Hrs, a FYVE finger protein localized to early endosomes, is implicated in vesicular traffic and required for ventral folding morphogenesis (original) (raw)
Abstract
Hrs is an early endosomal protein homologous to Vps27p, a yeast protein required for vesicular trafficking. Hrs has a FYVE double zinc finger domain, which specifically binds phosphatidylinositol(3)-phosphate and is conserved in several proteins involved in vesicular traffic. To understand the physiological role of Hrs, we generated mice carrying a null mutation of the gene. Hrs homozygous mutant embryos developed with their ventral region outside of the yolk sac, had two independent bilateral heart tubes (cardia bifida), lacked a foregut, and died around embryonic day 11 (E11). These phenotypes arise from a defect in ventral folding morphogenesis that occurs normally around E8.0. Significant apoptosis was detected in the ventral region of mutant embryos within the definitive endoderm, suggesting an important role of this germ layer in ventral folding morphogenesis. Abnormally enlarged early endosomes were detected in the mutants in several tissues including definitive endoderm, suggesting that a deficiency in vesicular transport via early endosomes underlies the mutant phenotype. The vesicular localization of Hrs was disrupted in cells treated with wortmannin, implicating Hrs in the phosphatidylinositol 3-kinase pathway of membrane trafficking.
Keywords: Hrs, FYVE finger, early endosome, ventral morphogenesis, cardia bifida, definitive endoderm
Endosomes are organelles involved in vesicular traffic between the plasma membrane and the lysosome and between the Golgi apparatus and the lysosome (for review, see Gruenberg and Howell 1989; Kornfeld and Mellman 1989). They are distributed among two morphologically and functionally distinct compartments—early and late endosomes. In receptor-mediated endocytosis, ligand/receptor complexes are first transported by coated vesicles to early endosomes, which function as a sorting station. Ligands and certain types of receptors such as growth factor receptors are then delivered to lysosomes for degradation via late endosomes. Other types of receptors such as the transferrin receptor and low-density lipoprotein receptor are recycled back to the plasma membrane from the early endosome via the recycling compartment. Newly synthesized lysosomal proteins are also delivered to lysosomes from the Golgi apparatus via endosomes.
Hrs is a 115-kD protein that has an amino-terminal double zinc finger (FYVE finger) domain (Stenmark et al. 1996), a proline-rich region, a coiled-coil domain, and a carboxy-terminal proline- and glutamine-rich region (Komada and Kitamura 1995). Hrs is thought to be involved in vesicular transport via endosomes for the following reasons. It is localized to the cytoplasmic surface of early endosomes (Komada et al. 1997). It has 23% sequence homology to the yeast protein Vps27p, with the highest homology (50%) in the FYVE finger domains. In _Vps27-_deficient cells, protein transport via endosomes is disrupted and endosomes are expanded as a result of accumulation of proteins destined to vacuoles, a yeast equivalent of mammalian lysosomes (Piper et al. 1995). Furthermore, Hrs associates in neuronal cells with SNAP-25, a protein involved in docking and fusion of endosome-derived synaptic vesicles with the presynaptic plasma membrane (Bean et al. 1997). It also inhibits neurotransmitter release from the synaptic vesicles when added to permeabilized neuronal cells (Bean et al. 1997).
The FYVE finger domains found in Hrs and Vps27p are highly homologous to those of several other proteins that are also implicated in vesicular transport, such as EEA1 (Mu et al. 1995), Fab1p (Yamamoto et al. 1995), Pep7p (Webb et al. 1997), and Vac1p (Weisman and Wickner 1992). They are also found in the signaling proteins FDG1, a guanine nucleotide exchange factor for the rho/rac family (Pasteris et al. 1994), and SARA, which recruits Smad2 to the TGFβ receptor (Tsukazaki et al. 1998). Recently, it was shown that the FYVE finger domain serves as a specific binding site for phosphatidylinositol(3)-phosphate [PI(3)-P; Burd and Emr 1998; Gaullier et al. 1998; Patki et al. 1998]. Because phosphatidylinositol 3-kinase (PI 3-kinase), which generates PI(3)-P, is required for vesicular transport both in yeast and mammalian cells (for review, see Shepherd et al. 1996), it has been suggested that the FYVE finger-containing proteins act as downstream effectors of PI 3-kinase in vesicular traffic.
Hrs was originally identified as a tyrosine-phosphorylated protein in cells stimulated with hepatocyte growth factor, platelet-derived growth factor, and epidermal growth factor (Komada and Kitamura 1995). It is also tyrosine phosphorylated in cells stimulated with cytokines such as IL-2 and GM-CSF (Asao et al. 1997). Hrs associates through its coiled-coil domain with STAM (signal-transducing adaptor molecule; Asao et al. 1997), a protein with a Src homology 3 (SH3) domain and an immunoreceptor tyrosine-based activation motif (ITAM) that itself is also tyrosine phosphorylated in response to growth factors and cytokines (Takeshita et al. 1996). Taken together, these observations suggest that Hrs is involved in cell growth signaling or ligand-induced endocytosis and down-regulation of growth factor and cytokine receptors.
To investigate the physiological function of Hrs, we have generated Hrs mutant mice by targeted disruption of the gene in embryonic stem (ES) cells. We show that Hrs homozygous mutant embryos exhibit a defect in ventral folding morphogenesis and die between embryonic day 10.5 (E10.5) and E11.5. Significant apoptosis was observed in the definitive endoderm of the mutant embryo. These results indicated that Hrs is required for ventral folding morphogenesis and the ectopic cell death in the endoderm may be responsible for the mutant phenotype. We also detected expansion of early endosomes in Hrs mutant cells and mislocalization of Hrs in wortmannin-treated cells, further implicating the factor in the PI 3-kinase pathway of vesicular trafficking.
Results
Embryonic Hrs expression and derivation of mutant mice
Expression of Hrs in wild-type embryos was examined by whole mount in situ hybridization with an antisense Hrs probe. Hrs was expressed ubiquitously at E7.5, E8.5, and E10.5 (data not shown). Sections through the E7.5 embryo showed Hrs expression in all three germ layers. Hrs message was also detected in blastocysts and ES cells by reverse-transcriptase-polymerase chain reaction (PCR) and Northern blot analysis, respectively, suggesting its expression at earlier stages of development (not shown). This broad expression pattern continues in the adult mouse and human (Komada and Kitamura 1995; Lu et al. 1998).
To disrupt the hrs locus, a gene-targeting construct was designed to replace a 2.1-kb hrs genomic sequence, which includes exons 2, 3, and 4 and encompasses the amino-terminal region of the protein (amino acids 14–93), with a neo cassette (Fig. 1A). Moreover, exon 1 and exon 5 are out of frame, so that a frameshift would occur in the eventuality of splicing between these two exons. The construct is therefore expected to lead to a null allele. Following electroporation of the construct into ES cells, 16 of 279 colonies scored positive for homologous recombination by a PCR assay. PCR-positive clones were further analyzed by Southern blots (Fig. 1B; data not shown). Germ-line chimeras were derived from three of the clones and were crossed to C57BL/6J or 129/Sv mice to derive mutant mice. Heterozygous offspring did not display an overt phenotype.
Figure 1.

Targeted disruption of the hrs gene. (A) Restriction map of the genomic locus. The restriction map of the targeting construct and of the wild-type and mutated allele is shown. The first five exons are shown in black boxes. Arrowheads indicate the position of PCR primers used to detect targeted insertions. 5′- and 3′-probes for Southern blotting are indicated by shaded boxes. B, D, E, and X represent restriction sites for _Bam_HI, _Dra_I, _Eco_RI, and _Xba_I, respectively. (B) Southern blot analysis of wild-type (+/+) and targeted (+/−) ES cell DNAs. _Bam_HI-digested and _Dra_I-digested DNAs were hybridized with the 5′- and 3′-probes shown in A, respectively. The size of hybridizing fragments are 8 and 6 kb (_Bam_HI), and 4 and 6 kb (_Dra_I), for wild-type and mutant alleles, respectively. (C) PCR- genotyping and immunoblot analysis of Hrs in embryonic cell lines derived from wildtype (+/+), heterozygous (+/−), and homozygous (−/−) mutant embryos. The length of PCR fragments are 408 bp and 554 bp for wild-type and mutant alleles, respectively. A 115-kD band of Hrs is not detected by immunoblotting in homozygous mutant cells.
To confirm the lack of Hrs expression in the mutant, immortalized cell lines were established from E9.5 wild-type, heterozygous, and homozygous mutant embryos that also carry a temperature-sensitive SV40 large T antigen (Jat et al. 1991). Hrs was immunoprecipitated from these cells with an anti-Hrs antibody raised against the central region of the protein (exon 5 and following exons) and immunoblotted with the same antibody. No immunoreactive bands were detected in the homozygous mutant cells (Fig. 1C), confirming that this mutation leads to a null allele.
