Man), gene function and homology with animal models of function and disease in order to prioritize the most likely candidate genes (Supplementary Table 3). Furthermore, we characterized the expression patterns of all the genes within a 1-Mb interval from the lead SNP in eight blood cell lines and endothelial cells using Illumina HumanWG-6 (v2) Expression BeadChip expression arrays (Supplementary Fig. 1 and Supplementary Note). Finally, for platelet loci, we also tested associations with transcript level in a panel of 35 platelet mRNAs. Although this effort provides supplementary evidence to prioritize a list of the most plausible candidates in each region, we note that more in-depth characterization will be required in order to conclusively associate genes with the observed phenotypic variation.
Table 1.
22 loci that reached genome-wide significance for association with eight hematological traits
| Trait | SNP | Chr(build36) | Pos(build 36) | Cytoband | Locus | Minorallele | MAFCEU | A1/A2a | Discovery (n = 4,627) | Replication (n = 9,316) | Combined (n = 13,943) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Beta (s.e.m.) | P | _I_2(%) | Beta (s.e.m.) | P | _I_2(%) | %variance | Beta (s.e.m.) | P | _I_2(%) | |||||||||
| MCH (pg) | rs5756506b | 22 | 35,797,338 | 22q12.3 | TMPRSS6 | C | 0.35 | C/G | 0.192 (0.040) | 1.2 × 10−6 | 0 | 0.111 (0.027) | 4.4 × 10−5 | 0 | 0.18 | 0.137 (0.022) | 9.5 × 10−10 | 0 |
| MCV (fl) | rs11970772 | 6 | 42,033,268 | 6p21.1 | BYSL/CCND3 | A | 0.15 | A/T | 0.591 (0.117) | 4.7 × 10−7 | 0 | 0.569 (0.078) | 2.7 × 10−13 | 50 | 0.51 | 0.575 (0.065) | 7.0 × 10−19 | 0 |
| rs1800562 | 6 | 26,201,120 | 6p21.3 | HFE | A | 0.04 | A/G | 1.319 (0.201) | 5.9 × 10−11 | 50 | 1.494 (0.197) | 3.1 × 10−14 | 0 | 0.94 | 1.408 (0.141) | 1.4 × 10−23 | 18 | |
| rs9609565 | 22 | 31,197,528 | 22q12–q13 | FBXO7 | A | 0.25 | G/A | 0.549 (0.111) | 8.2 × 10−7 | 0 | 0.301 (0.071) | 2.0 × 10−5 | 0 | 0.17 | 0.372 (0.060) | 4.3 × 10−10 | 20 | |
| rs9402686 | 6 | 135,469,510 | 6q23–q24 | HBS1L-MYB | A | 0.22 | A/G | 0.909 (0.109) | 9.1 × 10−17 | 14 | 0.777 (0.072) | 5.9 × 10−27 | 65 | 1.16 | 0.818 (0.060) | 7.4 × 10−42 | 37 | |
| RBC (1012/l), log | rs7385804 | 7 | 100,073,906 | 7q22 | TFR2 | C | 0.38 | C/A | 0.008 (0.002) | 4.7 × 10−6 | 0 | 0.005 (0.001) | 1.2 × 10−5 | 33 | 0.17 | 0.006 (0.001) | 4.9 × 10−10 | 0 |
| MPV (fl), log | rs10914144 | 1 | 170,216,373 | 1q24.3 | DNM3 | T | 0.17 | C/T | 0.016 (0.003) | 2.9 × 10−7 | 48 | 0.012 (0.002) | 7.3 × 10−9 | 0 | 0.34 | 0.013 (0.002) | 2.1 × 10−14 | 9 |
| rs11071720 | 15 | 61,129,049 | 15q22.1 | TPM1 | T | 0.37 | T/C | 0.013 (0.003) | 6.5 × 10−7 | 27 | 0.008 (0.003) | 3.1 × 10−3 | 8 | 0.18 | 0.011 (0.002) | 1.9 × 10−8 | 31 | |
| rs11602954 | 11 | 192,856 | 11p15.5 | BET1L | A | 0.23 | G/A | 0.014 (0.003) | 1.9 × 10−6 | 0 | 0.013 (0.002) | 1.4 × 10−9 | 25 | 0.41 | 0.013 (0.002) | 1.3 × 10−14 | 0 | |
| rs12485738 | 3 | 56,840,816 | 3p21–p13 | ARHGEF3 | A | 0.42 | A/G | 0.013 (0.002) | 1.5 × 10−8 | 71 | 0.016 (0.002) | 4.5 × 10−24 | 0 | 0.93 | 0.015 (0.001) | 5.5 × 10−31 | 46 | |
| rs1668873 | 1 | 203,502,613 | 1q32.1 | TMCC2 | A | 0.33 | G/A | 0.015 (0.002) | 3.3 × 10−10 | 0 | 0.011 (0.002) | 2.4 × 10−12 | 27 | 0.49 | 0.012 (0.001) | 1.4 × 10−20 | 24 | |
| rs2138852 | 17 | 24,727,475 | 17q11.2 | TAOK1 | C | 0.44 | T/C | 0.014 (0.002) | 2.5 × 10−9 | 57 | 0.018 (0.002) | 4.5 × 10−15 | 0 | 1.21 | 0.016 (0.002) | 1.4 × 10−22 | 34 | |
| rs2393967 | 10 | 64,803,162 | 10q21.2–q21.3 | JMJD1C | C | 0.37 | A/C | 0.014 (0.002) | 2.3 × 10−8 | 0 | 0.015 (0.002) | 2.3 × 10−14 | 0 | 0.68 | 0.014 (0.002) | 3.3 × 10−21 | 0 | |
| rs342293 | 7 | 106,159,455 | 7q22.3 | PIK3CG | G | 0.45 | G/C | 0.017 (0.002) | 6.8 × 10−13 | 22 | 0.015 (0.002) | 2.3 × 10−22 | 69 | 0.96 | 0.015 (0.001) | 1.6 × 10−33 | 48 | |
| rs6136489 | 20 | 1,871,734 | 20p13 | SIRPA | G | 0.26 | T/G | 0.012 (0.002) | 1.3 × 10−6 | 24 | 0.009 (0.002) | 7.6 × 10−6 | 0 | 0.25 | 0.010 (0.002) | 7.7 × 10−11 | 8 | |
| rs647316 | 2 | 31,318,333 | 2p21 | EHD3 | A | 0.25 | A/G | 0.013 (0.002) | 7.4 × 10−8 | 63 | 0.008 (0.002) | 2.8 × 10−5 | 55 | 0.39 | 0.010 (0.002) | 3.2 × 10−11 | 59 | |
