Formation of STIM and Orai complexes: puncta and distal caps (original) (raw)
. Author manuscript; available in PMC: 2011 Jun 8.
Summary
In the last few years, great progress has been made in understanding how stromal interacting molecule 1 (STIM1), a protein containing a calcium sensor that is located in the endoplasmic reticulum, and Orai1, a protein that forms a calcium channel in the plasma membrane, interact and give rise to store-operated calcium entry. Pharmacological depletion of calcium stores leads to the formation of clusters containing STIM and Orai that appear to be sites for calcium influx. Similar puncta are also produced in response to physiological stimuli in immune cells. In T cells engaged with antigen-presenting cells, clusters containing STIM and Orai accumulate at the immunological synapse. We recently discovered that in activated T cells, STIM1 and Orai1 also accumulate in cap-like structures opposite the immune synapse at the distal pole of the cell. Both caps and puncta are long-lived stable structures containing STIM1 and Orai1 in close proximity. The function of puncta as sites of calcium influx is clear. We speculate that the caps may provide a secondary site of calcium entry. Alternatively, they may serve as a source of preformed channel complexes that move to new immune synapses as T cells repeatedly engage antigen-presenting cells.
Keywords: STIM, Orai, T-cell activation, store-operated calcium entry, imaging
Introduction
In immune cells, increases in intracellular calcium control many aspects of activation (1–4). Cytosolic calcium first increases after antigen receptor engagement leads to the activation of phospholipase Cγ (PLCγ) by a well-defined signal transduction cascade. Active PLCγ cleaves phosphotidylinositol-3,4-bisphosphate to produce inositol-1,4,5-trisphosphate (IP3), which causes a spike of calcium release from the endoplasmic reticulum (ER) (5, 6). Short term responses such as decreased motility, spreading, and degranulation are induced by this transient increase in cytosolic calcium (2). Prolonged antigen receptor engagement leads to depletion of calcium from the ER lumen. Calcium depletion leads in turn to the opening of ion channels known as store-operated channels (SOC) in the plasma membrane (PM). The flow of extra-cellular calcium through these channels maintains elevated intracellular calcium for a long time, up to several hours in some cases. While SOC are found in most cell types and control a variety of important cellular responses, they play a critical role in the immune system (2, 7–9). Twenty years ago, the current produced by the primary SOC in lymphocytes and mast cells was described and named the calcium release-activated calcium (CRAC) current (10, 11). CRAC channels show low conductance, an inwardly rectifying current with a very positive reversal potential, and a high selectivity for calcium (7, 11, 12). These distinct biophysical properties allowed extensive study of the immune cell channels prior to the identification of their molecular components. Further studies established that CRAC channels are necessary for the sustained elevation of intracellular calcium that is required for proliferation, differentiation, and cytokine secretion (13). In T cells, the activation of many genes depends on this continued influx of calcium through CRAC channels. In particular, the activation of nuclear factor of activated T cells (NFAT), which controls the transcription of important genes such interleukin-2 (IL-2), requires sustained high intracellular calcium to maintain NFAT localization in the nucleus (4, 14). Jurkat T cells with defective CRAC channels show corresponding deficits in calcium-dependent gene transcription, while primary human T cells without functional CRAC channels are severely defective leading to severe combined immunodeficiency syndrome (SCID)(15–17). Pharmacological agents that deplete ER calcium stores activate CRAC channels and simulate T-cell activation, while CRAC channel blockers inhibit T-cell activation and proliferation (18).
Recently, two proteins required for CRAC channel activity were identified. RNA interference (RNAi) screens in both Drosophila and HeLa cells showed that suppression of stromal-interacting molecule 1 (STIM1) expression blocked CRAC channel activity and calcium influx (19, 20). STIM1 had been identified previously in a search for cell surface proteins on stromal cells and was reported to be involved in pre-B cell survival and tumor suppression. STIM is a 77 kDa single transmembrane protein with the N-terminus in the ER lumen and the C-terminus in the cytosol (21). Two isoforms of STIM (STIM1 and STIM2) exist in vertebrates, but decreasing STIM2 levels does not always reduce the CRAC current induced by store depletion (20, 22). Mammalian STIM1 is present in both the ER and PM, while mammalian STIM2 is found only in the ER (23). Several studies have indicated that STIM2 is involved in basal calcium influx, whereas STIM1 functions as the ER calcium sensor involved in CRAC channel activation (24–26). However, overexpression of STIM1 alone causes only a modest increase in CRAC current (19, 20).
A second protein required for CRAC channel activation was discovered using RNAi screening combined with linkage analysis in cell lines from patients with SCID lacking CRAC channel function. Originally named Orai in the first published report, the protein is sometimes referred to as CRAC-membrane (CRACM), based on the name given in a second study that also showed it was required for CRAC activity (27–29). Knockdown of Orai1 expression inhibited NFAT translocation to the nucleus, while expression of the wildtype protein in cells from patients with SCID syndrome restored CRAC channel activity. Orai1 is a 32.7 kDa, four-transmembrane spanning protein (27, 29). Mutations in Orai affect CRAC channel properties, demonstrating that it is the fundamental channel subunit (30, 31). Three isoforms of Orai (Orai1, Orai2, and Orai3) are found in mammalian cells, and they can form heteromeric complexes (27, 32, 33). Interestingly, overexpression of Orai1 can lead to decreased calcium influx (34).
Although overexpression of the single proteins, STIM1 or Orai1, does not increase calcium influx, combined overexpression of both proteins leads to a large current with electro-physiological properties that match those of native CRAC currents. This increased calcium entry depends on store depletion and is inhibited by CRAC channel blockers (29, 34–36). While most studies have focused on the role of Orai1, Orai2 can also produce substantial CRAC currents when coupled to STIM1 (35, 37). In contrast, overexpression of Orai3 and STIM1 generates only weak calcium influx (35, 37). However, in the absence of other Orai proteins, Orai3 is capable of serving as a channel (35). The exact function of the various isoforms is still being studied. Nonetheless, it is clear that Orai1 and STIM1 co-expression is sufficient to sense store depletion and allow calcium influx. This process depends on communication between two proteins that are found in two different subcellular compartments in unstimulated cells. Here, we review the movements and interactions of STIM1 and Orai1 leading to store-operated calcium entry (SOCE), highlighting the unexpected formation of distal cap structures in activated T cells.
