Signal-induced ubiquitination of IκBα by the F-box protein Slimb/β-TrCP (original) (raw)
Abstract
Signal-induced phosphorylation of IκBα targets this inhibitor of NF-κB for ubiquitination and subsequent degradation, thus allowing NF-κB to enter the nucleus to turn on its target genes. We report here the identification of an IκB–ubiquitin (Ub) ligase complex containing the F-box/WD40-repeat protein, β-TrCP, a vertebrate homolog of Drosophila Slimb. β-TrCP binds to IκBα only when the latter is specifically phosphorylated by an IκB kinase complex. Moreover, immunopurified β-TrCP ubiquitinates phosphorylated IκBα at specific lysines in the presence of Ub-activating (E1) and -conjugating (Ubch5) enzymes. A β-TrCP mutant lacking the F-box inhibits the signal-induced degradation of IκBα and subsequent activation of NF-κB-dependent transcription. Furthermore, Drosophila embryos deficient in slimb fail to activate twist and snail, two genes known to be regulated by the NF-κB homolog, Dorsal. These biochemical and genetic data strongly suggest that Slimb/β-TrCP is the specificity determinant for the signal-induced ubiquitination of IκBα.
Keywords: Phosphorylation, NF-κB, IκB, ubiquitin, SCF, Slimb, β-TrCP
Ubiquitin (Ub) is a small polypeptide that is covalently conjugated to protein substrates, thus committing these proteins for degradation (Ciechanover et al. 1978; Hershko et al. 1980; Wilkinson et al. 1980). Ub conjugation is catalyzed by an enzymatic cascade that begins with the ATP-dependent activation of Ub by a Ub-activating enzyme (E1) to form an E1–Ub thiolester. The activated Ub is then transferred to a Ub-conjugating enzyme (E2 or Ubc). Finally, in the presence of a Ub–protein ligase (E3), the carboxyl terminus of Ub is conjugated via an isopeptide bond to a lysine residue of the protein substrate. Processive conjugation of Ub to a previously conjugated Ub results in the formation of multiubiquitin chains that target the protein substrate for degradation by the 26S proteasome (Chau et al. 1989; for review, see Pickart 1997). In most cases, the specificity of protein degradation is determined by the identity of E3, which is operationally defined as a factor that binds to a specific protein substrate and facilitates its multi-ubiquitination.
Ub-dependent proteolysis plays a pivotal role in the regulation of many biological processes, including cell cycle progression, transcription, and signal transduction (for review, see Hochstrasser 1996; Hershko and Ciechanover 1998). The importance of the Ub pathway in the cell cycle is highlighted by recent studies on the degradation of cyclins and cyclin-dependent kinase (Cdk) inhibitors. Exit of cells from mitosis requires the degradation of mitotic cyclins, a step that is controlled by the activation of a 20S E3 complex known as anaphase-promoting complex (APC) or cyclosome (Hershko et al. 1994; King et al. 1995). Interestingly, the degradation of Cdk inhibitors such as Sic1, which triggers the entry of cell cycle into the S phase, is regulated by a distinct mechanism. In this case, phosphorylation of Sic1 at the end of G1 allows this inhibitor to bind to Cdc4, a protein that contains two structural motifs, an F-box at the amino terminus and seven WD40 repeats at the carboxyl terminus. Through the F-box, Cdc4 tethers phosphorylated Sic1 to Skp1, which in turn binds to Cdc53, which then recruits an E2, Cdc34, to ubiquitinate Sic1 (Feldman et al. 1997; Skowyra et al. 1997). This so-called SCF pathway is also responsible for the ubiquitination of several other substrates (for review, see Patton et al. 1998). In each case, the substrate specificity is determined by the presence of a distinct F-box protein. For example, the F-box/leucine-zipper protein Grr1 binds to phosphorylated Cln1 and Cln2, but not Sic1 (Skowyra et al. 1997).
An F-box/WD40-repeat-containing protein called Slimb was identified recently in a genetic screen for recessive mutations that alter adult patterning in Drosophila (Jiang and Struhl 1998; Theodosiou et al. 1998). Loss of function of slimb causes supernumerary limbs as a result of ectopic activation of the Hedgehog (Hh) and Wnt/Wingless (Wg) pathways. In the Hh pathway, the transcription factor Cubitus interruptus (Ci) is proteolytically processed to a truncated repressor form in the absence of signaling (Aza-Blanc et al. 1997). This processing depends on protein kinase A (PKA) activity, which is antagonized by Hh signaling. By analogy to the SCF pathway in yeast (Feldman et al. 1997; Skowyra et al. 1997) and to the processing of NF-κB1/p105 in mammals (Palombella et al. 1994), it was proposed that in the absence of Hh signaling, PKA phosphorylates Ci, thereby targeting Ci for Slimb-dependent processing via the Ub–proteasome pathway (Jiang and Struhl 1998). Similarly, the Wnt/Wg pathway is also regulated primarily through the stability of β-catenin/Armadillo (Arm), a putative cofactor of the transcriptional activator Lef1/TCF (Nusse 1997). In the absence of Wnt/Wg, β-catenin/Arm is phosphorylated by glycogen synthase kinase-3 (Gsk-3) or Zeste–White 3 (Zw3) and then degraded via the Ub–proteasome pathway (Aberle et al. 1997; Orford et al. 1997). Activation of the Wnt/Wg pathway leads to inhibition of Gsk-3/Zw3, thus allowing for the accumulation of β-catenin/Arm to turn on downstream genes in conjunction with Lef-1/TCF. The ectopic activation of the Wnt/Wg pathway and stabilization of Arm in slimb mutant cells suggests that Slimb may be required for the degradation of β-catenin/Arm.
The Gsk-3 phosphorylation sites on β-catenin are strikingly similar to those of IκB, a family of inhibitory proteins that sequester the transcription factor NF-κB in the cytoplasm of quiescent cells (for review, see Baldwin 1996; Baeuerle and Baltimore 1996). In response to a variety of stimuli, such as tumor necrosis factor α (TNFα), lipopolysaccharide (LPS) and ultraviolet light (UV), IκB proteins are phosphorylated rapidly at specific serine residues by a 700-kD protein kinase complex (for review, see Maniatis 1997; Stancovski and Baltimore 1997; Scheidereit 1998). Phosphorylation of IκBα at serines 32 and 36 targets this inhibitor for ubiquitination at lysines 21 and 22 (Chen et al. 1995; Scherer et al. 1995). Ubiquitinated IκBα is then degraded specifically by the 26S proteasome, allowing NF-κB to translocate into the nucleus. Two closely related E2s, Ubc4/5 and Ubch7/E2–F1, are capable of supporting the ubiquitination of IκBα in vitro (Alkalay et al. 1995; Chen et al. 1996). However, the E3 responsible for IκBα ubiquitination has remained unknown.
Recently, a human homolog of Slimb, h-βTrCP, was cloned in a yeast two-hybrid screen using human immunodeficiency virus (HIV) Vpu as a bait (Margottin et al. 1998). It was reported that h-βTrCP binds specifically to phosphorylated Vpu, which in turn binds to CD4 on T cells, resulting in the degradation of CD4 in the endoplasmic reticulum (ER). This study, however, did not reveal the function of h-βTrCP in normal cells (not infected with HIV). The structural and functional properties of Slimb/βTrCP described above led us to hypothesize that it is a component of IκB–Ub ligase (E3IκB). This hypothesis is strongly supported by the evidence presented in this report.
Results
βTrCP binds to phosphorylated IκBα
We cloned a mouse homolog of Slimb (mβTrCP, GenBank accession no. AF112979) based on two Slimb-related sequences from the mouse EST database (see Materials and Methods). mβTrCP is 79% (387/485) identical to Drosophila Slimb and 98% (560/569) identical to the recently cloned human βTrCP (hβTrCP; Margottin et al. 1998), whose normal physiological function was unknown. As an initial step in determining whether βTrCP binds to IκBα, we synthesized 35S-labeled IκBα and mβTrCP by in vitro translation. IκBα was phosphorylated by a MEKK1-activated IκB kinase complex (Lee et al. 1997) and then incubated with mβTrCP. The binding of mβTrCP to IκBα was determined by using a co-immunoprecipitation assay with an IκBα-specific antibody. As shown in Figure 1A, phosphorylated IκBα (p-IκBα) bound to mβTrCP, whereas unphosphorylated IκBα or the phosphorylation-defective IκBα mutant (S32A/S36A) was unable to bind to mβTrCP (Fig. 1A, lanes 1–3). The binding of p-IκBα to mβTrCP was detected under high stringency conditions (1% NP-40, 0.5% deoxycholate, and 0.1% SDS), suggesting a strong interaction. Similar results were obtained when IκBα was phosphorylated by recombinant IKKβ expressed from baculovirus-infected insect cells (data not shown). Drosophila Slimb and hβTrCP (kindly provided by Dr. Benarous, INSERM, Paris, France) also bound specifically to p-IκBα (data not shown), suggesting that Slimb/βTrCP functions are evolutionarily conserved.
