Interleukin-34 Selectively Enhances the Neuroprotective Effects of Microglia to Attenuate Oligomeric Amyloid-β Neurotoxicity (original) (raw)
Abstract
Microglia, macrophage-like resident immune cells in the brain, possess both neurotoxic and neuroprotective properties and have a critical role in the development of Alzheimer's disease (AD). We examined the function of Interleukin-34 (IL-34), a newly discovered cytokine, on microglia because it reportedly induces proliferation of monocytes and macrophages. We observed that the neuronal cells primarily produce IL-34 and that microglia express its receptor, colony-stimulating factor 1 receptor. IL-34 promoted microglial proliferation and clearance of soluble oligomeric amyloid-β (oAβ), which mediates synaptic dysfunction and neuronal damage in AD. IL-34 increased the expression of insulin-degrading enzyme, aiding the clearance of oAβ, and induced the antioxidant enzyme heme oxygenase-1 in microglia to reduce oxidative stress, without producing neurotoxic molecules. Consequently, microglia treated with IL-34 attenuated oAβ neurotoxicity in primary neuron-microglia co-cultures. In vivo, intracerebroventricular administration of IL-34 ameliorated impairment of associative learning and reduced oAβ levels through up-regulation of insulin-degrading enzyme and heme oxygenase-1 in an APP/PS1 transgenic mouse model of AD. These findings support the idea that enhancement of the neuroprotective property of microglia by IL-34 may be an effective approach against oAβ neurotoxicity in AD.
Amyloid-β peptide (Aβ) is a key molecule in the pathogenesis of Alzheimer's disease (AD), and fibrillar Aβ is a major constituent of the senile plaques in AD. Recent studies have demonstrated that, compared with fibrillar Aβ, soluble oligomeric Aβ (oAβ) exhibits greater neurotoxicity. Indeed, oAβ inhibits hippocampal long-term potentiation, facilitates long-term depression, and disrupts synaptic plasticity.1,2 In addition, oAβ induces neuronal reactive oxygen species (ROS) by means of a mechanism that requires NMDA (_N_-methyl-d-aspartate) receptor activation.3
Microglia, macrophage-like cells in the central nervous system, have a biphasic neurotoxic-neuroprotective role in the pathogenesis of AD. Insofar as its neurotoxic properties, microglia may be involved in the inflammatory component of AD.4 Both oAβ and fibrillar Aβ stimulate microglial secretion of proinflammatory cytokines, chemokines, complement components, and free radicals.5 Insofar as its neuroprotective properties, microglia produce neurotrophic factors and phagocytose and degrade Aβ.6–8 We have previously demonstrated that microglia activated with toll-like receptor (TLR) 9 ligand CpG attenuated oAβ neurotoxicity.9 CpG enhanced microglial phagocytosis of oAβ and induced higher levels of the antioxidant enzyme heme oxygenase-1 (HO-1) in microglia without inducing neurotoxic molecules.
The novel cytokine Interleukin-34 (IL-34) is broadly expressed in various organs including heart, brain, lung, liver, kidney, spleen, and colon.10 IL-34 stimulates proliferation of monocytes and macrophages through the colony-stimulating factor 1 receptor (CSF1R), which is also shared by colony-stimulating factor 1. However, the function of IL-34 in the central nervous system and its producing cells remains uncertain. Herein, we demonstrate that IL-34, primarily produced by neurons, promotes microglial proliferation and that microglia treated with IL-34 attenuate the neurotoxic effects of oAβ1-42 in vitro. Moreover, intracerebroventricular (ICV) administration of IL-34 ameliorates the impairment of associative learning in an APP/PS1 transgenic mouse model of AD.
Materials and Methods
Animals
All protocols for the animal experiments were approved by the Animal Experiment Committee of Nagoya University. APP/PS1 transgenic mice expressing mutant variants of human APP and PS1 [B6C3-Tg(APP695)3Dbo Tg(PSEN1)5Dbo/J] were purchased from Jackson Laboratory (Bar Harbor, ME). APP/PS1 transgenic mice used had been backcrossed with C57BL/6J mice for more than 10 generations.
Cell Culture
Primary neuronal cultures were prepared from the cortices of embryonic day 17 C57BL/6 mouse embryos as described previously.9 In brief, cortical fragments were dissociated into single cells in dissociation solution and resuspended in nerve culture medium (both from Sumitomo Bakelite Co., Ltd., Akita, Japan). Neurons were plated onto 12-mm polyethyleneimine-coated glass coverslips (Asahi Techno Glass Corp., Chiba, Japan) at a density of 5 × 104 cells per well in 24-well multidishes and incubated at 37°C in a humidified atmosphere containing 5% CO2. The purity of the cultures was more than 95% as determined by NeuN-specific immunostaining. Using the “shaking off” method described previously,11 microglia were isolated on day 14 in vitro from primary mixed glial cell cultures prepared from newborn C57BL/6 mice. Cultures were 97% to 100% pure, as determined at Fc receptor–specific immunostaining, and were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 5 μg/mL bovine insulin, and 0.2% glucose. Microglia were plated at a density of 7 × 104 cells per well in 8-well glass slides or at a density of 7 × 104 cells per well in 96-well multidishes. Neuron-microglia co-cultures were prepared as follows: 7 × 104 microglia in 50 μL neuronal medium were added to neuronal cultures (5 × 104 neuronal cells) on day 13 in vitro in 24-well multidishes.
Preparation of Aβ Solutions
oAβ1-42 was prepared as described previously.12 In brief, Aβ1-42 (Peptide Institute, Inc., Osaka, Japan) was dissolved in 100% HFIP (1,1,1,3,3,3-hexafluoro-2-propanol) to make a 1-mmol/L solution. HFIP was dried in a vacuum desiccator and resuspended to a concentration of 5 mmol/L in dimethyl sulfoxide. To form oligomers, amyloid peptide was diluted to a final concentration of 100 μmol/L in Ham's F-12 medium and incubated at 4°C for 24 hours, then immediately added to cultures at a final concentration of 5 μmol/L.
Real-Time RT-PCR
Total RNA was extracted from microglia, neurons, and astrocytes using an RNeasy Mini Kit (Qiagen KK, Tokyo, Japan). cDNA synthesis was performed using SuperScript II (Invitrogen Corp., Carlsbad, CA). Real-time PCR of the gene transcripts of mouse IL-34, CSF1R, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was performed using a Rotor-Gene Q (Quiagen, Inc., Valencia, CA) with a Power SYBR Green PCR Master Mix (Applied Biosystems, Inc., Foster City, CA). The following primers were used: for IL-34, forward 5′-CTTTGGGAAACGAGAATTTGGAGA-3′ and reverse 5′-GCAATCCTGTAGTTGATGGGGAAG-3′; for CSF1R, forward 5′-GCAGTACCACCATCCACTTGTA-3′ and reverse 5′-GTGAGACACTGTCCTTCAGTGC-3′; and for GAPDH, forward 5′-TGTGTCCGTCGTGGATCTGA-3′ and reverse 5′-CCTGCTTCACCACCTTCTTGA-3′.
BrdU Proliferation Assay
To assess the proliferation of microglia by IL-34, we used a BrdU (5′-bromo-2′-deoxyuridine) proliferation assay kit (Calbiochem, San Diego, CA). Microglia were plated at a density of 7 × 104 cells per well (200 μL) in 96-well multidishes and treated with 1, 10, 25, 50, or 100 ng/mL IL-34 (R&D Systems, Inc., Minneapolis, MN) or 100 ng/mL IL-34 plus 1 μmol/L c-Fms/CSF1R tyrosine kinase inhibitor GW2580 (Calbiochem). After 24 hours of incubation, BrdU was added, and cells were incubated for an additional 24 hours. The incorporated BrdU microglia were fixed, and DNA was denatured, after which BrdU was detected using anti-BrdU monoclonal antibody. Absorbance was measured using a spectrophotometric plate reader at dual wavelengths of 450 to 540 nm.