STIM1 and Orai1 interactions induced by calcium depletion
Although deficiencies in CRAC channel activity predominantly affect the functioning of the immune system, a great deal has been learned about SOCE from experiments in non-immune cell lines. Also, as many studies showed that the CRAC currents induced by pharmacological depletion of ER calcium stores were the same as those induced by physiologically relevant stimuli, store depletion has been used extensively to examine the roles of STIM1 and Orai1 in calcium influx. In particular, thapsigargin, which inhibits the SERCA [sarcoendoplasmic reticulum calcium adenosine triphosphatase (ATPase)] pump responsible for calcium uptake into the ER, is often used to deplete ER calcium (38). Immediately after STIM1 was identified as an essential element in CRAC channel activation, it was shown that thapsigargin-induced store depletion causes dramatic redistribution of STIM1 in cells expressing STIM1 conjugated to fluorescent proteins. In unstimulated cells, STIM1 is distributed diffusely throughout the ER membrane. Following store depletion, it clusters at sites near the PM. This redistribution was observed in immune cells such as Jurkat T cells and rat basophilic leukemia (RBL) mast cells as well as other cell lines including HeLa and HEK293. No systematic disruption of ER structure occurs during puncta formation (19, 39, 40). Mutating either the calcium-binding glutamate or aspartate residues in the STIM1 EF hand domain results in the loss of calcium binding. Either mutation causes STIM to localize constitutively in puncta with concomitant activation of the CRAC channel independent of ER calcium levels (19, 34, 35, 39). Other experiments showed that the STIM1 puncta correspond generally to the sites of calcium influx (41). Following the identification of Orai1 as the CRAC channel, it was demonstrated that Orai1 colocalizes with STIM1 in puncta after store depletion when both proteins are overexpressed in either immune or non-immune cells. In all the cell lines examined, if both wildtype STIM1 and Orai1 were over-expressed, then all puncta contained both proteins (40–43). No studies have shown significant differences between immune and non-immune cells in the clustering of STIM1, association with Orai1, or activation of calcium entry induced by store depletion.
Although the initial studies clearly showed that STIM1 moves to sites near the PM, they did not determine whether STIM1 was actually incorporated into the PM when activating Orai1 channels. Even though STIM1 was first identified as a PM protein, only 2% of the total pool of STIM1 was found in the PM with no increase after store depletion (42). Moreover, it is indisputable that STIM1 in the ER can activate channels, as labeled versions of STIM1 that cannot translocate to the PM generated normal CRAC currents (44). Electron microscopy studies indicated that most STIM1 remained in the ER following store depletion, and STIM1-containing membranes were observed 10–25 nm away from the PM (40). Furthermore, it was shown that a minimum distance between the ER and PM membranes was required to accommodate STIM1 and Orai1-containing puncta. If the membranes were forced closer together than 11–14 nm, STIM1 and Orai1 clusters were excluded from the apposed membranes (43). Thus, the consensus has developed that STIM1 is not incorporated into the PM following store depletion.
The required separation between the membranes containing STIM1 and Orai1 suggested that other molecules might be located between STIM1 and Orai1 and that channel activation might not require direct contact between them. The calculated distance needed to accommodate the two cytoplasmic domains of STIM1 and Orai1 was only 6 nm, but the ER and PM membranes had to be at least 11 nm apart for the clusters to fit between them, strongly suggesting that other proteins are present in the gap. However, fluorescence resonance energy transfer (FRET) studies in HEK293 and RBL mast cells showed significant energy transfer between fluorescently tagged versions of STIM1 and Orai1, demonstrating that the distance between them is <10 nm. This close association is dependent on store depletion (45, 46). So, it appears that STIM1 interacts directly with Orai1 but that other proteins might be involved in a complex that forms at the ER–PM junctions.
Other studies investigated the temporal ordering of STIM1 clustering, movement to the ER sites close to the PM, Orai1 recruitment, and the initiation of SOCE. Experiments monitoring STIM–STIM interactions by FRET in HEK293 cells showed that STIM1 molecules moved close together, within the 10 nm required for energy transfer, before the formation of visible STIM clusters (47). Chemically induced oligomerization of modified versions of STIM induced puncta formation and activated CRAC channels (48). Orai1 clustering did not occur if STIM1 was not overexpressed, so apparently Orai1 recruitment follows the formation of STIM1 puncta (41, 45). Presumably, the colocalization of STIM1 and Orai1 corresponds to the initiation of the STIM–Orai interactions that lead to channel opening. Careful kinetic analysis showed that CRAC current initiation lags behind STIM1 clustering (45, 47, 49). Accordingly, the simplest model to explain how STIM and Orai generate CRAC currents begins with a homo-oligomerization of STIM, as indicated by the increase in STIM–STIM FRET. Then, oligomers of STIM1 move to ER–PM junction sites, forming large clusters. Orai1 associates with these clusters and interactions between STIM1 and Orai1 cause the channels to open. This model predicts that any perturbation causing STIM aggregation will lead to SOCE independent of ER store status. Furthermore, any fragment of STIM1 that interacts properly with Orai1 could lead to channel opening without STIM aggregation, puncta formation or store depletion.
There are some results, however, that do not fit into this simple model. In Jurkat T cells, the loss of WAVE2 [WASP (Wiskott–Aldrich syndrome protein) family verprolin homologous protein], which nucleates actin polymerization, also significantly decreased calcium influx in thapsigargin-treated cells (50). Moreover, in DT40 B lymphocytes, the simultaneous loss of Lyn and Syk or inhibition of tyrosine kinase activity with PP2 blocked CRAC channel opening following store depletion (51). Thus, there are additional steps that lie between store depletion and channel opening that are not yet explained in a simple model where interaction between STIM1 and Orai1 determines calcium flow.
Interaction domains of STIM and Orai
Recent studies focusing on different domains of STIM and Orai have increased our understanding of which are needed for the formation of STIM–STIM oligomers and which contribute to interactions between STIM and Orai; the critical steps in SOCE. STIM can be roughly divided into two portions, the N-terminal ER luminal side and a cytoplasmic C-terminal side. In the luminal portion, the calcium binding EF hand is followed by a sterile α motif (SAM) domain (19, 52). Analysis of the crystal structure of a STIM1 fragment consisting only of the N-terminal EF hand and SAM domain revealed several hydrophobic contacts between these domains. When calcium was bound, this fragment of STIM1 was well folded and self-contained. In the absence of calcium, several hydrophobic residues became exposed to solvent, leading to formation of oligomers. In fact, any mutation that destabilized the EF hand or SAM domain caused exposure of the same hydrophobic amino acids and resulted in STIM aggregation (53).
