Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression (original) (raw)
. 2000 Apr 1;14(7):804–816.
Abstract
The E2F transcription factor plays a pivotal role in the timely activation of gene expression during mammalian cell cycle progression, whereas pRB and related proteins control cell growth in part through the ability to block the action of E2F. To identify physiologically important E2F-responsive promoters and to study their occupancy and histone acetylation state in vivo, we have taken advantage of a cross-linking approach in synchronized, living cells. We find that the pattern of E2F and pRB-related polypeptides recruited to these promoters changes in a strikingly dynamic fashion as cells progress from quiescence into G1 and S phase: Repression of each promoter in quiescent cells is associated with recruitment of E2F-4 and p130 and low levels of histone acetylation, but by late G1, these proteins are replaced largely by E2F-1 and E2F-3, in concert with acetylation of histones H3 and H4 and gene activation. These findings suggest that repression and activation of E2F-responsive genes may occur through distinct E2F heterodimers that direct the sequential recruitment of enzymes able to deacetylate and then acetylate core histones.
Keywords: E2F, activation, pRB, cell cycle progression
Transition through the mammalian cell cycle requires an interplay of transcription factors that coordinately induce or repress gene expression in a temporally defined manner. The E2F transcription factor is known to play a pivotal role in mediating gene expression during cell proliferation. E2F activity consists of a heterodimer containing one of six factors (E2F-1, E2F-2, E2F-3, E2F-4, E2F-5, and E2F-6) that pairs with a second subunit (DP-1 or DP-2; for review, see Dyson 1998). The transcriptional activation potential of E2F is counterbalanced by the retinoblastoma tumor suppressor protein (pRB) with which E2F tightly associates. E2F heterodimers not bound by the pRB family (free E2F) are thought to represent the active transcription factor, and this species may drive gene expression in cells entering S phase. It has been proposed that different E2F heterodimers might activate particular sets of growth-related gene targets, and a number of ectopic expression studies have suggested that this could be the case (DeGregori et al. 1995, 1997). These studies are also supported by experiments in which enhanced expression of E2F-1, E2F-2, and E2F-3, but not E2F-4 or E2F-5, efficiently induced cells to enter S phase, although E2F-1 was uniquely able to promote apoptosis (DeGregori et al. 1997).
The pRB family of inhibitors consists of pRB and the related proteins p107 and p130. pRB associates with each member of the E2F family, except E2F-5 and E2F-6, whereas p107 binds E2F-4 exclusively, and p130 binds both E2F-4 and E2F-5 (for review, see Dyson 1998). Complex formation between E2F and pRB families is cell cycle dependent: Although these proteins form tight physical interactions in early-to-mid-G1 phase, cyclin-dependent kinases phosphorylate the pRB family in late G1, liberating free E2F. Subsequent phosphorylation of specific E2F family members by cyclin A-associated kinases could down-regulate E2F activity after entry into S phase (Dynlacht et al. 1994, 1997; Krek et al. 1994).
Another aspect of E2F function—that of a transcriptional repressor—has emerged, reflecting the importance of E2F–pRB family complexes. A repressive role for E2F was suggested by studies in which mutation of an E2F site in several different promoters (B-Myb, Cdc2, cyclin E, and E2F-1) led to increased expression in quiescent and G1 cells (Dyson 1998 and references therein). Expression of these genes is therefore thought to result primarily from relief of repression (derepression) in G1 phase, although it is likely that other transcription factors also contribute to activation at the G1/S transition. Genomic footprinting experiments with the B-Myb, cyclin A, and Cdc2 promoters further support this notion because potential E2F-binding sites in each promoter are occupied in quiescent and early G1 phase cells, when the promoters are repressed, and largely unoccupied during the G1/S transition when the genes are actively transcribed (Tommasi and Pfeifer 1995; Huet et al. 1996; Zwicker et al. 1996). The observation that E2F-1 knockout mice develop tumors may further support this negative role for E2F and may be explained in part by the ability of E2F to act as a repressor of growth-related gene expression through the recruitment of pRB family members (Yamasaki et al. 1996).
The mechanisms by which the pRB family represses transcription have been the subject of considerable interest. Recently, it has been proposed that pRB repression is potentiated by recruitment of histone deacetylase (HDAC) activity to the promoter (Brehm et al. 1998; Luo et al. 1998; Magnaghi-Jaulin et al. 1998). Recruitment of this enzyme is thought to repress gene expression by altering chromatin structure, and decreased acetylation of histones is associated with transcriptionally inactive chromatin (for review, see Kornberg and Lorch 1999). The role of HDAC recruitment in repression by pRB may be promoter-specific, however, as HDAC is not strictly required for transcriptional inhibition of all promoters (Luo et al. 1998; Ross et al. 1999).
Although much progress has been made in understanding transcriptional control by E2F, the identification of those E2F and pRB family members, if any, that bind to and regulate potential target promoters under physiological conditions remains a central issue. The majority of studies aimed at addressing this point have made use of ectopically expressed E2F and pRB, whereby the abundance of these proteins far exceeds endogenous levels. Several recent studies have used genomic footprinting to address the issue of protein binding to cell cycle-regulated promoters (Zwicker et al. 1996; Le Cam et al. 1999). This technique is of great value in that it is able to distinguish those promoter elements that are occupied in vivo as cells progress through the cycle. However, the identities of _trans_-acting factors can only be inferred on the basis of sequence similarities, and this technique therefore does not allow for the identification of factors bound at occupied sites.
In an attempt to determine which genes are under the control of the E2F and pRB family of proteins under physiological conditions, we have begun to survey a number of genes thought to bind these proteins. Using in vivo cross-linking and chromatin immunoprecipitations, we document for the first time the binding of specific E2F family members to several growth-regulated genes in living human cells progressing synchronously through the cell cycle. We find that E2F-4 functions on these promoters as a repressor, most likely through the recruitment of p130 in quiescent cells. Interestingly, occupancy by E2F-4 markedly diminishes as cells progress through G1 and expression of each gene is induced. Other E2Fs, in particular E2F-1, E2F-2, and E2F-3, take the place of E2F-4 and are recruited to the promoter in late G1. Acetylation of histones H3 and H4 at these E2F-responsive promoters also changes dramatically during the cell cycle in ways that reflect both the recruitment of specific E2Fs and the activity of each promoter: E2F-4 and p130 occupancy correlates with decreased acetylation, whereas binding by other E2Fs in late G1 correlates with increased levels of acetylation.
Results
Use of chromatin immunoprecipitations to study E2F binding in vivo
An important challenge in the study of transcriptional control during cell cycle progression in mammalian cells has been the identification of proteins that bind and regulate promoters at each stage under physiological conditions. Here, we have taken advantage of an in vivo formaldehyde cross-linking technique that allows us to study promoter occupancy by E2F and pRB family members in living cells. This approach is delineated in Figure 1A and is based on a modification of an existing protocol (Parekh and Maniatis 1999). We have used this chromatin immunoprecipitation approach to investigate the p107, E2F-1, Cdc25A, Cdc6, B-myb, cyclin A, and Cdc2 promoters because each of these promoters has been implicated as a target of the E2F and pRB family on the basis of genetic and/or biochemical criteria. These promoters, many of which have several potential E2F sites, are diagrammed in Figure 1B.
Figure 1.