Measurement of HO-1, Matrix Metalloproteinase-9, Tumor Necrosis Factor-α, NO, and Glutamate
To measure factors produced by microglia treated with IL-34, microglia were plated at a density of 7 × 104 cells per well (200 μL) in 96-well multidishes and treated with 1, 10, 25, 50, or 100 ng/mL IL-34 with or without 5 μmol/L oAβ for 24 hours. Supernatants from microglia were assessed using enzyme-linked immunosorbent assay (ELISA) kits for tumor necrosis factor-α (BD Pharmingen, BD Biosciences, Franklin Lakes, NJ) and matrix metalloproteinase-9 (R&D Systems, Inc.). Cell extracts from microglia in extraction buffer (1% NP40 in PBS) were measured for HO-1 using an ELISA kit (Takara Bio, Inc., Mie, Japan). Measurement of NO was determined using the Griess reaction.13 To measure glutamate, the glutamate assay kit colorimetric assay (Yamasa Corp., Chiba, Japan) was used, as described previously.14
Measurement of Insulin-Degrading Enzyme
To measure the enzyme activity of insulin degrading enzyme (IDE), microglia were plated at a density of 7 × 104 cells per well (200 μL) in 96-well multidishes and treated with 1, 10, 25, 50, or 100 ng/mL IL-34 for 24 hours. Cell extracts from microglia were analyzed using an IDE immunocapture activity assay kit (InnoZyme; Calbiochem).
Measurement of ROS
To measure ROS in neuron-microglia co-cultures, we used the acetate ester form of H2DCFDA-AM (2′,7′-dichlorofluorescein diacetate) probe (Invitrogen Corp.). After neuron-microglia co-cultures were treated with or without 100 ng/mL IL-34 for 3 hours, cells were loaded with dye by replacing the medium with fresh nerve culture medium containing 5 μmol/L H2DCFDA-AM for 30 minutes. After washing, culture medium containing 5 μmol/L oAβ1-42 was added, and the fluorescence of the wells was measured using a Wallac 1420 ARVOMX (PerkinElmer Japan Co., Ltd., Yokohama, Japan).
Immunocytochemistry
Neuronal, microglial, and neuron-microglia co-cultures were fixed using 4% paraformaldehyde for 30 minutes at room temperature, blocked using 5% normal goat serum in PBS, and permeabilized using 0.3% Triton X-100. Neurons were stained using rabbit polyclonal anti–microtubule-associated protein 2 (MAP-2) antibody (1:500; Millipore Corp., Billerica, MA) and secondary antibodies conjugated to Alexa 488 (1:1000; Invitrogen Corp.). Synthetic Aβ was stained using a mouse monoclonal anti-Aβ antibody (4G8) (1:1000; Chemicon International, Inc., Temecula, CA) and secondary antibodies conjugated to Alexa 568 or Alexa 647 (1:1000; Invitrogen Corp.). Mouse IgG was used as a negative control. Microglia were stained using Alexa 488– or Alexa 647–conjugated rat anti-mouse CD11b monoclonal antibody (1:300; Invitrogen Corp.) before fixation. CSF1R was stained using rabbit polyclonal anti-Fms/CSF1R antibody (1:200; Millipore Corp.) and secondary antibodies conjugated to Alexa 488 (1:1000). Images were analyzed using a deconvolution fluorescent microscope system (BZ-8000; Keyence Corp., Osaka, Japan). To assess neuronal death induced by Aβ, purified neurons (5 × 104 cells per well) were plated in 24-well multidishes. oAβ1-42, 5 μmol/L, was added to the cultures on day 13 in vitro for 24 hours. To assess neuronal death in neuron-microglia co-cultures, 3 hours after treatment with or without IL-34, 5 μmol/L oAβ1-42 was added to cultures for 24 hours. Surviving neurons were identified by observing the cytoskeletal structure of neurons, as previously described.8 Viable neurons stained strongly with an anti–MAP-2 antibody, whereas damaged neurons stained more weakly. The number of MAP-2–positive neurons was counted in 10 random fields per well. More than 200 neurons were examined by a scorer blinded to the experimental condition (T.M.). The number of untreated viable neurons was normalized to 100%.
Western Blot Analysis
For detection of IL-34 protein, cell lysates of mouse microglia, neurons, and astrocytes were obtained from primary cultures. Recombinant mouse IL-34 was used as a positive control, and neuronal lysates in which IL-34 was knocked down by IL-34 small-interfering RNA (siRNA) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) were obtained. For detection of oAβ, neuronal cultures were treated with 5 μmol/L oAβ1-42 for 24 hours. Neuron-microglia co-cultures were pretreated with IL-34 for 3 hours before addition of 5 μmol/L oAβ1-42 for 24 hours. The supernatants of these cultures were collected. oAβ in 10-month-old APP/PS1 mouse brain was extracted from the soluble extracellular-enriched fraction as described previously.15 Hemi-forebrains were harvested in 500 μL solution containing 50 mmol/L Tris-HCl (pH 7.6), 0.01% NP-40, 150 mmol/L NaCl, 2 mmol/L EDTA, 0.1% SDS, and protease inhibitor cocktail (Sigma-Aldrich Corp., St. Louis, MO). Soluble extracellular-enriched proteins were collected from mechanically homogenized lysates after centrifugation for 5 minutes at 1000 × g. Collected samples were mixed with sample buffer (200 mmol/L Tris-HCl, 8% SDS, and 1% glycerol). Proteins were separated on a 5% to 20% Tris-glycine SDS-polyacrylamide gel and transferred to a Hybond-P polyvinylidene difluoride membrane (GE Healthcare UK Ltd., Little Chalfont, Buckinghamshire, England). Membranes were blocked using 1% skim milk in Tris-buffered saline solution containing 0.05% Tween 20. Blots were incubated in sheep anti-mouse IL-34 antibody (1:500; R&D Systems, Inc.) or mouse anti-Aβ monoclonal antibody (6E10) (1:1000; Chemicon International, Inc.) diluted in 1% skim milk overnight at 4°C. Subsequently, membranes were washed in TBS–Tween 20 for 3 × 5 minutes and incubated with a horseradish peroxidase–conjugated anti-sheep IgG (1:50,000; Invitrogen Corp.) or anti-mouse IgG (1:5000; GE Healthcare UK Ltd.) diluted in 1% skim milk for 1 hour. After washing in TBS–Tween 20 for 1 × 15 minutes and 2 × 5 minutes, and Tris-buffered saline solution for 1 × 5 minutes, signals were visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Inc., Rockford, IL). The intensity of the bands was calculated using a CS Analyzer 1.0 (Atto Corp., Tokyo, Japan).
IL-34 and oAβ in mouse brains were also measured using ELISA kits (USCN Life Science, Inc, Wuhan, China, and Immuno-Biological Laboratories Co., Ltd, Takasaki, Japan, respectively).
Cued and Contextual Fear Conditioning Tests in an APP/PS1 Mouse Model of AD
Female mice aged 10 months were used in behavioral experiments. Wild-type (WT) age-matched controls were littermates of the APP/PS1 mice. Mice were anesthetized using 50 mg/kg sodium pentobarbital i.p. before stereotaxic implantation of a microinjection cannula into the right lateral ventricle (anteroposterior −0.3 mm, mediolateral 1.0 mm from the bregma, and dorsoventral 2.5 mm from the skull) according to the method of Franklin and Paxinos.16 IL-34 was dissolved in PBS at a concentration of 3 μg/mL and injected in a volume of 3 μL for 3 minutes. The same volume of PBS was injected into the vehicle mice. One week after injection, behavioral testing was begun.
