The F-box protein Skp2 is a ubiquitylation target of a Cul1-based core ubiquitin ligase complex: evidence for a role of Cul1 in the suppression of Skp2 expression in quiescent fibroblasts (original) (raw)
Abstract
The ubiquitin protein ligase SCFSkp2 is composed of Skp1, Cul1, Roc1/Rbx1 and the F-box protein Skp2, the substrate-recognition subunit. Levels of Skp2 decrease as cells exit the cell cycle and increase as cells re-enter the cycle. Ectopic expression of Skp2 in quiescent fibroblasts causes mitogen-independent S-phase entry. Hence, mechanisms must exist for limiting Skp2 protein expression during the G0/G1 phases. Here we show that Skp2 is degraded by the proteasome in G0/G1 and is stabilized when cells re-enter the cell cycle. Rapid degradation of Skp2 in quiescent cells depends on Skp2 sequences that contribute to Cul1 binding and interference with endogenous Cul1 function in serum-deprived cells induces Skp2 expression. Furthermore, recombinant Cul1–Roc1/Rbx1–Skp1 complexes can catalyse Skp2 ubiquitylation in vitro. These results suggest that degradation of Skp2 in G0/G1 is mediated, at least in part, by an autocatalytic mechanism involving a Skp2-bound Cul1-based core ubiquitin ligase and imply a role for this mechanism in the suppression of SCFSkp2 ubiquitin protein ligase function during the G0/G1 phases of the cell cycle.
Keywords: cell cycle/cullin/proteasome/Skp2/ubiquitin
Introduction
Members of the SCF family of multi-component E3 ubiquitin protein ligase complexes play important roles in the regulation of diverse cellular processes including cell cycle progression (Krek, 1998; Patton et al., 1998a; Deshaies, 1999; Koepp et al., 1999). They function as adapters that interact simultaneously with both the substrate and the E2 ubiquitin conjugating enzyme, thereby allowing selectivity in ubiquitylation reactions that target selected substrates for multi-ubiquitylation and degradation by the 26S proteasome. Although SCF ubiquitin protein ligases regulate diverse physiological pathways little is known about their structure, mechanism of action and regulation.
Existing evidence suggests that SCF complexes contain four essential subunits: Skp1 (Bai et al., 1996), Cul1/Cdc53 (Kipreos et al., 1996; Mathias et al., 1996; Willems et al., 1996), the RING-H2 protein Roc1/Rbx1/Hrt1 (we refer to the mammalian versions of this protein Roc1/Rbx1 and to the yeast homologue Hrt1) (Kamura et al., 1999; Ohta et al., 1999a; Seol et al., 1999; Tan et al., 1999) and a member of the diverse F-box family of proteins (Bai et al., 1996). The latter function as substrate-specific receptors that bind through the F-box motif to Skp1 (Bai et al., 1996; Feldman et al., 1997; Skowyra et al., 1997), which in turn binds to the Cul1/Cdc53 protein (Michel and Xiong, 1998; Patton et al., 1998b). Cul1/Cdc53 in complex with Roc1/Rbx1/Hrt1 is thought to comprise a minimal core ubiquitin protein ligase subcomplex (Ohta et al., 1999a; Seol et al., 1999; Skowyra et al., 1999; Tan et al., 1999). In keeping with this view, human Cul1–Roc1/Rbx1 subcomplexes can catalyse polyubiquitin chain formation in the absence of substrate and it has been proposed that these ubiquitin polymers represent free, unattached ubiquitin chains (Ohta et al., 1999a; Tan et al., 1999). In addition, recombinant yeast Cdc53–Hrt1 subcomplexes are sufficient to stimulate the auto-ubiquitylation of the E2 enzyme Cdc34 in the absence of added substrate in vitro (Seol et al., 1999; Skowyra et al., 1999).
A key property of the SCF ubiquitin ligase system is that the specificity of SCF complexes can be programmed via an array of distinct substrate-specific receptors, the F-box proteins (Bai et al., 1996). Over the past few years, several members of the F-box protein family have been identified. Among these, the F-box protein Skp2 appears to play a particularly important role in the regulation of mammalian cell cycle progression. It was originally identified as a protein associated with the S-phase promoting kinase cyclin A–cyclin-dependent kinase 2 (Cdk2) (Zhang et al., 1995). Subsequently it has been shown that Skp2 assembles together with Skp1, Cul1 and Roc1/Rbx1 to form SCFSkp2 ubiquitin protein ligase complexes (Lisztwan et al., 1998; Lyapina et al., 1998; Michel and Xiong, 1998; Marti et al., 1999; Tan et al., 1999). Interestingly, levels of the SCFSkp2 components Skp1, Roc1/Rbx1 and Cul1 do not change significantly during the cell cycle (Lisztwan et al., 1998; Michel and Xiong, 1998; Ohta et al., 1999b), whereas the expression of Skp2 is cell cycle regulated (Lisztwan et al., 1998; Michel and Xiong, 1998). It can first be detected at the transition between the G1 and S phases, amounts of Skp2 then accumulate during S/G2 and drop as cells proceed through M phase. At present, little is known about the mechanisms underlying Skp2 periodicity.
There is evidence to suggest that interference with Skp2 function in cultured cells by microinjection of antibodies against Skp2 inhibits S-phase entry (Zhang et al., 1995). A requirement of Skp2 function for the initiation of DNA replication may be explained, at least in part, by the observation that SCFSkp2 has been implicated in ubiquitylation and degradation of the CDK inhibitor p27, a negative regulator of the G1–S transition in mammalian cells (Carrano et al., 1999; Sutterlüty et al., 1999; Tsvetkov et al., 1999). Consistent with this observation, adenovirus-mediated overexpression of Skp2 in serum-deprived rodent fibroblasts induces p27 degradation, cyclin A expression, cyclin–Cdk2 activation and S-phase entry. A mutant of Skp2 that fails to bind Cul1 is defective in these activities, suggesting that these activities of Skp2 represent SCF functions. In addition, coexpression of a degradation-resistant p27 mutant suppresses Skp2-induced cyclin–Cdk2 activation and S-phase entry, suggesting that the ability of Skp2 to promote p27 degradation is a key aspect of its S-phase promoting function (Sutterlüty et al., 1999). Taken together, these lines of experimental evidence suggest that Skp2 plays a prominent role in the progression of cells from G0 to S phase. Hence, specific mechanisms must exist for controlling Skp2 protein levels during G0/G1, failure of which could compromise the maintenance of the quiescent state and/or normal G0 to S phase progression.
Here we show that accumulation of Skp2 protein levels at the onset of S phase as cells emerge from G0, is brought about by changes in the half-life of Skp2 and not by fluctuations in the amounts of Skp2 mRNA present. We also show that the rapid turnover of endogenous Skp2 in quiescent fibroblasts is governed by proteasomal degradation and mutational analysis reveals that efficient interactions between Skp2 and Cul1 are required for proteolysis of Skp2 in serum-deprived cells. Consistent with a role for Cul1 in the regulation of Skp2 protein levels, antisense oligonucleotides that target Cul1 cause an accumulation of Skp2 protein in serum-deprived cells. In addition, a recombinant Cul1-based ligase complex can function as a potent E3 ubiquitin protein ligase for Skp2 in vitro. Cul1’s ability to support ubiquitylation of Skp2 within assembled SCFSkp2 complexes depends, at a minimum, on the availability of two discrete sequence elements within its primary structure that we refer to here as the A- and the B-boxes and which are independently required for Cul1 to interact with the E2 enzymes Cdc34 and Roc1/Rbx1, respectively. The A- and the B-box motifs of Cul1 are also independently required for Cul1 to complement the growth-defect of a yeast cdc53 temperature-sensitive (ts) mutant strain, suggesting that the A- and the B-boxes are both essential elements of Cul1 function. Interestingly, the A- and the B-boxes reside within the cullin homology (CH) region, which constitutes an ∼180 amino acid segment found in all known members of the cullin family as well as in the cullin-related anaphase-promoting complex/cyclosome (APC/C) subunit Apc2 (H.Yu et al., 1998; Zachariae et al., 1998).
Results
Rapid proteasome-dependent degradation of Skp2 in serum-deprived human cells
For many mammalian cell cycle proteins studied so far, a tight correlation has been found between the abundance of these proteins and changes in the mRNA present. To investigate whether the regulation of Skp2 abundance involves changes in Skp2 mRNA expression, human T98G cells were synchronized in G0/G1 by serum deprivation and released from the arrest by the addition of serum. Cell synchrony was monitored in parallel by flow cytometry. As seen by the northern analysis in Figure 1A, Skp2 mRNAs are readily detectable in G0/G1 of T98G cells and amounts increase only ∼2- to 3-fold as cells proceed into S phase. Consistent with our earlier data, Skp2 protein is absent in G0/G1 and accumulates rapidly at the onset of S phase (Lisztwan et al., 1998). Similar results were obtained with human diploid fibroblasts (HDFs) (Figure 1B). These results suggest that mechanisms other than transcriptional ones may contribute to a significant extent to Skp2 protein accumulation during G0 to S phase progression. Interestingly, Zhang et al. (1995) showed that in cycling HeLa cells, Skp2 mRNA expression is periodic. Thus, there might be fundamental differences with respect to the regulation of Skp2 mRNA expression between actively cycling cells and cells that emerge from quiescence.