In vivo detection of promoter occupancy by the E2F and pRB family using chromatin immunoprecipitations. (A) Outline of the in vivo cross-linking and chromatin immunoprecipitation (IP) protocol. (B) Promoters examined in this study. Solid ovals represent E2F-binding sites on the basis of previous studies; small arrows indicate primers used in PCR amplification reactions. Large arrows represent published major transcription start site. (C) Chromatin immunoprecipitations from asynchronously growing T98G cells. Input corresponds to PCR reactions containing 0.5% of total amount of chromatin used in immunoprecipitation reactions. Mock immunoprecipitations performed with irrelevant, control antibodies (anti-T Antigen pAb101) and control reactions lacking antibodies (No Ab) are shown. Amplified products were detected by autoradiography.
We focused our efforts on the human T98G cell line because of its utility in cell cycle synchronization experiments (see below). Using antibodies against various members of the E2F family, we initially immunoprecipitated chromatin from asynchronously growing cells. Interestingly, we detected a significant association between all E2F proteins, except E2F-5, and each promoter tested (Fig. 1C). We did not reproducibly observe significant binding by E2F-5 to the promoters tested. We did note some enrichment of individual E2Fs on certain promoters. For example, E2F-3 binding was enhanced on the p107 and E2F-1 promoters, whereas E2F-4 binding was somewhat enriched on the cyclin A promoter. Negligible quantities of chromatin were collected when an irrelevant control antibody was used or antibody was omitted altogether (Figure 1C).
We have also examined the occupancy of each promoter by the pRB family of proteins. Strikingly, each promoter was bound by the p107 and p130 proteins, although we failed to detect a significant enrichment of any promoter fragment using a panel of distinct antibodies against pRB (Fig. 1C; data not shown). We did notice weak but consistent binding of pRB to the p107 promoter, although the significance of this finding remains to be determined (see below). As before, we failed to detect amplified products when parallel immunoprecipitation reactions were performed in the absence of antibody or with an irrelevant antibody.
In addition to the controls listed above, we confirmed the specificity of our protocol by performing PCR amplification of identical immunoprecipitates with primers annealing to the actin promoter because transcription of this gene is not thought to be under the control of either the E2F or pRB family. Under no circumstances did we amplify significant levels of the actin promoter (Fig. 1C). As a further control, we have confirmed the specificity of each antibody using gel mobility shift and immunoprecipitation/Western blotting experiments (see below and data not shown). As a measure of the proficiency of this protocol, our calculated efficiency of immunoprecipitation ranged between 0.1% and 0.5% of input levels of chromatin, and this estimate is comparable with other published studies in yeast (Aparicio et al. 1997; Kuras and Struhl 1999).
Synchronization and cell cycle analyses
Next, we sought to address whether promoter binding by E2F and pRB polypeptides in vivo varied as a function of the cell cycle. Given the very high degree of sensitivity associated with PCR detection of chromatin immunoprecipitates, this analysis requires the isolation of highly synchronized, homogeneous populations of cells in each stage of the cell cycle. Human cell lines are notoriously difficult to synchronize by methods other than drug block. We therefore surveyed a number of cell lines and identified one, T98G, that could be efficiently synchronized by serum deprivation. Although it is a continuous cell line, T98G has retained growth arrest mechanisms characteristic of normal cells, including density-mediated growth inhibition and induction of quiescence in response to serum deprivation (Stein 1979). Importantly, T98G cells express abundant levels of E2F activity, and every member of the pRB family is functional (see below).
We have confirmed the utility of the T98G cell line as follows. First, serum starvation resulted in the accumulation of a quiescent population that synchronously progressed through G1 and S phase after serum stimulation (Fig. 2A), allowing for the isolation of populations in early G1 (4–8 hr), mid-G1 (12 hr), late G1 (16 hr), at the G1/S transition (20 hr), and in S phase (24 hr) (henceforth, all cell cycle phase designations refer to these time points). This cell line therefore satisfied the rigorous requirement for homogeneity of cell populations because each synchronized sample that we examined was highly pure.
Figure 2.

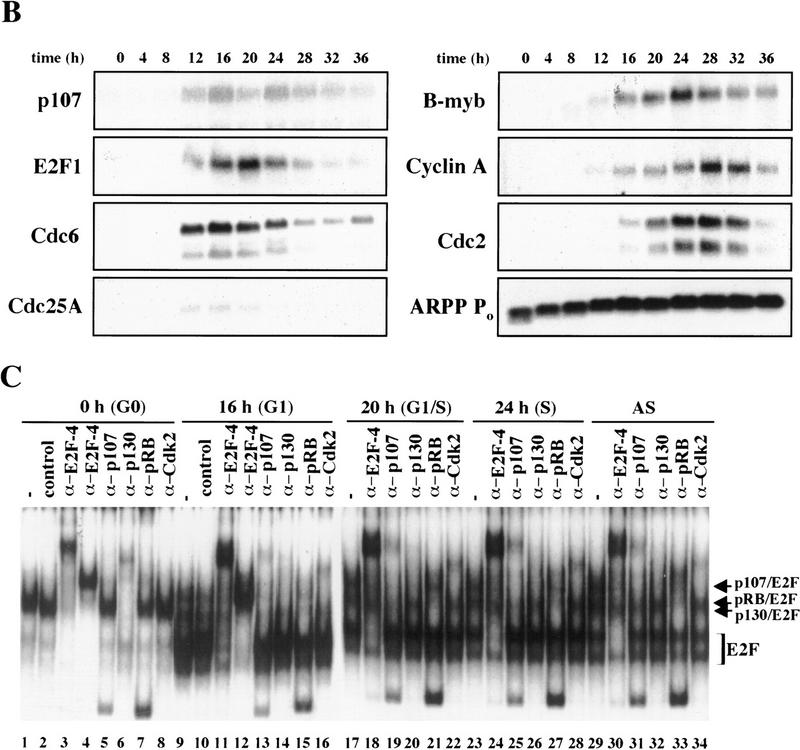
Cell cycle synchronization and gene expression analysis. (A) FACS analysis of T98G cells that were rendered quiescent by serum starvation (0 hr) and were subsequently restimulated with serum for the indicated lengths of time and allowed to enter the cell cycle. DNA content (propidium iodide intensity) is plotted vs. cell number. (B) Northern blot analysis of expression of the indicated genes during the synchronization experiment in A. To ensure equal loading of RNA, each blot was subsequently stripped and rehybridized with an ARPP P0 gene fragment, and a representative filter is shown. (C) Gel mobility shift analysis of E2F and pRB complexes. Whole cell extracts at each stage of the cell cycle (indicated at top) were incubated with a labeled probe containing an E2F site and each of the designated antibodies. The positions of E2F-pRB/p107/p130 complexes and free E2F (E2F) are shown at right. Each of these complexes was abrogated by a specific competitor oligonucleotide (data not shown). Asynchronous (AS) samples were prepared from cells entering a second cell cycle (with roughly equal percentages of G1, S, and G2 cells). Lanes marked − and control contained no antibody and 12CA5 (anti-flu hemagglutinin antibody), respectively. α-E2F-4 lanes contain two different monoclonal antibodies specific for E2F-4 (WUF3 and LLF4, respectively).
Second, as a measure of whether we could favorably compare our findings with those obtained using other human and murine cell types, we examined the gene expression profiles for several potential E2F-responsive genes by Northern blot analysis (Fig. 2B). p107, E2F-1, Cdc6, and Cdc25A were strongly induced 12 hr after serum stimulation, whereas B-myb, cyclin A, and Cdc2 were induced ∼4 hr later. Expression of the latter genes also peaked later (at 24 or 28 hr poststimulation vs. 16–20 hr). As an RNA loading control, we also examined expression of the acidic ribosomal phosphoprotein (ARPP P0), which varied modestly upon cell cycle re-entry, as expected (Hurford et al. 1997). In total, we have examined nine genes thought to be regulated by the E2F and pRB family, and each was shown to follow kinetics similar to those observed in other studies using both human and rodent cells (Jinno et al. 1994; Johnson et al. 1994; Desdouets et al. 1995; Lam et al. 1995; Tommasi and Pfeifer 1995; Hurford et al. 1997; Hateboer et al. 1998; Fig. 2B; data not shown). Thus, T98G cells display the same growth-related gene expression program observed in a variety of primary and continuous cells following cell cycle re-entry.