Cued and contextual fear conditioning tests were performed using a previously reported method,17 with minor modifications. For measuring basal levels of freezing response (preconditioning phase), mice were individually placed in a neutral cage (Plexiglas box with abundant wood chips, 30 cm wide × 30 cm long × 40 cm high) for 1 minute, then in the conditioning cage (transparent Plexiglas box, 30 × 30 × 40 cm) for 2 minutes. For training (conditioning phase), mice were placed in the conditioning cage, and a 15-second tone (80 dB) was delivered as a conditioned stimulus. During the last 5 seconds of the tone stimulus, a 0.6-mA shock to the foot was delivered as an unconditioned stimulus via a shock generator (Neuroscience Idea Co., Ltd., Osaka, Japan). This procedure was repeated four times, at 15-second intervals. Cued and contextual tests were performed at 1 day after fear conditioning. For the contextual test, mice were placed in the conditioning cage, and their freezing response was measured for 2 minutes in the absence of the conditioned stimulus. For the cued test, the freezing response was measured in the neutral cage for 1 minute in the presence of a continuous-tone stimulus identical to the conditioned stimulus.
Immunohistochemistry
Immunohistochemistry was performed on mouse brain tissue after the cued and contextual fear conditioning tests. Under deep anesthesia, mice were transcardially perfused using ice-cold borate-buffered 4% paraformaldehyde. After decapitation, their brains were rapidly removed. Brains were then postfixed overnight in periodate lysine paraformaldehyde, equilibrated in phosphate-buffered 20% sucrose for 48 hours, embedded in Tissue Tek O.C.T. compound (Sakura Finetechnical Co., Ltd., Tokyo, Japan), and frozen at −80°C overnight. Coronal brain sections (20 μm) were cut using a cryostat. The sections were permeabilized using 1% Triton X-100 after blocking with 10% normal goat serum for 30 minutes. The cell nucleus was stained using 1 μg/mL Hoechst 33342 (Invitrogen Corp.). Aβ was stained using a mouse monoclonal anti-Aβ antibody (4G8) (1:1000, Chemicon International, Inc.) and secondary antibodies conjugated to Alexa 488. Microglia were stained using a rat anti-mouse CD11b monoclonal antibody (1:1000; AbD Serotec Ltd., Oxford, UK) and secondary antibodies conjugated to Alexa 568. Similarly, IL-34 was stained using rabbit polyclonal anti–IL-34 antibody (1:500; ProSci, Inc., Poway, CA) and secondary antibodies conjugated to Alexa 488. Specificity of anti–IL-34 antibody has been validated previously.18 Neurons were stained using rabbit polyclonal anti–MAP-2 antibody (1:500) and secondary antibodies conjugated to Alexa 568. Images were collected and analyzed using a deconvolution fluorescent microscope system. Aβ load in immunostained tissue sections was quantified using a BZ-Analyzer (Keyence Corp.) as reported previously.19 Seven sections per animal were analyzed. The total Aβ burden was quantified for the hippocampus in coronal plane sections stained using the monoclonal antibody 4G8. Test areas (600 × 400 μm) were randomly selected, and the total Aβ burden was calculated as a percentage of the test area occupied by Aβ. The microglia load was also quantified for near plaques and in non–plaque-containing areas in the hippocampus of vehicle- and IL-34-treated APP/PS1 transgenic mice. Microglia were stained using a rat anti-mouse CD11b monoclonal antibody and secondary antibodies conjugated to Alexa 568. Test areas (450 × 300 μm) were randomly selected, and the total microglia burden was calculated as a percentage of the test area occupied by microglia.
Statistical Analysis
The statistical significance of the biochemical experiments and the behavioral data were assessed using the Student's _t_-test or one-way analysis of variance followed by Tukey's post hoc test using commercially available software (PRISM version 5.0; GraphPad Software, Inc., San Diego, CA).
Results
IL-34 Is Produced by Neuronal Cells and Promotes Microglial Proliferation
IL-34-producing cells and their target effector cells in the central nervous system were investigated. In primary neuron, microglia, and astrocyte cultures, IL-34 mRNA was expressed primarily in neurons and astrocytes but not in microglia, as determined using real-time RT-PCR (Figure 1A). However, using Western blog analysis, IL-34 protein was detected primarily in neurons (Figure 1B). Expression of IL-34 in neurons was decreased by siRNA knock-down (see Supplemental Figure S1, A and B, at http://ajp.amjpathol.org). mRNA for CSF1R, an IL-34 receptor,10 was expressed in microglia but not in neurons and astrocytes (Figure 1C). Microglial CSF1R expression was confirmed using immunocytochemistry (Figure 1D). Next examined was the effect of IL-34 on the proliferation of microglia. Results from immunocytochemistry and a BrdU proliferation assay revealed that treatment of IL-34 for 48 hours significantly enhanced microglial proliferation in a dose-dependent manner and that addition of 1 μmol/L c-Fms/CSF1R tyrosine kinase inhibitor GW2580 inhibited microglial proliferation by IL-34 (Figure 1, E and F).
Figure 1.
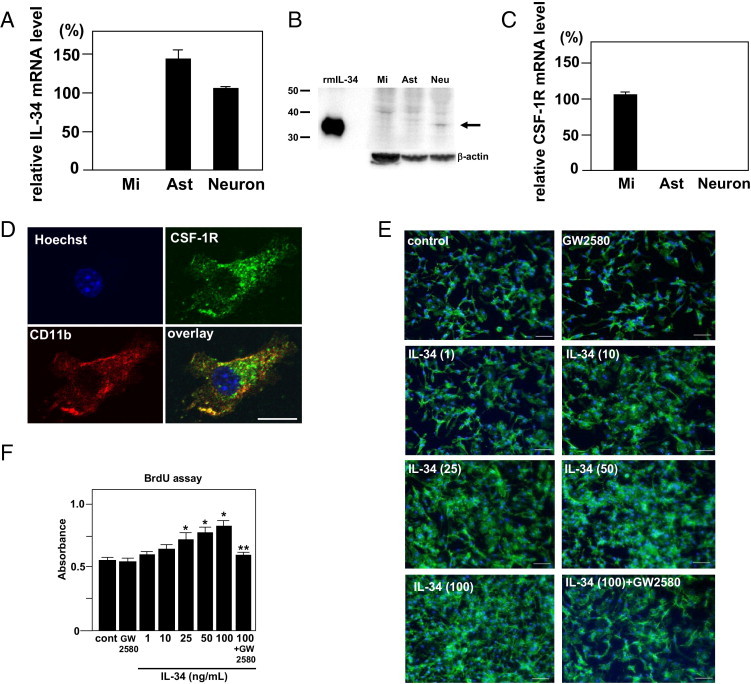
IL-34 is produced by neuronal cells and induces microglial proliferation. A: Expression of IL-34 mRNA in primary neuron, microglia (Mi), and astrocyte (Ast) cultures. GAPDH expression was used as control. B: Detection of IL-34 protein in cell lysates using Western blot analysis. IL-34 (arrow) was detected primarily in nontreated neuron (NT) but not in microglia (Mi) and astrocytes (Ast). The positive control was recombinant mouse IL-34 protein (rm IL-34). C: Expression of CSF1R mRNA in neuron, microglia, and astrocyte cultures. D: Detection of CSF1R protein in microglia at immunocytochemistry. Microglia were stained using rat anti-mouse CD11b monoclonal antibody (red), and the cell nuclei were stained using Hoechst 33342 (blue). CSF1R was stained using rabbit polyclonal anti-Fms/CSF1R antibody (green). Scale bar = 10 μm. E: Proliferative effect on microglia at various doses of IL-34 for 48 hours of treatment. Microglia were stained using Alexa 488–conjugated rat anti-mouse CD11b monoclonal antibody (green), and cell nuclei were stained using Hoechst 33342 (blue). Scale bar = 50 μm. F: The proliferative effect of IL-34 on microglia using a BrdU proliferation assay. *P < 0.05 as compared with untreated microglia; **P < 0.05 as compared with 100 ng/mL IL-34–treated microglia. Each column represents the mean ± SEM (n = 5).