Figure 1.



mβTrCP binds to phosphorylated IκBα. (A) In vitro binding. In vitro-translated 35S-labeled IκBα (lanes 1,2,4,5) or S32A/S36A mutant (lanes 3,6) was phosphorylated by a MEKK1-activated IκB kinase complex and incubated with 35S-labeled mβTrCP (lanes 1_–_3) or mβTrCPΔF (lanes 4_–_6) in RIPA buffer plus 0.1% SDS. Proteins associated with IκBα were coimmunoprecipitated with an IκBα-specific antibody and analyzed by SDS-PAGE. Lanes 7_–_10 show an aliquot of the in vitro-translated proteins, which is ∼40% of input in the immunoprecipitation experiments shown in lanes 1_–_6. (WT) Wild-type IκBα; (AA) S32A/S36A; (FL) full-length mβTrCP (wild type); (ΔF) F-box-deleted mutant of mβTrCP; (IP) immunoprecipitation; (IVT) in vitro translation. (B) mβTrCP forms a complex with phosphorylated IκBα, p65, and Skp1 in vivo. 293 cells were transfected with 3 μg of pcDNA3–Myc–mβTrCP (lanes 2,3,6,7,10,11) or pcDNA3-Myc-mβTrCPΔF (lanes 4,5,8,9,12,13). After incubating the cells with 20 μm of MG132 for 30 min, the cells were treated with (lanes 3,5,7,9,11,13) or without (lanes 2,4,6,8,10,12) calyculin A (0.1 μm) for 10 min. Cell extracts were immunoprecipitated with antibodies against IκBα (lanes 2_–_5), p65 (lanes 6_–_9), or Myc (lanes 10_–_13), respectively, and the precipitated proteins analyzed by immunoblotting with antibodies against Myc, Skp1, IκBα, and p65, respectively. Lane 1 is 10 μg of 293 cell extract that also expresses mβTrCP. (C) mβTrCP binds directly to p-IκBα. 293 cells were transfected with pcDNA3 containing Myc–mβTrCP, together with Flag–IκBα (F-IκBα, lanes 1,2) or Flag–IκBαΔN mutant (FΔN, lanes 3,4). Following treatment of the transfected cells with MG132 and calyculin A, the cell extracts were immunoprecipitated with an anti-Flag antibody (M2, Kodak), and the precipitated proteins analyzed by immunoblotting with antibodies against Myc, Skp1, IκBα and p65, respectively.
To determine whether mβTrCP binds to p-IκBα in vivo, 293 cells transfected with Myc-tagged mβTrCP were stimulated with calyculin A, a cell-permeable phosphatase inhibitor that allows for the accumulation of phosphorylated IκBα (Chen et al. 1995). To block the degradation of phosphorylated IκBα, cells were pretreated with the proteasome inhibitor MG132 before the addition of calyculin A. Cell extracts were then immunoprecipitated with an antibody against IκBα, followed by immunoblotting with anti-Myc. The presence of Myc–mβTrCP in the anti-IκBα precipitates was detected only when cells were stimulated with calyculin A (Fig. 1B, lanes 2,3). Conversely, when cells were immunoprecipitated with anti-Myc, only p-IκBα but not unphosphorylated IκBα was coprecipitated (Fig. 1B, lanes 10,11). Thus, mβTrCP binds specifically to p-IκBα in cells.
Both IκBα and p-IκBα are bound tightly to NF-κB, which is typically a heterodimer of p50 and p65 (for review, see Baldwin 1996). Similarly, the F-box protein βTrCP is likely to be part of a SCF complex that also includes Skp1 (Jiang and Struhl 1998; Margottin et al. 1998). Hence, our observation that p-IκBα binds to βTrCP raises the possibility that the NF-κB/p-IκBα complex might associate with a SCF complex that contains both βTrCP and Skp1. To test this possibility, we examined the presence of p65 and Skp1 in anti-IκBα and anti-Myc immunoprecipitates by immunoblotting with respective antibodies (Fig. 1B, lanes 2,3,10,11). Moreover, we immunoprecipitated calyculin A-stimulated cell extracts (see above) with a p65-specific antibody and then immunoblotted the precipitates with antibodies against Skp1, Myc, IκBα, and p65, respectively (Fig. 1B, lanes 6,7). In each case, when cells were stimulated, all four proteins (p-IκBα, p65, mβTrCP, and Skp1) were detected in the same precipitates (Fig. 1B, lanes 3,7,11), indicating that they are present in the same complex. In contrast, in the absence of calyculin A treatment, while IκBα remained bound to p65, neither IκBα nor p65 was present in the anti-Myc precipitates (Fig. 1B, lanes 2,6,10, top two panels). Likewise, when cells were not stimulated, mβTrCP remained bound to Skp1, but neither was found in the anti-IκBα or anti-p65 immunoprecipitates (Fig. 1B, lanes 2,6,10, bottom two panels). These results strongly suggest that phosphorylation of IκBα leads to the assembly of a multiprotein complex that contains minimally p-IκBα, p65, βTrCP, and Skp1.
It has been proposed that the F-box mediates the binding to Skp1 of several F-box proteins including Cdc4 (Feldman et al. 1997; Skowyra et al. 1997) and hβTrCP (Margottin et al. 1998). In an effort to generate a dominant-negative mutant of mβTrCP that might allow us to investigate its function in vivo, we deleted the F-box from mβTrCP and tested the ability of this mutant (mβTrCPΔF) to bind to p-IκBα and Skp1, respectively. Deletion of the F-box did not prevent the binding of mβTrCP to p-IκBα either in vitro (Fig. 1A, lane 5) or in vivo (Fig. 1B, lane 5,9,13, top panel). However, this deletion abolished the binding of mβTrCP to Skp1 (Fig. 1B, cf. lanes 10 and 12, bottom panel) and compromised the recruitment of p-IκBα/p65 to a complex containing Skp1 (Fig. 1B, cf. lanes 7 and 9, bottom panel).
Interestingly, both mβTrCP and mβTrCPΔF appeared to be phosphorylated when cells were stimulated with calyculin A (Fig 1B, lanes 11,13; also see Fig 1C, lane 2). Furthermore, calyculin A treatment leads to a weak but detectable binding of mβTrCPΔF to Skp1 (Fig. 1B, cf. lanes 12,13, bottom panel). It is possible that when it is phosphorylated, mβTrCP itself is recruited for ubiquitination by a SCF complex that includes Skp1. In this case, the F-box may be dispensable, as the binding can be mediated by the interaction between certain phosphorylation sites on βTrCP and a SCF complex. Ubiquitination and degradation of other F-box proteins such as Cdc4 and Grr1 within the SCF complexes has been reported recently (Zhou and Howley 1998). We also noted a low level of Skp1 in the anti-IκBα precipitates from mβTrCPΔF-expressing cells (Fig. 1B, cf. lanes 3 and 5, bottom panel). This may reflect a pool of endogenous p-IκBα/βTrCP/Skp1 complexes that were not displaced by mβTrCPΔF.