| rs7961894 | 12 | 120,849,966 | 12q24.31 | WDR66 | T | 0.12 | T/C | 0.036 (0.004) | 8.2 × 10−19 | 0 | 0.029 (0.003) | 1.3 × 10−27 | 0 | 1.39 | 0.031 (0.002) | 2.7 × 10−44 | 0 | |
| rs893001 | 18 | 65,667,825 | 18q22.3 | CD226 | A | 0.47 | C/A | 0.013 (0.002) | 8.3 × 10−8 | 0 | 0.009 (0.002) | 1.9 × 10−4 | 0 | 0.27 | 0.011 (0.002) | 1.4 × 10−10 | 0 | |
| PLT (109/l) | rs11065987 | 12 | 110,556,807 | 12q24 | ATXN2 | G | 0.34 | G/A | 7.521 (1.305) | 8.3 × 10−9 | 0 | 4.118 (0.815) | 4.4 × 10−7 | 0 | 0.23 | 5.073 (0.692) | 2.2 × 10−13 | 31 |
| rs11066301 | 12 | 111,355,755 | 12q24 | PTPN11 | G | 0.35 | G/A | 7.479 (1.251) | 2.3 × 10−9 | 0 | 3.467 (0.809) | 1.8 × 10−5 | 0 | 0.16 | 4.650 (0.680) | 7.7 × 10−12 | 45 | |
| rs210135 | 6 | 33,648,670 | 6p21.3 | BAK1 | T | 0.32 | A/T | 6.908 (1.342) | 2.6 × 10−7 | 0 | 4.380 (1.138) | 1.2 × 10−4 | 0 | 0.19 | 5.438 (0.868) | 3.7 × 10−10 | 0 | |
| rs385893 | 9 | 4,753,176 | 9p24.1–p24.3 | AK3 | T | 0.44 | C/T | 6.951 (1.389) | 5.6 × 10−7 | 47 | 5.979 (0.895) | 2.4 × 10−11 | 24 | 0.33 | 6.264 (0.753) | 8.5 × 10−17 | 26 | |
| WBC (109/l), log | rs17609240 | 17 | 35,364,215 | 17q12 | GSDMA/ORMDL3 | T | 0.26 | G/T | 0.030 (0.006) | 1.2 × 10−6 | 0 | 0.015 (0.004) | 2.1 × 10−4 | 11 | 0.12 | 0.019 (0.003) | 9.4 × 10−9 | 33 |
Red blood cell traits
Six independent regions were confirmed as strongly associated with red blood cell parameters with all exerting their main effect on MCV or RBC (Table 1). Among these regions were two well-characterized loci: the HBS1L-MYB region on 6q23–q24 (rs9402686, P = 7.4 × 10−42) and the C282Y amino acid change in HFE at 6p21.3 (rs1800562, P = 1.4 × 10−23). Rare nonsynonymous mutations in these genes have been associated with hereditary hemochromatosis and common SNPs with measures of iron status (Supplementary Table 3). Of the red cell loci, the HBS1L-MYB locus had the greatest pleiotropic effect, showing genome-wide significant associations with MCH (P = 4.5× 10−40), RBC (P = 1.6 × 10−29), PLT (P = 2.2 × 10−13) and, to a lesser extent, MCHC (P = 1.2× 10−5) and WBC (P = 6.3 × 10−5; Supplementary Table 4). Two other association signals were located near genes known to play a role in iron hemostasis (TMPRSS6 and TFR2). The serine protease matriptase-2, encoded by TMPRSS6 (lead SNP rs5756506, P = 9.5 × 10−10), regulates levels of the peptide hormone hepcidin, the master regulator of iron homeostasis in humans9. The rs5756506 SNP was the only red-blood-cell locus to be strongly associated with Hb levels (P = 3.4 × 10−8, Supplementary Table 4); the only other red blood cell locus with a nominal effect on Hb was HFE (P = 1.6 × 10−4). The signal on chromosome 7q22 (rs7385804, P = 4.9 × 10−10) is centered on the TFR2 gene, which encodes the type-2 transferrin receptor essential to cellular uptake of transferrin-bound iron10. Another likely candidate in this gene-dense region is EPO (erythropoietin), a growth factor critical for fate determination within the erythroid lineage11. Another newly identified MCV locus on chromosome 6p21.1 (rs11970772, P = 7.0 × 10−19) maps to a recombination interval near the BYSL and CCND3 genes./We found that five out of the seven genes in the _BYSL_-CCND3 region were abundantly transcribed in hematopoietic cells (Supplementary Fig. 1). Both BYSL and CCND3 have roles in hematopoiesis (Supplementary Table 3). BYSL (bystin) is a target of c-MYC mRNA, which is consistent with a role in rapid protein synthesis required for actively growing cells12. _Ccnd3_−/− mice show lethality due to heart abnormalities combined with severe anemia13. Finally, the association signal for MCV at 22q12–q13 overlaps with FBXO7 (rs9609565, P = 4.3 × 10−10), a gene highly expressed in erythroblasts (EBs), which are the precursors of red blood cells (Supplementary Fig. 1).
White blood cell counts
One association signal for the total number of leukocytes was identified on 17q12 near _GSDMA_-ORMDL3 (rs17609240, P = 9.4 × 10−9), a known susceptibility locus for childhood asthma14. Notably, this locus contains CSF3, which encodes colony stimulating factor 3, a cytokine controlling the production, differentiation and function of granulocytes15.