We have also examined the appearance and composition of E2F complexes in T98G cells as a function of the cell cycle by subjecting whole cell lysates of synchronized cells to gel mobility shift analysis, and we confirmed the identity of proteins bound to a consensus E2F-binding site oligonucleotide using antibodies specific for individual E2F and pRB family members (Fig. 2C). Here, the most prominent factor present in quiescent cells is E2F-4 (lanes 1–8). Nearly all of this protein is present as a p130–E2F complex, and very little free E2F-4 can be detected (cf. lanes 1, 3, 4, and 6). As cells traverse G1, this complex diminishes significantly and is replaced by free E2F and p107–E2F complexes that contain cdk2 (lanes 9–16). E2F activity at this stage is comprised of several family members (data not shown). pRB–E2F complexes appear at this stage and, together with p107–E2F, persist in later stages of the cell cycle (lanes 17–22). Importantly, the temporal pattern and composition of free E2F and E2F complexes with pRB family members were indistinguishable from those observed in primary human T cells and primary mouse embryo fibroblasts (MEFs) undergoing the G0-to-S transition (Vairo et al. 1995; Moberg et al. 1996; Hurford et al. 1997). These findings also confirm that each pRB family member is functional in T98G cells, insofar as each protein is capable of forming complexes with E2F and cyclin/Cdk2.
Promoter occupation by distinct E2F family members during cell cycle progression
Using chromatin immunoprecipitations, we have examined the occupancy of several promoters by the E2F family as a function of the cell cycle. We focused on examples of genes with slightly different cell cycle expression profiles (Fig. 2B). As before, we included several controls to confirm the specificity of our immunoprecipitation reactions: We performed parallel immunoprecipitations with protein A/G mixtures that either lacked antibody or had been bound to an irrelevant antibody and amplified the promoter region of the actin gene from identical immunoprecipitation reactions performed simultaneously.
Remarkably, E2F-4 was the predominant family member bound to each of these promoters during G0, although weaker binding of E2F-3 to the E2F-1, p107, and Cdc6 promoters was also observed (Fig. 3). E2F-4 binding persisted in cells entering G1 (early G1), but binding by this factor diminished dramatically by mid-G1 and had completely disappeared by late G1 (Fig. 3; data not shown). That E2F-4 is recruited to each promoter exclusively during G0 and early G1 is especially noteworthy, given that we were able to detect this protein in extracts of cells throughout G1 and S phase (Fig. 2C). This strongly suggests that E2F-4 is specifically recruited to its binding sites during G0 and early G1, a time when each promoter is transcriptionally inactive.
Figure 3.






The binding of E2F family of proteins changes during the cell cycle. Chromatin immunoprecipitations were performed with antibodies specific for individual E2F family members as indicated, and the resulting immunoprecipitates were amplified with primer pairs corresponding to the genes shown. G0, serum-starved cells; early G1, late G1, G1/S, and S-phase cells were stimulated with serum 4–8, 16, 20, and 24 hr, respectively. Antibodies tested in each case are listed in Materials and Methods.
Interestingly, the sudden disappearance of E2F-4 by late G1 coincides with the equally abrupt appearance of other E2F polypeptides (Fig. 3). In late G1, each promoter is occupied by a combination of E2F-1, E2F-2, and E2F-3, and E2F-3 binding is most prominent on the E2F-1, p107, cyclin A, and Cdc2 promoters (Fig. 3; data not shown). Binding by these E2F family members occurs at a time when each of these genes is induced (Fig. 2C). Still later, in S phase, E2F-1 and E2F-3 binding is consistently enriched on the E2F-1, p107, and Cdc2 promoters, whereas E2F-3 is most prominent on the Cdc6 promoter. Because we could weakly detect E2F-2 binding as well, we cannot rule out the possibility that residual binding may have arisen from a slight contamination by G1 phase cells (8% of cells; Fig. 1A). In contrast with these promoters, occupation of the B-myb promoter exhibited a unique pattern. Here, binding of all E2F family members disappeared by the G1/S transition, at a time when the B-myb gene was actively transcribed (Fig. 2B). These data complement genomic footprinting experiments that indicate the loss of protection of an E2F site during the G1-to-S phase transition, and they suggest that other factors may control the transcription of the B-myb gene during S phase (Zwicker et al. 1996). We were unable to detect significant binding of E2F-5 at any stage of the cell cycle (Fig. 3), in agreement with studies suggesting its low abundance, a tissue-specific role in differentiation rather than proliferation, and an inability of ectopically expressed E2F-5 to activate previously identified E2F responsive genes (DeGregori et al. 1997; Lindeman et al. 1998). As expected, we did not detect E2F binding to the actin control.
These findings strongly suggest that E2F-4, in contrast with other E2F proteins (namely E2F-1, E2F-2, and E2F-3), may function as a dedicated transcriptional repressor of a majority of E2F responsive genes. Given the potentially repressive role for this E2F family member, we investigated the possibility that such a regulatory function might require the simultaneous recruitment of pRB or related repressor proteins.
p130 recruitment correlates with transcriptional repression during G0 and early G1
We performed chromatin immunoprecipitations to analyze the occupancy of each promoter by pRB family members. Strikingly, we detected very strong binding by p130 during G0 and early G1 (Fig. 4). p130 binding dramatically diminished as cells entered mid-to-late-G1 and was no longer evident as cells progressed through the G1/S transition. This pattern was similar for each of the promoters we examined. Notably, this temporal pattern is reminiscent of that of E2F-4 binding observed on each promoter as well (Fig. 3). This suggests, but does not prove, that E2F-4 and p130 form a complex on these promoters and behave as a functional unit. Further studies will be required to test this idea. We have also detected very weak binding by the p107 protein to the E2F-1, p107, and cyclin A promoters during G0 and throughout G1 (Fig. 4). However, the intensity of binding was in each case greatly reduced in comparison with p130. On the basis of our ability to detect p107 binding to promoters during asynchronous growth, we surmise that p107 may play a less pronounced role in cells entering a new cell cycle after serum restimulation than in cells that are proliferating (see Discussion).
Figure 4.






Analysis of in vivo binding by the pRB family. Antibodies (listed in Materials and Methods) specific for each member of the pRB family were used to immunoprecipitate chromatin, and PCR was performed with primer pairs for each of the indicated genes. Cell cycle populations were identical to those in Fig. 3.
The absence of an association between pRB and any promoter surveyed during cell cycle progression was unexpected. Although this outcome might be explained by an inability of anti-pRB antibodies to detect this protein when bound to chromatin, we consider this explanation unlikely for the following reasons. First, we have performed chromatin immunoprecipitations with eight different polyclonal and monoclonal anti-pRB antibodies that recognize distinct regions of this protein, including both amino- and carboxy-terminal portions. Each of these antibodies failed to immunoprecipitate quantities of chromatin sufficient to yield an amplified product with the primer pairs we have tested (data not shown). Second, we have performed Western blotting experiments to determine whether our antibodies are able to immunoprecipitate pRB bound to chromatin. In this experiment, chromatin was isolated from asynchronous cultures of T98G cells and immunoprecipitated with antibodies against pRB family members. Western blots of these immunoprecipitates were then probed with antibodies against pRB, p107, and p130. To control for the possibility that these proteins were fortuitously present in the chromatin fraction but not associated with DNA, the protocol was also performed in parallel with chromatin from cells that had not been cross-linked with formaldehyde (Fig. 5A). Immunoprecipitation of chromatin isolated from cross-linked, but not control, cells resulted in the detection of each pRB family member in appropriate lanes (Fig. 5B). These experiments attest to the specificity of each antibody used in our chromatin immunoprecipitations and, more importantly, they suggest that each antibody, including those directed against pRB and p107, is capable of immunoprecipitating its target in the context of chromatin. We have also obtained identical chromatin immunoprecipitation results in two other human cell lines (data not shown).