Lethality of Hrs mutant embryos
No homozygous mutant mice were recovered following intercrossing of Hrs heterozygous parents, suggesting that the Hrs disruption leads to a recessive embryonic lethal phenotype (Table 1). Embryos of all genotypes were recovered according to Mendelian expectations between E7.5 and E10.5, however all the homozygotes recovered at E11.5 were dead and being resorbed (Table 1). Empty deciduas were also frequently found at E11.5. These results indicate that the Hrs homozygous embryos die between E10.5 and E11.5.
Table 1.
Genotype of offspring derived from hrs heterozygous parents
| | +/+ | +/− | −/− | | | ---------- | ------- | ------- | ----------------- | | At weaning | 97 | 151 | 0 (0%) | | E3.5 | 6 | 16 | 6 (21%) | | E7.5 | 15 | 27 | 10 (19%) | | E8.5 | 57 | 118 | 62 (26%) | | E9.5 | 48 | 75 | 43 (26%) | | E10.5 | 16 | 24 | 16 (29%) | | E11.5 | 15 | 25 | 9 (18%, all dead) |
At E7.5, homozygotes were always smaller than wild-type or heterozygous mutant littermates (Fig. 2A). Sections through the mutant embryo showed that the three germ layers were formed (not shown and Fig. 4C, below). By E8.5, homozygotes were morphologically abnormal, although embryonic axes and structures such as neural fold, amnion, and allantois were identified (Fig. 2B). At E9.5, wild-type embryos were completely enclosed by the yolk sac (Fig. 2C). In contrast, homozygous mutants were totally disorganized and the ventral region was exposed outside of the yolk sac (Fig. 2C). They continued to develop with their ventral region outside the yolk sac until their death around E11 (not shown). The head fold and the neural tube were wavy and not closed in the mutant (Fig. 2D,E). The allantois was dilated and never fused with the chorion (Fig. 2D). A notochord was visible in most of the mutants examined in sections (not shown). Somite segmentation was not observed by light microscopy as well as by in situ hybridization with Mox-1 (not shown). In addition, the mutants did not undergo the process of ‘turning’ in which the orientation of the germ layers is reversed. The same phenotype was observed in mutant strains derived from three independent ES clones, on congenic 129/Sv, mixed 129/Sv × C57BL/6J, and congenic C57BL/6J (>10-generation backcross) genetic backgrounds.
Figure 2.

Morphology of the Hrs mutant embryos. (A–C) Lateral view of the wild-type (+/+) and Hrs homozygous mutant (−/−) embryos at E7.5 (A), E8.5 (B), and E9.5 (C). The yolk sac was not removed from embryos in B and C. Dorsal (D) and ventral (E) view of the E9.5 Hrs mutant embryo from which the yolk sac was removed. Whole embryos (E7.5) and yolk sac (E8.5 and E9.5) were used for PCR genotyping. (Al) Allantois; (Am) amnion; (HF) head fold; (NT) neural tube; (YS) yolk sac.
Figure 4.
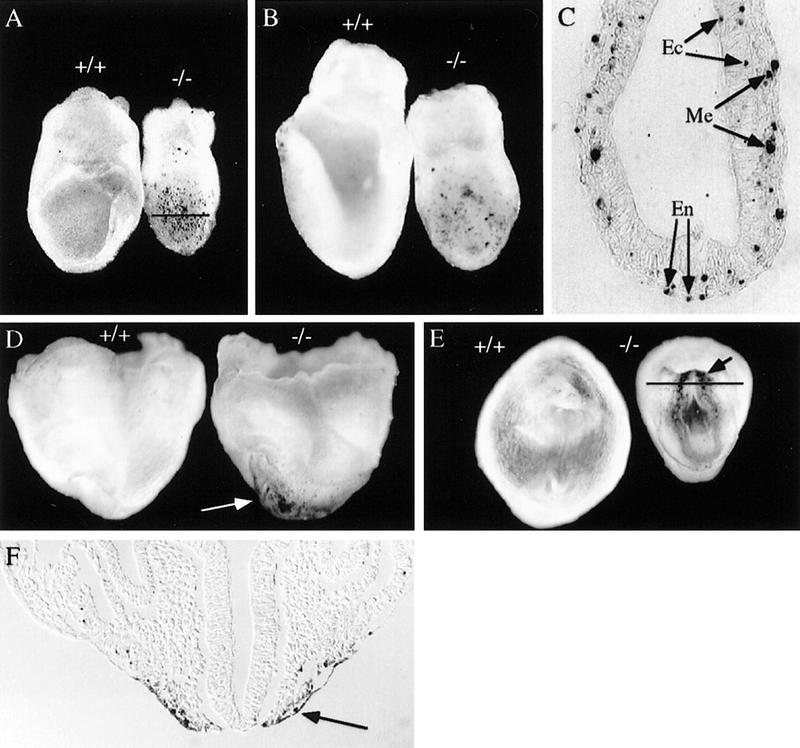
Apoptotic cell death in the Hrs mutant embryos, as detected by whole-mount TUNEL staining. (A,B) Lateral view of E7.5 (A) and E8.0 (B) wild-type (+/+) and Hrs homozygous mutant (−/−) embryos. (C) Transverse section of the E7.5 mutant embryo at the level of the line in A. Lateral (D) and ventral (E) view of E8.5 wild-type (+/+) and mutant (−/−) embryos. (F) Section of the E8.5 mutant at the level of the line in E. Arrows indicate TUNEL-positive apoptotic cells. Genotype was determined by PCR with embryo sections scraped from slides. (En) endoderm; (Me) mesoderm; (Ec) ectoderm.
Defects in ventral folding morphogenesis in Hrs mutant embryos
In mouse embryos, rostral-to-caudal and lateral-to-ventral folding occur at the anterior region around E8.0. The rostral-to-caudal folding allows the heart primordia, which develop bilaterally from the splanchnic mesoderm at the most anterior region, to become localized posterior to the head fold. The lateral-to-ventral folding permits the bilateral heart primordia, as well as the yolk sac derived from the visceral endoderm and extraembryonic mesoderm, to migrate and fuse at the ventral midline (Kaufman and Navaratnam 1981; DeRuiter et al. 1992; Fig. 3A). In this process, a single heart tube is formed, the foregut is generated by closing the definitive endoderm layer (Stalsberg and DeHaan 1968), and the anterior region of the embryo becomes positioned within the yolk sac. A defect in this process would therefore be predicted to lead to bilateral heart tubes (cardia bifida), an absence of foregut formation, and a failure of the ventral region of the embryo to be enclosed by the yolk sac. Because the ventral region of the Hrs mutant embryos was exposed outside of the yolk sac, we undertook marker analysis to examine if the phenotype is due to a defect in the ventral folding morphogenesis.
Figure 3.
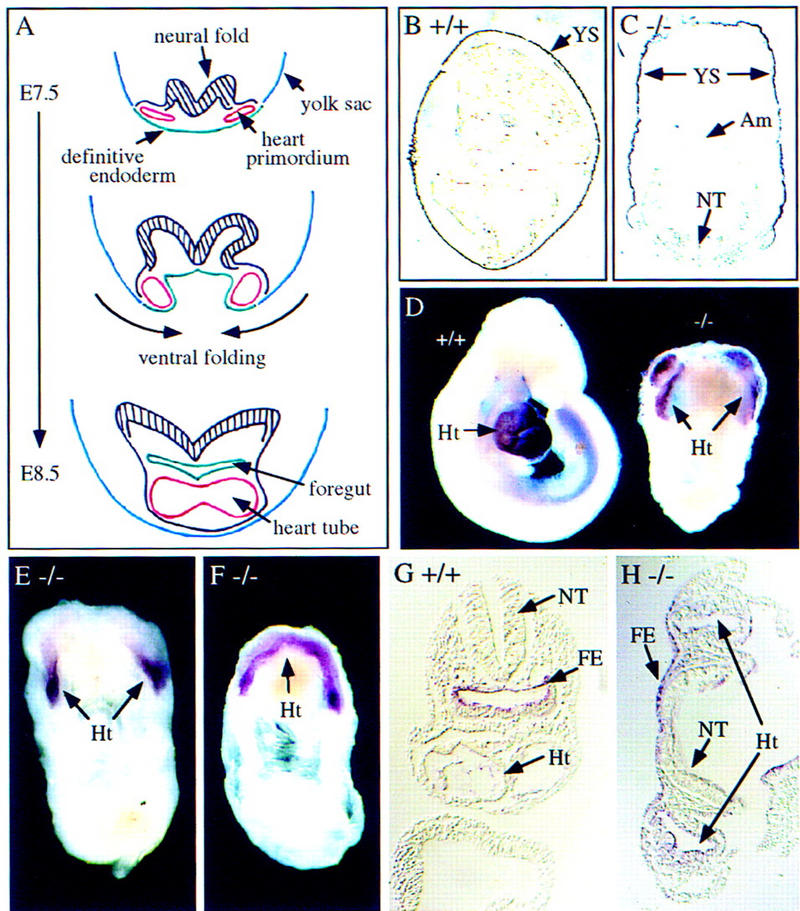
Abnormal yolk sac, heart, and foregut formation in the Hrs mutant embryos. (A) Schematic representation of ventral fusion morphogenesis during mouse embryogenesis. Transverse sections of a mouse embryo at E7.5–E8.5 are shown. The heart primordia and the heart tube are shown in red, the yolk sac are shown in blue, and the definitive endoderm are shown in green. The neural folds are shown as black hatched regions. (B,C) In situ hybrydization analysis of E9.5 wild-type (B) and Hrs homozygous mutant (C) embryos for α-fetoprotein. (D) Immunostaining of E9.5 wild-type (+/+) and mutant (−/−) embryos with anti-myosin heavy chain antibody. (E,F) In situ hybrydization analysis of E9.5 mutant (−/−) embryos for Nkx2.5. (G,H) In situ hybrydization analysis of E8.5 wild-type (G) and mutant (H) embryos for HNF-3α. (Am) Amnion; (FE) foregut endoderm; (Ht) heart; (NT) neural tube; (YS) yolk sac.