Platelet counts and mean platelet volume
In addition to the four loci associated with MPV (WDR66, ARHGEF3, TAOK1 and PIK3CG) previously described by our groups7,8, we detected eight new loci associated with MPV and the first three loci found to be associated with PLT (Table 1, Supplementary Fig. 1). Nine of the 12 MPV loci were also associated with PLT, of which 3 reached genome-wide significance in the combined sample. In all cases the MPV-raising alleles were associated with a decrease in PLT (Supplementary Table 4). Conditional analyses show however that all SNPs exerted their main effects through MPV (Online Methods).
Of the newly identified MPV-associated loci, the association signals on chromosome 1q24.3 (DNM3, rs10914144, P = 2.1 × 10−14) and 18q22.3 (CD226, rs893001, P = 1.5 × 10−10) contained two strong and highly plausible candidate genes with a known role in megakaryocyte (MK) development (Supplementary Table 3) and enhanced gene expression in MKs when compared with EBs (Supplementary Fig. 1). Four additional regions map in or near JMJD1C (rs2393967, P = 3.3 × 10−21), TPM1 (rs11071720, P = 1.9 × 10−8), SIRPA (rs6136489, P = 7.7 × 10−11) and EHD3 (rs647316, P = 3.2 × 10 11), which are candidates with indirect evidence for a role in hematopoiesis in humans (as discussed in Supplementary Table 3). Of these genes, JMJD1C (10q21) encodes a probable histone demethylase, with a possible function in hormone-dependent transcriptional activation. Mouse mutated at Jmjd1c (encoding Jumonji domain containing 1C) display increased proliferation of MK lineage cells16. TPM1 encodes tropomyosin I, which regulates the calcium-dependent interaction of actin and myosin, a key step in platelet formation. TPM1 was found to be highly downregulated in an individual with an unique mutation in RUNX1 (also called CBFA2) and a severe platelet function disorder17. Finally, two newly identified regions at 1q32.1 and 11p15.5 are gene rich, and further efforts will be required to identify the most likely gene candidates for association with MPV. The 11p15.5 signal maps to a region proximal to the genes BET1L, SIRT3 and PSMD13 (among others). In this region, we found evidence that the lead SNP rs11602954 affects expression of the two neighboring genes BET1L (Spearman’s test P = 3.1 × 10 5) and SIRT3 (P = 2.8 × 10 5) as well as PSMD13 to a lesser degree (P = 7.3 × 10 3, see Supplementary Note and Supplementary Fig. 1). A G477T variant in its promoter has been shown to co-regulate SIRT3 and PSMD13 and has been linked to longevity in humans18. Three independent loci with effects on PLT were identified. The association signal on the 6p21.3 locus was centered in the BAK1 gene (rs210135, P = 3.7 × 10−10), which encodes a protein with a strong proapoptotic effect that is known to control platelet lifespan19. Two further SNPs map to 12q24.12 (rs11065987, P = 2.2 × 10−13 and rs11066301, P = 7.7 × 10−12). The association signal on 12q24.12 spans ~1.6 Mb and harbors 15 genes including PTPN11, SH2B3 and BRAP. This region is discussed in more detail below. Finally, an association signal at 9p24.1–p24.3 (rs385893, P = 8.5 × 10−17) was found 400 kb upstream of JAK2, which is a key regulator of megakaryocyte maturation and is somatically mutated in half of the individuals with essential thrombocytosis20.
Multimarker scores
Overall, the fraction of genetic variance explained by each locus in regression models adjusted for sex and age was 8.6% for MPV traits, 0.5% for PLT traits, 3% for erythrocyte traits and 0.12% for the single validated WBC locus. We constructed a score to predict MPV levels from the joint model of the 12 validated MPV SNPs and the 6 validated SNPs associated with red blood cell traits as described in the Online Methods section (Fig. 3). The regression of mean on score indicates an average increase of MPV of 0.12 fl per copy of a MPV-increasing allele and 0.47 fl per copy of a MCV-increasing allele.
Figure 3.

Multimarker score tests for MPV and MCV. (a) MPV scores were calculated from the 12 validated MPV loci and are given for individuals with ≤ 7, 8 17 and ≥ 18 MPV-increasing alleles. (b) MCV scores were calculated from six validated red blood cell loci. MCV multimarker scores were calculated for males and females separately to account for substantial differences among sexes. Gray bars indicate the number of individuals in each score class, dots and triangles indicate the mean MPV and mean MCV levels in each class with bars showing the associated standard errors (blue for males and magenta for females); the lines are the linear regressions though these points. The regression indicates an average increase of MPV of 0.12 fl per copy of MPV-increasing allele, corresponding to a variation of between 8.25 and 9.59 fl for individuals carrying between 7 and 18 copies of MPV-increasing alleles, respectively. The corresponding average increase in MCV was 0.47 fl per allele (range 90.60 93.86 fl for individuals carrying ≤1 or ≥8 copies of MCV-increasing alleles) in males and 0.47 fl (range 89.23–92.49 fl for the same range of alleles) in females, respectively.
Associations with coronary artery disease
Because several of the hematological traits analyzed show an association with CAD or MI, we examined the association of the validated 23 SNPs (including the two associated SNPs on 12q24.12) with CAD. We used a two-stage approach to test for association with CAD (Fig. 1). First, we obtained association statistics for 4,021 affected individuals (cases) and 5,879 controls from three European CAD or MI case-control studies (Wellcome Trust Case Control Consortium (WTCCC)-CAD, German Myocardial Infarction Family Study (GerMIFS I and GerMIFS II) and calculated the pooled odds ratios. All studies included validated cases of premature MI or CAD as detailed in Supplementary Table 1b and the Supplementary Note. Two SNPs from one region (SNPs rs11066301 and rs11065987 on 12q24) had nominal significance (_P_≤ 0.05) in the stage 1 analysis (Supplementary Table 5). For these loci, we obtained summary statistics from an additional 5,458 cases and 4,648 controls from five further case-control collections, including the Ottawa Heart, MedSTAR, PennCATH, MIGen and the COROGENE studies. All samples had a validated diagnosis of CAD (including MI) compatible with the clinical criteria used in the stage 1 samples (See Supplementary Table 1b and Supplementary Note for case definition in the different studies).