Figure 5.

Antibodies against each pRB family member immunoprecipitate their chromatin-bound targets. (A) Schematic representing the position of chromatin in gradients relative to free DNA and protein under conditions in which cells were either fixed with formaldehyde (+ cross-linker) or left untreated (− cross-linker). Brackets indicate chromatin fractions selected for further analysis in B. (B) Fractions in A were immunoprecipitated with anti-pRB, anti-p107, and p130 antibodies as indicated and the resulting precipitates were probed with the same antibodies as indicated. (C) p107 and p130 are detected at similar levels in T98G cells entering a second cell cycle (32-hr time point in Fig. 2). Chromatin immunoprecipitations were performed with the indicated antibodies, and E2F-1, cyclin A, and actin promoter fragments were amplified as described in the legend to Fig. 1. Distinct monoclonal and polyclonal anti-pRB antibodies were tested as shown.
Nevertheless, when we compared the efficiency of precipitation among pRB family members, our p130 immunoprecipitations yielded significantly larger quantities of amplified promoter fragments than immunoprecipitations with either pRB or p107 antibodies (Fig. 4). We therefore considered the possibility that p130 might play a more prominent role than pRB or p107 in cells re-entering the cell cycle from quiescence, whereas binding of other pRB family members might be more evident in cells progressing through a second cell cycle after serum stimulation. Interestingly, this was the case, as p107 and p130 associated with each promoter to an equal extent in cells that had completed one cell cycle (after serum restimulation) and initiated another (Fig. 5C; data not shown). Likewise, this also appeared to be true in proliferating cells (Fig. 1C).
We conclude that p130 is the principal pRB family member bound to promoters in quiescent and early G1 phase cells and that pRB does not bind to the promoters that we have tested here, at least in cells that are re-entering the cell cycle from a quiescent state. Our data are also consistent with experiments performed in MEFs lacking p107 and p130 (Hurford et al. 1997). In that setting, loss of both of these genes led to derepression of B-myb, Cdc2, E2F-1, and cyclin A genes at the G0/G1 transition. Other genes that have not been tested here are likely to be physiological targets of pRB, and experiments designed to identify these genes are underway.
Histone acetylation of E2F-responsive genes correlates with transcriptional activation in late G1 cells
As a further probe of transcriptional regulatory events occurring at each E2F-responsive gene, we have used chromatin immunoprecipitations to investigate whether histone acetylation varies during cell cycle progression. In these experiments, we used antibodies specific for the acetylated forms of histones H3 and H4, as transcription factor-mediated acetylation of promoters correlates with gene activation (Chen et al. 1999; Parekh and Maniatis 1999). We found a good correlation between acetylation of histones H3 and H4 and activation of each E2F-responsive gene in late G1 at or near the G1/S transition: Core histones are under-acetylated in quiescent and early G1 phase cells at a time when each gene is transcriptionally silent, whereas acetylation of histones occurs concomitantly with gene activation (Fig. 6). Interestingly, we observed reproducible gene-specific differences in the cell cycle timing of acetylation. For example, levels of acetylation of the Cdc6 and E2F-1 genes were low during G0 and early G1, and they increased dramatically in mid-to-late G1 (seven- and sixfold for histones H3 and H4, respectively, for Cdc6), as transcription is induced (Figs. 2B and 6; data not shown). Acetylation levels for both genes decline rapidly in S phase, foreshadowing a sharp decrease in transcript levels (Fig. 2B). A second pattern is evident for the B-myb, p107, cyclin A, and Cdc2 genes. Here, gene activation and increased acetylation were evident in late G1, but unlike the Cdc6 and E2F-1 genes, increased histone acetylation levels were sustained in S phase, correlating with abundant levels of transcription for each of these genes well into G2 phase. For comparison, we have also probed histone acetylation levels for the actin gene, which is not thought to be under the control of E2F. In contrast with the E2F-responsive promoters, histone acetylation associated with the actin promoter increased only modestly as cells traversed G1, in agreement with observations that its transcriptional profile does not vary significantly during the cell cycle (Campisi et al. 1984).
Figure 6.
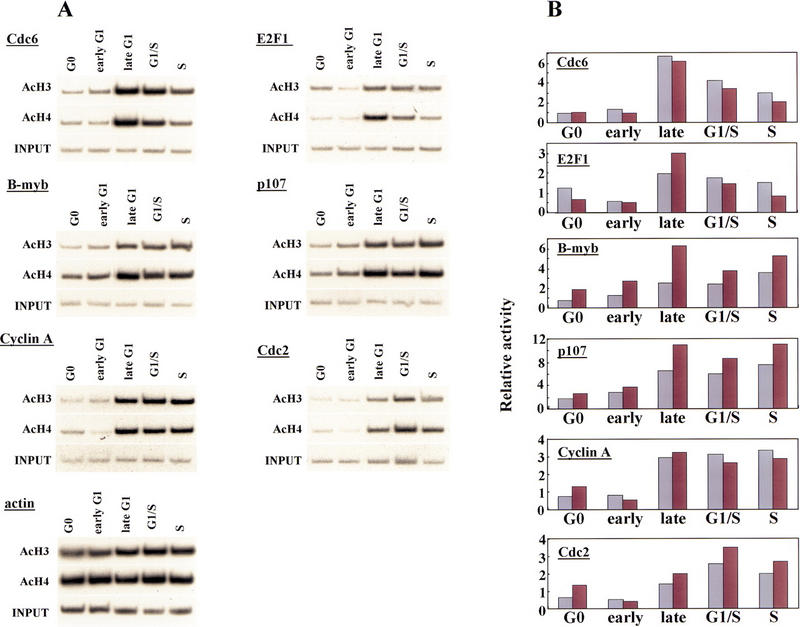
Histone acetylation levels of E2F-responsive genes change during the cell cycle. (A) Chromatin was prepared from synchronized T98G cells (as described in Fig. 2A) and immunoprecipitated with antibodies specific for acetylated histone H3 and acetylated H4 as described in the legend to Fig. 1. Cell cycle stages were identical to those described in Fig. 3. Parallel immunoprecipitations without antibody or with an irrelevant antibody control failed to yield detectable signals after an equivalent autoradiographic exposure (data not shown). (B) Data in A were quantitated by PhosphorImager analysis and normalized vs. input levels. The _y_-axis indicates fold acetylation (in arbitrary units). (Light-colored bars) acetylated H3; (dark bars) acetylated H4.
These findings suggest the intriguing possibility that HDAC recruitment to these promoters in G0/early G1 is then succeeded by recruitment of histone acetyltransferases (HATs) in late G1 near the G1/S transition. We are currently investigating the recruitment of specfic HDACs and HATs using our chromatin immunoprecipitation assay.