Yolk sac formation was examined by in situ hybridization of E9.5 embryos for α-fetoprotein, a specific marker for yolk sac endoderm. As shown in Figure 3, B and C, the yolk sac enclosed the wild-type embryo but not the ventral region of the mutant embryo. Heart formation was examined using two heart-specific markers, myosin heavy chain and Nkx2.5. Both markers detected myocardial primordia at both sides of the anterior region of the E9.5 mutants (Fig. 3D,E). In some cases, they fused at their anterior ends, making an arch-shaped single heart tube (Fig. 3F). Myocardial cells were functional in these bilateral and anteriorly fused heart tubes as they were beating when the embryos were isolated. Finally, foregut formation was examined with a gut-specific marker, HNF-3α, which specifically stained foregut endoderm in a wild-type embryo (Fig. 3G). In the mutant, a foregut was not formed because the definitive endoderm did not fold and fuse at the midline (Fig. 3H). These results indicate a failure of the yolk sac and the heart primordia to migrate and fuse at the ventral midline in the Hrs mutant, because of a defect in ventral folding morphogenesis. Mutant embryos also seemed to have a defect in rostral-to-caudal folding, which positions the heart posterior to the head fold, as the heart primordia retained a mostly anterior position (Fig. 3D–F).
Embryos lacking the transcription factor GATA4 are also defective in ventral folding and exhibit a phenotype similar to that of the Hrs mutants (Kuo et al. 1997; Molkentin et al. 1997). Therefore, we examined GATA4 expression in Hrs mutant embryos. By in situ hybridization, GATA4 was as highly expressed at E8.5 in the region corresponding to the anterior intestinal portal in the Hrs mutant as in the wild-type embryo (not shown; Molkentin et al. 1997), indicating that the Hrs mutant phenotype is not due to down-regulation of GATA4 expression. We also tested the expression of other GATA family members, GATA6 and GATA5, in the Hrs mutant as it has been suggested that they may be functionally redundant with GATA4 in cardiac development (Laverriere et al. 1994; Jiang and Evans 1996). However, their expression pattern was also unaffected in the mutant embryos (not shown).
Cell proliferation and apoptosis in Hrs mutant embryos
As many growth factors exert their effects on cells by promoting proliferation or survival, and because Hrs is implicated in growth factor signaling, we examined if these responses are affected in the mutant. Cell proliferation was examined by BrdU incorporation with embryos just before (E8.0) and after (E8.5) the onset of ventral fusion morphogenesis. In the E8.0 embryos, there was no difference in mitotic index in the neuroepithelium between the wild-type and the mutant (not shown). In the neuroepithelium of E8.5 embryos, however, the mitotic index was slightly higher in the mutant (72% ± 3%) than in the wild-type (61% ± 5%). In the definitive endoderm where significant apoptosis was detected in the mutant (see below), no difference was observed between the wild-type and the mutant (not shown). These results suggested that the defect in ventral folding in the mutant is not due to reduced cell proliferation.
Apoptotic cell death was examined by the whole mount TUNEL staining with E7.5, E8.0, and E8.5 embryos. At E7.5 and E8.0, many more apoptotic cells were observed in the mutants than in the wild-type embryos (Fig. 4A,B). Sections through the E7.5 mutant showed that the apoptotic cells were scattered and were present equally in the endoderm, mesoderm, and ectoderm (Fig. 4C). At E8.5, no obvious cell death was detected in the wild-type embryos. In contrast, significant number of apoptotic cells were observed in the ventral region of the mutants (Fig. 4D,E). More apoptotic cells were located in the anterior region than in the posterior region. Sections through the stained mutant showed that the apoptotic cells were mostly concentrated in the definitive endoderm (Fig. 4F), suggesting that the defect in ventral folding in the Hrs mutant is due to a failure in normal endoderm function.
Proliferation and differentiation of Hrs mutant embryonic cells in teratocarcinomas
The Hrs mutant phenotype and the observed apoptosis suggest a defect in definitive endoderm. However, the lethality of Hrs mutant embryos around E11 prevented us from testing whether Hrs is defective in all endodermal lineages. To test this possibility, E6.5 embryos derived from matings of heterozygous parents were transplanted under the kidney capsules of adult mice to form teratocarcinomas (Damjanov et al. 1987). Southern blot analysis showed that of 36 teratocarcinomas, 8 (22%) were wild-type, 22 (61%) were heterozygotes, and 6 (17%) were homozygotes. The size of the homozygous teratocarcinomas was about 0.5 cm in diameter, whereas that of the wild-type and heterozygous teratocarcinomas varied from 0.5 cm to 3 cm. These results indicated that the Hrs homozygous embryonic cells proliferate as teratocarcinomas, although to a lesser extent.
Cell types present in the teratocarcinomas were examined by histology. Derivatives of all three germ layers (ectoderm: neural tissue and keratinized epithelia; mesoderm: cartilage, pigmented cells, and smooth muscle; endoderm: gut) were found in the homozygous teratocarcinomas as well as in the wild type and heterozygotes (Fig. 5A–D, Table 2). However, skeletal muscle and fat cells, which were found in most of the wild type and heterozygotes (Fig. 5E), were never observed in the homozygous teratocarcinomas (Table 2). These results suggest that the Hrs mutant embryonic cells are capable of differentiating to gut endoderm and therefore Hrs is not inherently required in all endoderm lineages. These results also suggest that Hrs is required in certain types of mesodermal tissues, such as skeletal muscle and fat.
Figure 5.
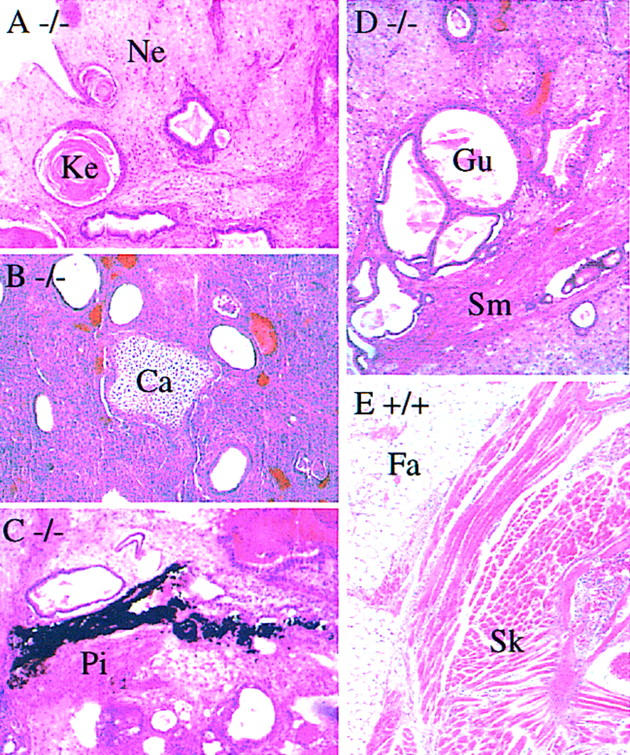
Histology of teratocarcinomas derived from the wild-type (+/+) and Hrs mutant (−/−) embryos. Sections of teratocarcinomas derived from Hrs mutant (A–D) and wild-type (E) embryos are shown. (Ca) cartilage; (Fa) fat cells; (Gu) gut; (Ke) keratinized epithelium; (Ne) neural tissue; (Pi) pigmented cells; (Sk) skeletal muscle; (Sm) smooth muscle.
Table 2.
