NUC-1, a Caenorhabditis elegans DNase II homolog, functions in an intermediate step of DNA degradation during apoptosis (original) (raw)
. 2000 Mar 1;14(5):536–548.
Abstract
One hallmark of apoptosis is the degradation of chromosomal DNA. We cloned the Caenorhabditis elegans gene nuc-1, which is involved in the degradation of the DNA of apoptotic cells, and found that nuc-1 encodes a homolog of mammalian DNase II. We used the TUNEL technique to assay DNA degradation in nuc-1 and other mutants defective in programmed cell death and discovered that TUNEL labels apoptotic cells only during a transient intermediate stage. Mutations in nuc-1 allowed the generation of TUNEL-reactive DNA but blocked the conversion of TUNEL-reactive DNA to a subsequent TUNEL-unreactive state. Completion of DNA degradation did not occur in the absence of cell-corpse engulfment. Our data suggest that the process of degradation of the DNA of a cell corpse occurs in at least three distinct steps and requires activities provided by both the dying and the engulfing cell.
Keywords: Caenorhabditis elegans, programmed cell death, DNA degradation, TUNEL, endonuclease, nuc-1
Apoptosis is important for the development of and homeostasis in metazoans, and both the morphological changes occurring in dying cells and the molecular mechanisms by which cells die are widely conserved (for review, see Ellis et al. 1991b; Steller 1995; Jacobson et al. 1997). One characteristic feature of apoptosis is the internucleosomal fragmentation of DNA into ∼180-bp repeats, often referred to as DNA laddering (Wyllie 1980). A number of candidate cell-death endonuclease activities have been characterized. CPAN/CAD (caspase-activated nuclease/caspase-activated DNase) and DFF45/ICAD (DNA fragmentation factor 45 kD/inhibitor of CAD) together constitute a DNase regulated by caspase cleavage (Liu et al. 1997; Enari et al. 1998; Halenbeck et al. 1998; Sakahira et al. 1998). Mutant mice that lack DFF45/ICAD are defective in chromatin condensation and DNA degradation during the cell-death process, but immune system development appears to be otherwise normal, suggesting that cells undergo programmed cell death even though cell-corpse DNA is not degraded normally (Zhang et al. 1998). Other candidate cell-death endonucleases include DNase I (Peitsch et al. 1992, 1993), DNase II (Barry and Eastman 1992, 1993; Odaka and Mizuochi 1999), inducible-lymphocyte Ca2+/Mg2+-dependent endonuclease (ILCME) (Khodarev and Ashwell 1996), and cyclophilins (Montague et al. 1997).
The TUNEL technique (Gavrieli et al. 1992) has been applied to studies of apoptosis in many organisms. Terminal deoxynucleotidyl transferase (TdT) is used to label DNA 3′-hydroxyl ends in situ with modified nucleotides detectable by fluorescence or immunohistochemistry. TUNEL specifically labels dying cells, because DNA degradation causes these cells to have more free DNA ends than do viable cells (Wyllie 1980; Oberhammer et al. 1993).
Fundamental questions about TUNEL remain to be answered. For instance, are all intermediates during DNA degradation TUNEL reactive, or does TUNEL label dying cells only during a specific stage of the death process? Time-course studies of DNA degradation in apoptotic mammalian epithelial cells by gel electrophoresis suggest that DNA degradation involves multiple steps (Oberhammer et al. 1993). Specifically, the chromosomal DNA of some apoptotic cells is cleaved at the chromatin loop domains, generating 50-kb fragments. The subsequent cleavage of DNA at the internucleosomal linker region produces the characteristic 180-bp DNA ladder.
Two findings suggest that the DNA fragments generated by DNA laddering may be those detected by TUNEL staining. First, in apoptotic lymphocytes, the DNA fragments generated by DNA laddering contain 3′-hydroxyl ends (Alnemri and Litwack 1990), which should be substrates for TdT. Second, the appearance of TUNEL-positive signals correlates temporally with the appearance of DNA laddering in dexamethasone-treated apoptotic thymocytes in culture (Gavrieli et al. 1992).
Programmed cell death in the nematode Caenorhabditis elegans is morphologically and molecularly similar to mammalian apoptosis (for review, see Metzstein et al. 1998). In C. elegans, cells undergoing programmed cell death adopt a refractile, raised button-like appearance as observed by Nomarski differential interference contrast optics (Sulston and Horvitz 1977). The time and place of death are known for each cell programmed to die and are essentially invariant from animal to animal. During the development of the hermaphrodite, 131 of the 1090 somatic cells generated undergo programmed cell death (Sulston and Horvitz 1977; Sulston et al. 1983). Most deaths (113/131) occur during embryogenesis, many (111) during a short period between 220 and 470 min after fertilization (Sulston et al. 1983).
The killing step of programmed cell deaths in C. elegans is controlled by the genes egl-1, ced-9, ced-4, and ced-3 [(egl) egg-laying defective; (ced) cell death abnormal]. Loss-of-function (lf) mutations in egl-1, ced-4, ced-3, or a gain-of-function (gf) mutation in ced-9 cause cells that normally die to survive. Each of these worm cell-death proteins has one or more mammalian counterparts that also have been implicated in cell death: EGL-1, BH3-only Bcl-2-family proteins; CED-9, Bcl-2 family; CED-4, Apaf-1; and CED-3, caspases (for review, see Metzstein et al. 1998).
Dying cells in C. elegans are swiftly engulfed by neighboring cells (for review, see Ellis et al. 1991b). Six genes, ced-1, ced-2, ced-5, ced-6, ced-7, and ced-10, important for cell-corpse engulfment have been characterized (Hedgecock et al. 1983; Ellis et al. 1991a). Mutations in any of these genes result in the persistence of many unengulfed cell corpses. In ced-1 and ced-2 mutants, Feulgen-reactive material is visible in the persistent cell corpses (Hedgecock et al. 1983), indicating that DNA degradation does not proceed to completion. Thus, degradation of the DNA in cell corpses requires the ced-1 and ced-2 genes and most likely the process of engulfment.
One C. elegans gene, nuc-1 (nuclease abnormal), is important for DNA degradation but does not appear to be involved in either the killing step of cell death or the engulfment of cell corpses (Sulston 1976). In nuc-1 mutants, both cell death and engulfment occur, but the pycnotic DNA of dead cells is not degraded and persists as a compact mass of Feulgen-reactive material (Sulston 1976; Hedgecock et al. 1983). The nuc-1 gene is also required to digest the DNA of the bacteria on which the animals feed: Persistent bacterial DNA can be detected in the intestinal lumina of nuc-1 mutants, but not in those of wild-type animals (Sulston 1976). An endonuclease activity present in protein extracts from wild-type animals is greatly reduced in nuc-1 mutants, suggesting that nuc-1 controls this activity (Hevelone and Hartman 1988).
To analyze the control of DNA degradation during programmed cell death, we developed a TUNEL protocol for C. elegans, used this protocol to assay DNA degradation in the wild-type embryo and in mutants defective in different aspects of the cell-death process, and cloned the nuc-1 gene.
Results
TUNEL labels a subset of dying cells in C. elegans
We developed a TUNEL protocol for C. elegans (see Materials and Methods) and used this protocol to stain wild-type embryos and count the number of TUNEL-positive nuclei. We chose to count nuclei in 1.5-fold stage embryos (∼420 min after fertilization) for two reasons. First, embryos progress through this easily recognizable stage in <20 min. Such a short time window should give highly reproducible TUNEL-staining patterns from embryo to embryo. Second, embryos at this stage contain an average of 14 refractile, dying cells (Table 1); prior to this stage, 68 cells have already died and been engulfed (Sulston et al. 1983). Therefore, at this developmental stage we could monitor DNA degradation both in cells that are dying and cells that died earlier and have been engulfed.
Table 1.
TUNEL specifically labels dying cells
| Genotype | No. of TUNEL-positive cellsa | Range of TUNEL-positive cells | No. of cell corpsesb |
|---|---|---|---|
| Wild type | 1.7 ± 1.3 | 0–4 | 14 ± 1 |
| ced-3(n717) | 0.1 ± 0.2 | 0–1 | 0.0 ± 0.0 |
| ced-4(n1162) | 0.0 ± 0.0 | 0–1 | 0.0 ± 0.2 |
| ced-9(n1950) | 0.1 ± 0.4 | 0–1 | 0.1 ± 0.3 |
We found that in labeled wild-type 1.5-fold embryos an average of 1.7 nuclei were TUNEL-positive (Table 1; Fig. 1A). TUNEL-positive signals colocalized with DNA as assayed by DAPI staining (Fig.1C,D; data not shown). The refractile appearance of dying cells by Nomarski optics was not retained during fixation, so we could not identify cell corpses by their morphology in these fixed embryos. To confirm that TUNEL staining was specific to dying cells, we examined TUNEL-staining patterns in embryos of ced-9(n1950gf) (Hengartner et al. 1992), ced-4(n1162), and ced-3(n717) (Ellis and Horvitz 1986) mutants, in which nearly all programmed cell deaths are blocked. We detected almost no TUNEL-reactive nuclei in these mutants (Table 1), confirming that the TUNEL-reactive nuclei in wild-type animals were those of dying cells.