Discussion
A central issue in the study of transcriptional regulation by the E2F and pRB families is the authenticity of previously defined targets and promoter selectivity of individual family members for these targets. It has been generally assumed that specific combinations of E2F heterodimers and pRB proteins might regulate distinct sets of genes (Dyson 1998). This issue has been difficult to resolve, however, because most techniques used to address this question have relied on ectopic expression of E2Fs, which can often lead to promiscuous promoter binding and secondary gene activation. We have circumvented this need for ectopic expression through the use of chromatin immunoprecipitations, and this technique has allowed us to study promoter occupancy in living cells entering a cell cycle from quiescence.
Using this technique, we find that individual E2F family members are not overtly involved in the regulation of particular subsets of genes in cells that are stimulated to enter a cell cycle from a quiescent state. Rather, it appears that E2F-4 plays a general and prominent role in the repression of transcription of multiple promoters during G0 and early G1. Later in G1 and in S phase, E2F-1, E2F-2, and E2F-3 bind to the majority of promoters tested, coincident with the activation of each of these genes. Thus, we propose that different E2F family members may not necessarily play a distinct role in expression of specific genes. Instead, individual E2Fs may play a role in distinguishing the timing of gene expression, in part through the recruitment of the pRB-related protein, p130, and histone modifying enzymes (Fig. 7; see below).
Figure 7.


Model for in vivo occupancy by E2F and pRB family members during cell cycle progression. (A) In vivo occupation of E2F-1, Cdc25A, cyclin A, Cdc6, and p107 promoters by the E2F and pRB family during cell cycle progression and transcriptional consequences. (B) In vivo binding of B-myb promoter. In A and B, promoter binding by E2F-4 correlates with transcriptional repression, whereas binding by E2F-1 and E2F-3 occurs at a time when each promoter is activated. However, the B-myb promoter is transcriptionally active after E2F is no longer bound, consistent with Zwicker et al. (1996). (D) HDAC activity; (H) nucleosomes; (Ac) acetylated histones; (HAT) histone acetyltransferase. These promoters are likely to be regulated by additional _trans_-activator proteins proximal to E2F, including Sp1. Recruitment of deacetylase and HAT activities could occur by direct interactions with E2F complexes or could require additional factors (X) yet to be defined.
Chromatin immunoprecipitation as a tool to study E2F regulation
The results of our chromatin immunoprecipitation experiments presented here are nevertheless remarkably consistent with two previous studies examining physiologically relevant levels of E2F and pRB family members in murine cells (Zwicker et al. 1996; Hurford et al. 1997). First, Hurford et al. (1997) examined the expression of potential E2F-responsive genes in MEFs deficient for each pRB family member as well as cells deficient for both p107 and p130 (Hurford et al. 1997). Here, expression of only two genes (of 21 examined), cyclin E and p107, was altered in the pRB-deficient fibroblasts, and even these differences were somewhat subtle. In sharp contrast, more significant alterations were uncovered in the p107−/−; p130−/− MEFs. In these cells, expression patterns of seven genes were deregulated. Among these genes, expression of B-myb, Cdc2, E2F-1, and cyclin A were strongly derepressed at the G0-to-G1 transition. These results agree with our own findings that p130 is associated with each of these promoters in G0 and early G1 phase cells at a time when all of these genes are transcriptionally inactive (Figs. 2, 4 and 5).
Furthermore, our results are in accordance with a second study examining the occupation of the B-myb promoter by E2F (Zwicker et al. 1996). In these experiments, in vivo footprinting suggested that occupation of a potential E2F site during G0 and early G1 is abolished as cells enter S phase. Likewise, our experiments show that neither E2F nor pRB family members are bound to B-myb as cells enter S phase, suggesting that transcription factors other than E2F (or the highly acetylated state of histones; Fig. 6) might maintain this promoter in an active state during S phase (Fig. 7; Zwicker et al. 1996). Overall, the agreement between our own observations and those of two independent studies performed in different cell types (with different techniques) reinforces the notion that our chromatin immunoprecipitations reflect the physiological binding by cell cycle regulatory proteins. Our findings therefore indicate the utility of this technique for studying the in vivo occupation of E2F-responsive promoters and suggest that it will continue to be an invaluable tool for deciphering the regulatory events involved in cell cycle-dependent gene expression.
Our experiments are also consistent with a number of transient expression studies suggesting an overlapping activation function for multiple E2F family members (DeGregori et al. 1997; Leone et al. 1998). For example, several of the promoters we examined were apparently occupied in vivo by several E2F polypeptides during late G1 phase, a period in which each promoter is active. We note that many of the E2F-responsive promoters examined in this study are thought to contain multiple E2F binding sites, as determined by sequence comparisons, in vitro binding analyses, and transient transfection. In addition, sister alleles could be bound by different E2F family members, as gene expression profiles may vary even within a given population of genetically identical cells (Fiering et al. 1990; Ko et al. 1990). This result is thought to stem from differences in accumulation of transcriptionally active preinitiation complexes within the population. At present, we cannot distinguish between each of these possibilities. In future experiments it should be possible for us to address this question using integrated copies of promoters bearing individually and combinatorially mutated E2F sites.
The apparent absence of pRB binding to the promoters we have tested by chromatin immunoprecipitation was surprising, given its well established role in cell cycle control. Although we have reproducibly observed pRB binding to the p107 promoter in asynchronous cells (Fig. 1), binding was not enriched in synchronized populations. Given our ability to clearly detect pRB binding to chromatin, however, it is very likely that bona fide targets of this repressor will be revealed by chromatin immunoprecipitations, and these experiments are currently underway. It is also important to point out that our study has focused exclusively on cells entering the first cell cycle after serum stimulation. The nonequivalence of total gene expression profiles of proliferating and serum-stimulated cells has been documented previously (Leone et al. 1998). For this reason, it will be particularly important to study the occupation of each promoter by E2F and pRB family members in synchronized cells at each stage of a second cell cycle or in proliferating cells released from a drug block. Although our initial analyses have already shown that p130 may play a prominent regulatory role in cells emerging from a quiescent state, an idea suggested by previous reports (Vairo et al. 1995; Moberg et al. 1996), both p130 and p107 occupy the same promoters to an equal extent in cycling cells (Figs. 1 and 6).
A model for E2F transcriptional regulation during the cell cycle
We propose the following model on the basis of the data presented and a consideration of previous work. Our experiments suggest that E2F-4 may function on certain promoters as a dedicated repressor in G0 and early G1 cells (Fig. 7) because we observe strong binding during this time, when each gene is transcriptionally silent, and negligible binding of E2F-4 in late G1 and S phase when transcription is induced. These results are consistent with the notion that E2F-4 localizes primarily to the nucleus during G0 and G1 and to the cytoplasm in S phase (Lindeman et al. 1997; Muller et al. 1997; Verona et al. 1997). Integrating our experimental findings with these localization data, we suggest that during G0 and G1, E2F-4 is localized to the nucleus, where it is able to bind promoters and recruit p130 (Fig. 7). Promoter binding by an E2F-4–p130 complex then results in diminished histone acetylation and transcriptional repression, possibly via the recruitment of HDACs.
Interestingly, there was a striking diminution of E2F-4 binding to all promoters tested as cells progressed through G1 (Fig. 3). Clearly, this could not be explained by a sudden decrease in the levels of E2F-4 protein because our gel mobility shift experiments indicated that this protein was present throughout G1 and S phase (Fig. 2C). The sudden disappearance of E2F-4 in mid-to-late G1 could result either from relocalization to the cytoplasm, which is known to occur at this time, or by an alternative, unknown mechanism. Such partitioning would prevent E2F-4 from directly affecting transcription in S phase, and we have not detected E2F-4 binding to any promoter during S phase. However, we have examined a relatively small number of promoters thus far, and therefore we cannot exclude the possibility that E2F-4 stimulates transcription from other promoters.