Tissues types in hrs mutant teratocarcinomas
| | +/+ | +/− | −/− | | | --------------------- | ------- | ------- | --- | | Ectoderm | | | | | Neural tissue | 4/4 | 12/12 | 6/6 | | Keratinized epithelia | 4/4 | 12/12 | 6/6 | | Mesoderm | | | | | Cartilage | 4/4 | 10/12 | 3/6 | | Pigmented cells | 4/4 | 12/12 | 5/6 | | Smooth muscle | 4/4 | 12/12 | 6/6 | | Skeletal muscle | 4/4 | 8/12 | 0/6 | | Fat cells | 4/4 | 12/12 | 0/6 | | Endoderm | | | | | Gut | 4/4 | 12/12 | 6/6 |
Abnormal morphology of early endosomes in Hrs mutant cells
In primary embryonic cell cultures derived from E9.5 embryos, vacuole-like structures were frequently detected under a phase contrast microscope in mutant cells (Fig. 6B, arrows), but rarely seen in wild-type cells (Fig. 6A). Endosomes have been reported to form similar phase-lucent swollen structures when PI 3-kinase activity is inhibited by wortmannin (Brown et al. 1995; Malide and Cushman 1997). Furthermore, a mutation in Vps27p, a yeast protein that is homologous to Hrs, causes atypically large endosomes because of a defect in vesicular transport via the organelle (Piper et al. 1995). Thus, we examined whether the vacuole-like structures in the mutant cells were expanded endosomes by immunofluorescence staining with endosome markers. Early endosomes were stained with an anti-transferrin receptor antibody. In the wild-type cells, the antibody detected punctate vesicular structures that were scattered throughout the cytoplasm (Fig. 6C). In the Hrs mutant cells, however, large vacuolar structures were stained in the perinuclear region (Fig. 6D,E, arrows). Phase contrast microscopy showed that these structures corresponded to the vacuolar structures shown in Figure 6B (not shown). Morphologically normal early endosomes were also detected, but to a lesser extent, throughout the cytoplasm in the mutant cells (Fig. 6D,E). Late endosomes and lysosomes were stained with an anti-Lamp1 antibody (Chen et al. 1985). Perinuclear vacuolar structures, as well as punctate cytoplasmic structures, were detected similarly in the wild-type (Fig. 6F) and the Hrs mutant (Fig. 6G) cells. The transferrin receptor-positive vacuolar structures in the mutant cells (Fig. 6B,D, arrows) were Lamp1-negative, indicating that the absence of Hrs does not affect the morphology of the late endosome/lysosome. These results showing that the morphological abnormality is specific to the early endosome were consistent with the previous observation that Hrs is specifically localized to this organelle (Komada et al. 1997).
Figure 6.
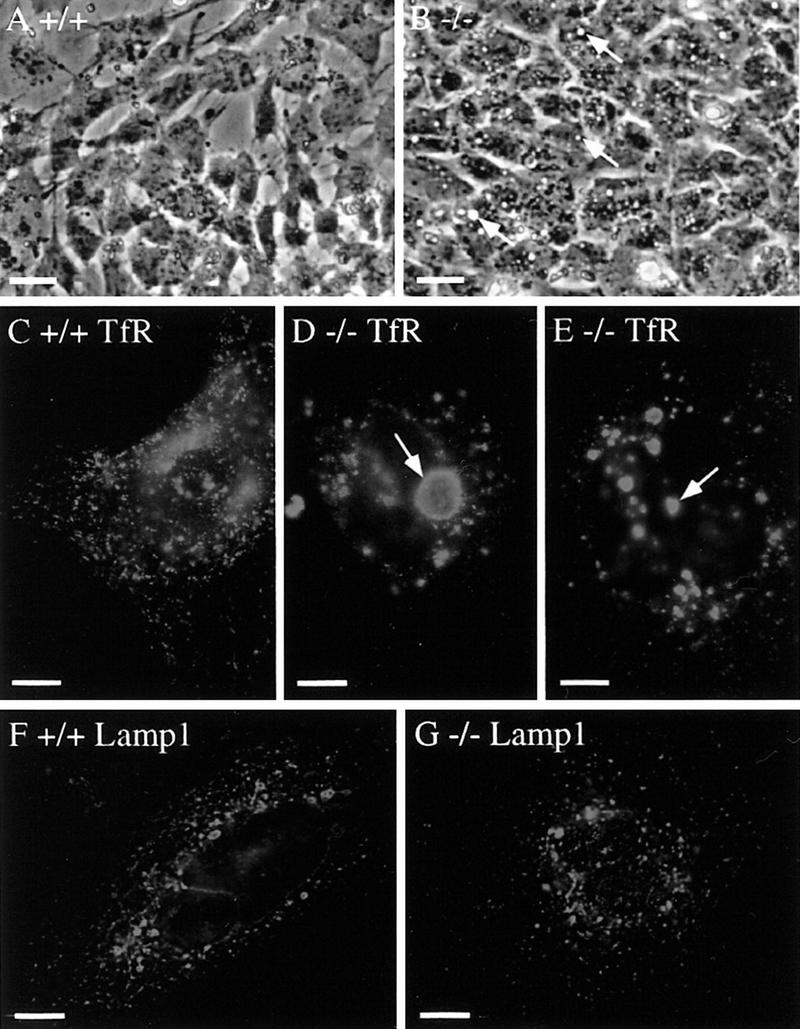
Morphology of endosomes in Hrs mutant cells. E9.5 wild-type (+/+) and Hrs mutant (−/−) embryos were isolated and trypsinized, and primary cells were put in culture. (A,B) Phase contrast image of the wild-type (A) and Hrs mutant (B) cells after a day of culture. Arrows indicate typical phase-lucent vacuolar structures in the Hrs mutant cells. (C–E) Immunofluorescence staining of the wild-type (C) and Hrs mutant (D,E) cells with an anti-transferrin receptor (TfR) antibody, a marker for early endosomes. Arrows indicate typical expanded early endosomes. (F,G) Immunofluorescence staining of the wild-type (F) and Hrs mutant (G) cells with an anti-Lamp1 antibody, a marker for late endosomes and lysosomes. Bars, 100 μm (A,B), 10 μm (C–G).
In situ detection of abnormal early endosomes in Hrs mutant embryos
To identify cell types in which the morphology of early endosomes was affected, E8.5 embryos were stained with the anti-transferrin receptor antibody by whole mount immunofluorescence staining. In wild-type embryos, punctate vesicular structures as well as the plasma membrane were stained in the definitive endoderm (Fig. 7A) as well as in the neuroepithelium (Fig. 7D), suggesting that the transferrin receptor is cycling between the early endosome and the plasma membrane. In Hrs mutant embryos, early endosomes were expanded both in the definitive endoderm (Fig. 7B,C) and neuroepithelium (Fig. 7E). Staining of the plasma membrane in these tissues was slightly weaker in the mutant than in the wild-type embryo. To confirm these results, staining for the transferrin receptor was also visualized with a peroxidase-conjugated secondary antibody and the morphology of early endosomes were observed in sections. In the mutant embryo, the antibody stained ring-shaped vacuolar structures in the neuroepithelium (Fig. 7F,I), definitive endoderm (Fig. 7G,J), and myocardium (Fig. 7H,K). These structures were not detected in the wild-type embryo (Fig. 7L–N), again indicating expansion of early endosomes in Hrs mutant embryos. Abnormal endosome morphology in the definitive endoderm cells suggested that the defect in ventral folding in the mutant is attributed to dysfunction of early endosomes in these cells.
Figure 7.