Figure 1.
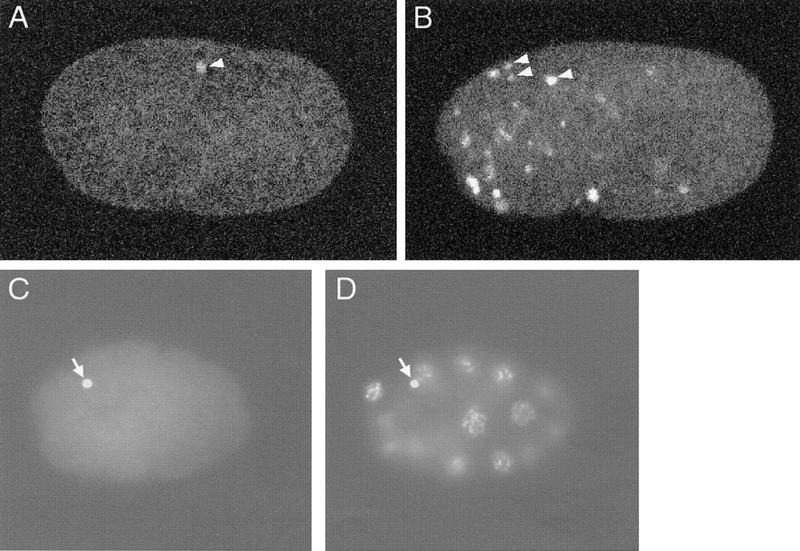
TUNEL-staining patterns of wild-type and nuc-1 embryos. (A) Wild-type 1.5-fold embryo stained with TUNEL. (B) nuc-1(e1392) 1.5-fold embryo stained with TUNEL. (C,D) Wild-type 16-cell embryo showing TUNEL (C) and DAPI staining (D). For A and B, each picture is a projection of eight serial confocal images. The white signals (some are indicated with white arrowheads) are TUNEL-positive nuclei. For C and D, arrows indicate the polar bodies.
TUNEL also labels polar bodies
The mature C. elegans oocyte is arrested at diakinesis of meiotic prophase I. Meiosis is completed after fertilization, and two polar bodies are generated (Hirsh et al. 1976). In wild-type embryos prior to the 1.5-fold stage, we occasionally observed TUNEL staining of polar bodies (Fig. 1C,D). Because the polar-body DNA stains with TUNEL in ced-3(n717), ced-4(n1162), and ced-9(n1950) embryos, these mutations do not prevent all TUNEL staining, but rather, that specifically resulting from programmed cell death.
nuc-1 embryos have more TUNEL-reactive nuclei than do wild-type embryos
In nuc-1 mutants, programmed cell death and cell-corpse engulfment still occur, but the DNA of dead cells is not completely degraded (Sulston 1976; Hedgecock et al. 1983). We observed many more TUNEL-reactive nuclei in nuc-1 embryos than in wild-type embryos (Table 2; Fig. 1B). Similar results were obtained with each of the three alleles of nuc-1, e1392, n334, and n887. All of these mutants contained the normal number of refractile corpses as compared with the wild type when observed by Nomarski microscopy (Table 2), suggesting that the killing step of programmed cell death is not affected in these mutants.
Table 2.
TUNEL labels persistent DNA in nuc-1 animals
| Genotype | No. of TUNELpositive cells (n = 45) | Range of TUNELpositive cells | No. of cell corpses (n = 15) |
|---|---|---|---|
| nuc-1(e1392) | 47.8 ± 4.8 | 38–64 | 14 ± 2 |
| nuc-1(n334) | 45.3 ± 6.2 | 36–60 | 14 ± 2 |
| nuc-1(n887) | 47.5 ± 5.3 | 35–59 | 14 ± 1 |
| ced-3(n717); nuc-1(e1392) | 0.4 ± 0.7 | 0–2 | 0.0 ± 0.0 |
| ced-4(n1162); nuc-1(e1392) | 0.2 ± 0.6 | 0–2 | 0.1 ± 0.2 |
| ced-9(n1950); nuc-1(e1392) | 4.2 ± 1.8 | 0–9 | 0.1 ± 0.4 |
To address whether the increase in TUNEL-reactive nuclei in nuc-1 embryos is specific to programmed cell death, we examined TUNEL-staining patterns of ced-9(n1950gf); nuc-1(e1392), ced-4(n1162); nuc-1(e1392), and ced-3(n717); nuc-1(e1392) embryos. These stained embryos had almost no TUNEL-positive signals (Table 2), showing that the presence of TUNEL-reactive DNA in nuc-1 embryos, just as in wild-type embryos, was dependent on the occurrence of programmed cell death.
DNA degradation is normally rapid and is slowed in nuc-1 mutants
Because we observed only an average of 1.7 TUNEL-positive cells in wild-type embryos, it was possible that our TUNEL technique was not sensitive enough to detect all dying cells. An alternative possibility was that DNA degradation occurs rapidly and that only certain transient intermediates are TUNEL reactive during the process of cell death. The latter hypothesis is supported by our finding that labeled nuc-1 embryos had many more TUNEL-positive signals than wild-type embryos. Time-course studies of the TUNEL-staining patterns in nuc-1 mutants (Fig. 2; data not shown) indicate that at least the majority of cell corpses become TUNEL reactive with wild-type kinetics in nuc-1 mutants (i.e., TUNEL-reactive cells appear with the same timing as cell corpses appear as visualized by Nomarski microscopy; Sulston et al. 1983), suggesting that the step of DNA degradation by which cell-corpse DNA becomes TUNEL reactive is not slowed in nuc-1 mutants. We hypothesize that nuc-1 mediates a subsequent step of the DNA degradation process involving either the masking or elimination of TUNEL-reactive DNA ends.
Figure 2.
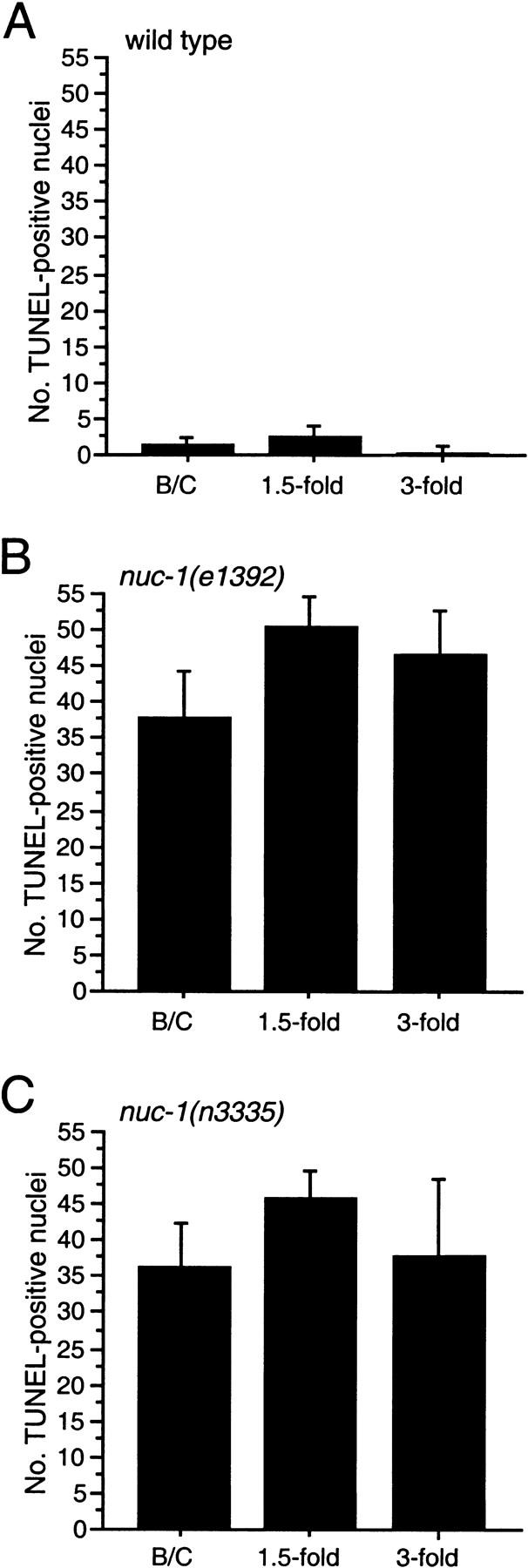
n3335 TUNEL staining is like that of nuc-1(e1392). Kinetics of TUNEL-reactive nuclei in wild-type (A), nuc-1(e1392) (B), and nuc-1(n3335) (C) mutant embryos. The _y_-axis indicates the number of TUNEL-positive nuclei in stained embryos at the bean and comma (B/C), 1.5-fold, or 3-fold stages. Error bars, s.d.