We noticed that phosphorylated p65 was also detected following calyculin A treatment (Fig. 1B, lane 7). It is not clear whether PKA or another cytokine-inducible kinase is responsible for the phosphorylation of p65 in this case (Zhong et al. 1997; Wang and Baldwin 1998). Notwithstanding this uncertainty, the observation that p65 is phosphorylated raises the possibility that phosphorylated p65 may bind to βTrCP directly, whereas p-IκBα binds to βTrCP indirectly through p65. To test this possibility, we transfected 293 cells with the Myc-tagged mβTrCP expression construct, together with Flag-tagged IκBα (F-IκBα) or a phosphorylation-defective IκBα mutant lacking the amino-terminal 36 residues (F-IκBαΔN; Brockman et al. 1995). Following treatment with MG132 and calyculin A, cell extracts were immunoprecipitated with a Flag-specific antibody and then immunoblotted with anti-Myc or anti-Skp1 antibodies (Fig. 1C). Like endogenous IκBα, transfected F-IκBα associated with Myc–mβTrCP and Skp1 when cells were stimulated with calyculin A (Fig. 1C, lane 2). We also observed a low but detectable level of binding between transfected F-IκBα and mβTrCP in the absence of calyculin A treatment (Fig. 1C, cf. lanes 1 and 2). This may be due to overexpression of F-IκBα, a small but significant fraction of which may be phosphorylated at the signaling sites even in the absence of calyculin A. In contrast to F-IκBα, transfected F-IκBαΔN did not co-immunoprecipitate with either Myc-mβTrCP or Skp1 even when cells were treated with calyculin A (Fig. 1C, lanes 3,4). However, F-IκBαΔN remained bound to p65. Therefore, p65 is recruited to a βTrCP/Skp1-containing complex by virtue of its association with p-IκBα, implying that p-IκBα binds to βTrCP directly. Further supporting this idea, unphosphorylated p65 was present in the anti-Myc immunoprecipitates, whereas only p-IκBα coprecipitated with Myc–mβTrCP (Fig. 1B, lanes 11,13).
We also found that the electrophoretic mobility of F-IκBαΔN was reduced slightly when cells were treated with calyculin A (Fig. 1C, cf. lanes 3 and 4), which is indicative of phosphorylation outside the inducible amino-terminal phosphorylation sites. However, phosphorylation at noninducible sites did not lead to the binding of IκBα to mβTrCP or Skp1, suggesting that the binding of mβTrCP to IκBα is strictly dependent on signaling and that mβTrCP does not simply bind to phosphorylated proteins indiscriminately.
Dominant-negative βTrCP blocks IκBα degradation and NF-κB activation
The ability of mβTrCPΔF to bind to p-IκBα, together with its inability to bind to Skp1, suggests that this mutant might be a dominant-negative inhibitor of IκBα degradation, provided that βTrCP is involved in this pathway. To test the in vivo function of mβTrCP, 293 cells were transfected with mβTrCPΔF, together with a Flag-tagged IκBα expression construct. The transfected cells were stimulated with TNFα for 30 min, and the degradation of transfected and endogenous IκBα analyzed by immunoblotting with antibodies specific for Flag or IκBα, respectively (Fig. 2). With increasing concentration of mβTrCPΔF, there was a concentration-dependent stabilization of the phosphorylated forms of both transfected and endogenous IκBα. High concentrations of wild-type mβTrCP also led to a slight stabilization of p-IκBα (data not shown). This finding may be explained if some of the overexpressed mβTrCP is not incorporated into a functional SCF complex but still binds to p-IκBα. The sequestered p-IκBα may be protected from ubiquitination.
Figure 2.

βTrCP is required for TNFα-induced degradation of IκBα. 293 cells were transfected with pCDNA3–Flag–IκBα (20 ng), together with increasing concentration of pcDNA3–mβTrCPΔF (lanes 1,2, vector only; lanes 3,4, 0.01 μg; lanes 5,6, 0.1 μg; lanes 7,8, 1 μg). After treatment of cells with or without TNFα (20 ng/ml) for 30 min, cell extracts were analyzed by immunoblotting with antibodies against Flag, IκBα, or Myc, respectively.
To investigate the role of βTrCP in NF-κB activation, we transfected 293 cells with a mβTrCPΔF expression construct, together with a luciferase reporter gene, which is under the control of three tandem repeats of NF-κB binding sites (Fig. 3A). As a control, we also examined the expression of a GAL4-dependent reporter gene. TNFα-induced expression of the NF-κB reporter, but not GAL4VP16-activated expression of the GAL4 reporter, was severely inhibited with increasing concentration of mβTrCPΔF. This result suggests that βTrCP is required for TNFα-induced activation of NF-κB.
Figure 3.
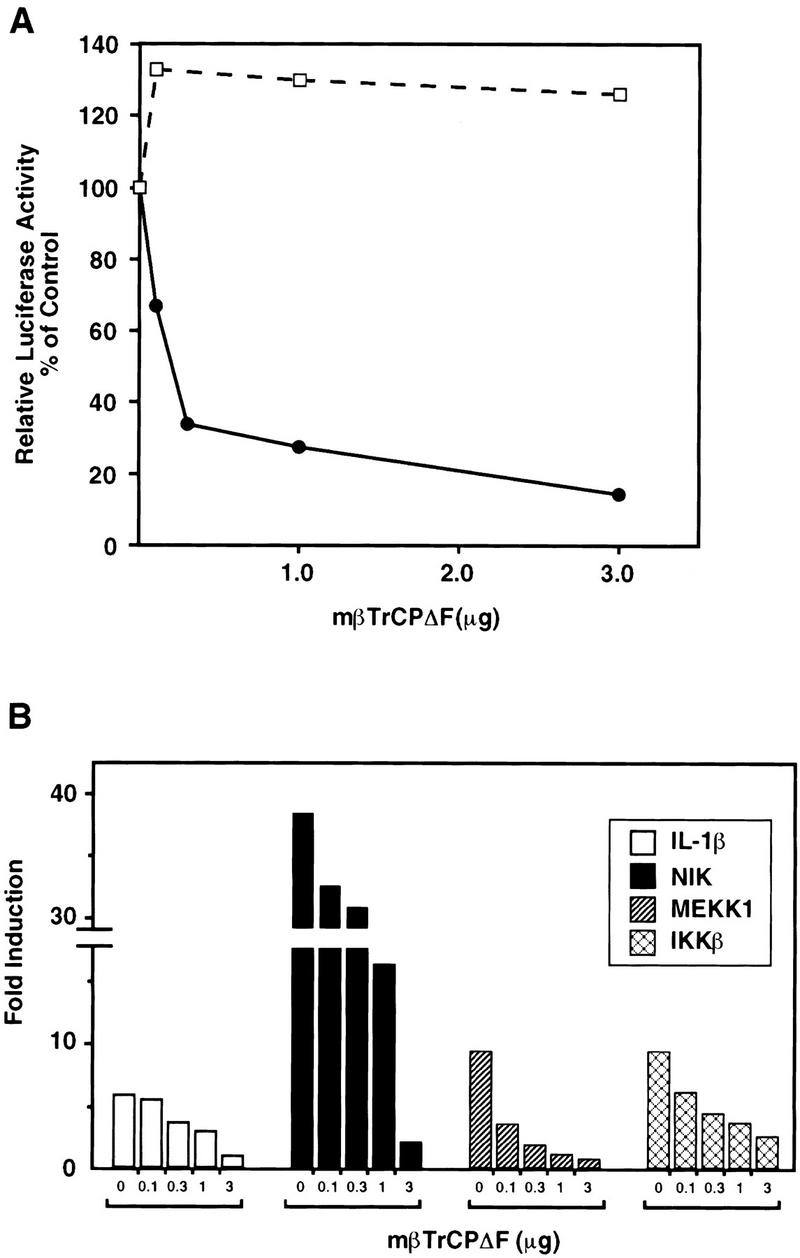
Dominant-negative βTrCP inhibits the activation of NF-κB-dependent transcription. (A) Effect of mβTrCPΔF on induction of the NF-κB-dependent reporter gene by TNFα. 293 cells were transfected with pcDNA3–mβTrCPΔF (0.01–3 μg as indicated), together with a NF-κB-dependent (●) or GAL4-dependent (□) luciferase reporter construct (p[κB]3–tk–Luc, or Gal4–E1b–Luc, 10 ng each). The NF-κB reporter gene was stimulated with TNFα (20 ng/ml) for 6 hr prior to harvest, whereas the GAL4 reporter gene was activated by cotransfection with the strong activator GAL4–VP16 (200 ng). Reporter activity is expressed as fold induction of normalized luciferase activity in stimulated cells relative to that of unstimulated cells. TNFα stimulated the NF-κB reporter by 14-fold, whereas the GAL4–VP16 activation was >3000-fold. (B) Effect of mβTrCPΔF on the activation of NF-κB by IL-1β, NIK, MEKK1, and IKKβ. 293 cells were transfected with pcDNA3–mβTrCPΔF and p[κB]3–tk–Luc as described in A, together with 100 ng of NIK, MEKK1, and IKKβ expression constructs, respectively. Stimulation of cells with IL-1β (10 ng/ml) was carried out for 6 hr prior to harvest. Luciferase activity was determined and normalized as described in Materials and Methods.