The cytoplasmic portion of STIM contains a coiled-coil domain overlapping a band 4.1/ezrin/radixin/moesin (FERM) domain as well as a serine/proline-rich domain and a polybasic, lysine-rich domain (19, 52, 54). Several reports have shown that a small portion of the coiled-coil domain within the FERM domain is responsible for Orai activation. When this piece was transfected into cells, it colocalized with and activated Orai1 regardless of the status of ER calcium stores (55–58). When it was deleted, there was no colocalization of the mutated STIM1 with Orai1 upon calcium depletion and thus no SOCE (57).
The serine/proline and polybasic domains at the C-terminus of STIM are required for localizing STIM to ER–PM junctions in the absence of Orai1 (47, 59). However, a STIM mutant lacking this domain was able to bind and activate Orai1 through interactions mediated by the coiled-coil domain (57, 60). There is some evidence that the polybasic region may also play an inhibitory role in channel activation through interactions with the N-terminal domain of Orai (56).
These studies indicate that oligomerization, puncta formation, and CRAC channel activation require different parts of the STIM molecule, but the functions of the various domains in the context of full-length STIM are more complicated. Although the EF hand and SAM domain are sufficient for oligomerization of the luminal domain, in full length STIM1, the coiled-coil domain also appears to be important for STIM1 homodimerization (45, 60). Moreover, deletion of the SAM domain did not lead to constitutive aggregation caused by exposure of the unprotected hydrophobic domains of the EF hand but instead prevented puncta formation and activation of Orai1 (61).
Both the N- and C-termini of Orai1 are located in the cytosol (29). The C-terminus contains a coiled-coil domain, while the Orai1 N-terminus contains an arginine-rich domain and two proline-rich domains (62, 63). If the Orai1 C-terminal coiled-coil is missing or mutated, no coupling to STIM1 or activation of Orai1 occurs, even with the C-terminal activating fragment of STIM1 (45). Negatively charged residues in this domain are important both for the interactions between STIM1 and Orai1 that lead to FRET and calcium influx (46).
Channel activity is not required for the association of STIM1 with Orai1. In humans, mutation of a single arginine near the first transmembrane helix of Orai1 eliminates calcium entry (27), but FRET measurements showed that this mutant still interacted with STIM1 in a store-dependent manner (45, 64). In addition, an Orai1 mutant with a deleted N-terminus still clustered with STIM but no longer fluxed calcium (45, 60).
Puncta formation in activated lymphocytes
In many cases, physiological stimulation and pharmacological store depletion leads to the same effects on STIM and Orai. However, a recent study showed some differences in the movement of STIM1 and Orai1 in response to these different stimuli. In RBL mast cells, STIM1 and Orai1 formed long-lived micrometer-sized puncta in response to thapsigargin treatment, but following antigen stimulation, the puncta were smaller and transient. Moreover, thapsigargin treatment resulted in longer interactions between STIM1 and Orai1, as monitored by FRET measurements. This disparity occurred despite a significant calcium flux after antigen stimulation (46). Inhibition of either store refilling or calcium entry in conjunction with antigen stimulation generated the same sustained FRET as that seen with thapsigargin treatment. The authors suggested that interactions between STIM1 and Orai1 require a sustained decrease in ER calcium, which is produced by thapsigargin treatment but not by antigen receptor signaling.
Signaling events in T cells leading to SOCE
In T cells, a series of molecular events have been identified that lead from stimulation of the T cell antigen receptor (TCR) to release of ER calcium (most of these events are bypassed in experiments using store depletion). Following receptor ligation, the CD3 and ζ subunits of the receptor are phosphorylated on tyrosine residues by the Src-family kinases, Lck and Fyn. These modifications allow the binding and activation of another tyrosine kinase, ζ-associated protein of 70 kDa (ZAP-70). Activated ZAP-70 phosphorylates important adapter molecules including linker for activation of T cells (LAT) and Src homology 2 (SH2) domain-containing leukocyte protein of 76 kDa (SLP-76). A complex of phosphorylated LAT and SLP-76 provides a platform for the recruitment of PLC-γ1, which then hydrolyzes PI-4,5-bisphosphate to IP3, allowing the initial release of calcium from the ER (Fig. 1). The remainder of this review focuses on the movements of STIM1 and Orai1 that are induced by TCR activation in T cells.
Fig. 1. Calcium signaling in T cells.

T-cell activation begins with binding of the TCR to a peptide conjugated with MHC on an antigen-presenting cell. Fyn and Lck kinases phosphorylate the CD3 and TCRζ subunits of the TCR. The kinase Zap70 (ζ-associated protein of 70 kDa) then binds to phosphorylated TCRζ and becomes active. Zap70 phosphorylates the adapter molecule LAT, producing docking sites for several downstream signaling molecules. Of particular importance in calcium signaling is the binding and activation of PLCγ1, which cleaves phosphatidylinositol biphosphate (PIP2) into IP3 and diacylglycerol (DAG). IP3 binds to the IP3 receptor on the ER, causing release of the ER calcium store. STIM1 senses the loss of calcium and induces Orai1 channel opening, thus producing the canonical ICRAC current. Sustained calcium flux activates calcineurin to dephosphorylate NFAT, allowing for its translocation to the nucleus where it activates transcription of critical effector molecules such as IL-2.
STIM–Orai interactions induced by receptor engagement in T cells
A recent study from our laboratory (65) showed that in Jurkat T cells activated by contact with a coverslip coated with anti-TCR antibodies, clusters containing STIM1-cyan fluorescence protein (CFP) and Orai1-yellow fluorescence protein (YFP) quickly formed at the stimulatory surface. As with puncta induced by store depletion, FRET analysis showed that STIM1 and Orai1 in stimulation-induced puncta were <10 nm apart and thus were close enough to directly interact. This interaction occurred quickly, as FRET could be observed in cells stimulated for only 2 min (65). Total internal reflection fluorescence (TIRF) images also showed many similarities between clusters observed near the coverslip, whether they were induced by receptor ligation or thapsigargin treatment. However, more mobile puncta were observed following TCR stimulation (Fig. 2A). High resolution confocal imaging showed that the clusters of STIM1-CFP and Orai1-YFP were distinct from microclusters containing TCR and other signaling molecules (65). STIM1 and Orai1 puncta link two separate membranes; thus, it is not surprising that these long-lived structures differ from the microclusters that rapidly translocate through the PM to the center of the forming immunological synapse (IS).
Fig. 2. Puncta and caps containing STIM1 in T cells.