How long does DNA persist in the TUNEL-reactive state in nuc-1 mutants? To address this question, we examined animals fixed and stained at later stages of embryogenesis. We observed many TUNEL-reactive nuclei in all nuc-1 3.5-fold elongated embryos (Fig. 2; data not shown). For example, in stained nuc-1(e1392) 3.5-fold embryos, we observed an average of 46 and a minimum of 30 TUNEL-positive cells. The 3.5-fold stage of embryogenesis corresponds to the period between ∼520 min after fertilization and hatching at ∼800 min; during this period, only two cells die (Sulston et al. 1983). Our data for early embryonic stages suggest that mutations in nuc-1 do not delay significantly the onset of TUNEL reactivity. Therefore, many nuclei apparently remain TUNEL reactive for at least several hours in nuc-1 animals.
nuc-1 encodes a DNase II homolog
To understand the nature of the nuc-1 activity, we cloned the nuc-1 gene. We mapped nuc-1 to the interval between the cloned genes ced-8 and egl-15 and near or to the left of unc-115 (Fig. 3A; see Materials and Methods). We observed that a predicted gene, C07B5.5, encoding a protein with strong similarity to DNase II (Baker et al. 1998; Krieser and Eastman 1998; Yasuda et al. 1998) lies within this region. The sequence of C07B5.5 is ∼30% identical to that of human DNase II and ∼16% identical to that of mouse DLAD (DNase II-like acid DNase; Shiokawa and Tanuma 1999). Structural features are conserved between C07B5.5 and the mammalian endonuclease proteins: each has a signal sequence, and the active site histidine is conserved (Baker et al. 1998; Krieser and Eastman 1998; Yasuda et al. 1998). Because the biochemical characteristics of DNase II (e.g., Baker et al. 1998) are similar to those of the putative nuc-1 activity (Hevelone and Hartman 1988), we sought to determine whether nuc-1 corresponds to C07B5.5.
Figure 3.
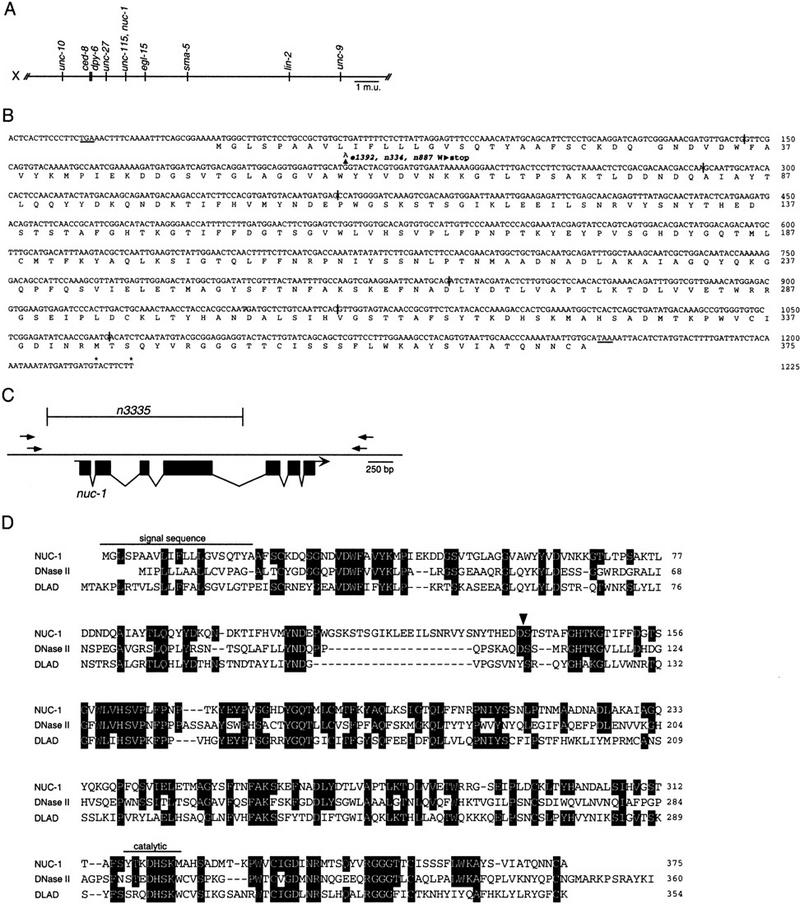
nuc-1 cloning. (A) Genetic map location of the nuc-1 region of the X chromosome (see Materials and Methods). (B) A composite nuc-1 transcript derived from genomic, cDNA, and 5′ RACE analysis. Intron positions are indicated by vertical bars. The deduced NUC-1 protein sequence is shown beneath. (Arrowhead) The base change in the three nuc-1 alleles. An in-frame stop codon upstream of the predicted ATG and the termination codon are underlined. (Asterisks) Locations of polyadenylation sites. (C) Deletion of C07B5.5 identified by PCR screening. The bracketed area indicates the extent of the deletion in n3335. (D) Alignment of NUC-1, human DNase II (Krieser and Eastman 1998), and mouse DLAD (Shiokawa and Tanuma 1999). Amino acids identical to NUC-1 are shaded. (Arrowhead) The site of cleavage in human DNase II. We have aligned the most similar nearby sequences in NUC-1 with the human DNase II cleavage site to indicate a possible cleavage site; it is not known whether the worm protein is processed analogously.
We first confirmed that the splicing pattern predicted for C07B5.5 by the C. elegans Genome Sequencing Consortium is correct by determining the sequences of cDNAs (generously provided by Yuji Kohara). We found that the cDNA yk434c9 contains the entire coding sequences of C07B5.5. We then determined the sequence of the C07B5.5 coding region and intron-exon junctions from the wild-type and the three nuc-1 alleles, e1392, n334, and n887. We found that all three mutants contain an identical change relative to the wild-type sequence, a G-to-A transition that is predicted to convert Trp59 to a TAG (amber) stop codon (Fig. 3B). The nature of this lesion is consistent with genetic data that indicate that e1392 is amber suppressible (Waterston and Brenner 1978). That all three mutants contain the same base change is consistent with biochemical assays (Hevelone and Hartman 1988) and our TUNEL assays (Table 2; data not shown), which revealed no phenotypic differences among these alleles.
That all three existing nuc-1 alleles contain the same base change in the C07B5.5 gene raised the possibility that this lesion represents a commonly occurring background mutation unrelated to the Nuc-1 phenotype. Therefore, we sought additional alleles of C07B5.5 using PCR to screen a library of mutagenized worms (Jansen et al. 1997) for animals carrying a deletion at the C07B5.5 locus. We identified the mutation n3335, a deletion of 2034 bp that extends from 363 bp before the predicted translation start of C07B5.5 to 166 bp before the start of exon 5 (Fig. 3C). n3335 also is associated with a 2-bp insertion (sequence TA) at the deletion site. Because most of the C07B5.5 coding region was eliminated in n3335, we expected that no functional protein should be produced.
We examined n3335 animals to determine whether they are phenotypically Nuc. In nuc-1 mutant animals, pycnotic DNA of dying cells persists (Sulston 1976; Hedgecock et al. 1983). We stained animals using the vital DNA-binding dye Syto 11 (see Materials and Methods) and scored for DNA persisting from the P-lineage cell deaths, which occur during the L1 and L2 stages in an approximately linear array along the ventral cord (Sulston and Horvitz 1977). As compared with wild-type animals, n3335 larvae contained extra Syto 11-positive DNA that appeared similar to that present in nuc-1(e1392) and nuc-1(n887) mutants (data not shown). Approximately one-fourth of the progeny of n3335/+ animals contained extra Syto 11-positive nuclei, indicating that this effect of n3335 is recessive. We also observed bright staining within the intestinal lumina of n3335 animals (data not shown). We then used TUNEL to stain n3335 embryos and found that the pattern of positive nuclei in n3335 embryos was indistinguishable from that caused by the nuc-1 allele e1392 (Fig. 2). Therefore, n3335 causes a Nuc phenotype like that of the known mutations in nuc-1.