Degradation of IκBα is required for NF-κB activation by many different stimuli (for review, see Baldwin 1996). If βTrCP is an obligatory component that mediates IκBα degradation, it is expected that interference of βTrCP function should compromise the activation of NF-κB by multiple stimuli. To address this possibility, we examined the effects of mβTrCPΔF on the induction of NF-κB reporter gene by interleukin-1β (IL-1β), NIK, MEKK1, and IKKβ, respectively (Fig. 3B). In each case, NF-κB activation was inhibited markedly by mβTrCPΔF, strongly suggesting that βTrCP participates in the activation of NF-κB by multiple signaling pathways.
Slimb is required for Dorsal-dependent activation of twist and snail in Drosophila embryos
Although the cell culture experiments shown above support the involvement of βTrCP/Slimb in NF-κB activation, it is imperative to determine whether the same conclusion can be reached in animal models that are amenable to genetic manipulations. In this regard, Drosophila embryos provide an excellent model system, not only because _slimb_-deficient embryos can be generated (Jiang and Struhl 1998) but also because there is a highly conserved signaling pathway in Drosophila that is analogous to that of NF-κB/IκB (for review, see Morisato and Anderson 1995). In Drosophila early embryos, dorsoventral patterning is established by a nuclear concentration gradient of the Dorsal morphogen, a homolog of NF-κB. In the ventral region of the Drosophila embryo, local activation of Toll, a homolog of mammalian IL-1 receptor, results in degradation of the IκB-like protein Cactus. Consequently, Dorsal translocates into the nucleus where it activates downstream genes that include twist (twi) and snail (sna) (for review, see Morisato and Anderson 1995). To explore the role of Slimb in Dorsal activation in vivo, we generated _slimb_-deficient embryos and examined the expression of twi and sna by whole-mount in situ RNA hybridization. As shown in Figure 4, wild-type embryos expressed twi and sna in the ventral region (Fig. 4A,C). In contrast, _slimb_-deficient embryos expressed markedly reduced levels of twi and sna in most of the ventral region (Fig. 4B,D). The residual expression of twi and sna at the posterior pole may be due to modification of the Dorsal/Cactus pathway by terminal signaling, such that reduced dosage of Dorsal is sufficient to activate the polar expression of twi and sna (Ray et al. 1991). This staining pattern in _slimb_-deficient embryos is reminiscent of what has been described in cactus gain-of-function mutant embryos (Roth et al. 1991), strongly suggesting that Slimb/βTrCP is required for Cactus/IκB degradation in vivo.
Figure 4.

Slimb is required for the Dorsal-dependent activation of twi and sna in Drosophila embryos. Wild-type (A,C) and slimb mutant (B,D) embryos were hybridized with twi (A,B) or sna (C,D) antisense RNA probes to visualize their expression. Embryos were oriented with anterior to the left and dorsal up. Wild-type embryos at the blastoderm stage express the Dorsal target genes twi and sna in the ventral region (A,C). In contrast, slimb mutant embryos at the same stage show diminished expression of both twi and sna in most of the ventral region, with residual expression detectable at the posterior pole (arrows in B and D).
Slimb/βTrCP is a component of IκB–Ub ligase (E3IκB)
To demonstrate directly that βTrCP is a component of E3IκB, we expressed Myc-tagged mβTrCP in 293 cells by transient transfection and then purified mβTrCP containing complex by immunoprecipitation using a Myc-specific antibody. The immunoprecipitates were used directly as the source of E3 in a reconstituted IκBα ubiquitination assay (Chen et al. 1995). The reconstituted system contains in vitro-translated 35S-labeled IκBα phosphorylated by the MEKK1-activated IκB kinase complex, recombinant p50/p65 (to form a complex with IκBα), purified E1, recombinant Ubch5 (which was shown previously to support the ubiquitination of IκBα in vitro), Ub, and ATP. The presence of Ubch5 in the reaction led to the formation of low molecular mass ubiquitinated IκBα even in the absence of any E3 (i.e., mono-ubiquitinated IκBα was evident in Fig. 5A, lanes 1 and 6, but not in lane 4). However, it has been shown that proteins bearing one or a few Ub molecules are poor substrates for the 26S proteasome (Chau et al. 1989). Only high molecular mass conjugates, whose synthesis usually requires E3s, can be degraded efficiently. As shown in Figure 5, addition of immunoprecipitates containing mβTrCP to the reconstituted system led to efficient multi-ubiquitination of phosphorylated IκBα. In contrast, the phosphorylation-defective IκBα mutant (S32A/S36A) was not ubiquitinated. A control IgG failed to immunoprecipitate any Ub ligase activity from the same extracts. Similarly, immunoprecipitates containing mβTrCPΔF were also unable to support the ubiquitination of p-IκBα, most likely because of the inability of this mutant to bind to Skp1 (Fig. 5A, bottom panel).
Figure 5.
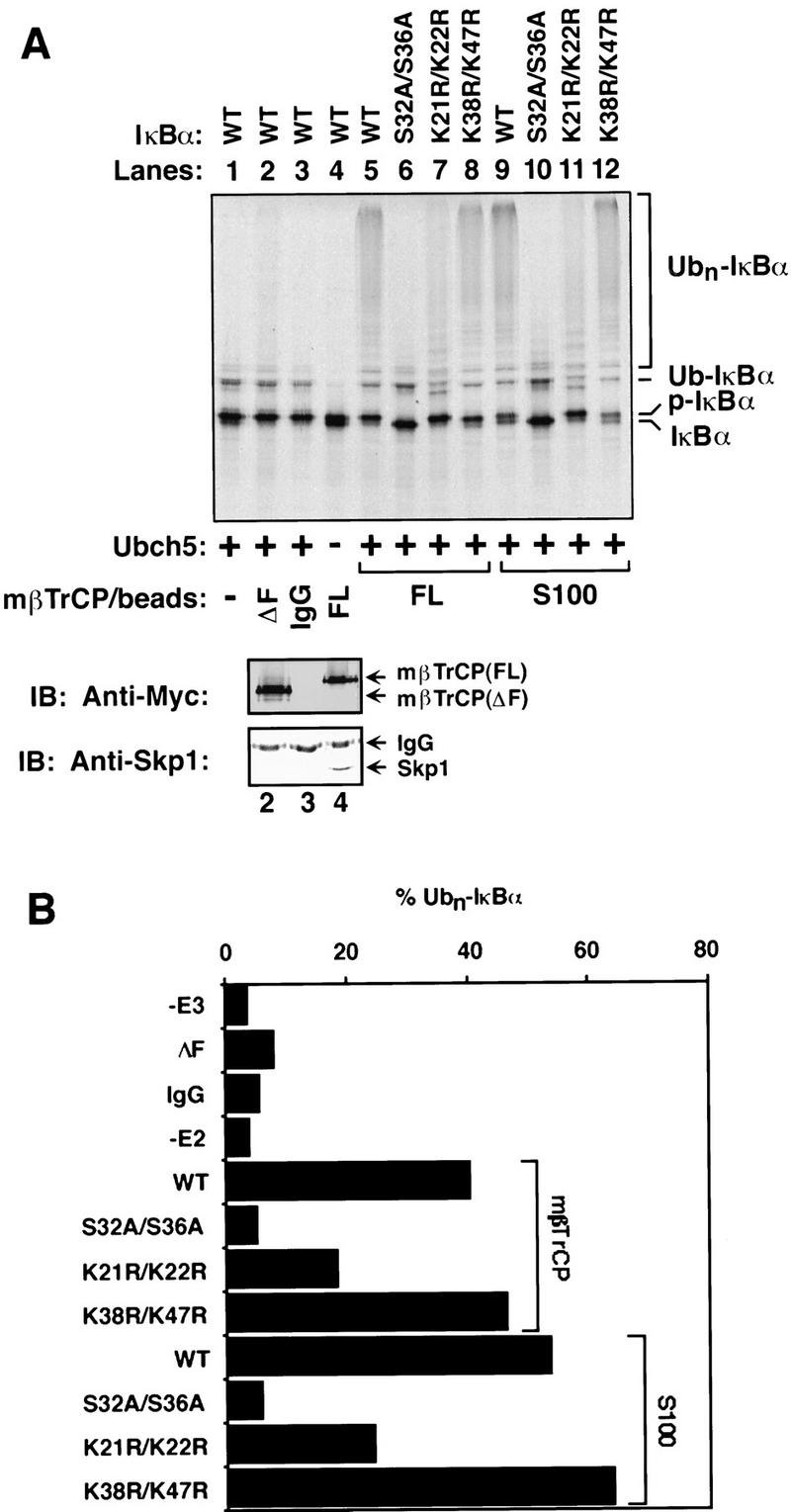
βTrCP promotes ubiquitination of phosphorylated IκBα in vitro. (A) Reconstitution of IκBα ubiquitination. 35S-labeled IκBα or mutants were incubated in an ATP-supplemented reconstitution system containing E1, Ubch5, Ub, p50/p65, IκB kinase and MEKK1Δ, as described in Materials and Methods. Ubiquitination was initiated by the addition of immunoprecipitated beads containing Myc–mβTrCP, which was expressed in 293 cells. Ubiquitinated products were analyzed by SDS-PAGE following immunoprecipitation with an anti-p65 antibody, which coprecipitated IκBα, as well as ubiquitinated IκBα (Chen et al. 1995). (Lane 1) No E3 was added; (lane 2) immunoprecipitated Myc–mβTrCPΔF was used in place of Myc–mβTrCP; (lane 3) normal rabbit IgG instead of Myc antibody was used to precipitate Myc–mβTrCP; (lane 4) no E2 was added; (lanes 5_–_8) Myc–mβTrCP (FL) beads were used to ubiquitinate wild-type and mutant IκBα as indicated; (lanes 9_–_12) 293 cell extracts (S100) were used to ubiquitinate wild-type and mutant IκBα as indicated. An aliquot of the immunoprecipitated beads used in the ubiquitination reaction was analyzed by western blotting using antibodies against Myc (middle panel) or Skp1 (bottom panel). (B) Quantitation of IκBα ubiquitination. Ubiquitinated IκBα shown in A was quantitated with the aid of PhosphorImager. Because only multiubiquitinated IκBα are degraded efficiently by the 26S proteasome, Ubn–IκBα containing more than two Ub units (n > 2) was quantitated and expressed as the percentage of total radioactivity in each lane.