Fig. 1. Expression of Skp2 mRNA during the G0 to S phase transition. (A) T98G cells were arrested by serum deprivation and then stimulated to re-enter the cell cycle by addition of fresh serum. At the indicated times thereafter, total RNAs were prepared and processed for northern blotting using 32P-labelled cDNAs encoding Skp2 (upper panel) or GAPDH (middle panel). Whole cell extracts were prepared at the same time points, equalized for protein content and processed for western blotting with antibodies against Skp2 (lower panel). The percentages of S-phase cells at various times after serum stimulation were calculated from FACS analysis during the ensuing cell cycle. (B) HDFs were synchronized and processed for FACS (upper panel), northern (middle panel) and western blot (lower panel) analysis as described in (A), except that equal loading of RNA was determined by staining of the 18S RNA by methylene blue following transfer of RNAs to the membrane. (C) Exponentially growing HDFs (lanes 1–3), HDFs arrested by serum deprivation (lanes 4–6) or HDFs induced to re-enter the cell cycle by addition of fresh serum (lanes 7–9) were either left untreated (lanes 1, 4 and 7), treated with DMSO (lanes 2, 5 and 8) or with LLnL (lanes 3, 6 and 9) for 5 h. Whole cell extracts were prepared and processed for immunoblotting using anti-(fl)Skp2 (upper panel) or anti-Cdk2 (lower panel) antibodies.
To examine whether failure of Skp2 protein expression in serum-deprived cells is due to active proteasome-dependent degradation of Skp2 in G0/G1, HDFs were deprived of serum mitogens and then treated for 5 h with the proteasome inhibitor LLnL or its solvent dimethylsulfoxide (DMSO). In parallel, we induced serum-deprived HDFs to enter S phase by the addition of serum prior to the addition of LLnL or DMSO. The amount of Skp2 protein in the relevant extracts was determined by immunoblotting. As shown in Figure 1C, addition of LLnL or MG132 (data not shown) induced the rapid accumulation of endogenous Skp2 protein (compare lane 6 with lanes 7–9). DMSO was inactive in this regard (lane 5). In contrast, LLnL treatment of either serum-stimulated or exponentially growing HDFs had little effect on Skp2 protein amounts compared with DMSO-treated cells (Figure 1C, compare lanes 9 and 3 with lanes 8 and 2, respectively). Reprobing of the same membrane with anti-Cdk2 antibody revealed that comparable quantities of proteins were present within each set of extracts (Figure 1C). The faster-migrating band detected with anti-Cdk2 antibody in serum-stimulated or exponentially growing HDFs represents the activated, threonine 161-phosphorylated form of Cdk2, consistent with the fact that cells are cycling. In parallel, cell synchrony was also monitored by flow cytometric analysis (data not shown). These results imply that a proteasome-dependent mechanism contributes to the suppression of Skp2 protein expression in quiescent cells.
Efficient proteasomal degradation of Skp2 in quiescent fibroblasts involves Skp2-bound Cul1
A definition of the structural elements of Skp2 that contribute to the observed instability of this F-box protein in serum-deprived cells requires an in vivo system that faithfully mimics the transcription-independent dynamics of Skp2 appearance and disappearance upon cell cycle entry and exit. The adenovirus-mediated Skp2 expression system that we have previously employed is not suitable for this purpose because it allows the accumulation of Skp2 in quiescent cells and induces mitogen-independent S phase progression (Sutterlüty et al., 1999). An alternative expression system may be provided by a retroviral expression system, which allows for the expression of very small amounts of protein. To test this directly, Rat1 fibroblasts were infected with retroviruses encoding untagged human Skp2(wt). This cell type was chosen because our anti-human Skp2 antibody does not recognize to a measurable extent the rat homologue of Skp2 (Sutterlüty et al., 1999). Stably infected cultures were selected as pools of puromycin-resistant cells (uncloned mass cultures). Immunoblot analysis of extracts of Skp2(wt)-infected Rat1 cell pools revealed the production of Skp2(wt) (Figure 2A, lane 7). No such signal was present in extracts of vector-infected Rat1 pools (Figure 2A, lane 5). Importantly, removal of serum mitogens from Skp2(wt)-infected Rat1 cells resulted in the exit of cells from the cell cycle as determined by flow cytometric analysis (data not shown) and the concomitant suppression of exogenous Skp2(wt) protein expression (Figure 2A, lane 3). Addition of LLnL to these serum-starved cultures resulted in the rapid accumulation of Skp2(wt) protein (Figure 2A, lane 4). In contrast, LLnL had only a moderate effect on Skp2 levels in the presence of serum (Figure 2A, compare lanes 8 and 7). Consistent with these observations, pulse–chase analysis revealed that the half-life of Skp2(wt) is significantly shorter in serum-starved cells than in cells grown under high serum concentrations (Figure 2B). Hence, retrovirally produced Skp2 undergoes dynamic changes in protein stability during the G0 to S phase progression much like endogenous Skp2.

Fig. 2. Determinants of Skp2 instability in quiescent fibroblasts. (A) Rat1 cells were infected with vector or Skp2(wt) retrovirus and, after selection in neomycin, cell pools of stably infected, neomycin-resistant cells were established. Vector- or Skp2-infected cell pools were growth-arrested by serum deprivation (lanes 1–4) or growth-arrested and then stimulated to re-enter the cell cycle by addition of fresh serum (lanes 5–8). Each of these cultures was treated with DMSO (lanes 1, 3, 5 and 7) or LLnL (lanes 2, 4, 6 and 8) for 5 h. Lysates were prepared from each cell pool, equalized for protein content and processed for western blotting with anti-(fl)Skp2 antibodies. (B) Skp2-infected Rat1 cells were growth-arrested (upper panel) or growth-arrested and stimulated to re-enter the cell cycle by addition of fresh serum (lower panel) and then pulse-labelled with [35S]methionine and chased for the indicated times. Denatured cell lysates were then processed for immunoprecipitation with anti-(fl)Skp2 antibodies. (C) Rat1 cells were infected with Skp2(wt), (ΔF6), (AxA) or (ΔF6/AxA) retrovirus and, after selection in neomycin, cell pools of stably infected, neomycin-resistant cells were established and either growth-arrested (lanes 1, 3, 5 and 7) or growth-arrested and stimulated to re-enter the cell cycle by addition of fresh serum (lanes 2, 4, 6 and 8). Lysates were prepared from each cell pool, equalized for protein content and processed for western blotting with anti-(fl)Skp2 (upper panel) and anti-Cdk2 (lower panels) antibodies. (D) Stably infected Rat1 cell pools as described in (C) were growth-arrested and then either left untreated (lanes 1, 4, 7 and 10) or were treated with DMSO (lanes 2, 5, 8 and 11) or MG132 (lanes 3, 6, 9 and 12) for 5 h and processed for western blotting as described in (C). (E) Stably infected Rat1 cell pools as described in (C) were first growth-arrested and then stimulated to re-enter the cell cycle by addition of fresh serum and were then either left untreated (lanes 1, 4, 7 and 10) or were treated with DMSO (lanes 2, 5, 8 and 11) or MG132 (lanes 3, 6, 9 and 12) for 5 h. Normalized lysates were processed for western blotting as described in (D). (F) HDFs were growth-arrested by serum deprivation for 64 h and were then either left untreated (lane 1) or were treated with transfection agent GS2888 alone (lane 2), antisense oligonucleotides (100 nmol) (lane 3) or control oligonucleotides (100 nmol) (lane 4) for 24 h. Whole cell extracts were prepared, equalized for protein content and processed for immunoblotting using anti-Cul1 (upper panel), anti-Cdk2 (middle panel) and anti-(fl)Skp2 (lower panel). A representative result from three independent experiments using different concentrations of oligonucleotides (100 or 200 nmol) is displayed.