The association results for the two SNPs rs11066301 and rs11065987 on 12q24 were strongly replicated in the stage 2 sample, providing independent confirmation for 12q24 as a risk locus for CAD. In the combined sample of 9,479 cases and 10,527 controls, the allelic odds ratios of rs11066301 (minor allele frequency (MAF) = 0.35) and rs11065987 (MAF = 0.34) were, respectively, 1.144 (95% CI 1.095–1.196, P = 2.52×10−9) and 1.152 (95% CI 1.104–1.202, P = 7.05×10−11 Fig. 4 and Supplementary Table 5a); the respective allelic odds ratios for a MI sub-analysis were 1.165 (95% CI 1.111–1.222], P = 3.43×10−10) and 1.177 (95%CI 1.124–1.231, P = 2.42×10−12; Supplementary Table 5b). For both SNPs, the minor allele was associated with increased PLT and risk of CAD and MI. The same association with CAD was recently reported by an independent study 21.
Figure 4.

Association of SNP rs11065987 with CAD. Pooled ORs and 95% CI were calculated in eight case-control studies of European origin under a fixed effects model, as there was no evidence for heterogeneity in associations at this locus. The remaining nine SNPs characterizing this haplotype are described in Supplementary Table 5.
Haplotype structure of the 12q24 locus, natural selection and pleiotropic effects in human disease
The SNPs rs11065987 and rs11066301 are located 799 kb apart and are in high LD (_r_2 = 0.82). Analysis of the PLT association plot shows that the signals map to two adjacent recombination intervals spanning approximately 1.6 Mb and containing 15 genes. The expression of such genes in blood lineages is shown in Figure 5. The haplotype structure of this region is shown in Figure 6. We analyzed the local LD pattern in three HapMap population panels (CEU, CHB+JPT, YRI). In the CEU panel, the region is characterized by extended LD. Ten common SNPs (MAF = 0.35–0.4) identify a common haplotype spanning the length of the associated interval (Table 2). Of the ten SNPs, one is an Arg262Trp nonsynonymous change in the gene SH2B3 (rs3184504), seven are intronic within four genes (ATXN2, C12orf30, C12orf51 and PTPN11) and two are intergenic. All of them display genome-wide significant association with PLT (Fig. 6a). We calculated the pooled summary statistics for associations with CAD in the same 1.6 Mb region using six of the eight case-control studies with available data. The ten SNPs had similarly elevated P values for association with CAD (Table 2, see also Supplementary Table 5), whereas the remaining SNPs in the region did not show strong association with CAD (Fig. 6b). The G allele at rs17696736 in C12orf30 is a known risk factor for type 1 diabetes (T1D)22,23. A second SNP on the same haplotype (rs3184504 in SH2B3) has been previously associated with celiac disease, whereby the CAD risk allele also increases risk for celiac disease24. We retrieved association data for T1D and celiac disease generated in previous studies (Supplementary Note) and plotted the association statistics for genotyped and imputed SNPs over the same interval (Fig. 6c,d). We observed a similar elevation of the association signals at the ten SNPs (where present), which suggests a pattern of association similar to PLT and CAD.
Figure 5.
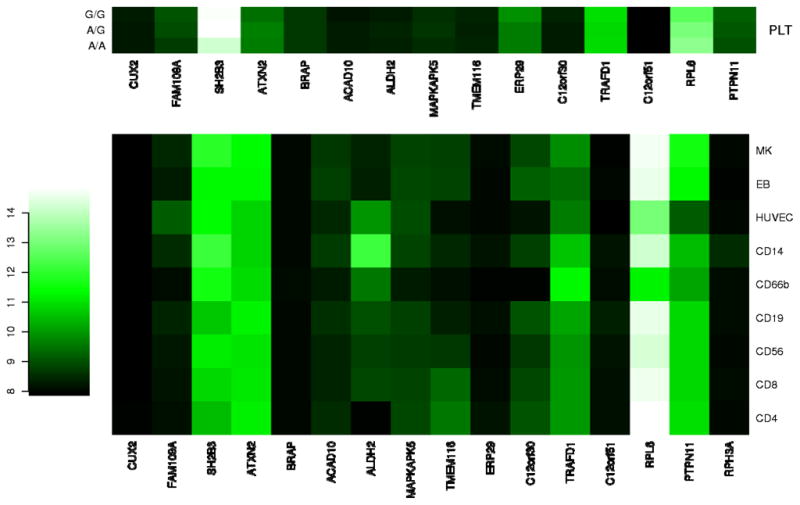
Heatmap of mRNA expression in the 12q24 region. For all genes contained within the 1.6-Mb interval, VST-transformed signal intensities from using Illumina HumanWG-6 (v2) Expression BeadChip expression arrays were median-normalized and values were averaged across biological replicates in stem cell-derived erythroblasts (EBs, n = 4), megakaryocytes (MK, n = 4), human umbilical vein endothelial cells (HUVECs, n = 3), CD4+ Th (CD4, n = 7) and CD8+ Tc lymphocytes (CD8, n = 7), CD14+ monocytes (CD14, n = 7), CD19+ B lymphocytes (CD19, n = 7), CD56+ natural killer cells (CD56, n = 7) and CD66b+ granulocytes (CD66, n = 7). For platelet-associated signals, levels of gene expression in 35 platelet mRNA were averaged based on genotype at the leading or proxy SNP. Signal intensities obtained with platelets were obtained using Illumina HumanWG-6 (v1) Expression BeadChip expression arrays and were normalized independently from the remaining blood cell lines.
Figure 6.