Microglia Treated with IL-34 Attenuate oAβ Neurotoxicity in Primary Neuron-Microglia Co-Cultures
To define the functional roles of IL-34, we evaluated the effects of IL-34 on neuronal survival against the neurotoxicity of oAβ1-42 in neuron-microglia co-cultures and neuron cultures. In unstimulated neuron-microglia co-cultures, neurons stained using anti–MAP-2 antibody exhibited no detectable morphologic abnormalities, and microglia stained using anti-CD11b antibody also appeared normal (Figure 2A). When the co-cultures were stimulated with 5 μmol/L oAβ1-42 for 24 hours, the neuronal cells were severely damaged, and the survival rate decreased to 51% (Figure 2, A and C). Treatment with 10, 25, 50, and 100 ng/mL IL-34 rescued approximately 68%, 71%, 73%, 75%, and 78% of neurons, respectively, from oAβ1-42 toxicity in the co-cultures. The effect was inhibited by 1 μmol/L GW2580. However, the protective effects of IL-34 were not observed in neuron cultures (Figure 2B), which suggests that the protective effects of IL-34 were mediated via microglia.
Figure 2.
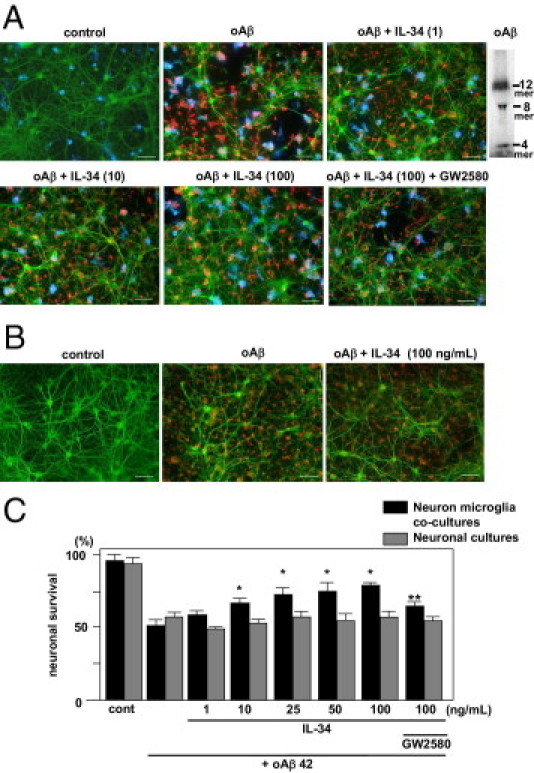
Protective effect of microglia treated with IL-34 against oAβ1-42 neurotoxicity. A: Representative deconvolution fluorescent images of neuron-microglia (1:2) co-cultures. Neurons were stained using anti–MAP-2 antibody (green), Aβ was stained using 4G8 (red), and microglia were stained using Alexa 647–conjugated anti-CD11b antibody (blue). Co-cultures were treated with 5 μmol/L oAβ1-42 or 5 μmol/L oAβ1-42 plus 1, 10, or 100 ng/mL IL-34 with or without GW2580. Scale bar = 50 μm. Upper right panel: Western blot data of oAβ1-42 used in the in vitro experiments. B: Representative deconvolution fluorescent images of neuronal cultures. Neuronal cultures were treated with 5 μmol/L oAβ1-42 or 5 μmol/L oAβ1-42 plus 100 ng/mL IL-34. C: Neuronal survival rate was quantified as the percentage of intact neurons after treatment relative to control wells in neuron-microglia co-cultures (black bars) and in neuronal cultures (gray bars). Viability of untreated neurons (control) was normalized to 100%. *P < 0.05 as compared with the survival rate of neuron-microglia co-cultures treated with oAβ1-42; **P < 0.05 as compared with 100 ng/mL IL-34–treated microglia. Each column represents the mean ± SEM (n = 9).
IL-34–Treated Microglia Increase Clearance of oAβ1-42 via Up-Regulation of Aβ-Degrading Enzyme IDE
To elucidate the mechanisms of neuroprotection by IL-34–treated microglia, we examined whether IL-34 increased microglial clearance of oAβ1-42, which is mediated by degradation and phagocytosis of oAβ.
Western blot analysis revealed that treatment with 100 ng/mL IL-34 decreased the amount of 4-, 8-, and 12-mer oAβ1-42 in neuron-microglia co-cultures, with the most significant decrease in 12-mer oAβ (Figure 3A). Semiquantification of oAβ1-42 was performed via densitometric analysis, and revealed that 12-mer of oAβ decreased significantly (Figure 3B). Next, the enzyme activity of zinc metalloprotease insulin IDE, known as Aβ-degrading enzyme, was analyzed using an InnoZyme IDE immunocapture activity assay kit. Treatment with 100 ng/mL IL-34 significantly enhanced enzyme activity of IDE in microglia. The effect was inhibited by 1 μmol/L GW2580. The addition of 100 μg/mL insulin, the competitive IDE substrate, in neuron-microglia co-cultures treated with oAβ and IL-34 canceled the reduction of oAβ1-42 by IL-34 (Figure 3C). Matrix metalloproteinase-9, another major Aβ-degrading enzyme, was not enhanced in IL-34–treated microglia (Figure 3D).
Figure 3.
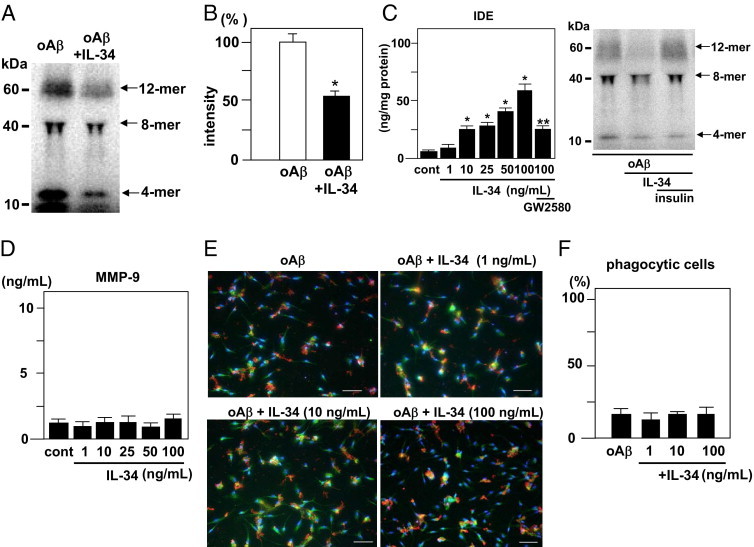
Clearance of oAβ1-42 by microglia treated with IL-34. A: Western blot analysis of oAβ1-42 in neuron-microglia co-cultures. At 24 hours after addition of 5 μmol/L oAβ1-42 or 5 μmol/L oAβ1-42 plus 100 ng/mL IL-34, oAβ1-42 present in the supernatants of these cultures was analyzed using Western blot analysis. B: Semiquantification of oAβ1-42 in A via densitometric analysis. The amount of oAβ1-42 in neuron-microglia co-cultures treated with oAβ1-42 was normalized to 100%. *P < 0.05 as compared with the intensity of oAβ1-42 in neuron-microglia co-cultures treated with oAβ1-42. Each column indicates the mean ± SEM (n = 7). C: The enzyme activity of IDE in microglia treated with IL-34. After 24 hours of treatment with 1, 10, or 100 ng/mL IL-34, the cell lysates of microglial cultures were analyzed using the InnoZyme IDE immunocapture activity assay kit. *P < 0.05 as compared with untreated controls; **P < 0.05 as compared with 100 ng/mL IL-34–treated microglia. Each column indicates the mean ± SEM (n = 6). Western blot analysis of oAβ1-42 in neuron-microglia co-cultures was performed. At 24 hours after addition of 5 μmol/L oAβ1-42 plus 100 ng/mL IL-34 or 5 μmol/L oAβ1-42 or 100 ng/mL IL-34 plus 100 μg/mL insulin, oAβ1-42 present in the supernatants of these cultures was analyzed. D: Production of MMP-9 by microglia treated with IL-34. After 24 hours, treatment with 1, 10, or 100 ng/mL IL-34, the supernatants of microglial cultures were analyzed. E: Immunocytochemical analysis of phagocytosis of oAβ by microglia treated with IL-34. Engulfed Aβ (red) was co-localized with Rab-7 (green) in phagocytic microglia. The cell nucleus of microglia was stained with Hoechst 33342 (blue). F: The ratio of phagocytic microglia to total counted microglia is not dependent on Il-34.