(A) TIRF images of STIM-CFP puncta in Jurkat T cells. TIRF images show fluorescence from the bottom 200–300 nm of the cell. Top panels show two time points from a sequence of images showing puncta formed in thapsigargin-treated Jurkat T cells attached to a non-stimulatory coverslip coated with anti-CD45. Bottom panels show two time points from a sequence of images showing puncta formed in Jurkat T cells plated onto a stimulatory coverslip coated with anti-CD3 antibodies. Scale is 10 μm. The puncta are quite similar, but during the 5 min of imaging, the puncta induced by TCR engagement showed more movement than the puncta induced by store depletion. (B) Three dimensional projections of fields of cells showing immunofluorescent staining of endogenous STIM1 (top panels) or endogenous Orai1 (bottom panels) caps in cells plated onto stimulatory coverslips. Caps formed in Jurkat T cells (left panels), human peripheral blood lymphocytes (middle panels), and mouse T cells (right panels). Arrowheads point to examples of cells with caps.
STIM1 and Orai1 accumulated at the IS formed between T cells and either Raji B cells or dendritic cells pulsed with superantigen. Superantigen stimulates T cells by interacting directly with the Vβ region of the TCR. In conjugates with dendritic cells, both STIM1 and Orai1 accumulated during the first 10 min after cell–cell contact, which was coincident with the initiation of calcium influx. However, the clustering and recruitment of STIM1 and Orai1 to the contact surface did not depend on calcium influx and occurred normally in cells transfected with non-functional mutants of Orai1 (65, 66). Presumably, the structures containing STIM1 and Orai1 at the IS are similar to the puncta observed at the stimulatory coverslip, as both were seen at the sites of TCR activation. Overall, these results suggested that store depletion and TCR activation cause the formation of similar punctate structures in T cells.
Unexpected cap-like structure containing STIM1 and Orai1
The localization of STIM1 and Orai1 to puncta near the site of TCR engagement was not unexpected, given the surface localization of puncta induced by store depletion. However, unlike pharmacological store depletion, TCR ligation produced an additional, unanticipated accumulation of the two proteins in a cap-like structure on either the side of the cell or at the pole farthest from the contact surface. In Jurkat T cells, caps containing STIM1-CFP and Orai1-YFP formed 5–15 min after the initial contact with a stimulatory surface. The two proteins colocalized and were observed moving together, around the nucleus to the top or side of the cell (65).
Caps were observed in a wide variety of T-cell systems. In Jurkat T cells overexpressing STIM1-CFP and Orai1-YFP, caps could be produced by stimulation with either plate-bound anti-TCR antibodies or Raji B cells pulsed with superantigen. Also, caps composed of endogenous STIM1 and Orai1 were observed in immunostained Jurkat T cells, human peripheral blood lymphocytes (PBLs), and in mouse T lymphocytes, all activated by contact with stimulatory coverslips (Fig. 2B). In Jurkat T cells, four phenotypes were observed following stimulation: about half of the cells showed no obvious rearrangement of endogenous STIM1, about one quarter of the cells contained both caps and puncta, and the remaining cells were evenly split between those with only caps and those with only puncta near the stimulatory surface. Finally, caps formed in TCR-restricted mouse lymphocytes stimulated with B cells pulsed with the appropriate peptide. Caps are not always solid structures but often have fenestrations or clear areas between the areas of densely packed STIM1 and Orai1. In immuno-stained samples, caps are generally much brighter than the puncta found at the site of TCR engagement, indicating that more STIM1 protein accumulates in the caps. Thus, the major reorganization of STIM1 following TCR stimulation is the unexpected formation of a distal cap, far away from the site of receptor activation and the presumed site of calcium entry in T cells.
Just as STIM1 remains in ER membranes when forming puncta in response to store depletion, STIM1 in a cap does not appear to leave the ER. Cap formation was not accompanied by a wholescale rearrangement of the ER. Although two ER markers, calnexin and M1-YFP, showed a slight accumulation in the caps, the bulk of these markers remained in ER membranes that appeared normal. In contrast, almost all of the STIM1 redistributed into the cap with very little remaining in the bulk ER (65). Overall, STIM1 caps are similar to puncta, in that they appear to result from rearrangement of STIM1 in ER membranes without any major change in the structure of the ER itself.
STIM1 also interacts with the cytoskeleton
It has been shown in HeLa cells that STIM1 plays a role in maintaining ER structure through an association with the ends of microtubules via STIM1 binding to the plus-end tracking proteins EB1 and EB3 (67). This association is responsible for the extension of ER tubules along microtubules and is likely to account for the colocalization of STIM1 with microtubules that has been observed in several cells lines (61, 68). The mobile tubular structures containing green fluorescence protein (GFP)-STIM1 observed in unstimulated DT40 B lymphocytes probably result from STIM1 interactions with the cytoskeleton (61). Following store depletion, the binding of STIM1 to EB1 is decreased. Therefore, it is thought that STIM1 cannot interact simultaneously with both EB1 and Orai1. During cap formation in T cells, EB1 and EB3 remained dispersed throughout the ER, and there was no indication that they were incorporated into caps or played a role in cap formation (V. A. Barr, unpublished data).
In HEK 293 cells, microtubule depolymerization decreased the CRAC current induced by store depletion but had little effect on STIM1 clustering (68). In contrast, in DT40 B cells, nocodazole treatment blocked formation of STIM1 puncta with little effect on calcium influx (61). In Jurkat T cells, depolymerization of microtubules with colchicine affected the shape of the caps, but STIM1-CFP and Orai1-YFP still clustered at the distal side of the cell. Without microtubules, caps were found in blebs that protruded away from the cell, as if the cell morphology had been affected while the overall association of cap proteins remained. Intriguingly, disruption of the actin cytoskeleton had a much larger effect on cap formation and structure. In the presence of low doses of latruculin, signaling microclusters still formed, indicating that signal transduction still occurred, but caps did not develop. Instead, small puncta containing both STIM1-CFP and Orai1-YFP appeared on the cell surface (65). Disruption of actin filaments after cap formation caused the cap to dissipate into these smaller puncta (V. A. Barr, unpublished data). Thus, an intact cytoskeleton was required for normal cap morphology, but the interactions between STIM1 and Orai1 were not compromised by disruption of the cytoskeleton.