We tested whether the n3335 mutation genetically complements the nuc-1 allele e1392, which is recessive (data not shown). We used Syto 11 to stain animals _trans_-heterozygous for the two alleles and found that, like either single mutant, these animals contained extra Syto 11-positive nuclei in the ventral cord (data not shown; see Materials and Methods). These data establish that n3335 is an allele of nuc-1 and confirm that nuc-1 corresponds to the DNase II-like gene C07B5.5.
Cell-corpse engulfment is not required for nuc-1 activity
The observation that unengulfed cell corpses in ced-1 and ced-2 mutants contain Feulgen-reactive DNA that is not pycnotic (Hedgecock et al. 1983) suggests that the engulfment process is necessary for complete DNA degradation during programmed cell death in C. elegans. To determine whether _nuc-1_-mediated conversion of TUNEL-positive to TUNEL-negative cell-corpse DNA is dependent on engulfment by neighboring cells, we examined TUNEL-staining patterns in mutants in which the engulfment of many cell corpses is delayed or blocked.
We found that ced-2, ced-5, ced-6, and ced-10 mutant embryos, although they contained many unengulfed cell corpses at the 1.5-fold stage, had no more TUNEL-reactive nuclei than did wild-type embryos (Table 3). ced-2; nuc-1, ced-5; nuc-1, ced-6; nuc-1, and ced-10; nuc-1 embryos had the same number of TUNEL-reactive nuclei as did nuc-1 embryos (Table 3). We obtained equivalent results for multiple alleles of ced-2, ced-5, and ced-6 and for the single described allele of ced-10. In addition, ced-2; nuc-1, ced-5; nuc-1, ced-6; nuc-1, and ced-10; nuc-1 embryos frequently displayed TUNEL staining in unengulfed cell corpses that had detached from embryos and had been shed into egg fluid (Fig. 4). These results indicate that the engulfment genes ced-2, ced-5, ced-6, and ced-10 are not required for the initial step of DNA degradation during which TUNEL-reactive DNA ends are generated or for the _nuc-1_-mediated step of DNA degradation during which TUNEL-reactive DNA ends are eliminated. Furthermore, engulfment per se is not required for these steps of DNA degradation; that is, engulfment blocks aspects of DNA degradation not because NUC-1 activity is blocked but, rather, because a DNase downstream of NUC-1 is blocked.
Table 3.
Mutations in ced-2, ced-5, ced-6 and ced-10 do not alter TUNEL staining
| Genotype | No. of TUNELpositive cells (n = 45) | Range of TUNELpositive cells | No. of cell corpses (n = 15) |
|---|---|---|---|
| Wild type | 1.7 ± 1.3 | 0–4 | 14 ± 1 |
| ced-2(e1752) | 1.5 ± 1.1 | 0–3 | 21 ± 3 |
| ced-2(n1994) | 1.7 ± 1.4 | 0–3 | 20 ± 3 |
| ced-5(n1812) | 1.5 ± 1.0 | 0–4 | 39 ± 5 |
| ced-5(n2691) | 1.4 ± 1.2 | 0–3 | 34 ± 4 |
| ced-6(n2095) | 1.4 ± 0.9 | 0–3 | 29 ± 4 |
| ced-6(n1813) | 1.6 ± 1.4 | 0–3 | 33 ± 5 |
| ced-10(n1993) | 1.4 ± 1.3 | 0–3 | 31 ± 4 |
| nuc-1(e1392) | 47.8 ± 4.8 | 38–64 | 14 ± 2 |
| ced-2(e1752); nuc-1(e1392) | 44.6 ± 4.6 | 37–51 | 23 ± 3 |
| ced-2(n1994); nuc-1(e1392) | 47.6 ± 4.9 | 41–56 | 21 ± 3 |
| ced-5(n1812); nuc-1(e1392) | 45.1 ± 5.8 | 39–59 | 38 ± 5 |
| ced-5(n2691); nuc-1(e1392) | 44.6 ± 4.6 | 35–56 | 35 ± 4 |
| ced-6(n2095); nuc-1(e1392) | 46.2 ± 4.5 | 33–54 | 30 ± 5 |
| ced-6(n1813); nuc-1(e1392) | 43.6 ± 4.6 | 35–52 | 32 ± 5 |
| ced-10(n1993); nuc-1(e1392) | 42.6 ± 4.3 | 37–51 | 33 ± 4 |
Figure 4.

Unengulfed cell corpses in engulfment-defective mutants are TUNEL positive. (A) A ced-6(n1813); nuc-1(e1392) embryo stained with TUNEL. (B) The same embryo observed using Nomarski microscopy. A TUNEL-positive unengulfed cell corpse, which is excluded from the embryo, is indicated by an arrow.
Completion of DNA degradation during programmed cell death requires engulfing cells
We stained ced-2, ced-5, ced-6, and ced-10 engulfment mutants using Syto 11. We found that Syto 11 labeled the DNA of both living and dying cells and that pycnotic nuclei of unengulfed cell corpses in all of these mutants were Syto 11-positive (Fig. 5; data not shown). Feulgen staining of persistent cell corpses of ced-2 mutants had indicated previously that DNA degradation is incomplete in ced-2 animals (Hedgecock et al. 1983). That DNA is present in unengulfed cell corpses suggests that some step(s) of DNA degradation must either require activities provided by engulfing cells or be activated by the engulfment process.
Figure 5.
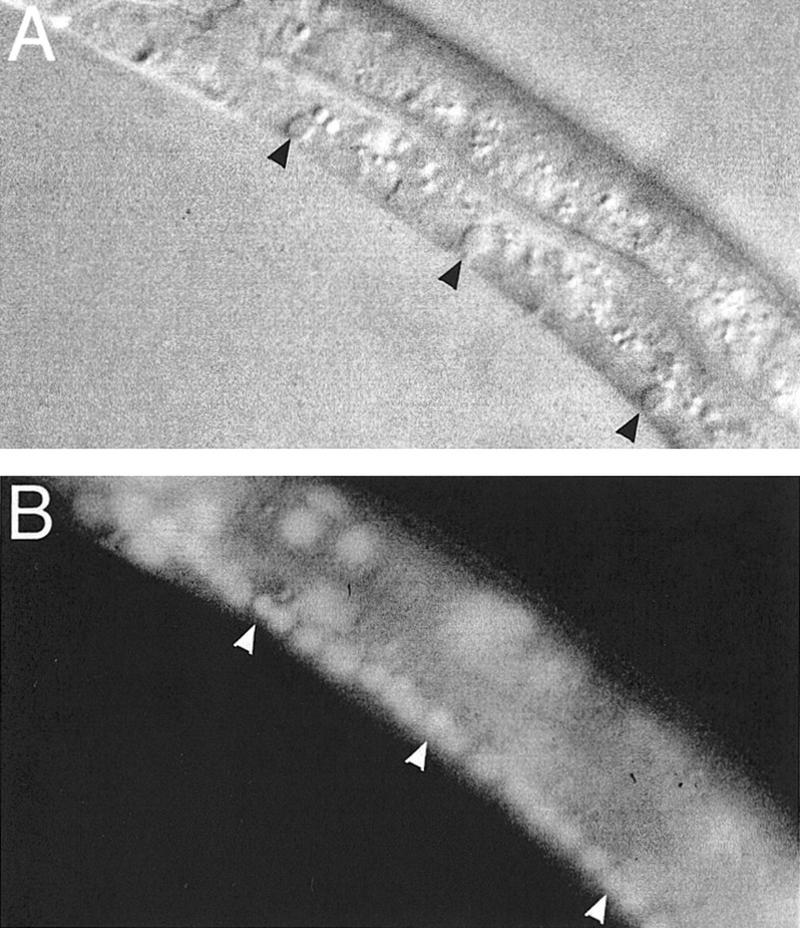
Unengulfed cell corpses stain with Syto 11. (A) A ced-6(n1813) L2 larva with unengulfed cell corpses (black arrowheads) in the ventral cord observed by Nomarski microscopy. (B) Syto 11 staining of the same larva; staining is visible within both live nuclei and those of the unengulfed corpses (white arrowheads).
The engulfment genes ced-1 and ced-7 are involved in the initiation of DNA degradation
Mutations in the genes ced-1 and _ced-7_—like mutations in ced-2, ced-5, ced-6, and _ced-10_—block or delay the engulfment of many cell corpses (Hedgecock et al. 1983; Ellis et al. 1991a). However, when we stained ced-1 and ced-7 embryos, we found a reduction in TUNEL-reactive nuclei in both of these mutants as compared with the wild type. ced-1 embryos had very few TUNEL-staining nuclei in either a wild-type or nuc-1 background (Table 4). Mutations in ced-7 also reduced the number of TUNEL-reactive nuclei but to a lesser degree. For example, ced-7; nuc-1 embryos had fewer than half the TUNEL-reactive nuclei of nuc-1 embryos (Table 4). We observed similar staining patterns using multiple alleles of ced-1 and ced-7. The reduction in TUNEL staining did not correlate with the strength of the engulfment defect (ced-1 mutants had a greater effect on TUNEL staining but a smaller effect on engulfment than did ced-7 mutants), suggesting that the DNA-degradation functions of ced-1 and ced-7 are not identical to their functions in cell-corpse engulfment.