Our studies further show that E2F-4 is rapidly replaced by E2F-1, E2F-2, and E2F-3 as cells enter mid-to-late G1. On the basis of these experiments, we conclude that these proteins, and in particular E2F-1 and E2F-3, play a pivotal role in the activation of transcription of several genes at the G1/S transition (Fig. 7). This is consistent with several reports linking the activity of these family members to induction of S phase (DeGregori et al. 1997; for review, see Dyson 1998; Leone et al. 1998). Moreover, antibody injection experiments showed that E2F-3, but not E2F-1, was necessary for S-phase entry in proliferating cells (Leone et al. 1998). On the basis of these observations and others, E2F-3 was thought to play an essential role in the expression of genes encoding products that are rate limiting for initiation of DNA replication, including the Cdc6 gene. Interestingly, we reproducibly observed enhanced binding of E2F-3 to the Cdc6 gene, relative to all other E2Fs, during the G1/S transition and S phase, when this gene is actively transcribed (Figs. 2B and 3).
Our results challenge some aspects of previous models that have explained the regulation of E2F-responsive promoters as a succession of events initiated by the binding of specific E2F–pRB complexes to a promoter, resulting in transcriptional inhibition. According to this model, subsequent phosphorylation of the pRB protein by cyclin/Cdk complexes results in the generation of a free form of the same E2F family member able to activate transcription. We propose (Fig. 7) instead, that in some instances, inhibition of one specific E2F family member, E2F-4, is followed by the binding of different family members (free E2F-1, E2F-2, and E2F-3) that are responsible for transcriptional activation. Further studies of additional E2F-responsive promoters are needed to address the generality of this mode of regulation.
Finally, our experiments also suggest that chromatin-modifying proteins may play a role in the cell cycle-dependent expression of E2F responsive genes in vivo (Fig. 7). Although a number of experiments have detected an association between the pRB family and HDAC activity, these experiments for the first time link the potential activities of HDACs and HATs to the transcriptional regulation of E2F-responsive genes in a physiological setting. Experiments designed to detect the recruitment of specific HDACs and HATs to these E2F-responsive promoters are currently underway.
Materials and methods
Cell lines, antibodies, cell cycle synchronization, and FACS analysis
T98G human glioblastoma cells were obtained from ATCC and were grown in DMEM containing 10% FBS. Cells were rendered quiescent by serum deprivation for 72 hr and stimulated with 20% FBS (final concentration) to allow cell cycle re-entry. Propidium iodide staining and cell cycle analysis using CellQuest and ModFit were performed exactly as described (Woo et al. 1997). Antibodies that recognize E2F-1 (sc-193), E2F-2 (sc-633x), E2F-3 (sc-878x), E2F-4 (sc-1082x), E2F-5 (sc-999, sc-968), p107 (sc-318), pRB (sc-50, G99-549, G3-245), p130 (sc-317, Rb2), cdk2 (sc-163), and 12CA5 anti-flu hemagglutinin were obtained from Santa Cruz Biotechnology, Neomarkers, Pharmingen, Transduction Labs, and BabCo. Antibodies specific for acetylated histones H3 and H4 were obtained from Upstate Biotechnology, Inc. Additional monoclonal antibodies against E2F-4 (LLF4, WUF3), p107 (mixture of SD2, SD4, SD6, SD9, SD15), and pRB (XZ77, XZ91, XZ104, XZ140) were gifts of J. Lees (MIT, Cambridge, MA), N. Dyson, C-L. Wu, and E. Harlow (all from MGH Cancer Center, Charlestown, MA).
Northern blot analysis
Total RNA was prepared from synchronized T98G cells using Trizol Reagent (Life Technologies) according to the manufacturer's protocol. A total of 10 μg of RNA from each cell cycle stage was electrophoresed through a 1% agarose-formaldehyde gel, transferred to a Nytran membrane (Schleicher & Schuell), and UV-cross-linked with a Stratalinker (Stratagene). Membranes were treated as described by the manufacturer. 32P-Labeled probes were prepared by random primer labeling (Life Technologies) of the following restriction fragments: (1) human E2F-1, a _Bam_HI fragment from pCMV-E2F-1 (Helin et al. 1993); (2) human cyclin A, a _Hin_dIII–_Xba_I fragment from pRc/CMV-CycA (gift of P. Hinds, Harvard Medical School, Boston, MA); (3) murine ARPP P0, an _Eco_RI–_Hin_dIII fragment from pARPP P0 [kind gift of F. Dick (MGH Cancer Center, Charlestown, MA) and N. Dyson]; (4) p107, an _Eco_RI–_Stu_I fragment from pCMV-p107 (Zhu et al. 1993); (5) Cdc2, a _Bam_HI–_Bam_HI fragment from pCMV–Cdc2DN (van den Heuvel and Harlow 1993). To generate probes for Cdc6, Cdc25A, and B-Myb, a HeLa cell cDNA library was amplified by standard PCR with primers corresponding to coding regions of each gene, and PCR fragments were cloned into the _Eco_RV site of pBluescript (Stratagene) and sequenced. Inserts were excised with _Bam_HI and _Hin_dIII and labeled. Equal loading of RNA was verified by probing identical blots with a 32P-labeled probe for murine ARPP P0 (acidic ribosomal phosphoprotein), which is highly conserved with the human homolog.
Gel mobility shift assays
Whole cell extracts of T98G cells were made by lysis in high salt buffer with one freeze-thaw. Equivalent amounts of protein (∼3 μg) from each cell cycle stage were incubated on ice for 30 min with 1 μg of salmon sperm DNA as a nonspecific competitor. In addition, where indicated, binding reactions also contained 1 or 2 μl of the specified polyclonal and monoclonal (tissue culture supernatant) antibodies (negative control anti-HA, 12CA5; anti-E2F-4, WUF3 and LLF4; anti-p107, mixture of SD2,4, 6,9, 15; anti-p130, sc-317; anti-pRB, mixture of XZ77, 91, 104, 140 mixture; anti-cdk2, sc-163), respectively. Reactions were further incubated at room temperature after addition of labeled double-stranded probe containing an E2F site, and electrophoretic mobility shifts were analyzed on 4% native acrylamide gels as described (Dynlacht et al. 1997).
Chromatin immunoprecipitations
We performed chromatin immunoprecipitations using a modification of previously published methods (Orlando et al. 1997; Parekh and Maniatis 1999). Approximately 1 × 108 T98G cells were grown on 15-cm2 dishes and cross-linked by addition of formaldehyde (to 1% final concentration) to attached cells. Cross-linking was allowed to proceed at room temperature for 10 min and was terminated with glycine (final concentration 0.125 m). Cells were washed with PBS, trypsinized, and scraped into PBS containing 10% FBS. Cells were collected by centrifugation and were rocked in buffer containing 50 mm HEPES (pH 7.5), 140 mm NaCl, 1 mm EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, and protease inhibitors (1 mm AEBSF, 1 mm benzamidine, 50 μg/ml TLCK, 50 μg/ml TPCK, 10 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin A), at 4°C for 10 min. Cells were collected by centrifugation at 4K rpm for 10 min, and the resulting pellet was resuspended in 200 mm NaCl, 1 mm EDTA, 0.5 mm EGTA, 10 mm Tris (pH 8), and protease inhibitors and incubated 10 min at room temperature. Nuclei were collected by centrifugation at 4K rpm for 10 min, resuspended in sonication buffer (1 mm EDTA, 0.5 mm EGTA, 10 mm Tris at pH 8, and protease inhibitors), and sonicated on ice to an average length of 700 bp. Samples were centrifuged in cesium chloride step gradients at 37K rpm in an SW41 rotor for 24 hr. Chromatin was collected and dialyzed against TE buffer (10 mm Tris-HCl at pH 8, 1 mm EDTA, 0.5 mm EGTA, 10% glycerol). Sedimentation of chromatin in given fractions was reproducible from one gradient to the next and did not vary with cell growth state. The equivalent of ∼107 cells were used per chromatin immunoprecipitation reaction.