It has been shown previously that mutation of lysines 21 and 22 of IκBα impair the ubiquitination and degradation of IκBα significantly, suggesting that these two lysines are the primary ubiquitination sites on IκBα (Scherer et al. 1995). We examined whether mβTrCP immunoprecipitates could ubiquitinate IκBα at specific sites in the reconstituted assay. As shown in Figure 5, the IκBα mutant in which lysines 21 and 22 were mutated to arginine (K21R/K22R) was only weakly ubiquitinated (∼40% of wild type; Fig. 5B), consistent with its weaker dominant-negative effect on NF-κB activation than the S32A/S36A mutant (Scherer et al. 1995). In contrast, mutation of the nearby lysines 38 and 47 (K38R/K47R) did not impair the ubiquitination of IκBα in the reconstituted system. Therefore, mβTrCP not only binds specifically to p-IκBα but also promotes the ubiquitination of phosphorylated IκBα at the physiologically relevant lysine residues in the presence of E1 and E2, thus fulfilling the major criteria for a bona fide E3IκB.
Discussion
In this report we have shown that βTrCP/Slimb exhibits several critical features expected of the substrate recognition subunit of IκB–Ub ligase (E3IκB). First, βTrCP binds specifically to phosphorylated, but not unphosphorylated, IκBα, both in vitro and in vivo. Second, a dominant-negative βTrCP mutant blocks the signalinduced degradation of IκBα and the activation of NF-κB. Third, _Drosophila slimb_-deficient embryos fail to activate twi and sna, two genes regulated by Dorsal, the Drosophila homolog of NF-κB. Finally, a βTrCP-containing complex ubiquitinates phosphorylated IκBα in the presence of E1 and Ubch5.
βTrCP/Slimb is an F-box protein and is likely to function within a SCF complex that serves as an E3 for IκB. Deletion of the F-box abolishes the binding of βTrCP to Skp1 and also abolishes its ability to support the ubiquitination of p-IκBα in vitro (Fig. 5A). Three F-box proteins involved in the ubiquitination of cell cycle proteins in yeast, including Cdc4, Grr1, and Met30, function as part of SCF complexes (Patton et al. 1998). In all three cases, the role of F-box is to mediate binding to Skp1, which in turn binds to Cdc53, which recruits an E2 (Cdc34) to the SCF complex. Based on evidence presented in this report and by analogy to the SCF complexes involved in cell cycle, we propose a model suggesting that a βTrCP-containing SCF complex is responsible for the signal-induced ubiquitination of IκBα (Fig. 6). In this model, βTrCP associates with Skp1, which in turn binds to a Cdc53-like protein whose identity remains to be determined. When cells are stimulated with NF-κB agonists, IκBα is phosphorylated at serines 32 and 36 by an IκB kinase complex. Phosphorylated IκBα is recruited to the SCFβTrCP complex through its binding to βTrCP. An E2, such as Ubch5, binds to the SCF complex and ubiquitinates the nearby IκBα at lysines 21 and 22. Because of the high-affinity binding of p-IκBα to βTrCP, ubiquitinated IκBα is not released from SCF and is processively multi-ubiquitinated to form a multi-Ub chain. Multi-Ub chains on IκBα recruit the 26S proteasome to degrade IκBα, allowing NF-κB to translocate into the nucleus, where it activates target genes.
Figure 6.

A proposed SCF pathway for the signal-induced ubiquitination of IκBα. βTrCP/Slimb associates with Skp1 and another protein (possibly Cdc53-like) to form a SCFβTrCP complex. Upon phosphorylation of IκB by an IκB kinase complex, βTrCP recruits IκBα to the SCF complex, allowing the associated E2, such as Ubch5, to ubiquitinate IκBα. Following ubiquitination, IκBα is rapidly degraded by the 26S proteasome, allowing NF-κB to translocate into the nucleus where it activates gene transcription. NF-κB is represented by a heterodimer of p50 and p65, whereas a multiubiquitin chain is represented by the branch structure on IκBα.
Two components in this SCFβTrCP pathway remain to be identified. First, what is the third component of the SCFβTrCP complex? It is now known that there is a large family of Cdc53-related proteins belonging to the Cullin family (Jackson 1996; Kipreos et al. 1996). Further work is needed to determine whether a mammalian homolog of Cdc53 or a distinct member of the family is involved in the assembly of the SCFβTrCP complex. Second, what is the identity of E2IκB? Although we and others have shown that Ubch5 or Ubch7 can ubiquitinate IκBα in vitro (Alkalay et al. 1995; Chen et al. 1996), this does not rule out the possibility that other E2s might also be involved. It remains to be determined which E2 (or E2s) functions in the signal-induced ubiquitination of IκBα in vivo.
Another important question concerns the dynamics of SCFβTrCP complex assembly and substrate binding. Coimmunoprecipitation experiments showed that βTrCP is associated with Skp1 regardless of signaling (Fig. 1), suggesting that SCFβTrCP is a preexisting complex in cells and that this complex binds further to p-IκBα/NF-κB to form a larger complex upon signaling. This dynamic assembly process is consistent with the previous report that IκBα eluted as part of a high molecular weight complex when TNFα-stimulated cell extracts were fractionated by gel filtration (Yaron et al. 1997). The dynamic nature of SCF complex assembly is underscored further by the recent finding that several F-box proteins in yeast are short-lived as a result of their own ubiquitination within SCF complexes (Zhou and Howley 1998). The rapid turnover of F-box proteins may provide an opportunity for different F-box proteins to compete for a limited pool of core SCF components such as Skp1. It is not known at present whether and how this combinatorial assembly of SCF complexes is regulated. The rapid degradation of F-box proteins also raises the possibility that these proteins might be limiting factors that control the rate of degradation of their target proteins. This may explain why overexpressed IκBα is not degraded efficiently in response to signals despite efficient phosphorylation (Traenckner et al. 1995).