Table 4.
Mutations in ced-1 and ced-7 reduce TUNEL staining
| Genotype | No. of TUNELpositive cells (n = 45) | Range of TUNELpositive cells | No. of cell corpses (n = 15) |
|---|---|---|---|
| Wild type | 1.7 ± 1.3 | 0–4 | 14 ± 1 |
| ced-1(e1735) | 0.5 ± 0.7 | 0–3 | 24 ± 3 |
| ced-1(n1995) | 0.3 ± 0.2 | 0–2 | 25 ± 4 |
| ced-7(n2094) | 0.7 ± 0.8 | 0–3 | 34 ± 3 |
| ced-7(n1892) | 0.5 ± 0.4 | 0–2 | 35 ± 5 |
| nuc-1(e1392) | 47.8 ± 4.8 | 38–64 | 14 ± 2 |
| ced-1(e1735); nuc-1(e1392) | 1.0 ± 1.0 | 0–3 | 26 ± 3 |
| ced-1(n1995); nuc-1(e1392) | 1.2 ± 1.0 | 0–5 | 27 ± 5 |
| ced-7(n2094); nuc-1(e1392) | 21.1 ± 1.3 | 15–28 | 38 ± 3 |
| ced-7(n1892); nuc-1(e1392) | 22.3 ± 3.2 | 14–29 | 37 ± 5 |
| ced-7(n1996); nuc-1(e1392) | 19.0 ± 1.7 | 16–26 | 38 ± 4 |
The reduction of TUNEL signals in ced-1 and ced-7 mutants was not caused by the disappearance of DNA in cell corpses, because unengulfed cell corpses in these mutants stain with Syto 11 (data not shown). Also, cell corpses in ced-1 mutants were shown previously to contain Feulgen-reactive DNA (Hedgecock et al. 1983). Therefore, TUNEL staining may be reduced in ced-1 and ced-7 mutants because the degradation of cell-corpse DNA is blocked prior to the generation of TUNEL-reactive ends. These data suggest that ced-1 and ced-7 are involved in the generation of TUNEL-reactive DNA ends, which our previous data indicate is likely to occur prior to engulfment of the dying cell.
Discussion
We have shown that the gene nuc-1, which is important for the process of DNA degradation during programmed cell death in C. elegans, encodes a protein similar to mammalian DNase II and the related DLAD protein. Previous results had indicated that an endonuclease activity detectable in wild-type C. elegans protein extracts is greatly reduced in or absent from protein extracts from nuc-1 mutants (Sulston 1976; Hedgecock et al. 1983; Hevelone and Hartman 1988). This endonuclease activity resembles that of mammalian DNase II in that neither requires Ca2+ or Mg2+ and they have a similar acidic pH optimum (Hevelone and Hartman 1988; Barry and Eastman 1992). DNase II has been suggested previously to act in DNA degradation during programmed cell death (Barry and Eastman 1992, 1993; Torriglia et al. 1995; Krieser and Eastman 1998). For instance, DNase II can induce DNA laddering in isolated nuclei (Barry and Eastman 1993) and has been localized to the nuclei of lens fibers undergoing an apoptosis-like death (Torriglia et al. 1995). However, many endonucleases have been suggested to function in apoptotic cells (Hughes and Cidlowski 1994). Our finding that nuc-1 encodes a DNase II-like protein provides in vivo evidence that such an endonuclease is important during programmed cell death. In addition, McIlroy et al. (2000) have obtained evidence suggesting that DNase II acts during the apoptotic death of mammalian cells.
We adapted the TUNEL technique, which labels dying cells, for use in C. elegans. We identified 47 TUNEL-reactive nuclei in 1.5-fold nuc-1 embryos, in which 68 cells have died and been engulfed and an additional 14 display the characteristic refractility of dying cells. In contrast, fewer than two nuclei were TUNEL reactive in 1.5-fold wild-type embryos. These data suggest that DNA degradation occurs more quickly in the wild-type than in nuc-1 mutants and that only certain normally transient intermediates are TUNEL reactive. Specifically, cell-corpse DNA appeared to become TUNEL reactive at the appropriate time in nuc-1 embryos, but was blocked or at least delayed in progressing to a subsequent TUNEL-unreactive state.
The enzymatic activity of DNase II is consistent with our suggestion that nuc-1 generates DNA fragments that are not TUNEL reactive. Under the reaction conditions used for TUNEL assays, the TdT polymerase extends 3′-hydroxyl ends of single-stranded or double-stranded DNA molecules with blunt, recessed, or overhanging ends (Roychoudhury et al. 1976). The cleavage of DNA by mammalian DNase II generates 5′-hydroxyl and 3′-phosphate ends, neither of which is a substrate for TdT.
How might a DNase II-like activity function to eliminate or mask TUNEL-reactive DNA ends? Perhaps NUC-1 cleaves TUNEL-reactive DNA to generate substrates that are rapidly further degraded by other enzymes. It is also possible that NUC-1 has an activity distinct from its presumptive DNase II-like activity.
Regulation of NUC-1 activity
How might the activity of nuc-1 be regulated? The _nuc-1_-dependent enzyme has an acidic pH optimum (Hevelone and Hartman 1988), so it is not expected to be active at the physiological pH of living cells. Some apoptotic mammalian cells undergo intracellular acidification (Barry and Eastman 1992). Such acidification, if it occurs during programmed cell death in C. elegans, could activate NUC-1.
NUC-1 activity also might be regulated at the level of either protein processing or subcellular localization. The maturation of human DNase II involves cleavage of the proprotein to produce fragments of ∼30 and 10 kD (Baker et al. 1998; Krieser and Eastman 1998; Yasuda et al. 1998). Although the larger subunit appears to have endonuclease activity on its own (Krieser and Eastman 1998), a biochemically purified form of DNase II has been found to be a heterodimer of 35 and 10 kD peptides (Liao 1985; Barry and Eastman 1993), which might correspond to the two cleavage products of pro-DNase II. The cleavage site of human DNase II lies after an aspartate residue (Baker et al. 1998; Krieser and Eastman 1998; Yasuda et al. 1998), raising the possibility that a caspase (Thornberry and Lazebnik 1998) might directly cleave and thereby activate DNase II in dying cells. However, the cleavage site (KAQDS in DNase II, which aligns with HEDDS in NUC-1) is not obviously conserved and the C. elegans site does not conform to the consensus cleavage site for caspases in general or the CED-3 caspase in particular (Xue et al. 1996; Talanian et al. 1997; Thornberry et al. 1997). Also, if cleavage by CED-3 were required for NUC-1 activation, then ced-3 mutants, like nuc-1 mutants (Sulston 1976), would have persistent bacterial DNA in the intestinal lumen, but they do not (Ellis and Horvitz 1986; Y.C. Wu, G.M. Stanfield, and H.R. Horvitz, unpubl.). Additional caspase genes in C. elegans have been identified recently and shown to encode functional proteases (Shaham 1998), so it is possible that one or more of these caspases, rather than CED-3, cleaves NUC-1. Alternatively, another protease might process NUC-1, at least in the intestine and possibly in dying cells as well. Once activated, NUC-1 must enter the nucleus to gain access to DNA; disassembly of the nuclear envelope, which is a feature of apoptosis (Rao et al. 1996), may be important for this process.
As noted above, nuc-1 is important not only for the degradation of the DNA of dying cells but also for the degradation of the DNA of bacteria ingested by the worm as food in the intestine (Sulston 1976). NUC-1 is presumably secreted into the intestinal lumen to act on bacterial DNA and could be secreted from dying and/or engulfing cells. Mammalian DNase II has been reported to be secreted into the medium when expressed in some mammalian cell types (Baker et al. 1998). NUC-1, like mammalian DNase II, has a predicted signal sequence at its amino terminus, consistent with the hypothesis that NUC-1 can be secreted.