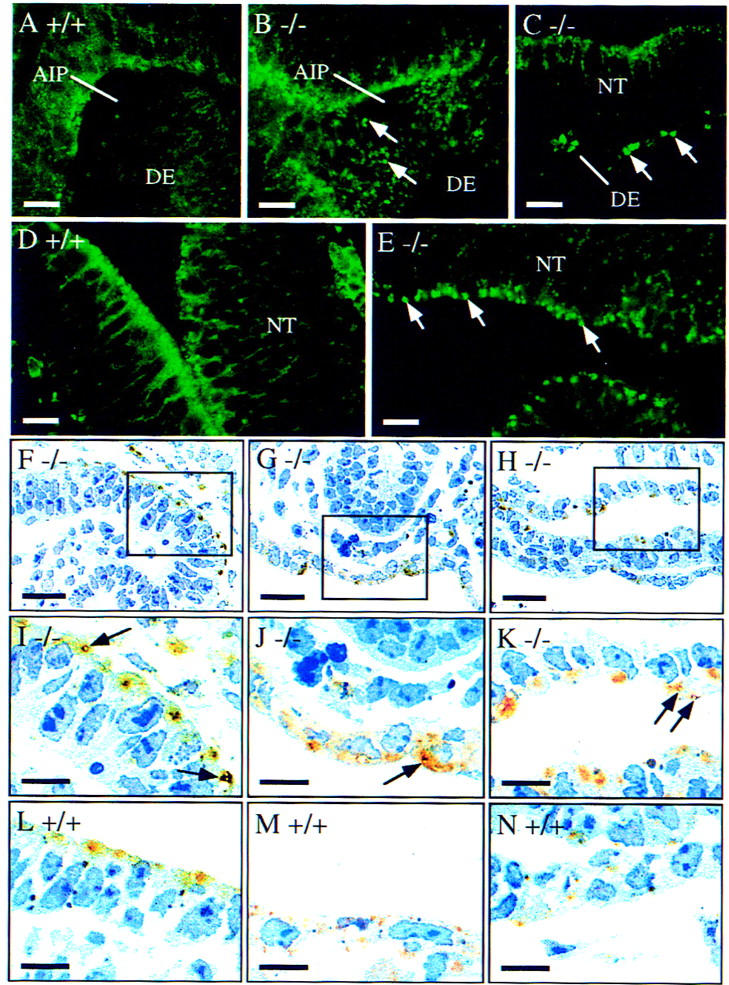
Morphology of early endosomes in the Hrs mutant embryos. E8.5 wild-type (+/+) and Hrs mutant (−/−) embryos were stained with a TfR antibody by whole mount immunofluorescence staining (A–E) or immunohistochemistry (F–N). Immunofluorescent images were captured by confocal microscopy and immunohistochemical staining was observed in sections. (A,B) Ventral view of the wild-type (A) and mutant (B) embryos around the anterior intestinal portal or the corresponding region showing the staining of the definitive endoderm. (C) Lateral view of the mutant embryo showing the staining of the neural tube and definitive endoderm. (D,E) Staining of the neural tube of the wild-type (D) and mutant (E) embryos. (F–K) Sections of the mutant embryo showing the neural tube (F), definitive endoderm (G), and myocardium (H). Regions in the boxes are shown at a higher magnification in I, J, and K, respectively. (L–N) Sections of the wild-type embryo showing the neural tube (L), definitive endoderm (M), and myocardium (N). Arrows indicate expanded early endosomes. (AIP) Anterior intestinal portal; (DE) definitive endoderm; (NT) neural tube. Bars, 50 μm (A–H); 25 μm (I–N).
Mislocalization of Hrs in cells treated with wortmannin
The similar expanded early endosome morphology seen in Hrs mutant cells and wortmannin-treated cells suggested that Hrs acts in the PI 3-kinase pathway of membrane trafficking. As the FYVE finger domain binds PI(3)-P, a role for PI 3-kinase in targeting FYVE finger domain-containing effectors to PI(3)-P-containing vesicle membranes has been suggested (Patki et al. 1997, 1998; Burd and Emr 1998; Gaullier et al. 1998). We therefore examined whether the membrane localization of Hrs, which has a FYVE finger domain, is regulated by PI 3-kinase using the inhibitor wortmannin. In NIH-3T3 cells that overexpress Hrs, it was detected on vesicular structures by immunofluorescence staining with an anti-Hrs antibody in the absence of wortmannin (Fig. 8A; Komada et al. 1997). However, when the cells were treated with wortmannin, it was mislocalized to the cytoplasm (Fig. 8B). Staining with an anti-transferrin receptor antibody showed that early endosomes were present in wortmannin-treated cells (Fig. 8D), although they were enlarged compared to those in untreated cells (Fig. 8C) as previously reported (Spiro et al. 1996; Malide and Cushman 1997).
Figure 8.
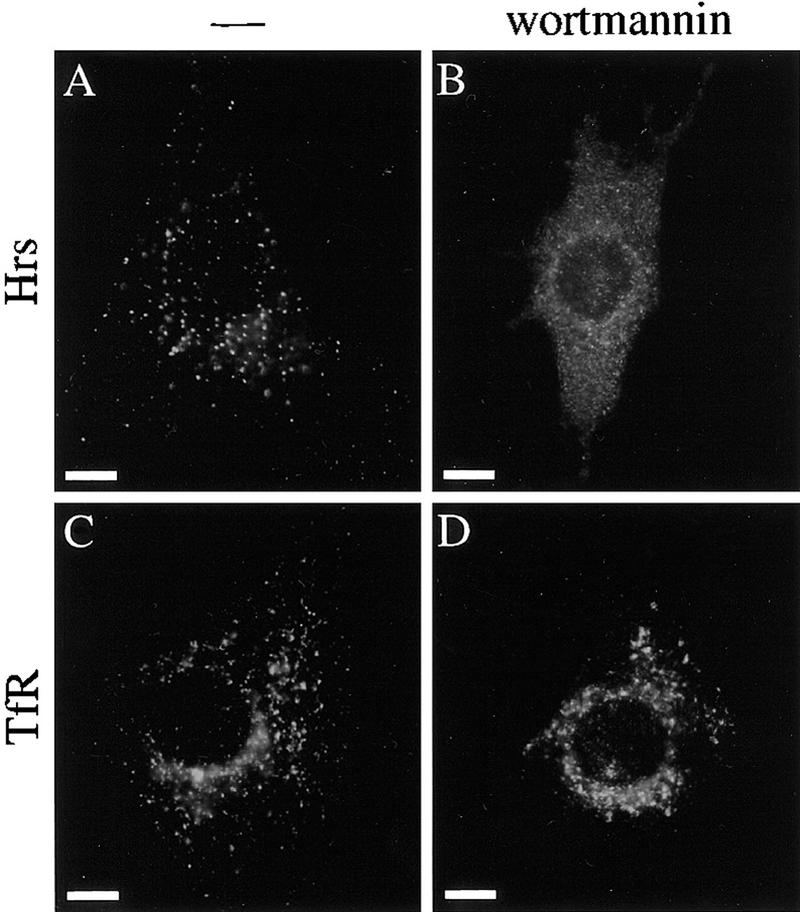
Mislocalization of Hrs in wortmannin-treated cells. NIH-3T3 cells overexpressing Hrs were treated without (A,C) or with (B,D) wortmannin, and stained with an anti-Hrs antibody (A,B) or a TfR antibody (C,D). Bar, 10 μm.
Discussion
Role of Hrs in embryonic development
Hrs homozygous mutant embryos exhibit a defect in ventral folding morphogenesis that normally occurs around E8. The defect is unlikely to be due to general developmental retardation as mutants were often as large as wild-type embryos and had normal-sized although disorganized neural fold at E8.5, and they had beating hearts at E9.5. Mutants never underwent the process of ‘turning’, but this is most likely a secondary defect as it must be physically impossible for embryos to turn without completing ventral morphogenesis. The vasculature of the yolk sac was well developed in the mutant, but the heart primordia, although beating, were totally disorganized at E9.5 and E10.5. These observations suggest that a primary cause of death of the mutant may be a defect in blood circulation caused by an impaired heart function. Additional defects in the circulatory system are expected as the allantois never fused with the chorion and thus umbilical vessels between the embryo and the chorioallantoic placenta would not form.
Targeted disruption of genes has identified several genes that are required for ventral morphogenesis in mice. Mutants for the proprotein convertase, furin, and fibronectin (when severely affected) are defective in fusion of heart tubes (George et al. 1993; Roebroek et al. 1998). However, ventral folding seems to progress quite far in these mutants as the two heart primordia almost stick to each other at the ventral midline and the ventral region is almost enclosed by the yolk sac. In contrast, GATA4 as well as Hrs mutants are defective in the folding process (Kuo et al. 1997; Molkentin et al. 1997). As a result, ventral regions are widely exposed outside the yolk sac and bilateral heart primordia stayed far apart from each other in these mutants. In the Hrs mutants, the heart primordia sometimes fused anteriorly and generated an arch-shaped single heart tube, suggesting that they are capable of fusion. In the GATA4 mutants, no cardiac muscle contraction is observed although a number of cardiac markers are expressed in the cardiomyocyte. As the Hrs mutant hearts were always beating at E9.5, this may suggest that GATA4 is also required for functional cardiac differentiation, or that embryonic development is less severely retarded in the Hrs mutants.