Next examined was the effect of IL-34 on the microglial phagocytosis of oAβ by means of immunostaining for the phagocytic marker Rab7 protein, which correlates with phagosome maturation. Engulfed Aβ was co-localized with Rab-7 in phagocytic microglia. Twenty percent of microglia were phagocytic in the unstimulated condition. IL-34 did not enhance microglial phagocytosis of oAβ1-42 (Figure 3, E and F). Phagocytosed intracellular oAβ was examined using an enzyme immunoassay. There was no significant change (data not shown).
IL-34–Treated Microglia Produce the Antioxidant Enzyme HO-1 and Release Fewer Neurotoxic Molecules
Because oxidative stress is involved in the neurotoxicity of oAβ1-42, we examined whether IL-34–treated microglia exhibited an antioxidant effect. IL-34 induced the antioxidant enzyme HO-1 in microglia in a dose-dependent manner, with a significant increase at 10, 25, 50, and 100 ng/mL (Figure 4A). The effect was inhibited by 1 μmol/L GW2580. ROS production was enhanced by treatment with 5 μmol/L oAβ in the neuron-microglia co-cultures. The addition of 100 ng/mL IL-34 significantly suppressed oAβ-induced ROS production (Figure 4B).
Figure 4.
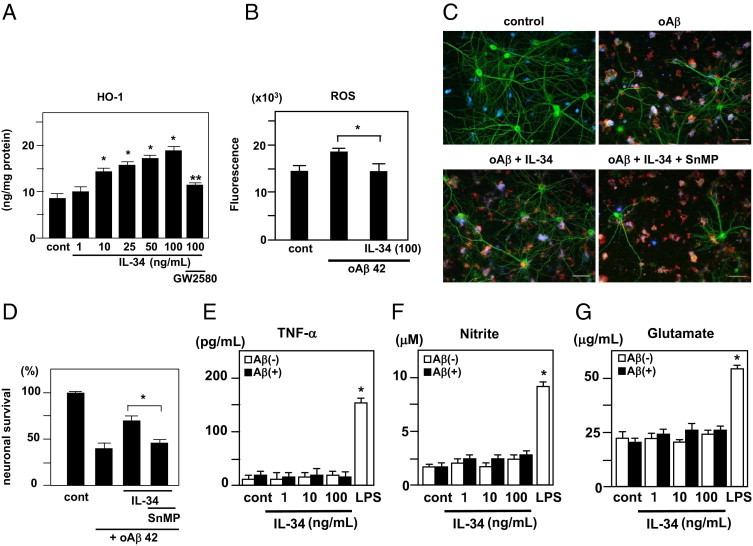
HO-1 production by microglia treated with IL-34. A: After 24 hours of treatment with 1, 10, or 100 ng/mL IL-34, the cell lysates of microglial cultures were analyzed. *P < 0.05 as compared with untreated controls; **P < 0.05 as compared with 100 ng/mL IL-34–treated microglia. Each column indicates the mean ± SEM (n = 5). B: The suppressive effect of IL-34 on oAβ-induced ROS production in the neuron-microglia co-cultures. *P < 0.05 as compared with unstimulated controls. C: Contribution of HO-1 to the neuroprotective effect of IL-34–treated microglia. Representative deconvolution fluorescent images of neuron (green)–microglia (blue) co-cultures. Co-cultures were treated with 5 μmol/L oAβ1-42 (red), oAβ plus100 ng/mL IL-34, or oAβ plus IL-34 in the presence of 10 μmol/L SnMP, a specific HO-1 inhibitor. D: Neuronal survival rate was quantified as the percentage of intact neurons after treatment relative to control wells in neuron-microglia co-cultures. *P < 0.05 as compared with co-culture cells treated with oAβ1-42 and IL-34. Each column indicates the mean ± SEM (n = 4). Tumor necrosis factor-α (E), nitrite (F), and glutamate (G) produced by microglia treated with IL-34 were measured. After 24 hours of treatment with 1, 10, or 100 ng/mL IL-34 with or without 5 μmol/L oAβ, supernatants of microglial cultures were analyzed. Treatment with 100 ng/mL Lipopolysaccharide for 24 hours was used as a positive control. *P < 0.05 as compared with untreated microglia. Each column indicates the mean ± SEM (n = 5).
To determine whether HO-1 contributes to the neuroprotective effects of IL-34–activated microglia, we applied the specific HO-1 inhibitor SnMP-9 (tin-mesoporphyrin IX). The neuroprotective effect of IL-34 was abolished by treatment with 10 μmol/L SnMP-9 (Figure 4, C and D). Also investigated was whether IL-34–treated microglia produce neurotoxic molecules such as tumor necrosis factor-α, nitrite, and glutamate. IL-34 did not induce these toxic molecules in microglia with or without oAβ stimulation (Figure 4, E–G).
ICV Injection of IL-34 Ameliorates Impairment of Associative Learning and Reduces oAβ Levels in an APP/PS1 Transgenic Mouse Model of AD
We examined the effect of IL-34 in vivo, specifically, whether ICV administration of IL-34 improves the cognitive function in the APP/PS1 transgenic mouse, a model of AD. The associative learning of mice was examined at age 10 months using the cued and contextual fear conditioning tests. In the contextual learning test, WT mice demonstrated a marked contextual freezing response at 24 hours after fear conditioning. Vehicle (PBS)–injected APP/PS1 transgenic mice exhibited less freezing response in the contextual tests, indicating impairment of associative learning. However, the single IL-34–injected APP/PS1 transgenic mice demonstrated a freezing response that was indistinguishable from that in WT mice (Figure 5A). Thus, IL-34 treatment significantly reversed the contextual freezing response as compared with that in vehicle-injected APP/PS1 transgenic mice (Figure 5A). In the cued (tone) learning test, there was a significant decrease in the freezing response in vehicle-injected APP/PS1 transgenic mice at 24 hours after fear conditioning as compared with that in WT mice. Again, injection of IL-34 reversed the cued freezing response (Figure 5B). No alterations in nociceptive response were observed in any of the mutant mice; there was no difference in the minimal current required to elicit flinching, running, jumping, or vocalization in the mice. Moreover, the effect of continuous intraventricular infusion of IL-34 using a micro-osmotic pump was examined.20 The results demonstrated similar effects of a single ICV injection of IL-34 (see Supplemental Figure S2 at http://ajp.amjpathol.org).
Figure 5.