Chromatin was pre-cleared with a mixture of protein A and protein G Sepharose (blocked previously with 1 mg/ml salmon sperm DNA and 1 mg/ml BSA) at 4°C for 4 hr two times. Pre-cleared chromatin was incubated with 2 μg of each antibody (E2F-1, sc-193; E2F-2, sc-633x; E2F-3, sc-878x; E2F-4, sc-1082x; E2F-5, sc-999; p107, sc-318; pRB, sc-50; p130, sc-317; anti-T Antigen pAb101, control antibody) in TE buffer containing 1% Triton X-100, 0.1% sodium deoxycholate (DOC), and protease inhibitors at 4°C overnight. Next, 20 μl of a 50% slurry of blocked protein A/G sepharose was added, and immune complexes were recovered. Immunoprecipitates were washed seven times with RIPA buffer (0.5 m LiCl, 50 mm HEPES at pH 7.5, 1 mm EDTA, 1% NP-40, 0.7% DOC, and protease inhibitors). Pellets were resuspended in 100 μl of TE and incubated at 55°C for 3 hr with 10 μg each of RNAse A and proteinase K. Cross-links were reversed by incubating samples at 65°C overnight, and samples were extracted with phenol:chloroform and ethanol precipitated. Pellets were resuspended in 100 μl of H2O and assayed by semiquantitative PCR.
Thirty cycles of PCR were performed in 25 μl with 5 μl of immunoprecipitated material, 10 pmole of each primer set, 0.125 units of Taq DNA polymerase (GIBCO), and 1 μCi of [α-32P]dCTP. To amplify E2F-responsive promoter regions, the following primer sets were used: E2F1, positions −102 to −79 and −2 to +22; B-myb, positions −119 to −101 and +21 to +39; p107, positions −56 to −33 and +82 to +105; Cdc25A, positions −120 to −98 and +40 to +62; Cdc6, positions −84 to −62 and +76 to +103; Cdc2, positions −193 to −170 and −18 to +5; cyclin A, positions −135 to −113 and +13 to +33; and β-actin, positions −212 to −190 and −72 to −48. We based our identification of E2F-binding sites and transcription start sites in each promoter on previously published reports (Johnson et al. 1994; Lam et al. 1995; Tommasi and Pfeifer 1995; Zhu et al. 1995; Bennett et al. 1996; Galaktionov et al. 1996; Liu et al. 1996; Hateboer et al. 1998; Ohtani et al. 1998; Chen and Prywes 1999). We have established in prior experiments that such PCR conditions are within the linear range of amplification. PCR products were electrophoresed on 8% polyacrylamide gels. Each experiment was performed at least three times, and representative data are shown.
Western blotting
To detect proteins by Western blotting of chromatin immunoprecipitates, immunoprecipitation reactions were performed as described above. After washing with RIPA buffer, the sample was boiled for 30 min in 100 μl of 1× sample buffer to reverse formaldehyde cross-links. One-fifth of the sample was electrophoresed on a 7% SDS–polyacrylamide gel and transferred to a membrane. To detect pRB family members, anti-pRB (G3-245), anti-p107 (sc-318), and anti-p130 (Rb2) antibodies were used.
Acknowledgments
We thank B. Parekh and T. Maniatis who enabled us to establish the chromatin immunoprecipitation protocol in our own laboratory. We thank I. Sanchez, R. Gregory, and J. Ross for helpful comments on the manuscript. We thank J. Lees, E. Harlow, and N. Dyson for the gift of various monoclonal antibodies and plasmids. J.B.R. is supported by a Howard Hughes Medical Institute pre-doctoral fellowship. This work was supported by an NCI grant (CA77245-02) to B.D.D. B.D.D. is also grateful for the support of the Pew Scholars Program in the Biomedical Sciences.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL dynlacht@biosun.harvard.edu; FAX (617) 496-1391.
References
- Aparicio OM, Weinstein DM, Bell SP. Components and dynamics of DNA replication complexes in S. cerevisiae: Redistribution of MCM proteins and Cdc45p during S phase. Cell. 1997;91:59–69. doi: 10.1016/s0092-8674(01)80009-x. [DOI] [PubMed] [Google Scholar]
- Bennett JD, Farlie PG, Watson RJ. E2F binding is required but not sufficient for repression of B-myb transcription in quiescent fibroblasts. Oncogene. 1996;13:1073–1082. [PubMed] [Google Scholar]
- Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzaride T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- Campisi J, Gray HE, Pardee AB, Dean M, Sonenshein GE. Cell-cycle control of c-myc but not c-ras expression is lost following chemical transformation. Cell. 1984;36:241–247. doi: 10.1016/0092-8674(84)90217-4. [DOI] [PubMed] [Google Scholar]
- Chen X, Prywes R. Serum-induced expression of the cdc 25A gene by relief of E2F-mediated repression. Mol Cell Biol. 1999;19:4695–4702. doi: 10.1128/mcb.19.7.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lin RJ, Xie W, Wilpitz D, Evans RM. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell. 1999;98:675–686. doi: 10.1016/s0092-8674(00)80054-9. [DOI] [PubMed] [Google Scholar]
- DeGregori J, Kowalik T, Nevins J. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol. 1995;15:4215–4224. doi: 10.1128/mcb.15.8.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci. 1997;94:7245–7250. doi: 10.1073/pnas.94.14.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desdouets C, Matesic G, Molina CA, Foulkes NS, Sassone-Corsi P, Brechot C, Sobczak-Thepot J. Cell cycle regulation of cyclin A gene expression by the cyclic AMP-responsive transcription factors CREB and CREM. Mol Cell Biol. 1995;15:3301–3309. doi: 10.1128/mcb.15.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dynlacht BD, Flores O, Lees JA, Harlow E. Differential regulation of E2F trans-activation by cyclin-cdk2 complexes. Genes & Dev. 1994;8:1772–1786. doi: 10.1101/gad.8.15.1772. [DOI] [PubMed] [Google Scholar]
- Dynlacht BD, Moberg K, Lees JA, Harlow E, Zhu L. Specific regulation of E2F family members by cyclin-dependent kinases. Mol Cell Biol. 1997;17:3867–3875. doi: 10.1128/mcb.17.7.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson N. The regulation of E2F by pRB-family proteins. Genes & Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- Fiering S, Northrop JP, Nolan GP, Mattila PS, Crabtree GR, Herzenberg LA. Single cell assay of a transcription factor reveals a threshold in transcription activated by signals emanating from the T-cell antigen receptor. Genes & Dev. 1990;4:1823–1834. doi: 10.1101/gad.4.10.1823. [DOI] [PubMed] [Google Scholar]
- Galaktionov K, Chen X, Beach D. Cdc25 cell cycle phosphatase as a target of c-myc. Nature. 1996;382:511–517. doi: 10.1038/382511a0. [DOI] [PubMed] [Google Scholar]
- Hateboer G, Wobst A, Petersen BO, Cam LL, Vigo E, Sardet C, Helin K. Cell cycle-regulated expression of mammalian CDC6 is dependent on E2F. Mol Cell Biol. 1998;18:6679–6697. doi: 10.1128/mcb.18.11.6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helin K, Harlow E, Fattaey AR. Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol Cell Biol. 1993;13:6501–6508. doi: 10.1128/mcb.13.10.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huet X, Rech J, Plet A, Vie A, Blanchard JM. Cyclin A expression is under negative transcriptional control during the cell cycle. Mol Cell Biol. 1996;16:3789–3798. doi: 10.1128/mcb.16.7.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurford RK, Cobrinik D, Lee M-H, Dyson N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes & Dev. 1997;11:1447–1463. [Google Scholar]