Multiple DNases may act in DNA degradation
Our data suggest that DNA degradation during programmed cell death involves at least three distinct steps and that nuc-1 is not the only DNase activity required for this process (Fig. 6). During the first step, TUNEL-reactive DNA ends are generated. nuc-1 is not required for this process, because cells become TUNEL reactive in nuc-1 mutants. However, the ced-1 gene is required for and the ced-7 gene facilitates this process, as mutations in ced-1 eliminate and in ced-7 reduce the number of TUNEL-staining cells. The ced-7(n2094) and ced-7(n1996) alleles are likely to be null, on the basis of the nature of the molecular lesions in these mutants (Wu and Horvitz 1998a). Therefore, the higher level of TUNEL staining in ced-7 embryos relative to that in ced-1 embryos probably is not a consequence of residual ced-7 activity. One possibility is that ced-7 acts with another gene or genes in a partially redundant way to regulate the generation of TUNEL-reactive DNA ends during programmed cell death.
Figure 6.

Model for DNA degradation during programmed cell death in C. elegans. (A) The pathway for DNA degradation, as defined by TUNEL and Syto 11 staining of wild-type and mutant embryos, involves three distinct steps. Each horizontal arrow represents one or more nuclease activities; blocking a step prevents or delays the occurrence of subsequent steps. The first step requires the activity of ced-1 and ced-7 and involves an unknown endonuclease that generates TUNEL-reactive DNA. The second step is mediated by the DNase II-like NUC-1 endonuclease. The third step requires cell-corpse engulfment, because it is blocked in unengulfed corpses of engulfment-defective mutants. (B) Schematic diagram of steps in the pathway of DNA degradation. Each solid arrow indicates the possible translocation of a nuclease. First, ced-1 and ced-7 act to promote the initiation of DNA degradation, possibly by promoting interaction(s) between the dying cell and the engulfing cell (indicated by open arrows). Second, nuc-1 DNase II-like activity mediates the degradation of cell-corpse DNA to a TUNEL nonreactive state. These first two degradation steps do not require cell-corpse engulfment and occur within the dying cell. Third, a nuclease activity, which is dependent on cell-corpse engulfment and therefore may well be provided by the engulfing cell, completely eliminates cell-corpse DNA. The small arrows inside the engulfing cell indicate the directions of cell-surface extension.
How might ced-1 and ced-7 be involved in generating TUNEL-reactive DNA? ced-1 and ced-7 could be involved in a process that promotes both cell-corpse recognition prior to engulfment and the activation of DNA degradation. In contrast, CED-2, CED-5, and CED-10 are likely to be involved in the extension of pseudopodia around the dying cell (Wu and Horvitz 1998b), which is a later event in the phagocytic process; by analogy ced-6, mutations which have a similar effect on DNA degradation, might also function in a later event. If ced-1 and ced-7 are involved in an intercellular communication that promotes both cell-corpse engulfment and DNA degradation, their sites of action could be in either the dying cell or the engulfing cell. The nature and site of action of the ced-1 gene product have not been reported. The ced-7 gene encodes a protein that is similar to ABC transporters and localized to the plasma membrane, and its activity is required in both dying and engulfing cells for efficient engulfment (Wu and Horvitz 1998a). On the basis of its localization, CED-7 is unlikely to be directly involved in DNA degradation in the nucleus, and the identification and characterization of substrate(s) transported by CED-7 should elucidate why this protein is important for both engulfment and DNA degradation. For instance, CED-7 might function to promote the acidification of dying cells, which could activate the NUC-1 enzyme, or CED-7 might be involved in exporting NUC-1 out of engulfing cells or importing NUC-1 into dying cells. However, we cannot preclude the possibility that mutations in ced-1 and ced-7 somehow render cell corpses insensitive to TUNEL reagents and result in decreased TUNEL staining for this reason.
The second step of DNA degradation involves the nuc-1 gene, which mediates the conversion of TUNEL-reactive DNA into TUNEL-unreactive DNA. By examining the TUNEL-staining patterns of engulfment-defective mutants, we attempted to determine whether nuc-1 activity was required in dying cells or in engulfing cells. We suggest that both the nuclease activity or activities important for generating TUNEL-reactive DNA ends and the NUC-1 nuclease act in dying cells rather than in engulfing cells, because blocking engulfment did not prevent a cell corpse from becoming TUNEL reactive or from having nuc-1 activity. Alternatively, these activities might be generated by engulfing cells and transported to dying cells in an engulfment-independent process, for example, by secretion and uptake of NUC-1, as described above.
The third step of DNA degradation involves the final destruction of TUNEL-unreactive DNA fragments and depends on the engulfment process. This step could involve either activities synthesized in engulfing cells or activities synthesized in dying cells and activated by engulfment. Because DNA persists within engulfed corpses in nuc-1 mutants, nuc-1 activity is apparently required for this step of degradation to occur or increases the rate at which this step of degradation occurs. Perhaps NUC-1 generates the substrate for the nuclease involved in this step of DNA degradation.
At least two other genes encoding DNase II-like proteins are present in the C. elegans genome (Krieser and Eastman 1998). Because nuc-1 mutations have a major effect on DNA degradation, these other DNase II-like genes either do not function in programmed cell death or play much less important roles. That DNase II-like activity is nearly absent from protein extracts in nuc-1 mutants (Hevelone and Hartman 1988) suggests that these other DNase II-like genes may be expressed at very low levels, if at all. However, the DNA of cell corpses does become TUNEL unreactive eventually in nuc-1 mutants, although the process is very slow. Perhaps residual endonuclease activity provided by the other DNase II-like genes degrades the DNA. Alternatively, the TUNEL-unreactive state generated by nuc-1 activity may not be an obligatory intermediate in the DNA degradation pathway, and DNA degradation may proceed in the absence of a DNase II-like activity, albeit at a slower rate. Further analysis of the DNA-degradation pathway should determine why _nuc-1_-mediated DNA degradation is a rate-limiting step in this process.
Mechanisms of DNA degradation in apoptosis may be conserved
McIlroy et al. (2000) found that an acidic endonuclease is involved in apoptotic DNA degradation in mammalian cells and that DNA degradation of mammalian apoptotic cells involves activities provided both by the dying cell and by the engulfing cell. We propose that their findings as well as the findings of others regarding apoptotic DNA degradation in mammalian cells can be interpreted in the framework of our model for the pathway of DNA degradation in C. elegans (Fig. 6). Specifically, CAD is a mammalian endonuclease that is active in dying cells and that can generate TUNEL-reactive DNA (McIlroy et al. 2000; Enari et al. 1998; Sakahira et al. 1998; Zhang et al. 1998); the unknown C. elegans endonuclease of step one could encode a CAD-like activity (although there is no obvious CAD-like sequence in the C. elegans genome; unpubl.; C. elegans Genome Sequencing Consortium 1998). An acidic endonuclease, which may be DNase II, promotes DNA degradation in mammalian cells (McIlroy et al. 2000); C. elegans NUC-1 is similar to DNase II in sequence and appears similar in activity (Hevelone and Hartman 1988). In apoptotic mammalian cells, phagocytes are involved in DNA degradation (McIlroy et al. 2000); in C. elegans a phagocytosis-dependent nuclease completes DNA degradation downstream of nuc-1. It is possible that DNase II acts in phagocytes during mammalian apoptosis, whereas NUC-1 acts in dying cells in C. elegans. In this case, a lysosomal endonuclease may have been independently adopted by both phagocytes and dying cells for DNA degradation during apoptosis. Alternatively, in both species, engulfing cells might synthesize a DNase II-like nuclease and supply it to dying cells in an interaction that is independent of phagocytosis per se (and in C. elegans requires the functions of the genes ced-1 and ced-7). If so, as for other steps in the pathway for apoptosis (Metzstein et al. 1998), the molecular mechanisms for the degradation of the DNA of apoptotic cells may be conserved from C. elegans to mammals.
Materials and methods
Nematodes
All strains were grown at 20°C on NGM agar seeded with Escherichia coli OP50 bacteria (Brenner 1974). The C. elegans Bristol N2 strain (Brenner 1974) was used as the wild-type strain. Alleles used were as follows: LGI, ced-1(e1735, n1995); LGIII, ced-6(n1813, n2095); ced-7(n1892, n1996, n2094); ced-4(n1162); ced-9(n1950); LGIV, ced-2(e1752, n1994); ced-5(n1812, n2691); ced-10(n1993), ced-3(n717); LGX, nuc-1(e1392, n334, n887, n3335); dpy-6(e14); unc-9(e101); unc-10(e102); ced-8(n1891); lin-2(e1309); sma-5(n678); unc-27(e155); egl-15(n484); unc-115(e2225). All mutations except ced-5(n2691), ced-1(n1995), and nuc-1(n3335) are described by Riddle et al. (1997); ced-5(n2691) is described by Wu and Horvitz (1998b), ced-1(n1995) is described by Ellis et al. (1991a), and nuc-1(n3335) is described in this study.
Cell-corpse counts
The number of refractile cell corpses in living embryos was counted by Nomarski optics (Ellis et al. 1991a).