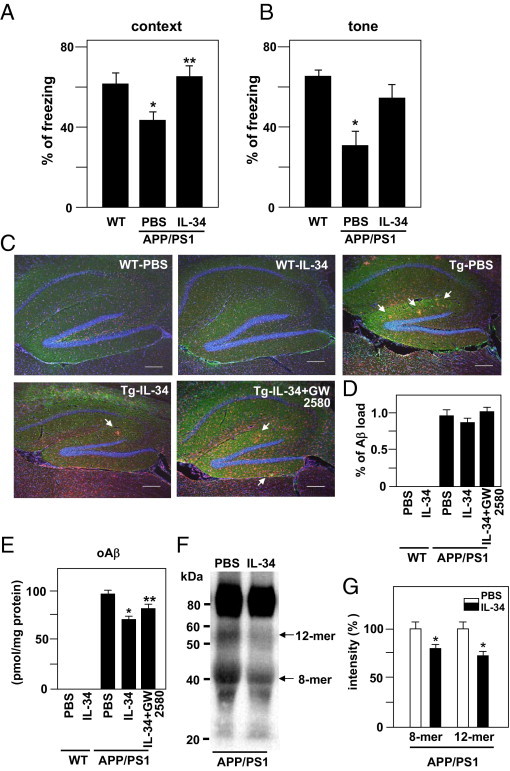
Effect of a single ICV injection of IL-34 on associative learning in the cued and contextual conditioning tests and Aβ clearance in APP/PS1 transgenic mice. The retention session was performed at 24 hours after the training. Context-dependent (A) and tone-dependent (B) freezing times were measured, and the percentage of freezing behavior was calculated. Each column indicates the mean ± SEM [WT mice, n = 10; vehicle (PBS)–treated APP/PS1 transgenic mice, n = 11; IL-34 (10 ng/3 μL)–treated APP/PS1 transgenic mice, n = 14]. *P < 0.05 as compared with WT mice; **P < 0.05 as compared with vehicle-treated APP/PS1 transgenic mice. C: Aβ deposits in the hippocampus. Aβ was stained using 4G8 (green), microglia were stained using anti-CD11b antibody (red), and cell nuclei were stained using Hoechst 33342 (blue). Arrows indicate Aβ deposits surrounded by microglia. Scale bar = 200 μm. D: Quantification of Aβ deposits in C. The ratio between the Aβ deposit area and the total area of the analyzed region was multiplied by 100. Each column indicates the mean ± SEM (n = 8). E: Measurement of oAβ via ELISA. Samples were extracted from the hemi-forebrains of WT and APP/PS1 transgenic mice. *P < 0.05 as compared with vehicle-treated APP/PS1 transgenic mice; **P < 0.05 as compared with IL-34-treated APP/PS1 transgenic mice. F: Western blot analysis of oAβ extracted from the hemi-forebrains of APP/PS1 transgenic mice. 8-mer and 12-mer oAβ was detected in vehicle-treated APP/PS1 transgenic mice, and was decreased in IL-34–injected transgenic mice. G: Semiquantification of 8-mer and 12-mer oAβ using densitometric analysis. The amount of 8-mer and 12-mer oAβ1-42 in vehicle-treated APP/PS1 transgenic mice was normalized to 100%. *P < 0.05 as compared with vehicle-treated APP/PS1 transgenic mice. Each column indicates the mean ± SEM (n = 8).
We then examined whether IL-34 decreased Aβ deposits in the hippocampus of APP/PS1 mice. Aβ deposits and clustered microglia were detected in the hippocampus of vehicle-injected APP/PS1 transgenic mice. ICV injection of IL-34 did not affect the number and size of Aβ deposits (Figure 5, C and D). However, measurement of oAβ using an ELISA revealed that IL-34 treatment decreased oAβ in the hemi-forebrains of APP/PS1 transgenic mice and that 1 μmol/L GW2580 inhibited the effect (Figure 5E). Next, 12-mer oAβ, which correlates with memory dysfunction in the mouse model of AD,15 and 8-mer in the soluble extracellular-enriched fractions of the hemi-forebrains of mice were analyzed using Western blot analysis. Compared with vehicle-injected APP/PSI transgenic mice, IL-34–injected mice demonstrated a significant decrease in 8-mer and 12-mer oAβ (Figure 5, F and G).
ICV Injection of IL-34 Enhances Microglial Accumulation and Production of HO-1 and IDE
Microglia cell numbers near plaques and in non–plaque-containing areas of vehicle- and IL-34–treated APP/PS1 transgenic mice were evaluated as microglia load. Microglia accumulated around the plaques in both vehicle- and IL-34–treated mice. However, microglia load was significantly increased near plaques and in non–plaque-containing areas in IL-34–treated mice compared with vehicle-treated mice (Figure 6,A and B). While IDE and HO-1 were decreased in vehicle-injected APP/PS1 transgenic mice as compared with WT mice, ICV injection of IL-34 significantly increased IDE and HO-1 production (Figure 6, C and D). Intrinsic IL-34 was detected in neuron cells stained using an anti–MAP-2 antibody in WT mice at immunocytochemistry (Figure 6E). IL-34 was measured in vehicle-treated WT and APP/PS1 transgenic mice using an ELISA. Intrinsic IL-34 was decreased in APP/PS1 transgenic mice as compared with WT mice (Figure 6F).
Figure 6.
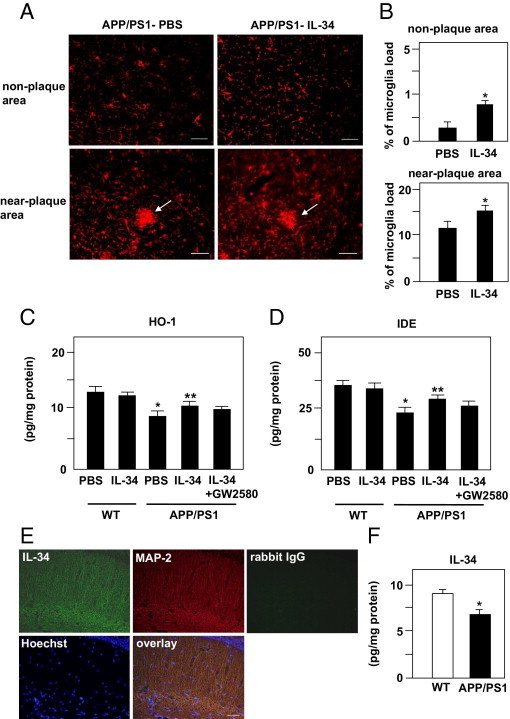
Effect of a single ICV injection of IL-34 on microglial accumulation and induction of HO-1 and IDE. A: Numbers of microglia near plaques and in non–plaque containing areas were evaluated in vehicle- or IL-34–treated APP/PS1 transgenic mice. Arrows indicate microglia accumulation around Aβ plaques. Scale bar = 50 μm. B: Quantification of microglia in A. *P < 0.05 as compared with vehicle-treated APP/PS1 transgenic mice. Each column indicates the mean ± SEM (n = 6). Production of HO-1 (C) and IDE (D) in brains of WT and APP/PS1 transgenic mice treated with or without IL-34. *P < 0.05 as compared with vehicle-treated WT mice; **P < 0.05 as compared with vehicle-treated APP/PS1 transgenic mice. Each column indicates the mean ± SEM (n = 8). E: Detection of IL-34 protein in neuronal cells of WT mice at immunocytochemistry. IL-34 was stained using rabbit anti-mouse IL-34 antibody (green). Neurons were stained using an anti–MAP-2 antibody (red), and cell nuclei were stained using Hoechst 33342 (blue). Rabbit IgG was used as a negative control. Scale bar = 20 μm. F: Measurement of intrinsic IL-34 via ELISA was performed in vehicle-treated WT and APP/PS-1 transgenic mice. **P < 0.05 as compared with vehicle-treated WT mice. Each column indicates the mean ± SEM (n = 5).
Discussion
The dimeric glycoprotein IL-34, discovered by means of functional screening of an extracellular proteome, is broadly expressed in various organs including the brain.10 We show herein for the first time that neurons produce IL-34. The major function of IL-34 is to stimulate the differentiation and proliferation of monocytes and macrophages via CSF1R. The study was focused on the monocytic lineage of microglia as target cells of IL-34 in the central nervous system. As expected, IL-34 enhanced proliferation of microglia that expressed CSF1R. Furthermore, neuronal cells primarily produced IL-34. Recent reports have suggested that neurons are not merely passive targets of microglia; rather, they control microglial activity by means of various signals including cytokines and chemokines.21 IL-34 may also be a neuronal cytokine that regulates microglial function.