Overview of the 12q24 region. (a–d) The −log10 P value for associations with platelet counts (a), coronary artery disease (b), type 1 diabetes (c) and celiac disease (d), expressed in −log10(P value), are shown for two consecutive recombination intervals in a 1.6-MB region on chromosome 12 (Build 36 pos 109,896,664–111,516,664). (e) The position of the 10 SNPs forming a high frequency (MAF 40%) haplotype is highlighted by gray bars; this also displays the evolutionarily ancestral (blue) and derived (red) alleles at the 10 SNPs. (f,g) Signatures of positive selection obtained from Haplotter, including a graphical display of haplotypes at different distances from the lead SNP rs11065987 (f) and a plot marking the decay of extended haplotype homozygosity at different distances from SNP rs11065987 (g).
Table 2.
Association with disease and signatures of natural selection at the 10 core SNPs in the 12q24 region
| SNP | Gene annotation | Platelet count association | CAD | Natural selection | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Increaserallele | Beta(s.e.m.)(109/l) | P | Riskallele | OR (95% CI) | P | Ancestral/derivedallele | DAFcCEU | DAFcYRI | DAFcCHB | StandardizediHS | FayandWu’s_H_+ | _F_STd | ||
| rs3184504a | SH2B3 (Arg262Trp) | T | 7.22 (1.28) | 1.6 × 10−8 | T | 1.186 (1.127–1.248) | 5.04×10−11 | C/T | 0.41 | 0 | 0 | −2.756 | −35.656 | 0.39** |
| rs4766578a | ATXN2 (intron) | T | 7.33 (1.28) | 1.0 × 10−8 | T | 1.169 (1.107–1.234) | 1.57×10−8 | A/T | 0.42 | – | – | −2.761 | −37.062 | – |
| rs10774625a | ATXN2 (intron) | A | 7.33 (1.28) | 9.9 × 10−9 | A | 1.169 (1.107–1.234) | 1.66×10−8 | G/A | 0.42 | 0 | 0 | −2.761 | −36.22 | 0.40** |
| rs653178a | ATXN2 (intron) | C | 7.25 (1.27) | 1.2 × 10−8 | C | 1.181 (1.123–1.241) | 6.30×10−11 | T/C | 0.41 | 0 | 0 | −2.882 | −34.185 | 0.39** |
| rs11065987 | Intergenic | G | 7.52 (1.31) | 8.3 × 10−9 | G | 1.177 (1.124–1.231) | 2.42×10−12 | A/G | 0.34 | 0 | 0 | −3.038 | −36.295 | 0.34* |
| rs17696736b | C12orf30 (intron) | G | 6.89 (1.24) | 3.1 × 10−8 | G | 1.164 (1.114–1.216) | 1.47×10−11 | A/G | 0.35 | 0 | 0 | −3.212 | −57.263 | 0.34* |
| rs17630235 | TRAFD1 (3 ′ of gene) | A | 7.07 (1.25) | 1.7 × 10−8 | A | 1.167(1.113–1.223) | 1.48×10−10 | G/A | 0.33 | 0 | 0 | −3.206 | −61.348 | 0.32* |
| rs11066188 | C12orf51 (intron) | A | 7.22 (1.25) | 8.5 × 10−9 | A | 1.171 (1.120–1.225) | 5.12×10−12 | G/A | 0.32 | 0.008 | 0 | −3.227 | −60.794 | 0.30* |
| rs11066301 | PTPN11 (intron) | G | 7.48 (1.25) | 2.3 × 10−9 | G | 1.165 (1.111–1.222) | 3.43×10−10 | A/G | 0.35 | 0.008 | 0 | −2.646 | −56.342 | 0.33* |
| rs11066320 | PTPN11 (intron) | A | 7.48 (1.25) | 2.3 × 10−9 | A | 1.169 (1.117–1.223) | 1.33×10−11 | G/A | 0.35 | 0 | 0 | −4.341 | −59.37 | 0.34* |
We retrieved the ancestral states by comparison with chimpanzee data from the University of California at Santa Cruz genome browser for each of the ten SNPs showing significant association with PLT (Table 2). The CAD-risk and PLT-raising alleles corresponded to the derived states. We retrieved the integrated haplotype score (iHS)25,26 and Fay and Wu’s H+ statistics for HapMap Phase II data (Table 2)25–27 to test the hypothesis that the long-range, evolutionarily derived haplotype in this region arose from a positive selection event—that is, a selective sweep. The 12q24 region showed a signature characteristic of a selective sweep, with highly negative iHS scores (−4.341 to −2.756, an extreme pattern compared to an empirical genome-wide threshold of −2 for positive selection25) and highly skewed Fay and Wu’s _H_+ statistics (Table 2). Accordingly, the extended haplotype homozygosity statistics28 show excess homozygosity on the evolutionarily derived haplotype over a 1.6-Mb interval (Fig. 6g). We estimated the age of the rs3184504 T-allele haplotype25 at approximately 3,400 years (Supplementary Note). Next, we compared the population differentiation statistics _F_ST at the 10 SNPs with the empirical distributions of frequency-matched HapMap SNPs (Table 2)29. The ancestral alleles at all SNPs were fixed in the YRI and CHB+JPT HapMap panels, yielding significant global differentiation (Table 2). Taken together, these results support the hypothesis of a selective sweep that increased the frequency of CAD, T1D and celiac disease risk alleles in Europeans and geographically nearby populations but not in East Asian or African populations.
DISCUSSION
This study represents, to our knowledge, the first GWA of hematological parameters to be completed in cohorts with large sample sizes. In a two-stage design with 4,627 discovery and 9,316 replication samples, we were able to confirm 22 independent loci as associated with 6 of the 8 traits at the genome-wide significance level. None of the loci selected from the meta-analysis of MCHC and Hb were replicated at genome-wide significance in our study. However, genome-wide significance for Hb was achieved for rs5756506 at locus TMPRSS6 in the combined analysis (Supplementary Table 4). The regions identified contain several plausible regulators of hematopoiesis in humans (see also Supplementary Table 3 for discussion on likely candidates). Associations with erythrocyte-related traits are dominated by two main effect loci, rs1800562 in _HBS1L_-MYB and the nonsynonymous change rs9402686 in HFE. Three loci (HFE, TFR2 and TMPRSS6) mapped to genes known to be associated with iron homeostasis. The nonsynonymous C282Y change in HFE (rs9402686) is a classic risk allele for hereditary hemochromatosis, but here we show for the first time that it also modifies MCV.