Ubiquitination of IκBα by the βTrCP-containing complex in vitro occurs primarily at lysines 21 and 22, and is strictly dependent on its phosphorylation at serines 32 and 36, thus recapitulating the in vivo setting (Fig. 5). Interestingly, the specificity of IκBα ubiquitination is compromised when it is not bound to NF-κB (Z.J. Chen, unpubl.). For example, free IκBα mutants (i.e., S32A/S36A) can be ubiquitinated in the reconstituted system, albeit much more weakly. It has been shown that free IκBα is a short-lived protein that can be stabilized by binding to NF-κB (Scott et al. 1993). The binding of NF-κB to IκBα is mediated primarily through the ankyrin repeats of IκBα, which encompasses the bulk of the molecule (Haskill et al. 1991). It is therefore possible that NF-κB masks the majority of the IκBα molecule except for the amino-terminal regulatory sequence, thus rendering the degradation of bound IκBα dependent on signaling. The binding of NF-κB to IκBα may also minimize the accessibility of lysine residues to ubiquitination enzymes, thereby facilitating the ubiquitination of IκBα at specific lysine residues. This may explain why IκBα is one of the few proteins in which ubiquitination sites can be defined (Scherer et al. 1995).
βTrCP/Slimb has now been implicated in the ubiquitination of several proteins, including IκB/Cactus, β-catenin/Arm, CD4 (through HIV Vpu), and Ci. However, IκBα is the only protein so far shown to be directly ubiquitinated by the βTrCP-containing complex in vitro. Strikingly, the phosphorylation sites among Vpu, IκB, and β-catenin are very similar, with a minimal consensus sequence of DSGΨ-S (Ψ represents a hydrophobic residue). Although the serine residues of these proteins appear to be phosphorylated by distinct kinases, the fates of these proteins after phosphorylation are likely to be the same in terms of their binding to βTrCP/Slimb and subsequent ubiquitination and degradation. The only exception is Vpu, which does not appear to be degraded. Instead, it was proposed that Vpu targets its cognate partner CD4 to the ER degradation pathway (Margottin et al. 1998). It will be important to determine whether Vpu or CD4 is ubiquitinated to understand why and how Vpu escapes degradation by the SCF pathway.
The potential involvement of βTrCP/Slimb in the NF-κB, Hh, and Wg raises the exciting possibility that these divergent pathways may in fact be interconnected and that βTrCP/Slimb may be deployed from one pathway to another to allow for integration of these pathways in response to signals. Further studies on βTrCP/Slimb may provide another avenue for modulating the activity of these pathways, all of which have been implicated in several human diseases, including cancer.
Materials and methods
Plasmids, antibodies, and chemicals
cDNAs encoding IκBα and mutants (S32/36A, ΔN, K21/22R, K38/47R) were gifts of Dr. Dean Ballard (Vanderbilt University, Nashville, TN) and have been described previously (Brockman et al. 1995; Chen et al. 1995; Scherer et al. 1995). A luciferase reporter construct containing three tandem repeats of NF-κB binding sites (p[κB]3–TK–Luc) was a gift of Dr. Shigeki Miyamoto (University of Wisconsin, Madison). Rabbit anti-IκBα (C15 and C21), anti-Myc (A14), anti-Skp1 (H163), and goat anti-p65 (C20G) polyclonal antibodies, as well as mouse anti-Myc monoclonal antibody (9E10), were purchased from Santa Cruz Biotechnology. Mouse anti-Flag antibodies (M2 and M5) were from Eastman Kodak. MG132 (z-Leu-Leu-Leu-H) was purchased from Calbiochem. Calyculin A and okadaic acid were purchased from Alexis.
Proteins
Ub was purchased from Sigma. Ub aldehyde (Ubal) was produced by periodate oxidation of Ubdiol, which was synthesized using the carboxylpeptidase Y method (Lam et al. 1997). Ubch5 was derived from GST–Ubch5 (Chen et al. 1996) by thrombin cleavage and then purified to apparent homogeneity by MonoS fast performance chromatography (FPLC, Pharmacia). E1 was purified from calf thymus by covalent affinity chromatography on Ub–Sepharose. Recombinant (His)6–MEKK1Δ was purified from baculovirus-infected Sf9 cells as described previously (Lee et al. 1997; baculovirus provided by Drs. Frank Lee and Tom Maniatis, Harvard University, Cambridge, MA). IκB kinase complex was partially purified as described previously, except that calf thymus instead of HeLa cells was used as the source of the kinase. The kinase activity from the Superdex fractions can be activated by ubiquitination (Chen et al. 1996), by MEKK1 (Lee et al. 1997), or by NIK (Z.J. Chen, unpubl.), and was used for in vitro phosphorylation of IκBα at serines 32 and 36. Further purification and characterization of the IκB kinase complex will be reported elsewhere. GST–p50 and p65–His6 were expressed from Escherichia coli and purified as described previously (Thanos and Maniatis 1992). To allow for the formation of p50/p65 heterodimer, these proteins were mixed in the presence of buffer A (20 mm Tris at pH 7.5, 0.5 mm DTT, 0.1 mm EDTA, 0.1 mm EGTA, 0.1 mm PMSF, 0.1 m NaCl) plus 6 m urea and then dialyzed against the same buffer overnight. The proteins were renatured by step-wise reduction of urea concentration to 4 m, 2 m, and finally no urea in buffer A. In some cases, the proteins were further purified by glutathione chromatography. The functionality of the renatured p50/p65 heterodimer was confirmed by its ability to coimmunoprecipitate in vitro-translated 35S-labeled IκBα.
cDNA cloning of mβTrCP and mβTrCPΔF
A BLAST search of the EST database for mammalian homologs of Slimb (Jiang and Struhl 1998) and Xenopus βTrCP (Spevak et al. 1993) identified two clones (AA197590 and AA033076) that together encompass ∼370 amino acids corresponding to the carboxyl terminus of the mouse homolog of Slimb/βTrCP (mβTrCP). PCR primers derived from these two clones (5′GCGGTCGACCGTCAGGACGGACTCTCTGTGG-3′ and 5′-AGTGCGGCCGCTTATCTGGAGATGTAGGTGTA-3′) were used to amplify a 1.1-kb fragment (mβTrCPΔN) from a mouse E8.5 (embryonic day 8.5) cDNA library in λZAPXR (Stratagene). The 5′-coding region of mβTrCP was amplified from the same library by PCR using T7 primer and a reverse mβTrCP primer (5′-AGTGCGGCCGCGAGCTTTTTCCACAGCATGCC-3′). A 650-bp fragment was subcloned into _Bam_HI and _Not_I sites of pBluescript (Strategene) and sequenced. This fragment contains the full-length 5′-coding sequence plus 60 bp of 5′-untranslated sequence. To generate a full-length mβTrCP that is fused in-frame with five tandem repeats of amino-terminal Myc epitopes, the amino terminus of mβTrCP was amplified by PCR using the following primers: 5′-CGCCCATGGACCCGGCAGAGGCGGTG-3′ and 5′-CGCTCTGCCAGGCCTCGCCACAGA-3′. A 600-bp fragment was subcloned into the _Nco_I and Stu_I sites of pSK–Myc–_slimb (Jiang and Struhl 1998) to replace the amino terminus of Slimb. The carboxyl terminus of Slimb was subsequently replaced with a _Stu_I–_Not_I fragment (1.1 kb) of mβTrCPΔN. Following partial digestion with _Kpn_I and _Not_I, the full-length Myc–mβTrCP fragment (∼1.9 kb) was subcloned into pcDNA3. The entire coding region was confirmed by DNA sequencing.
To delete the F-box from mβTrCP, the amino terminus of mβTrCP up to the F-box was amplified by PCR (5′-CGCCCATGGACCCGGCAGAGGCGGTG-3′ and 5′-GGCGACGTCGCAGTTATGAAATCCCTCTG-3′). The amplified fragment (450 bp) was then cloned into the _Nco_I–_Aat_II sites of pSK–Myc–mβTrCP to replace the amino terminus of mβTrCP, which includes almost the entire F-box (34 of approximately 40 residues; Bai et al. 1996). Following confirmation by DNA sequencing, the resulting mβTrCPΔF, including the amino-terminal Myc coding sequence, was subcloned into the _Kpn_I–_Not_I sites of pcDNA3.