As a first step towards defining the structural requirements of Skp2 degradation in quiescent cells, we asked whether Skp2 binding to Skp1 or Cul1 is important for degradation of Skp2 in quiescent cells. Retroviral expression plasmids encoding the following Skp2 mutants were generated: an F-box mutant, referred to as Skp2(ΔF6), in which six amino acids of the ∼40 amino acid residue long F-box domain were deleted; a cyclin A kinase binding-defective mutant p45SKP2(AxA), in which lysine 362 and leucine 364 were converted to alanines; and the double-mutant derivative, Skp2(ΔF6/AxA) (Lisztwan et al., 1998). A detailed biochemical characterization of these mutant Skp2 species with respect to their ability to bind Cul1 and Skp1 is shown in Figure 4A and B, respectively. In short, this analysis revealed that with the exception of Skp2(ΔF6/AxA), all Skp2 species under investigation retain, albeit to different extents, Cul1 binding properties. Stably infected mass cultures of Rat1 cells expressing untagged Skp2(wt) and the above-noted mutant species were generated. The various Skp2 species were synthezised in the corresponding stably infected Rat1 cultures under high serum concentrations as shown by western blotting (Figure 2C, lanes 2, 4, 6 and 8). Removal of serum mitogens from these cultures resulted in the elimination of Skp2(wt), (ΔF6) and (AxA) protein (Figure 2A, lanes 1, 3 and 5, respectively). Interestingly, the Cul1 binding- and SCF function-defective Skp2 species, Skp2(ΔF6/AxA), was resistant to this downregulation and remained abundantly expressed even under conditions of low serum concentration (Figure 2C, lane 7). Equal loading was confirmed by probing for Cdk2 (Figure 2C, lower panel). As shown in Figure 2D, addition of the proteasome inhibitor MG132 to the various serum-deprived cultures led to the rapid accumulation of Skp2(wt), (ΔF6) and (AxA) proteins (Figure 2D, lanes 3, 6 and 9, respectively). In contrast, MG132 failed to alter the levels of Skp2(ΔF6/AxA) (Figure 2D, compare lane 12 with lanes 10 and 11). As one would expect, MG132 had little effect on the abundance of these Skp2 species under conditions of high serum concentration (Figure 2E). Taken together, these results indicate that Skp2 degradation in serum-deprived cells is F-box independent, but Cul1 dependent. Interestingly, the very same structural requirements of Skp2 underlie its ability to promote the degradation of endogenous p27 and induce quiescent cells to enter S phase (Sutterlüty et al., 1999). Hence, Skp2 degradation in G0/G1 and Skp2’s S-phase promoting function each appear to be dependent on Skp2-associated Cul1 (see Discussion).

Fig. 4. Skp2(wt), (ΔF6) and (AxA) are ubiquitylated in vitro. (A) Sf9 insect cells were infected with baculovirus vectors that express GST–Skp1, HisCul1HA, PyRoc1/Rbx1 together with either HisSkp2(wt) (lanes 1 and 5), HisSkp2(ΔF6) (lanes 2 and 6), HisSkp2(AxA) (lanes 3 and 7) or HisSkp2(ΔF6/AxA) (lanes 4 and 8). Lysates were prepared and aliquots processed for immunoprecipitation using anti-(fl)Skp2 antibody followed by western blotting with anti-Cul1 or anti-(fl)Skp2 antibody (left panels). Note that similar amounts of Skp2 are present in each of the immunoprecipitates. Other aliquots were processed directly for western blotting with anti-GST, anti-(fl)Skp2 and anti-HA antibodies (right panels). (B) Sf9 insect cells were infected with baculovirus vectors expressing GST–Skp1 together with either HisSkp2(wt) (lanes 1 and 5), HisSkp2(ΔF6) (lanes 2 and 6), HisSkp2(AxA) (lanes 3 and 7) or HisSkp2(ΔF6/AxA) (lanes 4 and 8). Lysates were prepared and aliquots were incubated with GS beads and bound proteins were processed for western blotting with anti-(fl)Skp2 or anti-GST antibodies (left panels). Other aliquots were processed directly for western blotting with anti-GST and anti-(fl)Skp2 antibodies (right panels). (C) Recombinant SCFSkp2 complexes were produced in insect Sf9 cells with either HisSkp2(wt) (lanes 1, 2 and 7), HisSkp2(AxA) (lanes 3, 4 and 8) or HisSkp2(ΔF6) (lanes 5, 6 and 9). Lysates were prepared and aliquots incubated with GS beads. Beads were processed for in vitro ubiquitylation reaction in the presence (lanes 1, 3 and 5) or absence (lanes 2, 4 and 6) of ubiquitin and immunoblotted with anti-(fl)Skp2 antibodies. Other aliquots of cell lysates (lanes 7–9) were processed directly for western blotting with anti-GST (upper panel), anti-HA (second panel), anti-Py (third panel) or anti-(fl)Skp2 (lower panel) antibodies.
One prediction from the aforementioned results is that interference with Cul1 function in serum-deprived cells should cause an increase in Skp2 protein levels. To test this directly, we inhibited the expression of Cul1 in serum-deprived HDFs using antisense oligonucleotides that contain a phosphorothioate backbone and C-5 propyne pyrimidine substitutions. These antisense oligonucleotides have been reported previously to be effective in inhibiting the expression of human Cul1 (Z.K.Yu et al., 1998). Indeed, transfection of serum-starved HDFs with these antisense oligonucleotides resulted in a substantial decrease in Cul1 protein levels (Figure 2F, upper panel, lane 3). Control oligonucleotides or transfection agent alone had no such effect (Figure 2F, upper panel, lanes 4 and 2, respectively). Importantly, concomitant with a decrease in Cul1 protein, there was a significant increase in the levels of endogenous Skp2 protein (Figure 2F, lower panel, lane 3), while Cdk2 levels remained unaffected (Figure 2F, middle panel, lane 3). These results support the view that degradation of Skp2 in quiescent cells involves Cul1.
The F-box protein Skp2 is a ubiquitylation target of a recombinant Cul1-based core ubiquitin ligase complex in vitro
The aforementioned results raise the interesting possibility that Skp2 degradation in the G0/G1 phases of the cell cycle may be a result of its ubiquitylation mediated by a Cul1-based core ligase module that it binds to. To test directly whether Skp2 is a ubiquitylation target of recombinant Cul1-based core ligase complexes, glutathione _S_-trans ferase (GST)–Skp1, HisSkp2, PyRoc1/Rbx1 and HisCul1HA proteins were coexpressed in Sf9 insect cells (Figure 3A). SCFSkp2 complexes were retrieved on glutathione– Sepharose (GS) beads (Figure 3B) and added to reaction mixtures containing purified E1, purified human E2 enzyme Cdc34, ubiquitin and an ATP-regenerating system. The products of the reaction mixture were separated on SDS gels and analysed for the presence of Skp2 ubiquitin conjugates by anti-Skp2 immunoblotting. Indeed, extensive ubiquitin conjugation of Skp2 was observed, as defined by the appearance of more slowly migrating forms of Skp2 (Figure 3C, lane 1). The production of these forms was dependent on ubiquitin (lane 2), E1 (lane 3), Cdc34 (lane 4) and was blocked by mutant ubiquitin K48R (lane 5). These results suggest that SCFSkp2 complexes assembled in insect cells from recombinant proteins can catalyse the ubiquitylation of the F-box protein Skp2, which, in turn, suggests that Skp2 represents a potent ubiquitylation target of the bound ubiquitin ligase subcomplex in vitro.

Fig. 3. The F-box protein Skp2 is a ubiquitylation target of the bound Cul1-based core ubiquitin ligase complex. (A) Sf9 insect cells were infected with baculovirus vectors that express GST (lane 1) or GST–Skp1 (lane 2) together with HisSkp2, HisCul1HA and PyRoc1/Rbx1. Lysates were prepared and then aliquots immunoblotted with anti-GST (upper panel), anti-(fl)Skp2 (second panel), anti-HA (panel 3) or anti-Py (lower panel) antibodies. (B) Other aliquots prepared as in (A) were incubated with GS beads and bound proteins were processed for western blotting with indicated antibodies. Note that HisSkp2, HisCul1HA and PyRoc1/Rbx1 bound to GST–Skp1 (lanes 2, 4, 6, 8) but not to GST alone (lanes 1, 3, 5 and 7). (C) Recombinant SCFSkp2 complexes were produced in insect Sf9 cells and bound to GS beads as described in (B) and processed for the in vitro ubiquitylation reaction as described in Materials and methods. Lane 1, all components; lane 2, no ubiquitin; lane 3, no E1; lane 4, no Cdc34; lane 5, mutant K48R ubiquitin. Samples were processed for western blotting with anti-(fl)Skp2 antibodies. The bracket on the left marks ubiquitylated Skp2. (D) Sf9 insect cells were infected with various combinations of recombinant baculoviruses as indicated by the plus and minus signs at the top of the figure. Lysates were prepared and aliquots were incubated with GS beads. The corresponding Sepharose beads were either processed for in vitro ubiquitylation in the absence (lanes 2, 4, 6, 8, 10, 12, 14 and 16) or presence (lanes 1, 3, 5, 7, 9, 11, 13 and 15) of ubiquitin and western blotting with the antibodies described in (A). Asterisks in the upper panel (lanes 5 and 6) indicate a Skp2 signal that derives from spill-over from lane 4. (E) Other aliquots of cell lysates prepared as in (D) were processed directly for western blotting with the antibodies described in (A).
Next we determined whether Skp2 ubiquitylation within assembled SCFSkp2 complexes requires Cul1 and Roc1/Rbx1. SCFSkp2 complexes were purified on GS beads with or without HisSkp2, PyRoc1/Rbx1 or HisCul1HA from Sf9 insect cells infected with the relevant recombinant baculoviruses and added to reaction mixtures containing E1, Cdc34, ubiquitin and an ATP-regenerating system. As shown in Figure 3D, in the absence of Roc1/Rbx1, little Skp2 ubiquitylation was observed (lane 9). Similar results were obtained when Cul1 was lacking (lane 11). In contrast, the yield of Skp2 ubiquitylation was dramatically enhanced when both proteins were present within the complex (lane 15). Western blotting of GS precipitates showed that comparable quantities of GST–Skp1 were present and that Cul1, Roc1/Rbx1 and Skp2 copurified with GST–Skp1 where expected (Figure 3D, lower panels). In addition, the expression levels of each individual subunit in insect cell extracts were determined by immunoblotting with the corresponding antibody (Figure 3E). These results suggest that both Cul1 and Roc1/Rbx1 are required for catalysing Skp2 auto-ubiquitylation in the presence of the universally required components E1, E2 and ubiquitin.