TUNEL
Animals were washed from one to three 100 × 15-mm petri plates with water and treated with hypochlorite to obtain embryos (Wood et al. 1988). Embryos were fixed using a protocol modified from Finney and Ruvkun (1990) as follows. A total of 1 ml of fixation solution containing 80 mm KCl, 20 mm NaCl, 1.3 mm EGTA, 3.2 mm spermine, 7.5 mm sodium HEPES (pH 6.5), 25% methanol, 2% paraformaldehyde, and 0.4% glutaraldehyde was added to hypochlorite-treated embryos, which were immediately frozen in liquid nitrogen. The frozen embryos were thawed in a water bath at room temperature for 2 min and rocked at room temperature for 25 min. Fixed embryos were washed once in 1 ml of Tris–Triton buffer (1% Triton X-100, 100 mm Tris at pH 7.4) and three times in 1 ml of PBST (1× PBS containing 0.5% Triton X-100) for 10 min each. Approximately 3 μl of packed embryos were preincubated with 25 μl of TdT reaction buffer (200 mm sodium cacodylate, 25 mm Tris-HCl, 0.25 mg/ml BSA, 0.1% Triton X-100, 1.5 mm cobalt chloride at pH 6.6) for 5 min at room temperature. The TdT reaction buffer was then replaced with TdT reaction buffer containing 0.2 units of TdT (Boehringer Mannheim), 6.6 nm dUTP (Boehringer Mannheim) and 3.3 nm fluorescein-11-dUTP (Boehringer Mannheim), and incubated for 2 hr at 37°C. After incubation, embryos were washed in PBST three times for 10 min. For DAPI staining, fixed and stained embryos were incubated in 1 μg/ml DAPI in PBST for 10 min. Embryos were then mounted in Vectorshield mounting medium (Vector Laboratories) and visualized using either a Zeiss Axiophot microscope equipped for fluorescence microscopy or a Bio-Rad MRC-500 confocal microscope.
Syto 11 staining
We tested the vital DNA-binding dye Syto 11 (Molecular Probes) to determine whether its staining pattern appeared similar to that obtained with DNA-labeling reagents such as Feulgen (Sulston 1976; Hedgecock et al. 1983) or DAPI (unpubl.), which can be used to detect the pycnotic DNA of dead cells. Mixed-staged worms were washed from plates with M9 buffer (Wood et al. 1988), collected in a 1.5 ml of Eppendorf tube, and washed once in M9 buffer. The washed worms were then rocked at room temperature for 1.5 hr in 1 ml of 10 μm Syto 11 in M9 buffer. The stained worms were then washed once in M9 and transferred to a Petri plate seeded with OP50 to recover for 30 min to 2 hr. Alternatively, worms were picked into 50 μl of M9 buffer in a microtiter well, 50 μl of 20 μm Syto 11 was added and the worms were incubated at room temperature for 1.5 hr. Worms were mounted on 4% agar pads with 20 mm sodium azide and visualized using an FITC filter set.
In L3, L4, and young adult wild-type animals, Syto 11 labeled all nuclei. In Syto 11-stained nuc-1(e1392) or nuc-1(n887) mutant animals, undegraded DNA of cell corpses appeared as additional bright, condensed foci of staining that did not coincide with nuclei visible by Nomarski microscopy. Undegraded bacterial DNA within the intestinal lumen also stained brightly. These patterns of staining were similar to those seen when wild-type and nuc-1 animals are stained with Feulgen or DAPI, indicating that Syto 11 can be used as a stain for the Nuc phenotype in living animals.
Genetic mapping
nuc-1 was mapped previously to linkage group X between the genes dpy-7 and unc-9 (V. Ambros and H.R. Horvitz, unpubl.). We used three-factor mapping to further localize the nuc-1 gene to the region between the cloned genes ced-8 (Stanfield and Horvitz 2000) and egl-15 (DeVore et al. 1995). To score the nuc-1 phenotype, we obtained homozygous lines from recombinant animals and scored for persistent DNA of engulfed cell corpses in the ventral nerve cords of L3 and L4 larvae stained with Syto 11. We mapped nuc-1 between dpy-6 and unc-9; from dpy-6 unc-9/nuc-1(e1392) mothers, 7/8 Unc non-Dpy and 2/8 Dpy non-Unc lines were Nuc, and from dpy-6 unc-9/nuc-1(n887) mothers, 7/8 Unc non-Dpy and 0/6 Dpy non-Unc lines were Nuc. We mapped nuc-1 between ced-8 and lin-2; from unc-10 ced-8/nuc-1(e1392) lin-2 mothers, 8/12 Unc Lin lines were Ced-8 and 2/8 of these Ced-8 lines were Nuc. We mapped nuc-1 left or near to the right of sma-5; from sma-5 lin-2/nuc-1(e1392) mothers, 4/4 Lin non-Sma lines were Nuc, and from sma-5 lin-2/nuc-1(n887) mothers, 11/11 Lin non-Sma lines were Nuc. We mapped nuc-1 between unc-27 and egl-15; from unc-27 egl-15/nuc-1(e1392) mothers, 9/15 Egl non-Unc and 3/10 Unc non-Egl lines were Nuc. We mapped nuc-1 to the left or very near right of unc-115; from unc-115 egl-15/nuc-1(e1392) mothers, 13/13 Egl non-Unc and 0/14 Unc non-Egl lines were Nuc.
Complementation tests
n3335 males were mated to unc-27(e155) nuc-1(e1392) egl-15(n484) hermaphrodites, and non-Unc progeny at the L3 or older stage were stained with Syto 11 in a microtiter well. Fifty of fifty animals examined were Nuc, that is, they had extra staining nuclei in the ventral cord. The presence of the n3335 deletion was confirmed by PCR for 20 of these animals.
Molecular biology
Standard molecular biology methods were followed (Sambrook et al. 1989). The sequences of primers used for PCR amplification, DNA sequence determination, and 5′ RACE are available on request. We isolated RNA from a population of mixed-stage N2 animals using standard methods and performed poly(A) selection using Fast Track (Invitrogen). 5′-RACE reactions were performed using the 5′ RACE System 2.0 (GIBCO).
Deletion library screening
We used PCR to screen a library of chemically mutagenized N2 C. elegans and selected animals containing a deletion in C07B5.5, essentially as described in Jansen et al. (1997). The deletion library was constructed by the members of the Horvitz laboratory, as directed by R. Ranganathan and P. Reddien.
Acknowledgments
We thank R. Ranganathan and P. Reddien for engineering the Horvitz laboratory C. elegans deletion library and for providing screening advice and to K. Hill for suggesting the use of Syto 11 as a vital DNA-labeling reagent. We thank E. Castor for deletion library screening and for DNA sequence determinations, N. Tsung for assistance with nuc-1 mapping, and Y. Kohara for nuc-1 cDNAs. We thank D. McIlroy and S. Nagata for communicating their results prior to publication. We also thank S. Cameron, B. Hersh, M. Metzstein, P. Reddien, and Z. Zhou for comments concerning this manuscript. G.M.S. was supported by a Howard Hughes Medical Institute Predoctoral Fellowship. H.R.H. is an Investigator of the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL horvitz@mit.edu; FAX (617) 253-8126.