Ectopic apoptosis was detected exclusively in the definitive endoderm in the Hrs mutants at E8.5. Therefore, it is possible that the ventral defect originates from the definitive endoderm, as the cells undergoing apoptosis must be functionally affected at least several hours before E8.5 when ventral morphogenesis has not yet occurred. The mechanism of ventral folding morphogenesis is not well understood. However, as far as heart morphogenesis is concerned, several lines of evidence suggest that the definitive endoderm plays an inductive role in amphibians and avians (for review, see Nascone and Mercola 1996). It is suggested that the definitive endoderm is required for formation of the functional beating heart as well as cardiomyocyte differentiation. Therefore, the failure of normal heart morphogenesis caused by the endoderm defect in the Hrs mutant embryos may have prevented them from folding the ventral region correctly. Chimeric analysis with GATA4 homozygous mutant ES cells supports the idea that the endoderm is important for ventral morphogenesis, as in morphologically normal chimeras, _GATA4_-deficient ES cells were able to contribute to the cardiogenic mesoderm but not to the visceral and definitive endoderm (Narita et al. 1997). These observations raise questions as to how the definitive endoderm plays a role in ventral morphogenesis. The definitive endoderm folds and fuses to generate a foregut during ventral morphogenesis. Therefore, it may generate a mechanical force that drives the entire folding process, including folding of the cardiac mesoderm and yolk sac. Another possibility is that factor(s) secreted from the definitive endoderm are required for the cardiac mesoderm to drive ventral folding. It has been suggested for instance that transforming growth factor-β, fibroblast growth factors, and extracellular matrix proteins such as fibronectin, which are produced by the endoderm, play roles in the formation of the functional heart (for review, see Nascone and Mercola 1996). However, despite these observations implicating the definitive endoderm in ventral folding, we also can not exclude the possibility that the apoptosis in the Hrs mutant endoderm is just a result of the ventral folding defect.
Midline fusion of bilateral heart primordia is a common developmental process in vertebrates. Five mutants defective in this process (cardia bifida) have been identified in zebrafish by large-scale mutagenesis screens (Chen et al. 1996; Stainier et al. 1996). However, genes responsible for the mutant phenotypes remain to be identified. Because Hrs is evolutionarily conserved, it is possible that an Hrs homolog is mutated in one of these zebrafish mutations. The mouse hrs gene was mapped to the distal end of chromosome 11 (locus symbol Hgs in the Mouse Genome Database; M. Komada and P. Soriano, unpubl.), a region syntenic to human chromosome 17q25 where the human hrs gene was previously mapped (Asao et al. 1997). However, none of the zebrafish cardia bifida mutations for which chromosomal localization have been determined so far mapped to a syntenic region (D. Stainier, pers. comm.).
Hrs mutant embryos were smaller than wild-type littermates at E7.5. This phenotype is unlikely to be due only to a defect in the definitive endoderm because increased level of apoptosis was detected equally in the ectoderm, mesoderm, and endoderm at this stage. As growth of cells that are apoptotic at E7.5 is likely affected several hours earlier, and cell proliferation at this stage is very rapid (Snow 1977), the reduction in cell number attributable to the apoptosis may well be responsible for the smaller size of the mutant at E7.5. As apoptotic cell death was not noticed in all germ layers at later stages and embryos are of normal size at E8.5, it appears that Hrs-deficient embryos can overcome this first phase of a mutant phenotype. The fact that the embryos need to catch up may explain the higher mitotic index observed in the neuroepithelium of mutant embryos by E8.5.
Role of Hrs in function of early endosomes
Significantly enlarged early endosomes were detected in cells of the definitive endoderm in the Hrs mutant embryos. Taken together with the fact that ectopic apoptosis was observed exclusively in these cells at E8.5, this suggests that the defect in ventral folding in the mutant may be attributed to a cellular defect caused by an impaired endosome function in the endoderm and leading to their death. Abnormal early endosomes were also identified in the mutant neuroepithelium, where no apoptotic cells were detected at E8.5. Thus, survival of the definitive endoderm cells may be more susceptible to an impaired early endosome function than other cell types at this stage. It is also possible that ventral folding is directly regulated by some function of early endosomes, which is independent of cell survival or death.
Yeast mutants lacking Vps27p, the homolog of mammalian Hrs, exhibit a defect in vesicular traffic via endosomes. In these cells, endocytosed proteins and newly synthesized vacuolar proteins are not correctly transported to vacuoles but are accumulated in endosomes, leading to their expansion (Piper et al. 1995). In this work, we found that early endosomes were expanded also in Hrs mutant cells and embryos. These observations suggest that the mechanism of vesicular transport via endosomes is conserved between mammalian and yeast cells, and that Hrs and Vps27p play the same key role in the process. Expansion of early endosomes has also been reported in mammalian cells implicating other proteins. Overexpression of rab5, a small GTPase localized to early endosomes and involved in docking and fusion of early endosomes, causes expansion of the organelle (Bucci et al. 1992; Stenmark et al. 1994). The expansion can be inhibited by coexpression of the carboxy-terminal region of EEA1, an early endosomal protein that contains a FYVE finger domain and associates with rab5, suggesting that the EEA1 carboxy-terminal region acts dominant-negatively and EEA1 is critical for early endosome docking/fusion (Simonsen et al. 1998). It was reported that Hrs interacts with SNAP-25, a protein implicated in docking and fusion of endosome-derived synaptic vesicles with the presynaptic plasma membrane (Bean et al. 1997). These researchers also showed that Hrs, when added exogeneously, inhibits neurotransmitter release from the synaptic vesicles in permeabilized PC12 cells. Taken together with our finding that early endosomes were expanded in the absence of Hrs, these observations may suggest that Hrs negatively regulates membrane docking and/or fusion.
In addition to the expansion of early endosomes, plasma membrane staining for the transferrin receptor was noticeably weaker in the Hrs mutant embryos than in the wild-type embryos, suggesting a defect in vesicular traffic to recycle the transferrin receptor from the endosome back to the plasma membrane in the mutant cells. The same phenotype was reported for cells treated with wortmannin, a specific inhibitor of PI 3-kinase. Wortmannin causes the formation of phase-lucent swollen early endosomes and decrease in the number of cell surface transferrin receptors (Spiro et al. 1996; Malide and Cushman 1997). PI 3-kinase has been shown to play an essential role in multiple steps of vesicular traffic in mammalian cells as well as in yeast (for review, see Shepherd et al. 1996). Recently, it was shown that the FYVE finger domain, which is found in Hrs and several other proteins involved in vesicular traffic, specifically binds to PI(3)-P, a product of the PI 3-kinase activity (Burd and Emr 1998; Gaullier et al. 1998; Patki et al. 1998). Furthermore localization of Hrs as well as EEA1 to vesicle membranes is disrupted when PI 3-kinase activity is inhibited by wortmannin (this paper; Patki et al. 1997). These observations suggest a role for PI 3-kinase in providing a binding site for FYVE finger proteins on the target vesicle membrane. Taken together, these results strongly suggest that Hrs is a direct downstream effector of PI 3-kinase in the vesicular traffic pathway via early endosomes.
Materials and methods
Derivation of mutant mice
The hrs genomic locus was isolated from a 129/Sv genomic library using a 5′ region of Hrs cDNA. The targeting vector was constructed using a neo expression cassette (PGKneolox2DTA; Soriano 1997). This targeting construct replaces a 2.1-kb _Xba_I–_EcoR_I genomic fragment encompassing the amino-terminal region of Hrs, flanked by 3.6-kb (_Xba_I–_Xba_I) and 1.5-kb (_EcoR_I–_Xba_I) genomic sequences derived from the 129/Sv library. The construct was linearized and electroporated into 129/Sv-derived AK7 ES cells (Soriano 1997), and colonies were selected with G418. Homologous recombination events were screened by PCR as described (Soriano et al. 1991), using primers corresponding to the neo gene and a genomic sequence outside the targeting construct. Southern blots were done according to a standard procedure using hrs genomic probes (_Xba_I–_Dra_I 0.7 kb and _Bam_HI–_Xho_I 2.2 kb). The blots were rehybridized with neo to show that there was only a single insertion of the vector. Tissue culture and blastocyst injections were performed as described (Soriano et al. 1991).
PCR genotyping was performed on tail biopsies or yolk sac DNAs as described (Imamoto and Soriano 1993) using the following combination of primers: 5′-TTCTGGGCAGCATGAGTTGG-3′, 5′-ACTGGGAAAGGCTTCCAAGAAC-3′, and 5′-CAGCCATCTGTTGTTTGCCC-3′. This produces 408-bp and 554-bp diagnostic fragments for the wild-type and mutant alleles, respectively.
Isolation of embryonic cell lines
E9.5 embryos derived from crossing of heterozygous Hrs parents also carrying a temperature-sensitive SV40 large T antigen transgene (Jat et al. 1991) were incubated in 0.25% trypsin and 1 mm EDTA for 5 min at room temperature. The cell suspension was plated on 15-mm wells containing Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and 10 U/ml interferon-γ (GIBCO), and cultured in the same medium. Transmission of the large T antigen transgene was determined by PCR on yolk sac DNAs using the following primers: 5′-AGCGCTTGTGTCGCCATTGTATTC-3′ and 5′-GTCACACCACAGAAGTAAGGTTCC-3′.
Expression of Hrs was examined in these cells by immunoprecipitation and immunoblotting using an anti-Hrs antibody (Komada and Kitamura 1995).