AD begins with subtle alterations of hippocampal synaptic efficacy before obvious neuronal degeneration. The synaptic dysfunction is caused by diffusible oAβ.22 Although the precise molecular mechanisms of how oAβ disturbs neuronal function are unknown, calcium dysregulation, membrane disruption,23 and oxidative stress mediated by an NMDA receptor24 are involved in the pathogenesis of oAβ. Thus, decreasing or preventing formation of oAβ may be a potential therapeutic strategy against AD.
The present study revealed a novel function of IL-34, namely, microglial neuroprotection against oAβ toxicity. The neuroprotective effect of IL-34 was mediated via microglial clearance of oAβ and antioxidant functions. Western blot analysis revealed that microglia treated with IL-34 reduced the amount of oAβ present in the supernatant of neuron-microglia co-cultures. Clearance of oAβ is mediated primarily via degradation and phagocytosis. It was observed that IL-34 induced Aβ degrading enzyme IDE in microglia. The reduction of oAβ was inhibited by insulin, a competitive IDE substrate. Therefore, IDE induced by IL-34 may degrade oAβ. IDE activity is critical in determining the level of Aβ. Hippocampal IDE protein and activity are reduced in AD.25 The enhanced IDE activity in IDE and APP double-transgenic mice decreased Aβ levels and prevented formation of AD pathologic features.26 Whereas matrix metalloproteinase-9 is also a major protease that degrades Aβ, IL-34 did not increase the production of matrix metalloproteinase-9 in microglia. Previous studies have demonstrated that the TLR signaling pathways contribute to phagocytosis of Aβ. Specifically, TLR2,27 TLR4,28 and TLR99 enhance the phagocytic activity of microglia. In the present study, IL-34 did not enhance microglial phagocytosis of oAβ. However, it is possible that IL-34-treated microglia take up oAβ through fluid-phase macropinocytosis as reported.29
In addition, we observed that IL-34 is a potent inducer of the antioxidant enzyme HO-1 and that IL-34 suppressed oAβ-induced ROS. HO-1 oxidatively cleaves heme to produce biliverdin, CO, and iron.30 These end-products provide cellular and tissue protection through anti-inflammatory, anti-apoptotic, or antioxidative effects.31 Because the neuroprotective effect of IL-34 was abolished by treatment with the HO-1 inhibitor SnMP, up-regulation of HO-1 in microglia by treatment with IL-34 may lead to neuroprotection against oAβ toxicity through suppression of ROS. Moreover, less induction of neurotoxic molecules such as tumor necrosis factor-α, NO, and glutamate in microglia may also contribute to neuroprotection by IL-34.
The effect of IL-34 on oAβ1-42 neurotoxicity in vivo was examined. Impairment of associative learning in an APP/PS1 transgenic mouse model of AD was effectively suppressed by a single ICV injection of IL-34. At immunohistochemical analysis, intrinsic IL-34 was decreased in APP/PS1 transgenic mice. ICV injection of IL-34 increased the microglial load and production of HO-1 and IDE. Although Aβ deposits were not affected, ICV injection of IL-34 significantly decreased 8-mer and 12-mer oAβ in the soluble extracellular-enriched fractions of the hemi-forebrains of APP/PS1 transgenic mice. It has been reported that 12-mer oAβ impairs memory independent of plaques or neuronal loss and, furthermore, contributes to the cognitive deficits associated with AD.15 Aβ plaques are reported to form extraordinarily quickly, over 24 hours. Within 1 to 2 days of the appearance of a new plaque, microglia are activated and recruited to the site.32 Therefore, a single ICV injection of IL-34 may act directly on microglia, and microglia can rapidly eliminate oAβ by means of up-regulation of IDE and exert an antioxidant effect via HO-1.
CSF1, another ligand of CSF1R, is a macrophage-colony stimulating factor (M-CSF). M-CSF enables acidification of their lysosomes and, subsequently, degradation of internalized Aβ.33 Intraperitoneal injection of M-CSF prevented memory disturbance in APP/PS1 mice. Senile plaques were smaller, and microglia phagocytosed Aβ in the brain of mice treated with M-CSF.34 These data indicate that the mechanism of neuroprotection by IL-34 is different from that of M-CSF. Recent reports have demonstrated that IL-34 and M-CSF differ in their structure and the CSF1R domains that they bind, which causes different bioactivities and signal activation kinetics.35 IL-34 induces stronger but transient tyrosine phosphorylation of CSF1R and downstream molecules. The different spatiotemporal expression of IL-34 and CSF1 enables complementary activation of CSF1R in developing and adult tissues.36
Considered together, observations from the present study demonstrate that IL-34 drives microglia to a neuroprotective state. Enhancement of microglial neuroprotective properties is a useful therapeutic strategy in oAβ1-42 neurotoxicity in AD.
Footnotes
Supported in part by the Global Center of Excellence program “Integrated Functional Molecular Medicine for Neuronal and Neoplastic Disorders” funded by the Ministry of Education, Culture, Sports, Science and Technology of Japan and by the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NIBIO).
T.S. and Y.D. contributed equally to this work.
Supplementary data
Supplemental Figure S1
IL-34 protein expression in neurons and effect of siRNA knock-down. Expression of IL-34 in nontreated neurons (NT) was decreased by siRNA knock-down (siRNA). Intensity of IL-34 by siRNA knock-down was significantly decreased (n = 6). *P < 0.05 as compared with NT.
Supplemental Figure S2
Effect of continuous intraventricular infusion of IL-34 on associative learning in the cued and contextual conditioning tests in APP/PS1 transgenic mice. Mice were anesthetized using pentobarbital, and were placed in a stereotaxic apparatus. The infusion cannula (brain infusion kit 1; Alza Corp., Palo Alto, CA) was connected to a micro-osmotic pump (model 1002; total capacity, 90 μL), which was filled with vehicle (PBS) or IL-34 and implanted in the right ventricle [anteroposterior (AP) −0.3, mediolateral (ML) 1.0 from bregma, dorsoventral (DV) −2.5 from the skull, according to the method of Franklin and Paxinos16]. IL-34 was continuously infused into the cerebral ventricle at a dose of 6 ng/6 μL per day. Seven days after the start of infusion, the mice were subjected to the behavioral experiment. The retention session was performed at 24 hours after training. Context-dependent (A) and tone-dependent (B) freezing times were measured, and percentage of freezing behavior was calculated. Each column indicates the mean ± SEM. [WT mice, n = 10; vehicle (PBS)–treated APP/PS1 transgenic mice, n = 8; IL-34–treated APP/PS1 transgenic mice, n = 8]. *P < 0.05 as compared with WT mice; **P < 0.05 as compared with vehicle-treated APP/PS1 transgenic mice.