References
- Alnemri ES, Litwack G. Activation of internucleosomal DNA cleavage in human CEM lymphocytes by glucocorticoid and novobiocin. Evidence for a non-Ca2+-requiring mechanism(s) J Biol Chem. 1990;265:17323–17333. [PubMed] [Google Scholar]
- Baker KP, Baron WF, Henzel WJ, Spencer SA. Molecular cloning and characterization of human and murine DNase II. Gene. 1998;215:281–289. doi: 10.1016/s0378-1119(98)00280-7. [DOI] [PubMed] [Google Scholar]
- Barry MA, Eastman A. Endonuclease activation during apoptosis: the role of cytosolic Ca2+ and pH. Biochem Biophys Res Commun. 1992;186:782–789. doi: 10.1016/0006-291x(92)90814-2. [DOI] [PubMed] [Google Scholar]
- ————— Identification of deoxyribonuclease II as an endonuclease involved in apoptosis. Arch Biochem Biophys. 1993;300:440–450. doi: 10.1006/abbi.1993.1060. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C. elegans Genome Sequencing Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- DeVore DL, Horvitz HR, Stern MJ. An FGF receptor signaling pathway is required for the normal cell migrations of the sex myoblasts in C. elegans hermaphrodites. Cell. 1995;83:611–620. doi: 10.1016/0092-8674(95)90101-9. [DOI] [PubMed] [Google Scholar]
- Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- Ellis RE, Jacobson DM, Horvitz HR. Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans. Genetics. 1991a;129:79–94. doi: 10.1093/genetics/129.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis RE, Yuan JY, Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol. 1991b;7:663–698. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- Finney M, Ruvkun G. The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell. 1990;63:895–905. doi: 10.1016/0092-8674(90)90493-x. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halenbeck R, MacDonald H, Roulston A, Chen TT, Conroy L, Williams LT. CPAN, a human nuclease regulated by the caspase-sensitive inhibitor DFF45. Curr Biol. 1998;8:537–540. doi: 10.1016/s0960-9822(98)79298-x. [DOI] [PubMed] [Google Scholar]
- Hedgecock EM, Sulston JE, Thomson JN. Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans. Science. 1983;220:1277–1279. doi: 10.1126/science.6857247. [DOI] [PubMed] [Google Scholar]
- Hengartner MO, Ellis RE, Horvitz HR. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature. 1992;356:494–499. doi: 10.1038/356494a0. [DOI] [PubMed] [Google Scholar]
- Hevelone J, Hartman PS. An endonuclease from Caenorhabditis elegans: Partial purification and characterization. Biochem Genet. 1988;26:447–461. doi: 10.1007/BF02399412. [DOI] [PubMed] [Google Scholar]
- Hirsh D, Oppenheim D, Klass M. Development of the reproductive system of Caenorhabditis elegans. Dev Biol. 1976;49:200–219. doi: 10.1016/0012-1606(76)90267-0. [DOI] [PubMed] [Google Scholar]
- Hughes FM, Cidlowski JA. Apoptotic DNA degradation: Evidence for novel enzymes. Cell Death Differ. 1994;1:11–17. [PubMed] [Google Scholar]
- Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Jansen G, Hazendonk E, Thijssen KL, Plasterk RH. Reverse genetics by chemical mutagenesis in Caenorhabditis elegans. Nat Genet. 1997;17:119–121. doi: 10.1038/ng0997-119. [DOI] [PubMed] [Google Scholar]
- Khodarev NN, Ashwell JD. An inducible lymphocyte nuclear Ca2+/Mg2+-dependent endonuclease associated with apoptosis. J Immunol. 1996;156:922–931. [PubMed] [Google Scholar]
- Krieser RJ, Eastman A. The cloning and expression of human deoxyribonuclease II. A possible role in apoptosis. J Biol Chem. 1998;273:30909–30914. doi: 10.1074/jbc.273.47.30909. [DOI] [PubMed] [Google Scholar]
- Liao TH. The subunit structure and active site sequence of porcine spleen deoxyribonuclease. J Biol Chem. 1985;260:10708–10713. [PubMed] [Google Scholar]
- Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89:175–184. doi: 10.1016/s0092-8674(00)80197-x. [DOI] [PubMed] [Google Scholar]
- McIlroy, D., M. Tanaka, H. Sakahira, H. Fukuyama, M. Suzuki, K. Yamamura, Y. Ohsawa, Y. Uchiyama, and S. Nagata. 2000. An auxiliary mode of DNA fragmentation provided by phagocytes. Genes & Dev. (this issue). [PMC free article] [PubMed]
- Metzstein MM, Stanfield GM, Horvitz HR. Genetics of programmed cell death in C. elegans: Past, present and future. Trends Genet. 1998;14:410–416. doi: 10.1016/s0168-9525(98)01573-x. [DOI] [PubMed] [Google Scholar]
- Montague JW, Hughes FM, Jr, Cidlowski JA. Native recombinant cyclophilins A, B, and C degrade DNA independently of peptidylprolyl cis-trans-isomerase activity. Potential roles of cyclophilins in apoptosis. J Biol Chem. 1997;272:6677–6684. doi: 10.1074/jbc.272.10.6677. [DOI] [PubMed] [Google Scholar]
- Oberhammer F, Wilson JW, Dive C, Morris ID, Hickman JA, Wakeling AE, Walker PR, Sikorska M. Apoptotic death in epithelial cells: cleavage of DNA to 300 and/or 50 kb fragments prior to or in the absence of internucleosomal fragmentation. EMBO J. 1993;12:3679–3684. doi: 10.1002/j.1460-2075.1993.tb06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odaka C, Mizuochi T. Role of macrophage lysosomal enzymes in the degradation of nucleosomes of apoptotic cells. J Immunol. 1999;163:5346–5352. [PubMed] [Google Scholar]
- Peitsch MC, Hesterkamp T, Polzar B, Mannherz HG, Tschopp J. Functional characterization of serum DNase I in MRL-lpr/lpr mice. Biochem Biophys Res Commun. 1992;186:739–745. doi: 10.1016/0006-291x(92)90808-x. [DOI] [PubMed] [Google Scholar]
- Peitsch MC, Polzar B, Stephan H, Crompton T, MacDonald HR, Mannherz HG, Tschopp J. Characterization of the endogenous deoxyribonuclease involved in nuclear DNA degradation during apoptosis (programmed cell death) EMBO J. 1993;12:371–377. doi: 10.1002/j.1460-2075.1993.tb05666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao L, Perez D, White E. Lamin proteolysis facilitates nuclear events during apoptosis. J Cell Biol. 1996;135:1441–1455. doi: 10.1083/jcb.135.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle DL, Blumenthal T, Meyer BJ, Priess JR. C. elegans II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. [PubMed] [Google Scholar]
- Roychoudhury R, Jay E, Wu R. Terminal labeling and addition of homopolymer tracts to duplex DNA fragments by terminal deoxynucleotidyl transferase. Nucleic Acids Res. 1976;3:101–116. doi: 10.1093/nar/3.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Shaham S. Identification of multiple Caenorhabditis elegans caspases and their potential roles in proteolytic cascades. J Biol Chem. 1998;273:35109–35117. doi: 10.1074/jbc.273.52.35109. [DOI] [PubMed] [Google Scholar]
- Shiokawa D, Tanuma S. DLAD, a novel mammalian divalent cation-independent endonuclease with homology to DNase II. Nucleic Acids Res. 1999;27:4083–4089. doi: 10.1093/nar/27.20.4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfield, G.M. and H.R. Horvitz. 2000. The ced-8 gene controls the timing of cell deaths in C. elegans. Mol. Cell (In press). [DOI] [PubMed]
- Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- Sulston JE. Post-embryonic development in the ventral cord of Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1976;275:287–297. doi: 10.1098/rstb.1976.0084. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode Caenorhabditis elegans. Dev Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW. Substrate specificities of caspase family proteases. J Biol Chem. 1997;272:9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Lazebnik Y. Caspases: Enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- Torriglia A, Chaudun E, Chany-Fournier F, Jeanny JC, Courtois Y, Counis MF. Involvement of DNase II in nuclear degeneration during lens cell differentiation. J Biol Chem. 1995;270:28579–28585. doi: 10.1074/jbc.270.48.28579. [DOI] [PubMed] [Google Scholar]
- Waterston RH, Brenner S. A suppressor mutation in the nematode acting on specific alleles of many genes. Nature. 1978;275:715–719. doi: 10.1038/275715a0. [DOI] [PubMed] [Google Scholar]
- Wood WB the Community of C. elegans Researchers. The Nematode Caenorhabditis elegans. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- Wu YC, Horvitz HR. The C. elegans cell corpse engulfment gene ced-7 encodes a protein similar to ABC transporters. Cell. 1998a;93:951–960. doi: 10.1016/s0092-8674(00)81201-5. [DOI] [PubMed] [Google Scholar]
- ————— C. elegans phagocytosis and cell-migration protein CED-5 is similar to human DOCK180. Nature. 1998b;392:501–504. doi: 10.1038/33163. [DOI] [PubMed] [Google Scholar]
- Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284:555–556. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- Xue D, Shaham S, Horvitz HR. The Caenorhabditis elegans cell-death protein CED-3 is a cysteine protease with substrate specificities similar to those of the human CPP32 protease. Genes & Dev. 1996;10:1073–1083. doi: 10.1101/gad.10.9.1073. [DOI] [PubMed] [Google Scholar]
- Yasuda T, Takeshita H, Iida R, Nakajima T, Hosomi O, Nakashima Y, Kishi K. Molecular cloning of the cDNA encoding human deoxyribonuclease II. J Biol Chem. 1998;273:2610–2616. doi: 10.1074/jbc.273.5.2610. [DOI] [PubMed] [Google Scholar]
- Zhang J, Liu X, Scherer DC, van Kaer L, Wang X, Xu M. Resistance to DNA fragmentation and chromatin condensation in mice lacking the DNA fragmentation factor 45. Proc Natl Acad Sci. 1998;95:12480–12485. doi: 10.1073/pnas.95.21.12480. [DOI] [PMC free article] [PubMed] [Google Scholar]