Cell culture, transfection, and reporter assays
Human embryonic kidney 293 cells were grown on 0.1% gelatin-coated plates in DMEM supplemented with 10% fetal calf serum. Transfection of 293 cells was carried out by the calcium phosphate precipitation method (Sambrook et al. 1989), which yielded nearly 100% transfection efficiency (data not shown). For reporter gene assays, 293 cells were seeded into six-well plates (4 × 105 cells/well) and then transfected with 10 ng of p[κB]3–TK–Luc reporter construct, 10 ng of pCMV–lacZ, and various amounts of other expression constructs. The total DNA concentration was kept constant at 3 μg by supplementing with pcDNA3 vector. After 40 hr, cells were treated with or without TNFα (20 ng/ml) for 6 hr prior to harvest for luciferase assay (Promega). Transfection efficiencies were normalized by βgalactosidase activities, and the reporter gene activity was expressed as the mean of normalized luciferase activity from duplicated experiments.
In vitro translation, immunoprecipitation, and immunoblotting
35S-Labeled IκBα and its mutants were synthesized in wheat germ extracts programmed with the corresponding RNA using TNT in vitro translation kits (Promega). 35S-labeled mβTrCP and mβTrCPΔF were produced similarly, except that rabbit reticulocyte lysates were used in place of wheat germ extracts. To analyze the binding of IκBα to mβTrCP in vitro, 35S-labeled IκBα or S32A/S36A mutant was phosphorylated by MEKK1-activated IκB kinase as described previously (Lee et al. 1997) and incubated with equal amounts of 35S-labeled mβTrCP or mβTrCPΔF at 4°C in 0.15 ml of RIPA buffer (50 mm Tris-HCl at pH 8.0, 150 mm NaCl, 1% NP-40, 0.5% deoxycholate) plus 0.1% SDS. After 90 min of incubation, 0.1 μg of anti-IκBα antibody (C21, Santa Cruz) preadsorbed to protein A–Trisacryl (Pierce) was added, and the mixture was incubated further at 4°C for 1 hr with end-over-end rotation. The beads were then washed three times with 0.75 ml of RIPA/0.1% SDS and resuspended in Laemmli/SDS sample buffer. Proteins bound to the beads were resolved by 9% SDS-PAGE and analyzed with the aid of the PhosphorImager (Molecular Dynamics).
For analysis of IκBα binding to mβTrCP in vivo, 293 cells were plated on 10-cm dishes (106 cells/plate) and transfected with up to 10 μg of total DNA containing various expression constructs. Forty-eight hours after transfection, cells were pretreated with 20 μm of MG132 for 30 min, followed by stimulation with 0.1 μm calyculin A for 10 min. After washing with cold PBS, cells were resuspended in lysis buffer (20 mm Tris at pH 7.5, 150 mm NaCl, 0.5% NP-40, 50 mm NaF, 1 mm NaVO3, 0.5 mm PMSF, 5 μg/ml leupeptin, 0.2 mm EGTA) and lysed on ice for 15 min. Cell debris was removed by centrifugation, and the supernatant was used for immunoprecipitation with appropriate antibodies as described above, except that lysis buffer was used in place of RIPA buffer. Immunoprecipitated proteins were analyzed by immunoblotting and visualized by either colorimetric (Promega) or enhanced chemiluminescence (ECL, Amersham) methods.
In vitro ubiquitination assays
Myc-tagged mβTrCP was expressed in transiently transfected 293 cells and then purified by immunoprecipitation using anti-Myc polyclonal antibody (A14, Santa Cruz) bound to protein A–Sepharose. After extensive washing with lysis buffer, the beads were washed further with buffer A (10 mm Tris-HCl at pH 7.5, 0.5 mm DTT, 1.5 mm MgCl2, 10 mm KCl, 5 μg/ml leupeptin) and aliquoted into assay tubes to be used as E3 for ubiquitination reactions. The reaction conditions were modified from what was described previously (Chen et al. 1995; Scherer et al. 1995) to accommodate the reconstitution with E1, E2, and E3. Briefly, 35S-labeled IκBα or mutants were incubated with recombinant p50/p65 (50 ng) to form a complex in a reaction mixture (20 μl) containing an ATP-regenerating buffer (Chen et al. 1995), Ub (60 μm), Ubal (1.5 μm), okadaic acid (3 μm), MG132 (10 μm), MEKK1 (5 nm), IκB kinase complex (75 ng/μl), E1 (0.1 μm), and Ubch5 (0.6 μm). Reactions were initiated by adding the reaction mixture into the tubes containing mβTrCP beads. After 1 hr of incubation at 37°C, 150 μl of cold lysis buffer containing 0.4 μg of anti-p65 bound to protein A/G-Trisacryl was added to the reaction, and the mixture was incubated at 4°C for 1 hr with end-over-end rotation. The beads were washed once with 0.7 ml of lysis buffer, and the bound proteins analyzed by 9% SDS-PAGE followed by fluorography. Ubiquitinated IκBα was quantitated by PhosphorImager.
Generation of slimb mutant Drosophila embryos and whole-mount in situ hybridization
An EMS-induced allele of slimb, slimb2, was used to generate embryos deficient in both the maternal and zygotic slimb function. These mutant embryos were generated by the dominant female sterile technique (Chou and Perrimon 1992). Females of the genotype y, hs–flp121/y; FRT82 slimb2/FRT82 ovoD were subjected to multiple heat shock at pupal stages to induce germline clones, which were then mated with FRT 82 slimb2/TM2 males. Embryos were collected from such crosses for whole-mount in situ hybridization with twi or sna antisense RNA probes as described previously (Jiang et al. 1991).
Acknowledgments
We thank Drs. Dean Ballard, Tom Maniatis, Shigeki Miyamoto, and Richard Benarous for providing us with plasmids as indicated. We are grateful to Drs. Tom Maniatis, Joe Goldstein, Cecile Pickart, and Eric Olson for critically reading the manuscript. The excellent technical support of Ms. Wassila Amari is much appreciated. We also thank Ms. Alisha Tiznor for assistance with graphics. J.J. is a Leukemia Society Special Fellow and a Eugene McDermott Endowed Scholar in Medical Research. Z.J.C. is a Searle Scholar supported by the Chicago Community Trust.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Note
The GenBank accession number for the mouse βTrCP cDNA sequence reported in this paper is AF112979.
Footnotes
E-MAIL jchen@hamon.swmed.edu; FAX (214) 648-1196.