The 12 MPV loci showed similar per-allele effect sizes (Table 1) and jointly explain 8.6% of total genetic variance in MPV after adjusting for age and sex. We identified several key functional categories of genes implicated in the regulation of platelet counts and volume, including transcriptional activation (WDR66 and JMJD1C), intracellular signaling (PIK3CG, ARHGEF3, TAOK1 and SH2B3), protein transport and endocytosis (BET1L, DNM3 and EHD3), cell adhesion (SIRPA and CD226) and actin-myosin contraction and cell motility (TPM1) and apoptosis (BAK1). Of these, only a handful of genes encode proteins that had previously known roles in hematopoiesis in humans and mouse knockout models (_PIK3CG_-PRKAR2B, ARHGEF3, JMJD1C, CD226, BAK1, _SH2B3_-PTPN11 and SIRPA). SIRPA and CD226 both encode MK membrane proteins; results from cell biology studies in MK cells are strongly supportive of their candidacy for association to MPV (Supplementary Table 3). The marked overexpression of DNM3 in MKs compared with other blood cells and the increase in the TPM1 transcript level with MK polyploidization both support of the putative role of these proteins in MK and platelet biology, but further studies will be required to discern their precise role.
We also detected a greater number of loci for MPV than for red and, particularly, white blood cell traits. Measurements of WBC included all different white cell subtypes, thus adding to the overall noise in the association analysis and lowering power. It is possible that dissecting the WBC measurement into the main types of mononuclear cellular elements (lymphocytes, monocytes and granulocytes) may improve the ability to identify a large number of additional loci. A recent study identified an association of the Arg262Trp nonsynonymous change in the gene SH2B3 (rs3184504) and eosinophil counts and CAD (P = 8.6 × 10−8)21. The same locus was identified in our study as being strongly associated with PLT and CAD.
We extended knowledge of this locus by characterizing the association signal as a common (frequency ~40%) long-range haplotype (1.6 Mb) including the Arg262Trp site, seven intronic SNPs (in ATXN2, C12orf30, C12orf51 and PTPN11) and two intergenic SNPs. We obtained strong evidence suggesting that the haplotype at 12q24 has arisen from a selective sweep specific to Europeans and nearby populations beginning approximately 3,400 years ago, a period characterized by the expansion of high-density human settlements in this part of the world. The role of this region in T cell–mediated immune response is compatible with the notion of immunity being a strong selective force in human evolution28.
The 12q24 haplotype links risk alleles for T1D, CAD and celiac disease (carried on the derived haplotype) as well as a recently identified association with hypertension30, thus highlighting a remarkable example of disease pleiotropy at this locus. The functional validation of the effect of the Arg262Trp variant in SH2B3 and other variants on this haplotype will be important to clarify and dissect the underlying causes of such pleiotropy and also to establish whether variation in PLT and/or the Arg262Trp change are causal for CAD or whether they merely reflect a pleiotropic effect due to the persistence of multiple functional variants on the long-range haplotype. SH2B3 encodes Lnk, an important negative regulator of cell-signaling events originating from cell membrane activatory receptors such as the T-cell receptor and MPL, the receptor for thrombopoietin on MKs and platelets. Lnk-mediated regulation of Stat-5 activation regulates the crosstalk between integrin- and cytokine-mediated signaling31. Cells from Lnk-deficient mice show an increased sensitivity to several cytokines and altered activation of the RAS-MAPK pathway in response to IL3 and stem cell factor32. Using homology to mouse protein models, we mapped Arg262Trp to a putative pleckstrin homology domain (Supplementary Note and Supplementary Fig. 2). A possible functional effect could be caused by a charge reversal of this surface-exposed residue, affecting interaction with unidentified downstream signaling molecules. Pleckstrin homology domains form a structurally conserved family associated with several regulatory pathways through signal transduction or protein ligand recognition33.
Further functional assessment and in-depth analysis of the 12q24 region will be required to dissect the pleiotropic effects observed at this locus and, in particular, the causality relationship between platelet counts and CAD risk. We note that the region covered by the long-range haplotype contains a number of other candidate genes that may modify platelet phenotypes. The tyrosine-protein phosphatase non-receptor type 11 encoded by PTPN11 plays a regulatory role in a wide array of cell-signaling events involved in the control of cell functions, such as mitogenic activation, metabolic control, transcription regulation and cell migration. Mutations in PTPN11 are a cause of the mendelian disorder Noonan syndrome, which is characterized by platelet abnormalities34,35 and acute myeloid leukemias36,37. Also in this region, BRAP (encoding BRCA1-associated protein) was shown to interact in vitro and in vivo with p21 (encoded by CDKN1A), a regulator of cell cycle progression previously implicated in atherosclerosis38. Notably, a recent study in Japanese individuals has detected an association between common SNPs in BRAP and risk of CAD39. Such an effect, however, is not explained by the Arg262Trp variant in SH2B3, which is absent in East Asian populations.
An overarching scope of our analysis was to test whether blood cell loci, particularly those for platelets, are risk loci for cardiovascular disease. Apart from the association signal on 12q24, we found no overwhelming evidence for contribution of these loci to the risk of CAD or MI. Increased MPV represents a strong, independent predictor of post-event outcome in CAD6,40–42, and the new loci might contribute to survival and prognosis after a major CAD event. This possibility merits further investigation. Finally, the regions identified provide new targets to study in a range of other related diseases. For example, platelets are proposed as having a role in cancer progression and metastasis, which has largely been attributed to platelet-mediated enhancement of tumor cell survival, extravasation and angiogenesis. It has been proposed that platelet inhibition may slow the rate of tumor progression and metastasis. Further characterization of these loci will improve our understanding of key regulatory mechanisms of hematopoiesis in humans and may also lead to the discovery of new candidate genes that are somatically mutated in premalignant conditions such as essential thrombocytosis and polycythemia vera and in other hematological malignancies.