Cap formation depends on TCR signaling
TCR signaling is required for cap formation, while store depletion in the absence of TCR stimulation was not sufficient for rearrangement of STIM1 into caps. Neither calcium influx nor channel activity was needed for cap formation, as cap formation was normal in the absence of extracellular calcium or the presence of CRAC channel inhibitors (65). Blocking signal transduction either by inhibiting tyrosine phosphorylation or by removing essential proteins from the signaling cascade inhibited cap formation. Proteins in the signaling pathway upstream of calcium release from the ER, including Lck, SLP-76, and PLCγ, were required for cap formation, and re-expressing each individual protein in mutant cells lacking them restored caps. However, removing Vav1, which binds to SLP-76 but is part of a different downstream signaling pathway (69), did not affect cap formation (Fig. 3). Wherever it has been studied, the movement of STIM1 and clustering with Orai1 is independent of channel activity or calcium influx during the formation of structures containing both proteins, including thapsigargin-induced puncta as well as the accumulation of the two proteins at the IS and cap formation. However, only cap formation was completely dependent on signaling through the receptor and appeared to be completely independent of store depletion.
Fig. 3. Cap formation requires signaling through the TCR.
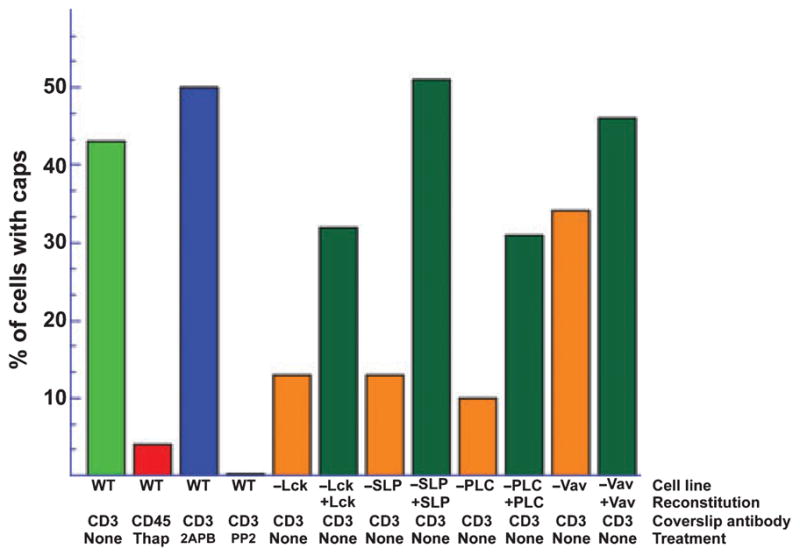
Graph showing the percentage of cells with caps following each treatment. Only cells plated on stimulatory coverslips (coverslip antibody anti-CD3) formed caps. Inhibiting channel activity with 2APB had no effect on cap formation, while inhibiting tyrosine phosphorylation with PP2 blocked cap formation. Mutant Jurkat T cells lacking Lck, SLP-76, or PLCγ made fewer caps than normal cells. Re-expressing the missing protein restored cap formation. Mutant cells lacking Vav1 showed relatively normal cap formation.
Protein–protein interactions in caps
Direct interaction is responsible, presumably, for the STIM1-induced opening of Orai1 channels, and FRET measurements have confirmed that they are in close proximity in puncta. FRET analysis in cells expressing STIM1-CFP and Orai1-YFP showed that the two proteins remained close enough for energy transfer during cap formation (65). Thus, it seems likely a functional coupling of STIM1 and Orai1 still exists in the caps. However, in cells with both puncta and caps, the FRET efficiency was higher in the caps (10.5% versus 5.9%) indicating that the nature of the interaction between STIM1 and Orai1 may not be identical in the two structures (65).
Although the properties of the clusters induced by store depletion have been studied in some detail, little is known about the structure of the caps. Following store depletion, puncta appear to be long lived and relatively stable. Puncta re-form at the same sites after several rounds of store refilling and emptying, indicating that the sites where STIM1 clusters at ER–PM junctions are also stable (70). These puncta span two opposing membranes, and proteins within them might be expected to be immobile and unable to diffuse within their respective membranes. In keeping with this view, studies in non-immune cells showed that YFP-STIM1 was not very mobile in ER membranes in unstimulated cells and that its mobility decreased further after store depletion, although it was never completely immobile (47). Fluorescence recovery after photobleaching (FRAP) analysis of STIM1-CFP and Orai1-YFP showed that cap formation also led to decreases in the mobilities of the proteins. The mobile fraction of STIM1-CFP in caps was half that seen in ER membranes of unstimulated cells. Orai1-YFP showed a similar but somewhat smaller loss of mobility in caps compared with Orai1-YFP in the PM of unstimulated cells (65). Still, the proteins in caps showed some movement within their respective membranes. Thus, these FRAP experiments allowed the possibility that STIM1-CFP molecules in the caps might be in equilibrium with molecules in either ER membranes or puncta. Fluorescence loss induced by photobleaching (FLIP) indicated that there is probably only one pool of STIM1 in activated T cells. Jurkat T cells conjugated to Raji B cells are often oriented such that it is possible to bleach either the cap or synapse area. In a cell with an orientation conducive to photobleaching, a portion of the cap or of the contact area between the two cells was repetitively bleached for the amount of time needed to bleach a substantial amount of the pool of ER STIM-CFP in unstimulated cells (about 10 min). After repeated bleaching within the cap, STIM-CFP fluorescence was decreased throughout the whole cell, including puncta present at the synapse. Similarly, repeated bleaching of the synapse induced bleaching of both the cap and whole cell (V. A. Barr, unpublished data). These results indicate that there is only a single pool of STIM1 molecules in the cell and that while STIM1 mobility is low, there is some exchange of molecules between puncta and caps. Of course, the areas being bleached undoubtedly included some ER, and it may be that STIM1 molecules must first leave the cap or puncta and return to a diffuse distribution in bulk ER membranes before being incorporated into another structure. Overall, the physical attributes of puncta and caps are similar. Both are long-lived structures containing STIM1 and Orai1 in close proximity. The mobility of STIM1 is decreased by store depletion or when STIM1 is incorporated into a cap, yet there is enough mobility for a single STIM1 molecule to move between caps and puncta.