References
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. β-Catenin is a target for the Ub-proteasome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkalay I, Yaron A, Hatzubai A, Orian A, Ciechanover A, Ben-Neriah Y. Stimulation-dependent IκBα phosphorylation marks the NF-κB inhibitor for degradation via the Ub-proteasome pathway. Proc Natl Acad Sci. 1995;92:10599–10603. doi: 10.1073/pnas.92.23.10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aza-Blanc P, Ramirez-Weber FA, Laget MP, Schwartz C, Kornberg TB. Proteolysis that is inhibited by hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell. 1997;89:1043–1053. doi: 10.1016/s0092-8674(00)80292-5. [DOI] [PubMed] [Google Scholar]
- Baeuerle P, Baltimore D. NF-κB: Ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- Baldwin AS. The NF-κB and IκB proteins: New discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Brockman JA, Scherer DC, McKinsey TA, Hall SM, Qi X, Lee WY, Ballard DW. Coupling of a signal response domain in IκBα to multiple pathways for NF-κB activation. Mol Cell Biol. 1995;15:2809–2818. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- Chen ZJ, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T. Signal-induced site-specific phosphorylation targets IκBα to the ubiquitin–proteasome pathway. Genes & Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IκBα by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–862. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- Chou T-B, Perrimon N. Use of a yeast site-specific recombinase to produce female germline chimeras in Drosophila. Genetics. 1992;131:643–653. doi: 10.1093/genetics/131.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Hod Y, Hershko A. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem Biophys Res Commun. 1978;81:1100–1105. doi: 10.1016/0006-291x(78)91249-4. [DOI] [PubMed] [Google Scholar]
- Feldman RM, Correll CC, Kaplan KB, Deshaies RJ. A complex of Cdc4p, Skp1p, and Cdc53p/Cullin catalyzes Ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell. 1997;9:221–230. doi: 10.1016/s0092-8674(00)80404-3. [DOI] [PubMed] [Google Scholar]
- Haskill S, Beg AA, Tompkins SN, Morris JS, Yurochko AD, Sampson-Johannes A, Mondal K, Ralph P, Baldwin AS. Characterization of an immediate-early gene induced in adherent monocytes that encodes IκB-like activities. Cell. 1991;65:1281–1289. doi: 10.1016/0092-8674(91)90022-q. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A, Heller H, Haas AL, Rose IA. Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc Natl Acad Sci. 1980;77:1783–1786. doi: 10.1073/pnas.77.4.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, Ganoth D, Sudakin V, Dahan A, Cohen LH, Luca FC, Ruderman JV, Eytan E. Components of a system that ligates cyclin to ubiquitin and their regulation by the protein kinase cdc2. J Biol Chem. 1994;269:4940–4946. [PubMed] [Google Scholar]
- Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- Jackson PK. Cell cycle: cull and destroy. Curr Biol. 1996;6:1209–1212. doi: 10.1016/s0960-9822(96)00697-5. [DOI] [PubMed] [Google Scholar]
- Jiang J, Struhl G. Regulation of the Hedgehog and Wingless signaling pathways by the F-box/WD40-repeat protein Slimb. Nature. 1998;391:493–496. doi: 10.1038/35154. [DOI] [PubMed] [Google Scholar]
- Jiang J, Kosman D, Ip YT, Levine M. The dorsal morphogen gradient regulates the mesoderm determinant twist in early Drosophila embryos. Genes & Dev. 1991;5:1881–1891. doi: 10.1101/gad.5.10.1881. [DOI] [PubMed] [Google Scholar]
- Kipreos ET, Lander LE, Wing JP, He WW, Hedgecock EM. cul-1 is required for cell cycle exit in C. elegans and identifies a novel gene family. Cell. 1996;85:829–839. doi: 10.1016/s0092-8674(00)81267-2. [DOI] [PubMed] [Google Scholar]
- King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 1995;81:279–288. doi: 10.1016/0092-8674(95)90338-0. [DOI] [PubMed] [Google Scholar]
- Lam YA, DeMartino GN, Pickart CM, Cohen RE. Specificity of the ubiquitin isopeptidase in the PA700 regulatory complex of 26 S proteasomes. J Biol Chem. 1997;272:28438–28446. doi: 10.1074/jbc.272.45.28438. [DOI] [PubMed] [Google Scholar]
- Lee FS, Hagler J, Chen Z, Maniatis T. Activation of the IκBα kinase complex by MEKK1, a kinase of the JNK pathway. Cell. 1997;88:213–222. doi: 10.1016/s0092-8674(00)81842-5. [DOI] [PubMed] [Google Scholar]
- Maniatis T. Catalysis by a multiprotein IκB kinase complex. Science. 1997;278:818–819. doi: 10.1126/science.278.5339.818. [DOI] [PubMed] [Google Scholar]
- Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, Thomas D, Strebel K, Benarous R. A novel human WD protein, h-βTRCP, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-Box motif. Mol Cell. 1998;1:565–574. doi: 10.1016/s1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- Morisato D, Anderson KV. Signaling pathways that establish the dorsal-ventral pattern of the Drosophila embryo. Annu Rev Genet. 1995;29:371–379. doi: 10.1146/annurev.ge.29.120195.002103. [DOI] [PubMed] [Google Scholar]
- Nusse R. A versatile transcriptional effector of Wingless signaling. Cell. 1997;89:321–323. doi: 10.1016/s0092-8674(00)80210-x. [DOI] [PubMed] [Google Scholar]
- Orford K, Crockett C, Jensen JP, Weissman AM, Byers SW. Serine phosphorylation-regulated ubiquitination and degradation of β-catenin. J Biol Chem. 1997;272:24735–24738. doi: 10.1074/jbc.272.40.24735. [DOI] [PubMed] [Google Scholar]
- Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- Patton E, Willems AR, Tyers M. Combinatorial control in ubiquitin-dependent proteolysis: Don’t Skp the F-box hypothesis. Trends Genet. 1998;14:236–243. doi: 10.1016/s0168-9525(98)01473-5. [DOI] [PubMed] [Google Scholar]
- Pickart CM. Targeting of substrates to the 26S proteasome. FASEB J. 1997;11:1055–1066. doi: 10.1096/fasebj.11.13.9367341. [DOI] [PubMed] [Google Scholar]
- Ray RP, Arora K, Nüsslein-Volhard C, Gelbart WM. The control of cell fate along the dorsal-ventral axis of the Drosophila embryo. Development. 1991;113:35–54. doi: 10.1242/dev.113.1.35. [DOI] [PubMed] [Google Scholar]
- Roth S, Hiromi Y, Godt D, Nüsslein-Volhard C. cactus, a maternal gene required for proper formation of the dorsoventral morphogen gradient in Drosophila embryos. Development. 1991;112:371–388. doi: 10.1242/dev.112.2.371. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Scheidereit C. Signal transduction. Docking IκB kinases. Nature. 1998;395:225–226. doi: 10.1038/26121. [DOI] [PubMed] [Google Scholar]
- Scherer DC, Brockman JA, Chen ZJ, Maniatis T, Ballard D. Signal-induced degradation of IκBα requires site-specific ubiquitination. Proc Natl Acad Sci. 1995;92:11259–11263. doi: 10.1073/pnas.92.24.11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott ML, Fujita T, Liou HC, Nolan GP, Baltimore D. The p65 subunit of NF-κB regulates IκB by two distinct mechanisms. Genes & Dev. 1993;7:1266–1276. doi: 10.1101/gad.7.7a.1266. [DOI] [PubMed] [Google Scholar]
- Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Spevak W, Keiper BD, Stratowa C, Castanon MJ. Saccharomyces cerevisiae cdc15 mutants arrested at a late stage in anaphase are rescued by Xenopus cDNAs encoding N-ras or a protein with β-transducin repeats. Mol Cell Biol. 1993;13:4953–4966. doi: 10.1128/mcb.13.8.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancovski I, Baltimore D. NF-κB activation: the IκB kinase revealed? Cell. 1997;91:299–302. doi: 10.1016/s0092-8674(00)80413-4. [DOI] [PubMed] [Google Scholar]
- Thanos D, Maniatis T. The high mobility group protein HMG I(Y) is required for NF-κB dependent virus induction of the human IFN-β gene. Cell. 1992;71:777–789. doi: 10.1016/0092-8674(92)90554-p. [DOI] [PubMed] [Google Scholar]
- Theodosiou NA, Zhang S, Wang WY, Xu T. slimb coordinates wg and dpp expression in the dorsal-ventral and anterior-posterior axes during limb development. Development. 1998;125:3411–34166. doi: 10.1242/dev.125.17.3411. [DOI] [PubMed] [Google Scholar]
- Traenckner EB, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. Phosphorylation of human IκBα on serines 32 and 36 controls IκBα proteolysis and NF-κB activation in response to diverse stimuli. EMBO J. 1995;14:2876–2883. doi: 10.1002/j.1460-2075.1995.tb07287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Baldwin AS., Jr Activation of nuclear factor-κB-dependent transcription by tumor necrosis factor-α is mediated through phosphorylation of RelA/p65 on serine 529. J Biol Chem. 1998;273:29411–29416. doi: 10.1074/jbc.273.45.29411. [DOI] [PubMed] [Google Scholar]
- Wilkinson KD, Urban MK, Haas AL. Ubiquitin is the ATP-dependent proteolysis factor I of rabbit reticulocytes. J Biol Chem. 1980;255:7529–7532. [PubMed] [Google Scholar]
- Yaron A, Gonen H, Alkalay I, Hatzubai A, Jung S, Beyth S, Mercurio F, Manning AM, Ciechanover A, Ben-Neriah Y. Inhibition of NF-κB cellular function via specific targeting of the IκB-ubiquitin ligase. EMBO J. 1997;16:6486–6494. doi: 10.1093/emboj/16.21.6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. The transcriptional activity of NF-κB is regulated by the IκB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
- Zhou P, Howley P. Ubiquitination and degradation of the substrate recognition subunits of SCF ubiquitin-protein ligases. Mol Cell. 1998;2:571–580. doi: 10.1016/s1097-2765(00)80156-2. [DOI] [PubMed] [Google Scholar]