ONLINE METHODS
Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturegenetics/.
Note: Supplementary information is available on the Nature Genetics website.
Supplementary Material
Supplemental Data
Acknowledgments
The Wellcome Trust; EU (HEALTH-F2-2008-ENGAGE, QLG2-CT-2002-01254), NIHR (TwinsUK); The Wellcome Trust (076113/C/04/Z), Juvenile Diabetes Research Foundation (WT061858), National Institute of Health Research of England (UKBS-CC1); Helmholtz Zentrum München, German Research Center for Environmental Health, Neuherberg, Germany, the German Federal Ministry of Education and Research (BMBF), the German National Genome Research Network (NGFN), Munich Center of Health Sciences (MC Health) (KORA); Federal Ministry of Education and Research (grants no. 01ZZ9603, 01ZZ0103, and 01ZZ0403), Ministry of Cultural Affairs, Social Ministry of the Federal State of Mecklenburg-West Pomerania,Deutsche Forschungsgemeinschaft (grant SFB TR 19), the Federal Ministry of Education and Research (grant no. 03ZIK012); a joint grant from Siemens Healthcare, Erlangen, Germany and the Federal State of Mecklenburg- West Pomerania (SHIP); The National Institute for Health Research to CBMRC and NHSBT, the Wellcome Trust and Juvenile Diabetes Research Foundation International (CBR); Deutsche Forschungsgemeinschaft, the German Federal Ministry of Education and Research (BMBF) (NGFN-2 and NGFN-plus), EU (LSHM-CT-2006-037593) (GerMIFS I and II); BHF and the UK MRC, the Wellcome Trust, Leicester NIHR Biomedical Research Unit in Cardiovascular Disease (WTCCC-CAD); Cardiovascular Institute (University of Pennsylvania), GlaxoSmithKline, MedSTAR Research Institute (PennCATH/MedSTAR); US National Institutes of Health (NIH) and National Heart, Lung, and Blood Institute (STAMPEED), National Center for Research Resource (U54 RR020278) (MIGen); Canadian Institutes of Health Research, Canada Foundation for Innovation and Ontario Research Foundation (OHGS); Finnish Heart Foundation, Sigrid Juselius Foundation (COROGENE); Juvenile Diabetes Research Foundation/Wellcome Trust (T1D).
Footnotes
AUTHOR CONTRIBUTIONS
Manuscript preparation: N.S., M.M., A.R., W.H.O., T.D.S., P.D., N.J.S., C.G. Main data analysis: N.S., C.G., B.K., A.R., A.T., R.A.L., Y.X., C.T.-S.
Intermediate trait analysis cohorts. Study design and biobanking: T.D.S. (TwinsUK), J.R.B., W.E., S.F.G., J.S.-C., J. Sambrook, N.A.W., W.H.O. (UKBS-CC1 and CBR), C.G., T.I., H.-E.W. (KORA F3 and F4), M.N., U.V., H.V. (SHIP). Phenotype assessment: S.M., M.F., S.L.T., T.D.S. (TwinsUK), A.D., C.M. (KORA F3 and F4), A.G. (SHIP). Genotyping: R.G., S.C.P., C.M.R., P.D. (TwinsUK), S.B., M.J.R.G., R.G., N.H., J.Stephens (CBR), H.P., T.I. (KORA F3 and F4). Statistical analysis: N.S. (TwinsUK), N.S. (CBR), N.S. (UKBS-CC1), C.G, B.K. (KORA F3 and F4), A.T. (SHIP), A.R., P.B. (Transcriptomics).
CAD/MI cohorts. GerMIFS I and GerMIFS II: C.H., I.R.K, S.S., K.S., C.W., H.-E.W., C.W., J.E., H.S. WTCCC-CAD: N.J.S., A.H.G., A.S.H., B.W., J.R.T. Ottawa Heart Study: L.C., R.M., R.R., G.A.W., A.F.R.S. PennCATH/MedSTAR: M.L., M.S.B., J.D., S.E., H.H.H., D.J.R., M.P.R., V.M., C.W.K. MIGEN: S.K., B.F.V., S.M.S., V.S., R.E., O.M., C.J.O., L.P., D.S.S., D.A. COROGENE: M.P., P.S., V.S., L.P., I.S., J.Sinisalo, M.S.N.
Celiac disease. D.A.V.H.