STIM1 caps are similar to yet distinct from the distal pole complex
The location and general morphology of caps is reminiscent of another important structure induced by T-cell activation, the distal pole complex (DPC). This complex is defined by the localization of ERM domain proteins and their associated cargo proteins to a region across the cell from the IS, often forming a tight cap-like structure. The DPC contains many proteins associated with cell polarity, and the complex itself may be important in establishing T-cell polarity. Active ERM proteins bind most cargo molecules via a conserved N-terminal FERM domain (71, 72). STIM proteins also contain a FERM domain, so it seemed plausible that STIM1 and Orai1 could be components of the DPC. However, although STIM1 caps and distal poles certainly exist in the same part of the cell, the two sets of proteins do not mingle. In human PBLs, ezrin and STIM1 formed distinct patches, with ezrin appearing in the fenestrations of the cap. A similar segregation was seen between STIM1 and several polarity markers found in the DPC such as scribble, numb, and PKCζ (65).
Studies using dominant negative FERM domains have shown that the activity of ERM domain proteins is needed for DPC formation. Cap formation might also depend on this ERM protein activity. Indeed, a recent study showed that ezrin−/− T cells were defective in SOCE. When moesin expression was reduced with short interfering RNA (siRNA) in ezrin−/− cells, the resulting T cells showed very low levels of total ERM protein, and calcium influx was severely inhibited (73). However, when STIM1 cap formation was assessed in these ERM protein-deficient cells, only a slight defect was observed (Fig. 4A). In ezrin+/+ T-cell blasts transfected with control siRNA, which had no effect on moesin immunostaining, about 60% of the cells produced STIM1 caps when the cells were activated on stimulatory coverslips at either 48 h or 72 h after transfection. In ezrin−/− T-cell blasts transfected with moesin siRNA, the amount of moesin immunostaining decreased significantly at both timepoints. Nonetheless, about 50% of the cells produced STIM1 caps. Moreover, there were no differences in the area of the caps or in the amount of STIM1 recruited to the caps as monitored by the overall brightness of the caps (Fig. 4B). Thus, it appears that ERM proteins are not needed for cap formation. One caveat to this interpretation comes from a recently published study showing that T-cell blasts without ezrin and with severely reduced moesin could still form DPCs as monitored by the segregation of CD43. Furthermore, the amount of phosphorylated ERM proteins remained at 50% of control levels, indicating that substantial ERM protein activity remained despite low levels of the proteins themselves. However, several lines of evidence suggest that cap formation is not the same as DPC formation. There was clear cap formation in cells with no detectable moesin, while some cells with residual moesin that had DPCs failed to produce caps (Fig. 4C). It had been noted in earlier studies that Jurkat T cells do not form well-defined DPCs; nonetheless, caps containing STIM1 and Orai1 form in these cells (71, 74). We conclude that cap formation does not require the same ERM protein activity as DPC formation, despite the similarities between caps and DPCs in morphology and cellular localization.
Fig. 4. Cap formation occurs in ERM protein-deficient T-cell blasts.

(A) Three dimensional projections of fields of cells showing immunofluorescent staining of endogenous STIM1 in wildtype (WT) mouse cells (left panels) transfected with control siRNA for 48 h (top panels) or 72 h (bottom panels). There are fewer cells at the 72 h timepoint but almost the same percentage of cells produce caps. The right panels show similar views of endogenous STIM1 in ezrin−/− mouse cells transfected with moesin siRNA for 48 h (top panels) or 72 h (bottom panels). The percentage of cells forming caps is only slightly reduced. (B) Graphs showing efficient suppression of moesin expression by siRNA treatment, relatively normal cap formation in all treatment groups, no differences in cap area between treatment groups and no differences in STIM1 accumulation as monitored by cap brightness. Green bars: WT mouse cells transfected with control siRNA; yellow bars: ezrin−/− mouse cells transfected with moesin siRNA. (C) Three dimensional projection showing four ezrin−/− cells treated with moesin siRNA for 72 h. Three of the cells have clear STIM1 caps; the yellow arrow marks a cell with no moesin staining. The cell marked with a red arrow shows a cell with an accumulation of moesin but no STIM1 cap.
Caps are dynamic structures
Another unexpected feature of caps was their dynamic behavior in T cells stimulated by contact with B cells pulsed with superantigen. Puncta induced by either store depletion or by contact with stimulatory coverslips are relatively stable long-lived structures (Fig. 2A). While an individual cap can persist for many minutes in a T cell interacting with a B cell, often the whole cap moved rapidly to a new location. STIM1 and Orai1 in a cap sometimes moved en masse to the contact surface, either back to the original IS or to a newly forming IS after contacting another B cell. When a single T cell interacted with more than one B cell, STIM1-CFP and Orai1-YFP sometimes aggregated and then moved together from side to side before forming a cap in a stable location. During these dynamic changes, the cell seems to treat the cap as a single entity, either moving it or releasing it to the IS (65). One explanation for this behavior is that the cap may sequester channels already docked to STIM1 to generate rapid T-cell responses when encountering additional stimulation.
We presume that during formation of both caps and puncta, the first step is homo-oligomerization of STIM1. However, when puncta form, it appears that STIM1 moves first to the ER–PM junctions followed by the association of Orai1 with the clustered STIM1. In contrast, when caps form, STIM1 and Orai1 first interact and then this complex moves to the back of the cell. Nothing is currently known about the domain requirements for cap formation; however, regions that were identified as important for interactions between STIM1 and Orai1 during puncta formation are probably also involved in bringing both proteins to the caps.
Conclusion
Considerable progress has been made in understanding how STIM1 and Orai1 generate SOCE, a critical element in immune cell activation. The aggregation of STIM1 initiated by calcium loss from the ER leads to interactions between the coiled-coil domain of STIM1 and the C-terminus of Orai1, resulting in channel activation and calcium influx. In both non-immune cells and immune cells, STIM1 and Orai1 cluster in puncta at ER–PM junctions in response to pharmacological calcium store depletion. Similar puncta form in response to physiological stimulation of immune cells, although in Jurkat T cells the puncta formed after TCR ligation are more mobile than those induced by store depletion alone. In T cells engaged by antigen-presenting cells, puncta containing STIM1 and Orai1 accumulate at the IS. It is believed that these puncta containing interacting molecules of STIM1 and Orai1 are sites of calcium entry.