- Jinno S, Suto K, Nagata A, Igarashi M, Kanaoka Y, Nojima H, Okayama H. Cdc25A is a novel phosphatase functioning early in the cell cycle. EMBO J. 1994;13:1549–1556. doi: 10.1002/j.1460-2075.1994.tb06417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DG, Ohtani K, Nevins JR. Autoregulatory control of E2F-1 expression in response to positive and negative regulators of cell cycle progression. Genes & Dev. 1994;8:1514–1525. doi: 10.1101/gad.8.13.1514. [DOI] [PubMed] [Google Scholar]
- Ko MS, Nakauchi H, Takahashi N. The dose dependence of glucocorticoid-inducible gene expression results from changes in the number of transcriptionally active templates. EMBO J. 1990;9:2835–2842. doi: 10.1002/j.1460-2075.1990.tb07472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- Krek W, Ewen ME, Shirodkar S, Arany Z, Kaelin WG, Livingston D. Negative regulation of the growth-promoting transcription factor E2F-1 by a stably bound cyclin A-dependent protein kinase. Cell. 1994;78:161–172. doi: 10.1016/0092-8674(94)90582-7. [DOI] [PubMed] [Google Scholar]
- Kuras L, Struhl K. Binding of TBP to promoters in vivo is stimulated by activators and requires PolII holoenzyme. Nature. 1999;399:609–613. doi: 10.1038/21239. [DOI] [PubMed] [Google Scholar]
- Lam EW, Bennett JD, Watson RJ. Cell-cycle regulation of human B-myb transcription. Gene. 1995;160:277–281. doi: 10.1016/0378-1119(95)00184-8. [DOI] [PubMed] [Google Scholar]
- Le Cam L, Polanowska J, Fabbrizio E, Olivier M, Phillips A, Eaton EN, Classon M, Geng Y, Sardet C. Timing of cyclin E gene expression depends on the regulated association of a bipartite repressor element with a novel E2F complex. EMBO J. 1999;18:1878–1890. doi: 10.1093/emboj/18.7.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone G, DeGregori J, Yan Z, Jakoi L, Ishida S, Williams RS, Nevins JR. E2F3 activity is regulated during the cell cycle and is required for the induction of S phase. Genes & Dev. 1998;12:2120–2130. doi: 10.1101/gad.12.14.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeman GJ, Gaubatz S, Livingston DM, Ginsberg D. The subcellular localization of E2F-4 is cell-cycle dependent. Proc Natl Acad Sci. 1997;94:5095–5100. doi: 10.1073/pnas.94.10.5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeman GJ, Dagnino L, Gaubatz S, Xu Y, Bronson RT, Warren HB, Livingston DM. A specific, nonproliferative role for E2F-5 in choroid plexus function revealed by gene targeting. Genes & Dev. 1998;12:1092–1098. doi: 10.1101/gad.12.8.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Lucibello FC, Zwicker J, Engeland K, Muller R. Cell cycle-regulated repression of B-myb transcription: cooperation of an E2F site with a contiguous corepressor element. Nucleic Acids Res. 1996;24:2905–2910. doi: 10.1093/nar/24.15.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo RX, Postigo A, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92:463–473. doi: 10.1016/s0092-8674(00)80940-x. [DOI] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, Troalen F, Trouche D, Harel-Bellan A. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- Moberg K, Starz MA, Lees JA. E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol Cell Biol. 1996;16:1436–1449. doi: 10.1128/mcb.16.4.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller H, Moroni MC, Vigo E, Petersen BO, Bartek J, Helin K. Induction of S-phase entry by E2F transcription factors depends on their nuclear localization. Mol Cell Biol. 1997;17:5508–5520. doi: 10.1128/mcb.17.9.5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani K, Tsujimoto A, Ikeda M, Nakamura M. Regulation of cell growth-dependent expression of mammalian CDC6 gene by the cell cycle transcription factor E2F. Oncogene. 1998;17:1777–1785. doi: 10.1038/sj.onc.1202105. [DOI] [PubMed] [Google Scholar]
- Orlando V, Strutt H, Paro R. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods. 1997;11:205–214. doi: 10.1006/meth.1996.0407. [DOI] [PubMed] [Google Scholar]
- Parekh BS, Maniatis T. Virus infection leads to localized hyperacetylation of histones H3 and H4 at the IFN-beta promoter. Mol Cell. 1999;3:125–129. doi: 10.1016/s1097-2765(00)80181-1. [DOI] [PubMed] [Google Scholar]
- Ross JF, Liu X, Dynlacht BD. Mechanism of transcriptional repression of E2F by the retinoblastoma tumor suppressor protein. Mol Cell. 1999;3:195–205. doi: 10.1016/s1097-2765(00)80310-x. [DOI] [PubMed] [Google Scholar]
- Stein GH. T98G: An anchorage-independent human tumor cell line that exhibits stationary phase G1 arrest in vitro. J Cell Physiol. 1979;99:43–54. doi: 10.1002/jcp.1040990107. [DOI] [PubMed] [Google Scholar]
- Tommasi S, Pfeifer GP. In vivo structure of the cdc2 promoter: Release of a p130-E2F-4 complex from sequences immediately upstream of the transcription initiation site coincides with induction of cdc2 expression. Mol Cell Biol. 1995;15:6901–6913. doi: 10.1128/mcb.15.12.6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vairo G, Livingston DM, Ginsberg D. Functional interaction between E2F-4 and p130: Evidence for distinct mechanisms underlying growth suppression by different retinoblastoma protein family members. Genes & Dev. 1995;9:869–881. doi: 10.1101/gad.9.7.869. [DOI] [PubMed] [Google Scholar]
- van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- Verona R, Moberg K, Estes S, Starz M, Vernon JP, Lees JA. E2F activity is regulated by cell cycle-dependent changes in subcellular localization. Mol Cell Biol. 1997;17:7268–7282. doi: 10.1128/mcb.17.12.7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo MS-A, Sanchez I, Dynlacht BD. p130 and p107 use a conserved domain to regulate cellular cyclin-dependent kinase activity. Mol Cell Biol. 1997;17:3566–3579. doi: 10.1128/mcb.17.7.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson NJ. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell. 1996;85:537–548. doi: 10.1016/s0092-8674(00)81254-4. [DOI] [PubMed] [Google Scholar]
- Zhu L, van den Heuvel S, Helin K, Fattaey A, Ewen M, Livingston D, Dyson N, Harlow E. Inhibition of cell proliferation by p107, a relative of the retinoblastoma protein. Genes & Dev. 1993;7:1111–1125. doi: 10.1101/gad.7.7a.1111. [DOI] [PubMed] [Google Scholar]
- Zhu L, Zhu L, Xie E, Chang L-S. Differential roles of two tandem E2F sites in repression of the human p107 promoter by retinoblastoma and p107 proteins. Mol Cell Biol. 1995;15:3552–3562. doi: 10.1128/mcb.15.7.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwicker J, Liu N, Engeland K, Lucibello CF, Muller R. Cell cycle regulation of E2F site occupation in vivo. Science. 1996;271:1595–1597. doi: 10.1126/science.271.5255.1595. [DOI] [PubMed] [Google Scholar]