Since Cul1-dependent Skp2 ubiquitylation can be readily demonstrated in vitro, we asked whether mutants of Skp2 that are unstable in vivo (see Figure 2C) can be ubiquitylated in vitro. To this end, we first assessed the Cul1 and Skp1 binding activities of these Skp2 mutants. Skp2(wt), Skp2(ΔF6), (AxA) or (ΔF6/AxA) were coexpressed with GST–Skp1, HisCul1HA and PyRoc1/Rbx1 in Sf9 insect cells and assayed for their ability to bind Cul1. As shown in Figure 4A, anti-(fl)Skp2 antibodies immunoprecipitated Cul1 from Skp2(wt)- and Skp2(AxA)- containing lysates (Figure 4A, lanes 1 and 3, respectively). Much less Cul1 was recovered from Skp2(ΔF6)-containing lysates (lane 2). In contrast, no Cul1 was present in Skp2 immunoprecipitates from Skp2(ΔF6/AxA)-containing lysates (lane 4). Importantly, similar amounts of the various Skp2 species were present in each of these immunoprecipitates (Figure 4A, lower panel, lanes 1–4) and Cul1 was produced to equivalent levels in each of the infected cultures (Figure 4A, right panels, lanes 5–8). Because GST–Skp1 comigrated with the IgG band, it was necessary to determine the Skp1 binding activities of these Skp2 species in a separate experiment. GST–Skp1 was coexpressed with Skp2(wt) or the above-noted mutants in Sf9 insect cells and cell lysates incubated with GS beads. Skp2(wt) bound abundantly to GST–Skp1 (Figure 4B, lane 1). Skp2(ΔF6) and (ΔF6/AxA) failed to bind (Figure 4B, lanes 2 and 4, respectively), while Skp2(AxA) displayed very little Skp1 binding activity (Figure 4B, lane 3). Western blotting confirmed that similar amounts of GST–Skp1 were recovered by GS beads and that equivalent levels of GST–Skp1 and the relevant Skp2 species were produced in each infected cell culture (Figure 4B, right panels). In summary, the following conclusions can be drawn: (i) the F-box mutant Skp2(ΔF6) lacks detectable Skp1 binding function but retains measurable F-box-independent Cul1 binding activity; (ii) Skp2(AxA) displays residual Skp1 binding but interacts efficiently with Cul1, much like the wild-type Skp2 protein; and (iii) the double mutant derivative, Skp2(ΔF6/AxA), is able to associate to a measurable extent neither with Skp1 nor with Cul1, suggesting that the (AxA) mutations affect the F-box-independent Cul1 bind ing function of Skp2. The observation that Skp2(ΔF6) and (AxA) exhibit Cul1 binding activity is entirely consistent with the fact that they are each able to promote the degradation of endogenous p27 and induce quiescent cells to enter S phase. The double-mutant derivative, Skp2(ΔF6/AxA), which lacks Cul1 binding activity, is inactive in this regard (Sutterlüty et al., 1999). Hence, there is close correlation between the ability of Skp2 to bind Cul1, its S-phase promoting activity and its instability in G0/G1 (see Figure 2C).
To learn whether Skp2(AxA) and (ΔF6), which display Cul1 binding activity, are also ubiquitylated in vitro, Skp2(wt), (AxA) or (ΔF6) was expressed together with GST–Skp1, HisCul1HA and PyRoc1/Rbx1 in Sf9 insect cells. Complexes were retrieved on GS beads and assayed for their ability to support ubiquitylation of bound Skp2. Skp2(wt) was extensively ubiquitin conjugated (Figure 4C, lane 1). Skp2(AxA) showed a wild-type ubiquitylation profile although fewer ubiquitin conjugates formed (Figure 4C, lane 3). In the case of Skp2(ΔF6), ubiquitin conjugates also formed (Figure 4C, lane 5), but we did not observe the smearing associated with branched polyubiquitylated chains (Figure 4C, compare lane 5 with 3 and 1). These results suggest that Skp2 ubiquitylation in vitro can occur in an F-box-independent but Cul1-dependent manner and establish a link between Skp2 degradation in G0/G1 and its ubiquitylation in vitro.
Identification of sequence elements within Cul1’s primary structure required for stimulating ubiquitylation of Skp2 in vitro
To probe further the mechanism underlying a requirement of Cul1 for SCFSkp2 ubiquitin ligase activity directed towards the bound F-box protein Skp2, we constructed a series of Cul1 mutants (Figure 5). Cul1(wt) or relevant mutants of it were then coexpressed with GST–Skp1, HisSkp2 and PyRoc1/Rbx1 in Sf9 insect cells. The SCFSkp2 complexes assembled with Cul1(wt) or Cul1 mutant subunits were retrieved on GS beads, added to reaction mixtures containing E1, CDC34, ubiquitin and an ATP-regenerating system and assayed for their ability (i) to support ubiquitylation of Skp2 (Figure 5A and C), (ii) to bind the RING-H2 protein Roc1/Rbx1 (Figure 5A and C) and (iii) to bind Skp1 (Figure 5A and C). The expression levels of each individual subunit in insect cell extracts were determined by immunoblotting with the respective antibodies (Figure 5B). Two major conclusions can be drawn from these experiments. First, the capacity of Cul1 to bind Roc/Rbx1 depends, at the minimum, on the availability of amino acid residues 555–561 of Cul1’s primary structure since Cul1(ΔB/S) and (Δ6), mutants that are lacking amino acid residues 539–603 and 555–561, respectively, failed to bind Roc1/Rbx1 although they were still able to form complexes with Skp1 and Skp2 (Figure 5A and C). The domain comprising amino acid residues 539–603 is referred to here as the B-box motif. B-box mutants of Cul1 were also defective in promoting Skp2 ubiquitylation (Figure 5A and C). These results imply that Cul1’s function as a required component for SCFSkp2 ubiquitin protein ligase activity directed towards Skp2 depends, at a minimum, on its ability to bind Roc1/Rbx1. Secondly, complex formation of Cul1 with Roc1/Rbx1, although necessary, is not sufficient for generating Skp2 ubiquitylation mediated by SCFSkp2 ubiquitin protein ligase, since Cul1(ΔC63) and (Δ26), two mutants that lack amino acid residues 452–518 and 452–478, respectively, were wholly capable of binding to Roc1/Rbx1 and assembling into quaternary complexes, but were clearly defective in supporting Skp2 ubiquitylation (Figure 5A and C). We refer to this specific segment that comprises amino acid residues 452–518 of Cul1 as the A-box motif, whose structural integrity is independent of the B-box, required for Cul1 function. Importantly, a deletion mutant of Cul1, Cul1(ΔN63), which lacks amino acid resides 251–314, behaved as wild-type Cul1 in this assay, suggesting that the aforementioned deletion mutants of Cul1 selectively affect key residues within Cul1’s primary structure important for its function. Finally, assembly of Roc1/Rbx1 into quaternary SCFSkp2 complexes is strictly dependent on Cul1, since Roc1/Rbx1 failed to copurify with Skp1 in the presence of a Cul1 mutant that was defective in Skp1 binding (Figure 5A and C).


Fig. 5. The availability of A- and B-box sequences of Cul1 primary structure are independently required for Cul1 to support Skp2 ubiquitylation. (A) Recombinant SCFSkp2 complexes were produced in insect Sf9 cells with either Cul1(wt) or indicated mutant derivatives of it. Lysates were prepared and aliquots were incubated with GS beads. The corresponding Sepharose beads were either processed for in vitro ubiquitylation in the absence (lanes 2, 4, 6, 8, 10, 12 and 14) or presence (lanes 1, 3, 5, 7, 9, 11 and 13) of ubiquitin and western blotting with the antibodies described in the legend to Figure 3A, except that anti-His antibodies were used to detect Cul1. (B) Other aliquots of cell lysates prepared in (A) were processed directly for western blotting with the antibodies described in the legend to Figure 3A, except that anti-His antibodies were used to detect Cul1. (C) Schematic representation of the Cul1 deletion mutants tested and their corresponding Roc1 binding and Skp2 ubiquitylation activity (plus versus minus). (D) Sequence alignment of the CH region of cullin members and Apc2 from different species. The alignment was made with the ClustalX method and the Boxshade program. Black bars on top of the alignment indicate the A- and the B-box. Black lines on the bottom of the alignment represent in-frame deletion mutants of Cul1. Identical residues are indicated by white letters on red boxes and functionally similar residues are in white letters on blue boxes.