In situ procedures
In situ hybridization and immunohistochemistry were performed essentially as described (Hogan et al. 1994). The following probes were used for in situ RNA hybridization: a mouse Hrs cDNA 0.8-kb _Sca_I–_Xho_I fragment; α-fetoprotein (from Shirley Tilghman, Princeton University, NJ); Nkx2.5 and HNF-3α (from Eric Olson, University of Texas Southwestern Medical Center, Dallas, TX); Mox1 (from Albert Candia, Stanford University, CA); and GATA4, GATA5, and GATA6 (from Mona Nemer, University of Montreal, Quebec, Canada). Anti-myosin heavy chain monoclonal antibody MF-20 was obtained from Atsushi Asakura, McMaster University, Hamilton, Ontario, Canada; and Steve Tapscott, Fred Hutchinson Cancer Research Center, Seattle, WA. Proliferating cells were detected in sections of embryos labeled for 1 hr by injecting pregnant females with 5-bromo-2′-deoxyuridine (BrdU; 100 μg/gram body weight) intraperitoneally, using anti-BrdU antibody (Becton Dickinson; Soriano 1997). The whole mount TUNEL assay (Conlon et al. 1995) was used to examine apoptosis in embryos. For sections, embryos were embedded in paraffin and cut at 5 μm.
Generation of embryo-derived teratocarcinomas
E6.5 embryos were isolated from 129/Sv congenic matings and were transplanted under the kidney capsules of 129/Sv adult mice. After 6 weeks, teratocarcinomas were removed, fixed in Bouin’s solution, and embedded in paraffin for sections. The sections were stained with hematoxylin and eosin. Genomic DNA was extracted from a small portion of teratocarcinomas before fixation for genotyping by Southern blot analysis.
Immunofluorescence staining of culture cells
Immunofluorescence staining of cells in culture was performed as described (Komada et al. 1997) except that cells were permeabilized with phosphate-buffered saline (PBS) containing 0.1% saponin for staining with an anti-Lamp1 antibody. Primary antibodies used were a rat anti-transferrin receptor monoclonal antibody R17-217 (4 μg/ml; a gift of Suhaila White and Ian Trowbridge, The Salk Institute, La Jolla, CA), a rat anti-Lamp1 monoclonal antibody 1D4B (1:10; a gift of Thomas August, Johns Hopkins University, Baltimore, MD), and a rabbit anti-Hrs polyclonal antibody (1:2000; Komada and Kitamura 1995). The secondary antibodies are fluorescein-conjugated anti-rat and anti-rabbit immunoglobulin G (IgG) antibodies (1:100; Jackson Immunoresearch). Fluorescent images were captured and processed using the Deltavision microscope (Applied Precision).
To examine the effect of wortmannin on Hrs localization, Hrs was overexpressed in NIH-3T3 cells using a pBabe retrovirus transducing the Hrs cDNA (Morgenstern and Land 1990). The cells were cultured in DMEM containing 0.1% calf serum for 3 hr, treated with 1 μm wortmannin (Sigma) for 30 min, and used for immunofluorescence staining.
In situ detection of early endosomes
Embryos were fixed in 4% paraformaldehyde in PBS for 30 min and stained with the anti-transferrin receptor antibody (10 μg/ml) as described (Hogan et al. 1994). Secondary antibodies were the fluorescein- and horseradish peroxidase-conjugated anti-rat IgG antibodies (1:400). Fluorescent images were captured with a confocal microscope (MRC-600, Bio Rad). The peroxidase activity was visualized with the DAB peroxidase substrate kit (Vector Laboratories). Embryos stained for the peroxidase activity were post-fixed with 1% OsO4 for 2 hr, dehydrated with a graded series of ethanol, and embedded in epoxy resin. Sections were cut at 1 μm and counterstained with toluidine blue.
Acknowledgments
We thank Chris Auger, Phil Capp, and Karin Weismann for help with genotyping of mice and embryos; Jon Cooper and our laboratory colleagues for critical reading of the manuscript; Tim Knight for help with figure preparation; Adrian Quintanilla and Maureen Ryan for help with immunofluorescence staining; and Atsushi Asakura, Thomas August, Albert Candia, Mona Nemer, Eric Olson, Steve Tapscott, Shirley Tilghman, Ian Trowbridge, Suhaila White, and Chris Wright for in situ probes and antibodies. M.K. was supported by fellowships from the Human Frontier Science Program and the Uehara Memorial Life Science Foundation. This work was supported by grant HD24875 from National Institute of Child Health and Development to P.S.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL psoriano@fhcrc.org; FAX (206) 667-6522.
References
- Asao H, Sasaki Y, Arita T, Tanaka N, Endo K, Kasai H, Takeshita T, Endo Y, Fujita T, Sugamura K. Hrs is associated with STAM, a signal-transducing adaptor molecule. J Biol Chem. 1997;272:32785–32791. doi: 10.1074/jbc.272.52.32785. [DOI] [PubMed] [Google Scholar]
- Bean AJ, Seifert R, Chen YA, Sacks R, Scheller RH. Hrs-2 is an ATPase implicated in calcium-regulated secretion. Nature. 1997;385:826–829. doi: 10.1038/385826a0. [DOI] [PubMed] [Google Scholar]
- Brown WJ, DeWald DB, Emr SD, Plutner H, Balch WE. Role for phosphatidylinositol 3-kinase in the sorting and transport of newly synthesized lysosomal enzymes in mammalian cells. J Cell Biol. 1995;130:781–796. doi: 10.1083/jcb.130.4.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci C, Parton RG, Mather IH, Stunnenberg H, Simons K, Hoflack B, Zerial M. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell. 1992;70:715–728. doi: 10.1016/0092-8674(92)90306-w. [DOI] [PubMed] [Google Scholar]
- Burd CG, Emr SD. Phosphatidylinositol(3)-phosphate signaling mediated by specific binding to ring FYVE domains. Mol Cell. 1998;2:157–162. doi: 10.1016/s1097-2765(00)80125-2. [DOI] [PubMed] [Google Scholar]
- Chen J-N, Haffter P, Odenthal J, Vogelsang E, Brand M, van Eeden FJM, Furutani-Seiki M, Granato M, Hammerschmidt M, Heisenberg C-P, Jiang Y-J, Kane DA, Kelsh RN, Mullins MC, Nüsslein-Volhard C. Mutations affecting the cardiovascular system and other internal organs in zebrafish. Development. 1996;123:293–302. doi: 10.1242/dev.123.1.293. [DOI] [PubMed] [Google Scholar]
- Chen JW, Murphy TL, Willingham MC, Pastan I, August JT. Identification of two lysosomal membrane glycoproteins. J Cell Biol. 1985;101:85–95. doi: 10.1083/jcb.101.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon RA, Reaume AG, Rossant J. Notch1 is required for the coordinate segmentation of somites. Development. 1995;121:1533–1545. doi: 10.1242/dev.121.5.1533. [DOI] [PubMed] [Google Scholar]
- Damjanov I, Damjanov A, Solter D. Production of teratocarcinomas from embryos transplanted to extra-uterine sites. In: Robertson EJ, editor. Teratocarcinomas and embryonic stem cells: A practical approach. Washington, D.C.: Oxford; 1987. pp. 1–18. [Google Scholar]
- DeRuiter MC, Poelmann RE, VanderPlas-de Vries I, Mentink MMT, Gittenberger-de Groot AC. The development of the myocardium and endocardium in mouse embryos. Anal Embryol. 1992;185:461–473. doi: 10.1007/BF00174084. [DOI] [PubMed] [Google Scholar]
- Gaullier J-M, Simonsen A, D’Arrigo A, Bremnes B, Stenmark H, Aasland R. FYVE fingers bind PtdIns(3)P. Nature. 1998;394:432–433. doi: 10.1038/28767. [DOI] [PubMed] [Google Scholar]
- George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119:1079–1091. doi: 10.1242/dev.119.4.1079. [DOI] [PubMed] [Google Scholar]
- Gruenberg J, Howell KE. Membrane traffic in endocytosis: Insights from cell-free assays. Annu Rev Cell Biol. 1989;5:453–481. doi: 10.1146/annurev.cb.05.110189.002321. [DOI] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the mouse embryo: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- Imamoto A, Soriano P. Disruption of the csk gene, encoding a negative regulator of Src family tyrosine kinases, leads to neural tube defects and embryonic lethality in mice. Cell. 1993;73:1117–1124. doi: 10.1016/0092-8674(93)90641-3. [DOI] [PubMed] [Google Scholar]