Unexpectedly, TCR activation in T cells also causes the accumulation of STIM1 and Orai1 in a dense cap-like structure on the distal side of the cell. Like puncta, caps are long-lived structures composed of STIM1 and Orai1 in close proximity. In both structures, Orai1 channels remain in the PM while STIM1 remains part of the ER. There is some equilibrium between the two structures, as individual STIM1 molecules apparently move between caps and puncta and the entire cap can be recruited to an IS. While the role of the puncta in calcium influx seems clear, much remains to be discovered about the behavior and function of the prominent caps. As STIM1 and Orai1 within the caps are close enough to interact, the channels could be active, making the caps another site of calcium entry. This putative influx would be close to the nucleus and might be designed to modulate nuclear calcium levels. Alternatively, caps might sequester preassembled complexes of STIM1 and Orai1 to allow rapid activation of T cells when encountering multiple antigen-presenting cells. Given their dependence on TCR-mediated signaling rather than calcium flux, it is reasonable to presume that caps reflect T-cell-specific roles for STIM1 and Orai1.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute, Center for Cancer Research and by NIH grant P01 CA093615 to J. K. B.
References
- 1.Vig M, Kinet JP. Calcium signaling in immune cells. Nat Immunol. 2009;10:21–27. doi: 10.1038/ni.f.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oh-hora M, Rao A. Calcium signaling in lymphocytes. Curr Opin Immunol. 2008;20:250–258. doi: 10.1016/j.coi.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewis RS. The molecular choreography of a store-operated calcium channel. Nature. 2007;446:284–287. doi: 10.1038/nature05637. [DOI] [PubMed] [Google Scholar]
- 4.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 5.Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7:690–702. doi: 10.1038/nri2152. [DOI] [PubMed] [Google Scholar]
- 6.Samelson LE. Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu Rev Immunol. 2002;20:371–394. doi: 10.1146/annurev.immunol.20.092601.111357. [DOI] [PubMed] [Google Scholar]
- 7.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 8.Lewis RS. Calcium signaling mechanisms in T lymphocytes. Annu Rev Immunol. 2001;19:497–521. doi: 10.1146/annurev.immunol.19.1.497. [DOI] [PubMed] [Google Scholar]
- 9.Iezzi G, Karjalainen K, Lanzavecchia A. The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity. 1998;8:89–95. doi: 10.1016/s1074-7613(00)80461-6. [DOI] [PubMed] [Google Scholar]
- 10.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 11.Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci USA. 1993;90:6295–6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prakriya M, Lewis RS. CRAC channels: activation, permeation, and the search for a molecular identity. Cell Calcium. 2003;33:311–321. doi: 10.1016/s0143-4160(03)00045-9. [DOI] [PubMed] [Google Scholar]
- 13.Hogan PG, Rao A. Dissecting ICRAC, a store-operated calcium current. Trends Biochem Sci. 2007;32:235–245. doi: 10.1016/j.tibs.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 14.Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–484. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- 15.Feske S, Prakriya M, Rao A, Lewis RS. A severe defect in CRAC Ca2+ channel activation and altered K+ channel gating in T cells from immunodeficient patients. J Exp Med. 2005;202:651–662. doi: 10.1084/jem.20050687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fanger CM, Hoth M, Crabtree GR, Lewis RS. Characterization of T cell mutants with defects in capacitative calcium entry: genetic evidence for the physiological roles of CRAC channels. J Cell Biol. 1995;131:655–667. doi: 10.1083/jcb.131.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Partiseti M, Le Deist F, Hivroz C, Fischer A, Korn H, Choquet D. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J Biol Chem. 1994;269:32327–32335. [PubMed] [Google Scholar]
- 18.Quintana A, Griesemer D, Schwarz EC, Hoth M. Calcium-dependent activation of T-lymphocytes. Pflugers Arch. 2005;450:1–12. doi: 10.1007/s00424-004-1364-4. [DOI] [PubMed] [Google Scholar]
- 19.Liou J, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roos J, et al. STIM1, anessentialandconserved componentofstore-operatedCa2+channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manji SS, et al. STIM1: a novel phosphoprotein located at the cell surface. Biochim Biophys Acta. 2000;1481:147–155. doi: 10.1016/s0167-4838(00)00105-9. [DOI] [PubMed] [Google Scholar]
- 22.Wedel B, Boyles RR, Putney JW, Jr, Bird GS. Role of the store-operated calcium entry proteins Stim1 and Orai1 in muscarinic cholin-ergic receptor-stimulated calcium oscillations in human embryonic kidney cells. J Physiol. 2007;579:679–689. doi: 10.1113/jphysiol.2006.125641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soboloff J, et al. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ Entry. Curr Biol. 2006;16:1465–1470. doi: 10.1016/j.cub.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 24.Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131:1327–1339. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parvez S, et al. STIM2 protein mediates distinct store-dependent and store-independent modes of CRAC channel activation. FASEB J. 2008;22:752–761. doi: 10.1096/fj.07-9449com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stathopulos PB, Zheng L, Ikura M. Stromal interaction molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J Biol Chem. 2009;284:728–732. doi: 10.1074/jbc.C800178200. [DOI] [PubMed] [Google Scholar]
- 27.Feske S, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 28.Vig M, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang SL, et al. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci USA. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vig M, et al. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol. 2006;16:2073–2079. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443:226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gwack Y, et al. Biochemical and functional characterization of Orai proteins. J Biol Chem. 2007;282:16232–16243. doi: 10.1074/jbc.M609630200. [DOI] [PubMed] [Google Scholar]
- 33.Lis A, et al. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr Biol. 2007;17:794–800. doi: 10.1016/j.cub.2007.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soboloff J, Spassova MA, Tang XD, Hewavi-tharana T, Xu W, Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281:20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 35.Mercer JC, et al. Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem. 2006;281:24979–24990. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peinelt C, et al. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat Cell Biol. 2006;8:771–773. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeHaven WI, Smyth JT, Boyles RR, Putney JW., Jr Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release-activated calcium channels. J Biol Chem. 2007;282:17548–17556. doi: 10.1074/jbc.M611374200. [DOI] [PubMed] [Google Scholar]
- 38.Lytton J, Westlin M, Hanley MR. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem. 1991;266:17067–17071. [PubMed] [Google Scholar]