References
- 1.Garner C, et al. Genetic influences on F cells and other hematologic variables: a twin heritability study. Blood. 2000;95:342–346. [PubMed] [Google Scholar]
- 2.Evans DM, Frazer IH, Martin NG. Genetic and environmental causes of variation in basal levels of blood cells. Twin Res. 1999;2:250–257. doi: 10.1375/136905299320565735. [DOI] [PubMed] [Google Scholar]
- 3.Ensrud K, Grimm RH. The white blood cell count and risk for coronary heart disease. Am Heart J. 1992;124:207–213. doi: 10.1016/0002-8703(92)90942-o. [DOI] [PubMed] [Google Scholar]
- 4.Danesh J, Collins R, Appleby P, Peto R. Association of fibrinogen, C-reactive protein, albumin, or leukocyte count with coronary heart disease: meta-analyses of prospective studies. J Am Med Assoc. 1998;279:1477–1482. doi: 10.1001/jama.279.18.1477. [DOI] [PubMed] [Google Scholar]
- 5.Hoffman M, Blum A, Baruch R, Kaplan E, Benjamin M. Leukocytes and coronary heart disease. Atherosclerosis. 2004;172:1–6. doi: 10.1016/s0021-9150(03)00164-3. [DOI] [PubMed] [Google Scholar]
- 6.Boos CJ, Lip GYH. Assessment of mean platelet volume in coronary artery disease what does it mean? Thromb Res. 2007;120:11–13. doi: 10.1016/j.thromres.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Meisinger C, et al. A genome-wide association study identifies three loci associated with mean platelet volume. Am J Hum Genet. 2009;84:66–71. doi: 10.1016/j.ajhg.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soranzo N. A novel variant on chromosome 7Q22.3 associated with mean platelet volume, counts, and function. Blood. 2009;113:3831–3837. doi: 10.1182/blood-2008-10-184234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silvestri L, et al. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008;8:502–511. doi: 10.1016/j.cmet.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wallace DF, Subramaniam VN. Non-HFE haemochromatosis. World J Gastroenterol. 2007;13:4690–4698. doi: 10.3748/wjg.v13.i35.4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elliott S, Pham E, Macdougall IC. Erythropoietins: a common mechanism of action. Exp Hematol. 2008;36:1573–1584. doi: 10.1016/j.exphem.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Fukuda MN, Miyoshi M, Nadano D. The role of bystin in embryo implantation and in ribosomal biogenesis. Cell Mol Life Sci. 2008;65:92–99. doi: 10.1007/s00018-007-7302-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kozar K, et al. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004;118:477–491. doi: 10.1016/j.cell.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 14.Moffatt MF, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 15.Hollard D, Berthier R, Douady F. Granulopoiesis and its regulation. Sem Hop. 1975;51:643–651. [PubMed] [Google Scholar]
- 16.Kitajima K, Kojima M, Kondo S, Takeuchi T. A role of jumonji gene in proliferation but not differentiation of megakaryocyte lineage cells. Exp Hematol. 2001;29:507–514. doi: 10.1016/s0301-472x(00)00686-x. [DOI] [PubMed] [Google Scholar]
- 17.Sun L, Gorospe JR, Hoffman EP, Rao AK. Decreased platelet expression of myosin regulatory light chain polypeptide (MYL9) and other genes with platelet dysfunction and CBFA2/RUNX1 mutation: insights from platelet expression profiling. J Thromb Haemost. 2007;5:146–154. doi: 10.1111/j.1538-7836.2006.02271.x. [DOI] [PubMed] [Google Scholar]
- 18.Bellizzi D, et al. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics. 2005;85:258–263. doi: 10.1016/j.ygeno.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 19.Mason KD, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128:1173–1186. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 20.Wickrema A, Crispino JD. Erythroid and megakaryocytic transformation. Oncogene. 2007;26:6803–6815. doi: 10.1038/sj.onc.1210763. [DOI] [PubMed] [Google Scholar]
- 21.Gudbjartsson DF, et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat Genet. 2009;41:342–347. doi: 10.1038/ng.323. [DOI] [PubMed] [Google Scholar]
- 22.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Todd JA, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunt KA, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. 2008;40:395–402. doi: 10.1038/ng.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kudaravalli S, Veyrieras JB, Stranger BE, Dermitzakis ET, Pritchard JK. Gene expression levels are a target of recent natural selection in the human genome. Mol Biol Evol. 2009;26:649–658. doi: 10.1093/molbev/msn289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sabeti PC, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 29.Xue Y, et al. Adaptive evolution of UGT2B17 copy-number variation. Am J Hum Genet. 2008;83:337–346. doi: 10.1016/j.ajhg.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newton-Cheh C, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41:666–676. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takizawa H, et al. Growth and maturation of megakaryocytes is regulated by Lnk/Sh2b3 adaptor protein through crosstalk between cytokine- and integrin-mediated signals. Exp Hematol. 2008;36:897–906. doi: 10.1016/j.exphem.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 32.Velazquez L, et al. Cytokine signaling and hematopoietic homeostasis are disrupted in Lnk-deficient mice. J Exp Med. 2002;195:1599–1611. doi: 10.1084/jem.20011883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blomberg N, Baraldi E, Nilges M, Saraste M. The PH superfold: a structural scaffold for multiple functions. Trends Biochem Sci. 1999;24:441–445. doi: 10.1016/s0968-0004(99)01472-3. [DOI] [PubMed] [Google Scholar]
- 34.Tartaglia M, et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007;39:75–79. doi: 10.1038/ng1939. [DOI] [PubMed] [Google Scholar]
- 35.Tartaglia M, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29:465–468. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 36.Hugues L, et al. Mutations of PTPN11 are rare in adult myeloid malignancies. Haematologica. 2005;90:853–854. [PubMed] [Google Scholar]
- 37.Tartaglia M, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–150. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 38.Merched AJ, Chan L. Absence of p21Waf1/Cip1/Sdi1 modulates macrophage differentiation and inflammatory response and protects against atherosclerosis. Circulation. 2004;110:3830–3841. doi: 10.1161/01.CIR.0000148681.01282.89. [DOI] [PubMed] [Google Scholar]
- 39.Ozaki K, et al. SNPs in BRAP associated with risk of myocardial infarction in Asian populations. Nat Genet. 2009;41:329–333. doi: 10.1038/ng.326. [DOI] [PubMed] [Google Scholar]
- 40.Martin JF, Bath PM, Burr ML. Influence of platelet size on outcome after myocardial infarction. Lancet. 1991;338:1409–1411. doi: 10.1016/0140-6736(91)92719-i. [DOI] [PubMed] [Google Scholar]
- 41.Huczek Z, et al. Mean platelet volume on admission predicts impaired reperfusion and long-term mortality in acute myocardial infarction treated with primary percutaneous coronary intervention. J Am Coll Cardiol. 2005;46:284–290. doi: 10.1016/j.jacc.2005.03.065. [DOI] [PubMed] [Google Scholar]
- 42.Yang A, Pizzulli L, Luderitz B. Mean platelet volume as marker of restenosis after percutaneous transluminal coronary angioplasty in patients with stable and unstable angina pectoris. Thromb Res. 2006;117:371–77. doi: 10.1016/j.thromres.2005.04.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data