- Jat PS, Noble MD, Ataliotis P, Tanaka Y, Yannoutsos N, Larsen L, Kioussis D. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci. 1991;88:5096–5100. doi: 10.1073/pnas.88.12.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Evans T. The Xenopus GATA-4/5/6 genes are associated with cardiac specification and can regulate cardiac-specific transcription during embryogenesis. Dev Biol. 1996;174:258–270. doi: 10.1006/dbio.1996.0071. [DOI] [PubMed] [Google Scholar]
- Kaufman MH, Navaratnam V. Early differentiation of the heart in mouse embryos. J Anat. 1981;133:235–246. [PMC free article] [PubMed] [Google Scholar]
- Komada M, Kitamura N. Growth factor-induced tyrosine phosphorylation of Hrs, a novel 115-kilodalton protein with a structurally conserved putative zinc finger domain. Mol Cell Biol. 1995;15:6213–6221. doi: 10.1128/mcb.15.11.6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komada M, Masaki R, Yamamoto A, Kitamura N. Hrs, a tyrosine kinase substrate with a conserved double zinc finger domain, is localized to the cytoplasmic surface of early endosomes. J Biol Chem. 1997;272:20538–20544. doi: 10.1074/jbc.272.33.20538. [DOI] [PubMed] [Google Scholar]
- Kornfeld S, Mellman I. The biogenesis of lysosomes. Annu Rev Cell Biol. 1989;5:483–525. doi: 10.1146/annurev.cb.05.110189.002411. [DOI] [PubMed] [Google Scholar]
- Kuo CT, Morrisey EE, Anandappa R, Sigrist K, Lu MM, Parmacek MS, Soudais C, Leiden JM. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes & Dev. 1997;11:1048–1060. doi: 10.1101/gad.11.8.1048. [DOI] [PubMed] [Google Scholar]
- Laverriere AC, MacNeill C, Mueller C, Poelmann RE, Burch JBE, Evans T. GATA-4/5/6, a subfamily of three transcription factors transcribed in developing heart and gut. J Biol Chem. 1994;269:23177–23184. [PubMed] [Google Scholar]
- Lu L, Komada M, Kitamura N. Human Hrs, a tyrosine kinase substrate in growth factor-stimulated cells: cDNA cloning and mapping of the gene to chromosome 17. Gene. 1998;213:125–132. doi: 10.1016/s0378-1119(98)00184-x. [DOI] [PubMed] [Google Scholar]
- Malide D, Cushman SW. Morphological effects of wortmannin on the endosomal system and GLUT4-containing compartments in rat adipose cells. J Cell Sci. 1997;110:2795–2806. doi: 10.1242/jcs.110.22.2795. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Lin Q, Duncan SA, Olson EN. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes & Dev. 1997;11:1061–1072. doi: 10.1101/gad.11.8.1061. [DOI] [PubMed] [Google Scholar]
- Morgenstern JP, Land H. Advanced mammalian gene transfer: High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu F-T, Callaghan JM, Steele-Mortimer O, Stenmark H, Parton RG, Campbell PL, McCluskey J, Yeo J-P, Tock EPC, Toh B-H. EEA1, an early endosome-associated protein. J Biol Chem. 1995;270:13503–13511. doi: 10.1074/jbc.270.22.13503. [DOI] [PubMed] [Google Scholar]
- Narita N, Bielinska M, Wilson DB. Wild-type endoderm abrogates the ventral developmental defects associated with GATA-4 deficiency in the mouse. Dev Biol. 1997;189:270–274. doi: 10.1006/dbio.1997.8684. [DOI] [PubMed] [Google Scholar]
- Nascone N, Mercola M. Endoderm and cardiogenesis: New insights. Trends Card Med. 1996;6:211–216. doi: 10.1016/S1050-1738(96)00086-2. [DOI] [PubMed] [Google Scholar]
- Pasteris NG, Cadle A, Logie LJ, Porteous MEM, Schwartz CE, Stevenson RE, Glover TW, Wilroy RS, Gorski JL. Isolation and characterization of the faciogenital dysplasia (Aarskog-Scott syndrome) gene: A putative rho/rac guanine nucleotide exchange factor. Cell. 1994;79:669–678. doi: 10.1016/0092-8674(94)90552-5. [DOI] [PubMed] [Google Scholar]
- Patki V, Virbasius J, Lane WS, Toh B-H, Shpetner HS, Corvera S. Identification of an early endosomal protein regulated by phosphatidylinositol 3-kinase. Proc Natl Acad Sci. 1997;94:7326–7330. doi: 10.1073/pnas.94.14.7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patki V, Lawe DC, Corvera S, Virbasius JV, Chawla A. A functional PtdIns(3)P-binding motif. Nature. 1998;394:433–434. doi: 10.1038/28771. [DOI] [PubMed] [Google Scholar]
- Piper RC, Cooper AA, Yang H, Stevens TH. VPS27 controls vacuolar and endocytic traffic through a prevacuolar compartment in Saccharomyces cerevisiae. J Cell Biol. 1995;131:603–617. doi: 10.1083/jcb.131.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roebroek AJM, Umans L, Pauli IGL, Robertson EJ, van Leuven F, Van de Ven WJM, Constam DB. Failure of ventral closure and axial rotation in embryos lacking the proprotein convertase Furin. Development. 1998;125:4863–4876. doi: 10.1242/dev.125.24.4863. [DOI] [PubMed] [Google Scholar]
- Shepherd PR, Reaves BJ, Davidson HW. Phosphoinositide 3-kinases and membrane traffic. Trends Cell Biol. 1996;6:92–97. doi: 10.1016/0962-8924(96)80998-6. [DOI] [PubMed] [Google Scholar]
- Simonsen A, Lippé R, Christoforidis S, Gaullier J-M, Brech A, Callaghan J, Toh B-H, Murphy C, Zerial M, Stenmark H. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 1998;394:494–498. doi: 10.1038/28879. [DOI] [PubMed] [Google Scholar]
- Snow MHL. Gastrulation in the mouse: Growth and regionalization of the epiblast. J Embryol Exp Morphol. 1977;42:293–303. [Google Scholar]
- Soriano P. The PDGFα receptor is required for neural crest cell development and for normal patterning of the somites. Development. 1997;124:2691–2700. doi: 10.1242/dev.124.14.2691. [DOI] [PubMed] [Google Scholar]
- Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- Spiro DJ, Boll W, Kirchhausen T, Wessling-Resnick M. Wortmannin alters the transferrin receptor endocytic pathway in vivo and in vitro. Mol Biol Cell. 1996;7:355–367. doi: 10.1091/mbc.7.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stainier DYR, Fouquet B, Chen J-N, Warren KS, Weinstein BM, Meiler SE, Mohideen M-APK, Neuhauss SCF, Solnica-Krezel L, Schier AF, Zwartkruis F, Stemple DL, Malicki J, Driever W, Fishman MC. Mutations affecting the formation and function of the cardiovascular system in the zebrafish embryo. Development. 1996;123:285–292. doi: 10.1242/dev.123.1.285. [DOI] [PubMed] [Google Scholar]
- Stalsberg H, DeHaan RL. Endodermal movements during foregut formation in the chick embryo. Dev Biol. 1968;18:198–215. doi: 10.1016/0012-1606(68)90043-2. [DOI] [PubMed] [Google Scholar]
- Stenmark H, Parton RG, Steele-Mortimer O, Lütcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994;13:1287–1296. doi: 10.1002/j.1460-2075.1994.tb06381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H, Aasland R, Toh B-H, D’Arrigo A. Endosormal localization of the autoantigen EEA1 is mediated by a zinc-binding FYVE finger. J Biol Chem. 1996;271:24048–24054. doi: 10.1074/jbc.271.39.24048. [DOI] [PubMed] [Google Scholar]
- Takeshita T, Arita T, Asao H, Tanaka N, Higuchi M, Kuroda H, Kaneko K, Munakata H, Endo Y, Fujita T, Sugamura K. Cloning of a novel signal-transducing adaptor molecule containing an SH3 domain and ITAM. Biochem Biophys Res Commun. 1996;225:1035–1039. doi: 10.1006/bbrc.1996.1290. [DOI] [PubMed] [Google Scholar]
- Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL. SARA, a FYVE domain protein that recruits Smad2 to the TGFβ receptor. Cell. 1998;95:779–791. doi: 10.1016/s0092-8674(00)81701-8. [DOI] [PubMed] [Google Scholar]
- Webb GC, Zhang J, Garlow SJ, Wesp A, Riezman H, Jones EW. Pep7p provides a novel protein that functions in vesicle-mediated transport between the yeast Golgi and endosome. Mol Biol Cell. 1997;8:871–895. doi: 10.1091/mbc.8.5.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisman LS, Wickner W. Molecular characterization of VAC1, a gene required for vacuole inheritance and vacuole protein sorting. J Biol Chem. 1992;267:618–623. [PubMed] [Google Scholar]
- Yamamoto A, DeWald DB, Boronenkov IV, Anderson RA, Emr SD, Koshland D. Novel PI(4)P 5-kinase homologue, Fab1p, essential for normal vacuole function and morphology in yeast. Mol Biol Cell. 1995;6:525–539. doi: 10.1091/mbc.6.5.525. [DOI] [PMC free article] [PubMed] [Google Scholar]