Given the consequences of disrupting the integrity of the A- and B-boxes for Cul1 function, we asked whether Cul1 residues comprising the A- and the B-boxes bear sequence similarity to other proteins, in particular members of the cullin family. Remarkably, database searches revealed that the A- and B-boxes reside within the so-called CH region, which constitutes an ∼180 amino acid segment found in members of the cullin family as well as in the APC/C subunit Apc2. Sequence alignment of the CH regions from 26 different proteins revealed that the amino acid residues comprising the A- and B-boxes are highly conserved among all proteins listed (Figure 5D). Notably, the A- and the B-boxes appear to be separated by a very short stretch of amino acid residues bearing very little sequence homology, in keeping with the notion that they represent discrete elements of Cul1 independently required for its function in vitro.
The availability of A-box sequences of Cul1 is important for Cdc34 E2 enzyme binding
Since A-box mutants of Cul1 are capable of assembling into quarternary SCFSkp2 complexes much like the wild-type Cul1 protein but are nevertheless defective in ubiquitin ligase activity directed towards Skp2, we tested whether A-box mutants were compromised in their ability to bind the E2 enzyme Cdc34. To this end, Cdc34 was expressed alone or in combination with HisCul1HA (wt) or selected A- and B-box mutant derivatives in Sf9 insect cells and Cul1–Cdc34 complex formation was evaluated by anti-HA immunoprecipitation analysis. As shown in Figure 6A, anti-HA immunoprecipitates from insect cells expressing Cdc34 and Cul1 contained Cdc34 (lane 3). No Cdc34 was detectable in anti-HA immunoprecipitates derived from insect cells expressing Cdc34 alone (lane 1). Importantly, Cdc34 did not coimmunoprecipitate with Cul1(ΔC63), an A-box mutant (Figure 6B, lane 3), but it did coimmunoprecipitate with Cul1(Δ6), a B-box mutant (Figure 6B, lane 4). Cdc34 coimmunoprecipitated also with Cul1(ΔN63) (Figure 6B, lane 5), which behaved like wild-type Cul1 in its ability to support Skp2 ubiquitylation (see Figure 5A). Western blotting confirmed that similar amounts of the relevant Cul1 species were present in the corresponding anti-His immunoprecipitates and that Cdc34 and Cul1 species were expressed to similar levels in Sf9 cells (Figure 6A and B, lower panels). Thus, it appears that A-box sequences of Cul1 are involved in mediating, in a Roc1/Rbx1-independent manner, an interaction of Cul1 with Cdc34.
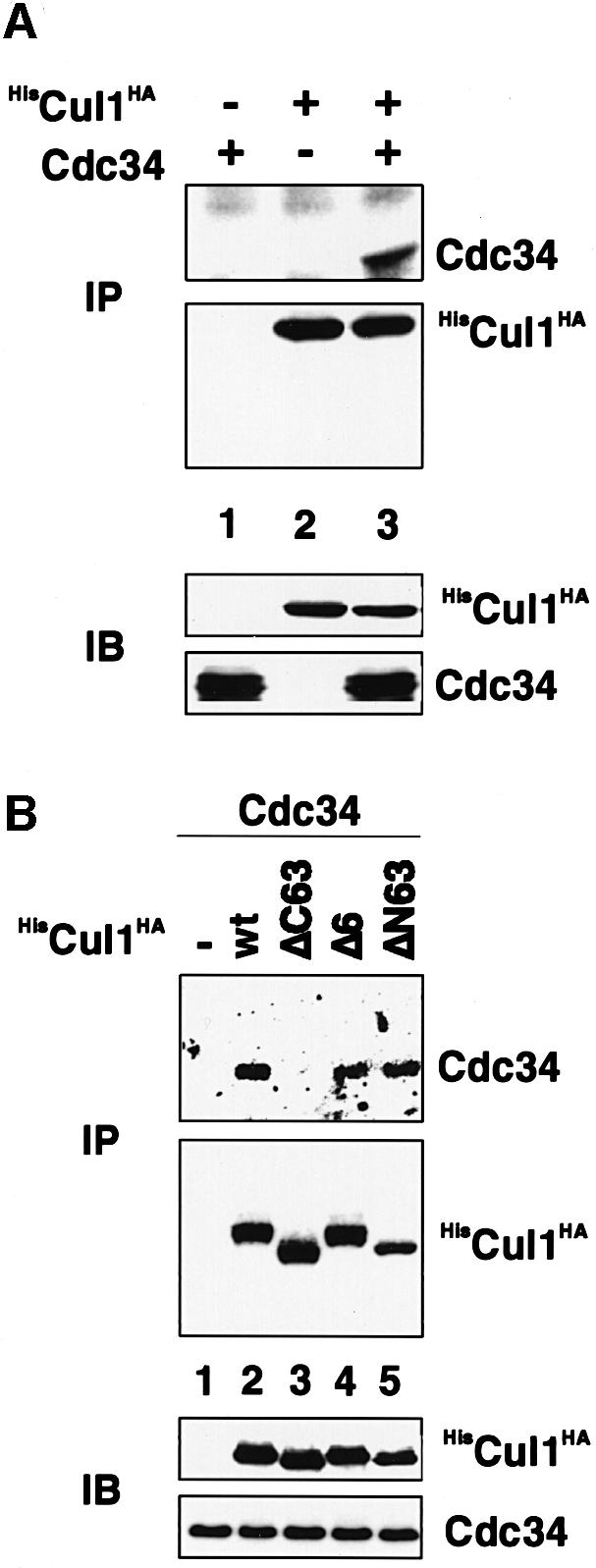
Fig. 6. A-box mutants of Cul1 are defective in Cdc34 E2 enzyme binding. (A) Sf9 insect cells were infected with baculovirus vectors encoding Cdc34 (lane 1), HisCul1HA (lane 2) or Cdc34 and HisCul1HA (lane 3). Lysates were prepared and aliquots processed for immuno precipitation using anti-HA antibody followed by western blotting with anti-Cdc34 (upper panel) or anti-HA antibody (second panel). Other aliquots were processed directly for western blotting with anti-HA (third panel) or anti-Cdc34 (lower panel) antibodies. (B) Sf9 insect cells were infected with baculovirus vectors encoding Cdc34 (lane 1) or Cdc34 and HisCul1HA-wt (lane 2), ΔC63 (lane 3), Δ6 (lane 4) or ΔN63 (lane 5). Lysates were prepared and aliquots processed for immunoprecipitation using anti-His antibody followed by western blotting with anti-Cdc34 (upper panel) or anti-HA antibody (second panel). Other aliquots were processed directly for western blotting with anti-HA (third panel) or anti-Cdc34 (lower panel) antibodies.
Complementation of the growth defect of a yeast cdc53ts mutant by human Cul1 depends on the integrity of both the A- and B-boxes
The results presented thus far point to a critical role of Cul1’s A- and B-boxes in the ubiquitin ligase activation process in vitro. To establish the general importance of these sequence elements for Cul1 function, we determined the ability of selected A- and B-box mutants of human Cul1 to rescue the growth defect of the cdc53ts strain. Previous work has demonstrated that human Cul1 can complement the cdc53ts mutant phenotype (Lyapina et al., 1998). To this end, human Cul1 and relevant mutant derivatives of it were introduced under the control of the constitutive GPD promoter into a yeast strain carrying a ts mutation in the CDC53 gene. Individual transformants were plated on glucose media at the permissive (25°C) and restrictive (34°C) temperatures (Figure 7A). Trans formants that expressed Cul1(wt) or Cul1(ΔN63) were able to grow at the restrictive temperature (Figure 7A). In contrast, both A- and B-box mutants failed to complement the cdc53ts mutant phenotype (Figure 7A), although they were abundantly expressed as determined by anti-HA immunoblotting (Figure 7B). Hence, the ability of Cul1(wt) or mutant derivatives to support SCFSkp2 ubiquitin protein ligase activity in vitro correlates with the ability to rescue the yeast cdc53ts mutant phenotype in vivo.

Fig. 7. A- and B-box mutants of Cul1 fail to restore growth of cdc53-2 yeast cells. (A) Individual transformants of the cdc53-2 strain (YMP740) containing the plasmids indicated were plated on selective media and grown either at the permissive (25°C, right plate) or restrictive (34°C, left plate) temperature. Photographs were taken after 3 days. (B) The protein levels of wild-type or mutant Cul1 were analysed by immunoblotting of cdc53-2 cells grown at the restrictive temperature (25°C).