- 39.Zhang SL, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol. 2006;174:803–813. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol. 2006;174:815–825. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu P, Lu J, Li Z, Yu X, Chen L, Xu T. Aggregation of STIM1 underneath the plasma membrane induces clustering of Orai1. Biochem Biophys Res Commun. 2006;350:969–976. doi: 10.1016/j.bbrc.2006.09.134. [DOI] [PubMed] [Google Scholar]
- 43.Varnai P, Toth B, Toth DJ, Hunyady L, Balla T. Visualization and manipulation of plasma membrane-endoplasmic reticulum contact sites indicates the presence of additional molecular components within the Stim1–Orai1 complex. J Biol Chem. 2007;282:29678–29690. doi: 10.1074/jbc.M704339200. [DOI] [PubMed] [Google Scholar]
- 44.Hauser CT, Tsien RY. A hexahistidine-Zn2+-dye label reveals STIM1 surface exposure. Proc Natl Acad Sci USA. 2007;104:3693–3697. doi: 10.1073/pnas.0611713104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muik M, et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem. 2008;283:8014–8022. doi: 10.1074/jbc.M708898200. [DOI] [PubMed] [Google Scholar]
- 46.Calloway N, Vig M, Kinet JP, Holowka D, Baird B. Molecular clustering of STIM1 with Orai1/CRACM1 at the plasma membrane depends dynamically on depletion of Ca2+ stores and on electrostatic interactions. Mol Biol Cell. 2009;20:389–399. doi: 10.1091/mbc.E07-11-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA. 2007;104:9301–9306. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454:538–542. doi: 10.1038/nature07065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stathopulos PB, Li GY, Plevin MJ, Ames JB, Ikura M. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: an initiation mechanism for capacitive Ca2+ entry. J Biol Chem. 2006;281:35855–35862. doi: 10.1074/jbc.M608247200. [DOI] [PubMed] [Google Scholar]
- 50.Nolz JC, et al. The WAVE2 complex regulates actin cytoskeletal reorganization and CRAC-mediated calcium entry during T cell activation. Curr Biol. 2006;16:24–34. doi: 10.1016/j.cub.2005.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chung SC, Limnander A, Kurosaki T, Weiss A, Korenbrot JI. Coupling Ca2+ store release to Icrac channel activation in B lymphocytes requires the activity of Lyn and Syk kinases. J Cell Biol. 2007;177:317–328. doi: 10.1083/jcb.200702050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Williams RT, et al. Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem J. 2001;357:673–685. doi: 10.1042/0264-6021:3570673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stathopulos PB, Zheng L, Li GY, Plevin MJ, Ikura M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell. 2008;135:110–122. doi: 10.1016/j.cell.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 54.Williams RT, Senior PV, Van Stekelenburg L, Layton JE, Smith PJ, Dziadek MA. Stromal interaction molecule 1 (STIM1), a transmembrane protein with growth suppressor activity, contains an extracellular SAM domain modified by N-linked glycosylation. Biochim Biophys Acta. 2002;1596:131–137. doi: 10.1016/s0167-4838(02)00211-x. [DOI] [PubMed] [Google Scholar]
- 55.Zhang SL, et al. Store-dependent and -independent modes regulating Ca2+ release-activated Ca2+ channel activity of human Orai1 and Orai3. J Biol Chem. 2008;283:17662–17671. doi: 10.1074/jbc.M801536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 2009;11:337–343. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park CY, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muik M, et al. A cytosolic homomerization and a modulatory domain within STIM1 C terminus determine coupling to ORAI1 channels. J Biol Chem. 2009;284:8421–8426. doi: 10.1074/jbc.C800229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang GN, et al. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat Cell Biol. 2006;8:1003–1010. doi: 10.1038/ncb1454. [DOI] [PubMed] [Google Scholar]
- 60.Li Z, Lu J, Xu P, Xie X, Chen L, Xu T. Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activation. J Biol Chem. 2007;282:29448–29456. doi: 10.1074/jbc.M703573200. [DOI] [PubMed] [Google Scholar]
- 61.Baba Y, et al. Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc Natl Acad Sci USA. 2006;103:16704–16709. doi: 10.1073/pnas.0608358103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cahalan MD, Zhang SL, Yeromin AV, Ohlsen K, Roos J, Stauderman KA. Molecular basis of the CRAC channel. Cell Calcium. 2007;42:133–144. doi: 10.1016/j.ceca.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takahashi Y, et al. Essential role of the N-terminus of murine Orai1 in store-operated Ca2+ entry. Biochem Biophys Res Commun. 2007;356:45–52. doi: 10.1016/j.bbrc.2007.02.107. [DOI] [PubMed] [Google Scholar]
- 64.Navarro-Borelly L, Somasundaram A, Yamashita M, Ren D, Miller RJ, Prakriya M. STIM1-Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J Physiol. 2008;586:5383–5401. doi: 10.1113/jphysiol.2008.162503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barr VA, et al. Dynamic movement of the calcium sensor STIM1 and the calcium channel Orai1 in activated T-cells: puncta and distal caps. Mol Biol Cell. 2008;19:2802–2817. doi: 10.1091/mbc.E08-02-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lioudyno MI, et al. Orai1 and STIM1 move to the immunological synapse and are up-regulated during T cell activation. Proc Natl Acad Sci USA. 2008;105:2011–2016. doi: 10.1073/pnas.0706122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grigoriev I, et al. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr Biol. 2008;18:177–182. doi: 10.1016/j.cub.2007.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smyth JT, DeHaven WI, Bird GS, Putney JW., Jr Role of the microtubule cytoskeleton in the function of the store-operated Ca2+ channel activator STIM1. J Cell Sci. 2007;120:3762–3771. doi: 10.1242/jcs.015735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu J, Motto DG, Koretzky GA, Weiss A. Vav and SLP-76 interact and functionally cooperate in IL-2 gene activation. Immunity. 1996;4:593–602. doi: 10.1016/s1074-7613(00)80485-9. [DOI] [PubMed] [Google Scholar]
- 70.Smyth JT, Dehaven WI, Bird GS, Putney JW., Jr Ca2+-store-dependent and -independent reversal of Stim1 localization and function. J Cell Sci. 2008;121:762–772. doi: 10.1242/jcs.023903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cullinan P, Sperling AI, Burkhardt JK. The distal pole complex: a novel membrane domain distal to the immunological synapse. Immunol Rev. 2002;189:111–122. doi: 10.1034/j.1600-065x.2002.18910.x. [DOI] [PubMed] [Google Scholar]
- 72.Burkhardt JK, Carrizosa E, Shaffer MH. The actin cytoskeleton in T cell activation. Annu Rev Immunol. 2008;26:233–259. doi: 10.1146/annurev.immunol.26.021607.090347. [DOI] [PubMed] [Google Scholar]
- 73.Shaffer MH, et al. Ezrin and moesin function together to promote T cell activation. J Immunol. 2009;182:1021–1032. doi: 10.4049/jimmunol.182.2.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roumier A, et al. The membrane-microfilament linker ezrin is involved in the formation of the immunological synapse and in T cell activation. Immunity. 2001;15:715–728. doi: 10.1016/s1074-7613(01)00225-4. [DOI] [PubMed] [Google Scholar]