Discussion
The results presented in this report provide experimental evidence that a major determinant of Skp2 periodicity during the G0 to S phase transition is protein stability and that a key event in the process underlying G0/G1-specific proteasome-dependent Skp2 degradation may be a specific interaction of the F-box protein Skp2 with Cul1. In support of this view, in quiescent cells induced to exit G0 and enter the cell cycle, the amounts of Skp2 mRNA increase only modestly but Skp2 protein accumulated dramatically at the onset of S phase. Addition of proteasome inhibitors to quiescent cells led to the rapid accumulation of endogenous or retrovirally produced Skp2 protein. The same inhibitors had little effect on Skp2 levels in cycling cells. In keeping with these observations, the half-life of retrovirally produced Skp2 was significantly prolonged in cells that had entered the cell cycle compared with quiescent cells. Hence, Skp2 can switch from being unstable to stable as a result of cell cycle entry. Genetic analysis reveals that efficient degradation of Skp2 in quiescent cells is, like Skp2’s function as a p27-degrading and S-phase promoting element, F-box independent but Cul1 binding dependent, implying that each of these processes may depend on Skp2-bound Cul1-based ligase activity. Indeed, a specific role for Cul1 in Skp2 degradation is underscored by the observation that interference with its function by antisense oligonucleotides causes a stabilization of Skp2 in quiescent cells. Moreover, a recombinant Cul1-based ligase subcomplex can catalyse the ubiquitylation of bound Skp2 in vitro. This activity of the ligase module depended on the ability of the Cul1 subunit to recruit Cdc34 and Roc1/Rbx1 through specific sequence elements, the A- and the B-box motifs, which comprise the CH region. The notion that A- and B-box sequences are indeed important elements of Cul1 function as a core ligase component is further supported by the failure of the corresponding Cul1 mutant species to complement the yeast cdc53ts mutant phenotype. Taken together, these observations suggest a model in which degradation of Skp2 in serum-deprived, quiescent cells is mediated by a Cul1-based ubiquitin ligase module, which in turn implies a role for Cul1 in G0/G1 exit control. Thus, we hypothesize that Cul1 serves at least two distinct functions during the G0 to S phase progression pathway with respect to Skp2. In the G0/G1 phases, Cul1 functions as an inhibitor of SCFSkp2 ubiquitin ligase by mediating the degradation of the bound F-box protein. At the onset of S phase, Cul1 arms the F-box protein Skp2 with a potent ligase activity allowing the latter to proceed with the ubiquitylation and degradation of key cell cycle regulatory proteins. Both of these activities of Cul1 may require the integrity of the A- and the B-boxes of Cul1, because, as shown here, these elements are involved in recruiting key regulatory and catalytic subunits, in particular Roc1/Rbx1 and the E2 enzyme Cdc34, required for Cul1-dependent E3 ligase function.
There are certain notable considerations concerning the biological significance of degradation of Skp2 for normal cell proliferation. Skp2 is a potent S-phase promoting element and it is now clear that the higher the accumulation of Skp2, the greater its potential to interfere with cell cycle exit or the maintenance of a quiescent state (Sutterlüty et al., 1999). Thus, one might imagine that efficient degradation of this protein contributes to the normal regulation of the dynamics of cell cycle entry and exit of mammalian cells. A critical element in the pathway of Skp2 degradation during the G0 and G1 phases of the cell cycle may be a Skp2-bound Cul1-based ubiquitin ligase complex. This conclusion is based on the observation that there is a close correlation between the structural requirements of Skp2 to function as SCF and its instability in G0/G1. Reinforcing this proposal is the finding that a recombinant Cul1-based ligase complex can function as a potent E3 able to ubiquitylate bound Skp2. Accordingly, complex formation of Skp2 with a Cul1-based ubiquitin ligase in G0/G1 would destabilize Skp2 during a phase of the cell cycle when Skp2 must be absent. Viewed in this way, it is possible that a Cul1-dependent core ubiquitin ligase functions in negative regulation of G0/G1 exit by promoting the ubiquitylation and degradation of Skp2. Since Skp2 is stabilized at the onset of S phase following serum stimulation, one might predict that the negative effect of this Cul1-dependent core ubiquitin ligase on Skp2 is cancelled, the end product of which is a rapid accumulation of Skp2. As a result of this, Cul1 becomes a positive regulator of the cell cycle by collaborating with complexed Skp2 in the ubiquitylation of Skp2 substrates such as the Cdk inhibitor p27 (Carrano et al., 1999; Sutterlüty et al., 1999; Tsvetkov et al., 1999). The specific mechanism involved in switching the activity of the Cul1-based core ubiquitin ligase from Skp2 to Skp2-bound substrates at the onset of S phase remains to be determined.
An intriguing finding was that destabilization of Skp2 in quiescent cells was not dependent on the integrity of the F-box but was dependent on the availability of Skp2 sequences that contribute to Cul1 binding. Notably, the capacity of Skp2 to induce p27 degradation and S-phase entry is, likewise, F-box independent but Cul1 binding dependent (Sutterlüty et al., 1999). We cannot exclude a participation of Skp1 in these processes because Cul1 itself possesses a domain dedicated to Skp1 binding and, thus, Skp1 might join this complex via Cul1. In Saccharomyces cerevisiae, the instability of the yeast F-box proteins Cdc4 and Grr1 was shown to be dependent on their F-box (Zhou and Howley, 1998; Galan and Peter, 1999), whereas Met30 is subject to F-box-independent, but Cdc53 (the yeast Cu1l homologue)-dependent degradation (Roullion et al., 2000), much like Skp2 reported here. Thus, it appears that depending on the nature of the F-box protein, complex formation with Cul1/Cdc53 can occur F-box dependently and/or independently. Yet, in both cases, complex formation with Cul1 can lead to the degradation of the relevant F-box protein. Therefore, it seems reasonable to link Cul1–Skp2 complex formation to Skp2 degradation in serum-deprived, quiescent cells.
Major participants in Cul1-based ubiquitin ligase activity directed towards Skp2 appear to be Roc1/Rbx1 and the E2 enzyme Cdc34. From what is apparent, one can argue that deletions of sequences comprising either the A- or the B-box in Cul1, greatly compromise the efficiency of Skp2 ubiquitylation. Of course, a role for yet other Cul1 sequences in governing the Skp2 ubiquitylation process, in particular those extending to the very C-terminus is not excluded. Irrespective of that, it is interesting to note that Cul1 mutants specifically affected in the integrity of the A-box motif assembled as efficiently as wild-type Cul1 into SCFSkp2 complexes but were nevertheless defective in catalysing Skp2 ubiquitylation. Hence, quaternary complex formation is not sufficient for ubiquitin ligase activity to develop when A-box sequences are not available. The notion that A-box sequences are indeed biologically important is further supported by the fact that A-box mutants of Cul1 failed to complement the yeast cdc53ts mutant phenotype. Although we cannot formally exclude the possibility that A-box mutants have defects that prevent, for example, an efficient transfer of activated ubiquitin to the substrate, we found that A-box mutants are defective in Cdc34 E2 enzyme binding. Amino acid residues 452–518, comprising the A-box, display a significant degree of sequence similarity between cullin family members. Hence, one might argue that the A-box could serve as an interaction site for the relevant E2 enzyme in other members of the cullin family. In keeping with this observation, yeast Cdc53–Cdc34 complex formation is disrupted by the cdc53-1 mutation (R488A) (Patton et al., 1998b), which lies within the equivalent region of the the A-box of human Cul1 (see Figure 5D). Unlike A-box mutants, B-box mutants of Cul1 failed to bind Roc1/Rbx1 but were wholly competent in Cdc34, Skp1 and Skp2 binding, supporting the view that elimination of B-box sequences selectively affects Roc1/Rbx1 binding. Since B-box mutants of Cul1 were deficient in supporting Skp2 ubiquitylation in vitro and in functionally complementing the yeast cdc53ts growth defect, Cul1– Roc1/Rbx1 complex formation is likely to be a necessary step in core ligase activation. Also, the amino acid residues comprising the B-box display a high degree of sequence conservation among members of the cullin family. Of additional interest is the fact that the A- and the B-boxes reside immediately adjacent to one another, separated only by a very short stretch of amino acid residues, which displays very little overall sequence similarity among cullin family members. Based on previous analyses of large truncation mutants of Cul1/Cdc53, it has been proposed that Cdc34 and Roc1/Rbx1 bind to a region of Cul1/Cdc53 involving the CH region (Patton et al., 1998b; Wu et al., 2000) and that Roc1/Rbx1 recruits Cdc34 to Cul1. Our analysis suggest that the CH region may contribute two distinct, but separable activities important for Cul1 function—one includes the binding of the E2 enzyme and the other one RING-H2 protein binding. Accordingly, the CH region may be viewed as a platform that brings together relevant regulatory and catalytic components essential for ubiquitin ligase function. We hypothezise that the A- and the B-box motifs of the CH region may fulfil a similar role in other cullin-based ubiquitin protein ligases, including the VCB [von Hippel Lindau (VHL), Elongin C, Elongin B)–Cul2 (Krek, 2000) and the APC/C (Zachariae and Nasmyth, 1999) complexes.
In conclusion, results presented here point to distinct roles of Cul1 in the G0 to S phase progression pathway in mammalian cells. One role of Cul1 appears to be linked to its ability to bind Skp2 in G0/G1 and in this manner it contributes to the suppression of Skp2 expression in this period of the cell cycle. Another function of Cul1 develops at the onset of S phase, at which time it collaborates with Skp2 in the degradation of Skp2 substrates allowing S phase to proceed.
Materials and methods
Plasmid constructions
Retroviral expression plasmids were constructed by cloning cDNAs encoding untagged Skp2(wt), (ΔF6), (AxA) and (ΔF6/AxA) as _Sma_I–_Xho_I fragments into the _Hpa_I–_Xho_I sites of pCMV(neo-retro). Full-length human Roc1/Rbx1 coding sequence was amplified from a HeLa cDNA library by a one-step PCR using two oligonucleotide primers that were designed on the basis of the previously published sequence (Tan et al., 1999). _Bam_HI and _Xba_I restriction sites were incorporated into the sense and anti-sense primers, respectively. Following digestion of the amplified DNA with _Bam_HI and _Xba_I, the fragment was subcloned into the _Bam_HI–_Xba_I sites of pcDNA3 (Invitrogen). The PCR-derived region was confirmed by direct sequencing. Details of the construction of baculovirus expression constructs [based on the pAcSG2 plasmid (Pharmingen)] encoding GST, GST–Skp1, HisSkp2, HisCul1(wt)HA, HisCul1(ΔC_Eco_RI), HisCul1(ΔC_Bst_E), HisCul1(Δ26)HA, HisCul1(ΔC63)HA, HisCul1(Δ127)HA, HisCul1(ΔB/S), HisCul1(Δ6)HA, HisCul1(ΔN63)HA, HisCul1(ΔNCla)HA, PyRoc1/Rbx1, Cdc34 and of yeast expression plasmids 415GPD–Cul1(wt)HA and mutant derivatives are available from the authors on request.
Antibodies, metabolic labelling, western blotting, immunoprecipitation and northern analysis
The mouse monoclonal antibodies 12CA5 and HA11 recognizing the HA-epitope were purchased from Boehringer Mannheim and BAbCo, respectively. Anti-Ubi-1 antibody was purchased from Zymed or Babco. Anti-GST antibodies, anti-His monoclonal antibody and anti-Cdk2 monoclonal antibody M2 were obtained from Sigma, Clontech and Santa Cruz. Anti-polyoma (Py) monoclonal antibodies recognizing the polyoma epitope were a kind gift from M.Peter (ISREC, Lausanne, Switzerland). Affinity-purified anti-(fl)Skp2 antibodies have been described before (Lisztwan et al., 1998). Anti-Cdc34 antibodies are described elsewhere (Reymond et al., 2000). Radioisotopic labelling was performed on indicated Rat1 pools by methionine starvation for 30 min followed by growth in 3 ml of methionine-free Dulbecco’s modified Eagle’s medium (DMEM) supplemented with [35S]methionine (250 µCi) and 10% dialysed fetal calf serum (FCS) for 30 min and chase for 1, 2 and 3 h at 37°C in a humidified 10% CO2 atmosphere. Immunoblotting was performed as described (Lisztwan et al., 1998). Blots were processed by enhanced chemiluminescence (Amersham) according to the manufacturer’s instructions. Immunoprecipitations were performed essentially as described previously (Lisztwan et al., 1998). Total RNA was isolated from T98G cells and HDF cells by the guanidinium thyocyanate–phenol/choroform extraction method and 20 µg of total RNA were resolved by electrophoresis on 1% agarose gel containing formaldehyde and transferred to nylon membrane (Hybond-N; Amersham). The same membrane was successively hybridized with 32P-labelled probes derived from human Skp2 and GAPDH cDNAs.
Cell culture, cell cycle analysis, retroviral infection and antisense experiments
Human T98G, HDFs, BOSC23 and Rat1 cells were maintained in DMEM supplemented with 10% FCS (Gibco). T98G, Rat1 and HDF cells were growth-arrested by incubation of cells for ∼72 h in DMEM containing 0.5% serum. Cells were serum stimulated by addition of FCS to a final concentration of 20%. LLnL and MG132 (1000× stock in DMSO) were added to HDFs or Rat1 cells at a final concentration of 50 µM. BOSC23 were transfected with a total of 30 µg of pCMV(neo-retro) plasmid DNA or its derivatives (containing the indicated Skp2 alleles) by calcium phosphate coprecipitation as described (Krek et al., 1995). Medium containing the retrovirus was harvested 24–30 h following removal of the precipitate and used to infect Rat1 cells at 30% confluency. Uncloned pools of neomycin-resistant Rat1 were generated as described using 1 mg/ml neomycin (Xu et al., 1995). For cell cycle analysis, cells were harvested by trypsinization at the indicated times and processed for FACS analysis as described (Lisztwan et al., 1998). Antisense experiments were performed as described (Z.K.Yu et al., 1998). The sequence of antisense oligodeoxynucleotides targeting Cul1 mRNA and control oligodeoxy nucleotides were identical to those described by Z.K.Yu et al. (1998) and were delivered into serum-starved HDFs using Cytofectin (GS2888) transfection agent (Glen Research) according to the manufacturer’s instructions.
Yeast complementation assay
YMP740 (Mata, ade2-1, trp1-1, leu2-3,112, his3-11,15, ura3, cdc53-2) was transformed with an empty control plasmid (415GPD; Mumberg et al., 1995) or with vectors expressing either wild-type or various mutant forms of Cul1 from the constitutive GPD promoter. Individual transformants were plated on selective medium and grown at either 25°C (permissive temperature) or at 34°C (restrictive temperature) for 3 days.
To monitor expression of wild-type or mutant forms of Cul1p, transformants were grown to early log phase in selective media at restrictive temperature (25°C), and then shifted to 34°C for 9 h. Cells were pelleted, lysed using standard procedures (Brown et al., 1997), and the protein levels determined by immunoblotting using anti-HA antibodies.
Expression of recombinant proteins in Sf9 insect cells and ubiquitylation assay
Insect Sf9 cells were cultivated in complete Grace’s medium containing 10% heat-inactivated FCS. Recombinant baculoviruses were generated using the BaculoGold Transfection Kit (Pharmingen), following the manufacturer’s instructions. The E1 activating enzyme used in ubiquitylation assays was from Affinity Inc. (Exeter, UK). Human E2 enzyme Cdc34 was expressed in Escherichia coli and purified as described (Banerjee et al., 1993). Both enzymes were tested for thioester formation capacity before use. For one ubiquitylation reaction GST or GST–Skp1 fusion protein was affinity purified from insect cell cultures that had been infected previously with the recombinant baculoviruses indicated or a combination thereof using 30 µl of a 50% slurry of GS beads per reaction and washed four times in Ubiquitination Assay Lysis (UAL) buffer [20 mM HEPES pH 7.4, 100 mM NaCl, 0.1% Triton X-100, 5 mM MgCl2, 5 mM EDTA, 0.2 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 µg/ml aprotinin]. Ubiquityl ation reactions were carried out in a total volume of 25 µl containing 15 µl of either GS beads loaded with indicated GST fusion proteins and 10 µl assay buffer containing 1 µl of 10× reaction buffer (40 mM MgOAc, 5 mM DTT and 1 mM PMSF), 1 µl of 10× ATP-regenerating system (20 mM HEPES pH 7.4, 10 mM ATP, 10 mM MgOAc, 300 mM creatine phosphate, 0.5 mg/ml creatine phosphokinase), 1 µl of 1× HEPES– KOAc buffer (20 mM HEPES pH 7.4, 100 mM KOAc, 0.5 mM DTT), 5 pmol fo ubiquitinaldehyde (Affiniti Inc.) (in 50 mM HEPES pH 6.9), 6 µg of ubiquitin (Sigma) (in 1× HEPES–KOAc buffer without DTT), 1 µg of purified human CDC34 (in 20 mM HEPES pH 7.4, 100 mM KOAc, 1 mM DTT and 10% glycerol) and 0.2 µg of purified E1 protein. The reaction mixture was incubated at 25°C for 1.5 h. To stop the reaction, 70 µl of sample buffer were added. Samples were boiled for 10 min, analysed by SDS–PAGE and processed for western blotting using the indicated antibody.
Acknowledgments
Acknowledgements
We wish to thank members of our laboratory for helpful discussions and advice. We thank Dr R.Deshaies (Caltech) for the cdc53(ts) yeast strain and P.Müller for synthesis of oligonucleotides. Special thanks go to J.Lisztwan (FMI) and Martin Scheffner (University of Köln) for critical reading of the manuscript. C.W. and F.R. are supported by the Novartis Research Foundation. H.S. is supported by a grant from the Roche Foundation. M.G. is supported by a grant from the Swiss Cancer League. M.B. is supported by a long-term EMBO fellowship. M.P. is a START fellow and is supported by the Swiss National Science Foundation and the Horton Foundation. W.K. is a START fellow and is supported by the Swiss National Science Foundation and by the Novartis Research Foundation.
References
- Bai C., Sen,P., Hofmann,K., Ma,L., Goebl,M., Harper,J.W. and Elledge,S.J. (1996) SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell, 86, 263–274. [DOI] [PubMed] [Google Scholar]
- Banerjee A., Gregori,L., Xu,Y. and Chau,V. (1993) The bacterially expressed yeast CDC34 gene product can undergo autoubiquitination to form a multiubiquitin chain-linked protein. J. Biol. Chem., 268, 5668–5675. [PubMed] [Google Scholar]
- Brown J.L., Jaquenoud,M. Gulli,M.-P. Chant,J. and Peter,M. (1997) Novel Cdc42 binding proteins Gic1 and Gic2 control cell polarity in yeast. Genes Dev., 11, 2972–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrano A., Eytan,E., Hershko,A. and Pagano,M. (1999) SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nature Cell Biol., 1, 193–199. [DOI] [PubMed] [Google Scholar]
- Deshaies R.J. (1999) SCF and cullin/Ring-H2-based ubiquitin ligases. Annu. Rev. Cell Dev. Biol., 15, 435–467. [DOI] [PubMed] [Google Scholar]
- Feldman R.M., Correll,C.C., Kaplan,K.B. and Deshaies,R.J. (1997) A complex of Cdc4p, Skp1p and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell, 91, 221–230. [DOI] [PubMed] [Google Scholar]
- Galan J.M. and Peter,M. (1999) Ubiquitin-dependent degradation of multiple F-box proteins by an autocatalytic mechanism. Proc. Natl Acad. Sci. USA, 96, 9124–9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamura T. et al. (1999) Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science, 284, 657–661. [DOI] [PubMed] [Google Scholar]
- Kipreos E.T., Lander,L.E., Wing,J.P., He,W.W. and Hedgecock,E.M. (1996) cul-1 is required for cell cycle exit in C.elegans and identifies a novel gene family. Cell, 85, 829–839. [DOI] [PubMed] [Google Scholar]
- Koepp D.M., Harper,J.W. and Elledge,S.J. (1999) How the cyclin became a cyclin: regulated proteolysis in the cell cycle. Cell, 97, 431–434. [DOI] [PubMed] [Google Scholar]
- Krek W. (1998) Proteolysis and the G1/S transition: the SCF connection. Curr. Opin. Genet. Dev., 8, 36–42. [DOI] [PubMed] [Google Scholar]
- Krek W. (2000) VHL takes HIF’s breath away. Nature Cell Biol., 2, E121–E123. [DOI] [PubMed] [Google Scholar]
- Krek W., Xu,G. and Livingston,D.M. (1995) Cyclin A-kinase regulation of E2F-1 DNA binding function underlies suppression of an S phase checkpoint. Cell, 83, 1149–1158. [DOI] [PubMed] [Google Scholar]
- Lisztwan J., Marti,A., Sutterlüty,H., Gstaiger,M., Wirbelauer,C. and Krek,W. (1998) Association of human CUL-1 and ubiquitin-conjugating enzyme CDC34 with the F-box protein p45(SKP2): evidence for evolutionary conservation in the subunit composition of the CDC34–SCF pathway. EMBO J., 17, 368–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyapina S.A., Correll,C.C., Kipreos,E.T. and Deshaies,R.J. (1998) Human CUL1 forms an evolutionarily conserved ubiquitin ligase complex (SCF) with SKP1 and an F-box protein. Proc. Natl Acad. Sci. USA, 95, 7451–7456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti A., Wirbelauer,C., Scheffner,M. and Krek,W. (1999) Interaction between the ubiquitin-protein ligase SCFSKP2 and E2F-1 underlies the regulation of E2F-1 degradation. Nature Cell Biol., 1, 14–19. [DOI] [PubMed] [Google Scholar]
- Mathias N., Johnson,S.L., Winey,M., Adams,A.E., Goetsch,L., Pringle,J.R., Byers,B. and Goebl,M.G. (1996) Cdc53p acts in concert with Cdc4p and Cdc34p to control the G1-to-S-phase transition and identifies a conserved family of proteins. Mol. Cell. Biol., 16, 6634–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel J.J. and Xiong,Y. (1998) Human Cul-1, but not other cullin family members selectively interacts with SKP1 to form a complex with SKP2 and cylin A. Cell Growth Differ., 9, 435–449. [PubMed] [Google Scholar]
- Mumberg D., Muller,R. and Funk,M. (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene, 156, 119–122. [DOI] [PubMed] [Google Scholar]
- Ohta T., Michel,J.J., Schottelius,A.J. and Xiong,Y. (1999a) ROC1, a homolog of APC11, represents a family of cullin partners with an associated ubiquitin ligase activity. Mol. Cell, 3, 535–541. [DOI] [PubMed] [Google Scholar]
- Ohta T., Michel,J. and Xiong,Y. (1999b) Association with cullin partners protects ROC proteins from proteasome-dependent degradation. Oncogene, 18, 6758–6766. [DOI] [PubMed] [Google Scholar]
- Patton E.E., Willems,A.R. and Tyers,M. (1998a) Combinatorial control in ubiquitin-dependent proteolysis: don’t Skp the F-box hypothesis. Trends Genet., 14, 236–243. [DOI] [PubMed] [Google Scholar]
- Patton E.E., Willems,A.R., Sa,D., Kuras,L., Thomas,D., Craig,K.L. and Tyers,M. (1998b) Cdc53 is a scaffold protein for multiple Cdc34/Skp1/F-box protein complexes that regulate cell division and methionine biosynthesis in yeast. Genes Dev., 12, 692–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond F., Wirbelauer,C. and Krek,W. (2000) Association of human ubiquitin-conjugating enzyme CDC34 with the mitotic spindle in anaphase. J. Cell Sci., 113, 1687–1694. [DOI] [PubMed] [Google Scholar]
- Roullion A., Barbey,R., Patton E.E., Tyers,M. and Thomas,D. (2000) Feedback-regulated degradation of the transcriptional activator Met4 is triggered by the SCFMet30 complex. EMBO J., 19, 282–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seol J.H. et al. (1999) Cdc53/cullin and the essential Hrt1 RING-H2 subunit of SCF define a ubiquitin ligase module that activates the E2 enzyme Cdc34. Genes Dev., 13, 1614–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra D., Craig,K.L., Tyers,M., Elledge,S.J. and Harper,J.W. (1997) F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell, 91, 209–219. [DOI] [PubMed] [Google Scholar]
- Skowyra D., Koepp,D.M., Kamura,T., Conrad,M.N., Conaway,R.C., Conaway,J.W., Elledge,S.J. and Harper,J.W. (1999) Reconstitution of G1 cyclin ubiquitination with complexes containing SCFGrr1 and Rbx1. Science, 284, 662–665. [DOI] [PubMed] [Google Scholar]
- Sutterlüty H., Chatelain,E., Marti,A., Wirbelauer,C., Senften,M., Muller,U. and Krek,W. (1999) p45SKP2 promotes p27KIP1 degradation and induces S phase in quiescent cells. Nature Cell Biol., 1, 207–214. [DOI] [PubMed] [Google Scholar]
- Tan P., Fuchs,S.Y., Chen,A., Wu,K., Gomey,C., Ronai,Y. and Pan,Z.-Q. (1999) Recruitment of a ROC1-CUL1 ubiquitin ligase by Skp1 and HOS to catalyze the ubiquitination of IκBα. Mol. Cell, 3, 527–533. [DOI] [PubMed] [Google Scholar]
- Tsvetkov L.M., Yeh,K.H., Lee,S.Y., Sun,H. and Zhang,H. (1999) p27 ubiquitination and degradation is regulated by the SCFSkp2 complex through phosphorylated thr187 in p27. Curr. Biol., 9, 661–664. [DOI] [PubMed] [Google Scholar]
- Willems A.R., Lanker,S., Patton,E.E., Craig,K.L., Nason,T.F., Mathias,N., Kobayashi,R., Wittenberg,C. and Tyers,M. (1996) Cdc53 targets phosphorylated G1 cyclins for degradation by the ubiquitin proteolytic pathway. Cell, 86, 453–463. [DOI] [PubMed] [Google Scholar]
- Wu K., Fuchs,S.Y., Chen,A., Tan,P., Gomez,C., Ronai,Z. and Pan,Z.-Q. (2000) The SCFHoS/b–TRCP-Roc1 E3 ubiquitin protein ligase utilizes two distinct domains within Cul1 for substrate targeting and ubiquitin ligation. Mol. Cell. Biol., 20, 1382–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G., Livingston,D.M. and Krek,W. (1995) Multiple members of the E2F transcription factor family are the products of oncogenes. Proc. Natl Acad. Sci. USA, 92, 1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Peters,J.-M., King,R.W., Page,A., Hieter,P. and Kirschner,M.W. (1998) Identification of a cullin homology region in a subunit of the anaphase-promoting complex. Science, 279, 1219–1222. [DOI] [PubMed] [Google Scholar]
- Yu Z.K., Gervais,J.L. and Zhang,H. (1998) Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc. Natl Acad. Sci. USA, 95, 11324–11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariae W. and Nasmyth,K. (1999) Whose end is destruction: cell division and the anaphase promoting complex. Genes Dev., 13, 2039–2058. [DOI] [PubMed] [Google Scholar]
- Zachariae W., Shevchenko,A., Andrews,P.D., Ciosk,R., Galova,M., Stark,M.J., Mann,M. and Nasmyth,K. (1998) Mass spectrometric analysis of the anaphase-promoting complex from yeast: identification of a subunit related to cullins. Science, 279, 1216–1219. [DOI] [PubMed] [Google Scholar]
- Zhang H., Kobayashi,R., Galaktionov,K. and Beach,D. (1995) p19Skp1 and p45Skp2 are essential elements of the cyclin A-CDK2 S phase kinase. Cell, 82, 915–925. [DOI] [PubMed] [Google Scholar]
- Zhou P. and Howley,P.M. (1998) Ubiquitination and degradation of the substrate recognition subunits of SCF ubiquitin-protein ligases. Mol. Cell, 2, 571–580. [DOI] [PubMed] [Google Scholar]