References
- 1.Walsh D.M., Klyubin I., Fadeeva J.V., Cullen W.K., Anwyl R., Wolfe M.S., Rowan M.J., Selkoe D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 2.Li S., Hong S., Shepardson N.E., Walsh D.M., Shankar G.M., Selkoe D.J. Soluble oligomers of amyloid beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Felice F.G., Velasco P.T., Lambert M.P., Viola K., Fernandez S.J., Ferreira S.T., Klein W.L. Aβ oligomers induce neuronal oxidative stress through an N-methyl-d-aspartate receptor–dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- 4.Meda L., Cassatella M.A., Szendrei G.I., Otvos L., Jr, Baron P., Villalba M., Ferrari D., Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 5.McGeer P.L., McGeer E.G. Inflammation, autotoxicity and Alzheimer disease. Neurobiol Aging. 2001;22:799–809. doi: 10.1016/s0197-4580(01)00289-5. [DOI] [PubMed] [Google Scholar]
- 6.Mizuno T., Kurotani T., Komatsu Y., Kawanokuchi J., Kato H., Mitsuma N., Suzumura A. Neuroprotective role of phosphodiesterase inhibitor Ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology. 2004;46:404–411. doi: 10.1016/j.neuropharm.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 7.Richard L., Filali M., Préfontaine P., Rivest S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta 1–42 and delay the cognitive decline in a mouse model of Alzheimer's disease. J Neurosci. 2008;28:5784–5793. doi: 10.1523/JNEUROSCI.1146-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan P., Hu Xi, Song H., Yin K., Bateman R.J., Cirrito J.R., Xiao Q., Hsu F.F., Turk J.W., Xu J., Hsu C.Y., Holtzman D.M., Lee J.M. Matrix metalloproteinase-9 degrades amyloid-β fibrils in vitro and compact plaques in situ. J Biol Chem. 2006;281:24566–24574. doi: 10.1074/jbc.M602440200. [DOI] [PubMed] [Google Scholar]
- 9.Doi Y., Mizuno T., Maki Y., Jin S., Mizoguchi H., Ikeyama M., Doi M., Michikawa M., Takeuchi H., Suzumura A. Microglia activated with the toll-like receptor 9 ligand CpG attenuate oligomeric amyloid β neurotoxicity in in vitro and in vivo models of Alzheimer's disease. Am J Pathol. 2009;175:2121–2132. doi: 10.2353/ajpath.2009.090418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin H., Lee E., Hestir K., Leo C., Huang M., Bosch E., Halenbeck R., Wu G., Zhou A., Behrens D., Hollenbaugh D., Linnemann T., Qin M., Wong J., Chu K., Doberstein S.K., Williams L.T. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science. 2008;320:807–811. doi: 10.1126/science.1154370. [DOI] [PubMed] [Google Scholar]
- 11.Suzumura A., Mezitis S.G., Gonatas N.K., Silberberg D.H. mHC antigen expression on bulk isolated macrophage-microglia from newborn mouse brain: induction of Ia antigen expression by gamma-interferon. J Neuroimmunol. 1987;15:263–278. doi: 10.1016/0165-5728(87)90121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dahlgren K., Manelli A.M., Stine W.B., Jr, Baker L.K., Krafft G.A., LaDu M.J. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 13.Pollock J.S., Forstermann U., Mitchell J., Warne T.D., Schmidt H.H.H.W., Nakane M., Murad F. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc Natl Acad Sci USA. 1991;88:10480–10484. doi: 10.1073/pnas.88.23.10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takeuchi H., Mizuno T., Zhang G., Wang J., Kawanokuchi J., Kuno R., Suzumura A. Neuritic beading induced by activated microglia is an early feature of neuronal dysfunction toward neuronal death by inhibition of mitochondrial respiration and axonal transport. J Biol Chem. 2005;280:10444–10454. doi: 10.1074/jbc.M413863200. [DOI] [PubMed] [Google Scholar]
- 15.Lesné S., Koh M.T., Kotilinek L., Kayed R., Glabe C.G., Yang A., Gallagher M., Ashe K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 16.Franklin K.B.J., Paxinos G. Academic Press; San Diego, CA: 1997. The Mouse Brain in Stereotaxic Coordinates; pp. 69–89. [Google Scholar]
- 17.Mouri A., Noda Y., Hara H., Mizoguchi H., Tabira T., Nabeshima T. Oral vaccination with a viral vector containing Abeta cDNA attenuates age-related Abeta accumulation and memory deficits without causing inflammation in a mouse Alzheimer model. FASEB J. 2007;21:2135–2148. doi: 10.1096/fj.06-7685com. [DOI] [PubMed] [Google Scholar]
- 18.Baud'huin M., Renault R., Charrier C., Riet A., Moreau A., Brion R., Gouin F., Duplomb L., Heymann D. Interleukin-34 is expressed by giant cell tumours of bone and plays a key role in RANKL-induced osteoclastogenesis. J Pathol. 2010;221:77–86. doi: 10.1002/path.2684. [DOI] [PubMed] [Google Scholar]
- 19.Scholtzova H., Kascsak R.J., Bates K.A., Boutajangout A., Kerr D.J., Meeker H.C., Mehta P.D., Spinner D., Wisniewski T. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer's disease-related pathology. J Neurosci. 2009;29:1846–1854. doi: 10.1523/JNEUROSCI.5715-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mizoguchi H., Yamada K., Mouri A., Niwa M., Mizuno T., Noda Y., Nitta A., Itohara S., Banno Y., Nabeshima T. Role of matrix metalloproteinase (MMP) and tissue inhibitor of MMP (TIMP) in methamphetamine-induced behavioral sensitization and reward: implications for dopamine receptor downregulation and dopamine release. J Neurochem. 2007;102:1548–1560. doi: 10.1111/j.1471-4159.2007.04623.x. [DOI] [PubMed] [Google Scholar]
- 21.Biber K., Neumann H., Inoue K., Boddeke H.W. Neuronal “on” and “off” signals control microglia. Trends Neurosci. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 22.Selkoe D.J. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 23.Demuro A., Mina E., Kayed R., Milton S.C., Parker I., Glabe C. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- 24.Shankar G.M., Bloodgood B.L., Townsend M., Walsh D.M., Selkoe D.J., Sabatini B.L. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor–dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Z., Xiang Z., Haroutunian V., Buxbaum J.D., Stetka B., Pasinetti G.M. Insulin degrading enzyme activity selectively decreases in the hippocampal formation of cases at high risk to develop Alzheimer's disease. Neurobiol Aging. 2007;28:824–830. doi: 10.1016/j.neurobiolaging.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 26.Leissring M.A., Farris W., Chang A.Y., Walsh D.M., Wu X., Sun X., Frosch M.P., Selkoe D.J. Enhanced proteolysis of β-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–1093. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- 27.Chen K., Iribarren P., Hu J., Chen J., Gong W., Cho E.H., Lockett S., Dunlop N.M., Wang J.M. Activation of toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease–associated amyloid beta peptide. J Biol Chem. 2006;281:3651–3659. doi: 10.1074/jbc.M508125200. [DOI] [PubMed] [Google Scholar]
- 28.Tahara K., Kim H.D., Jin J.J., Maxwell J.A., Li L., Fukuchi K. Role of toll-like receptor signalling in Aβ uptake and clearance. Brain. 2006;129:3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mandrekar S., Jiang Q., Lee C.Y., Koenigsknecht-Talboo J., Holtzman D.M., Landreth G.E. Microglia mediate the clearance of soluble Aβ through fluid phase macropinocytosis. J Neurosci. 2009;29:4252–4262. doi: 10.1523/JNEUROSCI.5572-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maines M.D. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 31.Morse D., Lin L., Choi A.M., Ryter S.W. Heme oxygenase-1, a critical arbitrator of cell death pathways in lung injury and disease. Free Radic Biol Med. 2009;47:1–12. doi: 10.1016/j.freeradbiomed.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyer-Luehmann M., Spires-Jones T.L., Prada C., Garcia-Alloza M., de Calignon A., Rozkalne A., Koenigsknecht-Talboo J., Holtzman D.M., Bacskai B.J., Hyman B.T. Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer's disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Majumdar A., Cruz D., Asamoah N., Buxbaum A., Sohar I., Lobel P., Maxfield F.R. Activation of microglia acidifies lysosomes and leads to degradation of Alzheimer amyloid fibrils. Mol Biol Cell. 2007;18:1490–1496. doi: 10.1091/mbc.E06-10-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boissonneault V., Filali M., Lessard M., Relton J., Wong G., Rivest S. Powerful beneficial effects of macrophage colony-stimulating factor on beta-amyloid deposition and cognitive impairment in Alzheimer's disease. Brain. 2009;132:1078–1092. doi: 10.1093/brain/awn331. [DOI] [PubMed] [Google Scholar]
- 35.Chihara T., Suzu S., Hassan R., Chutiwitoonchai N., Hiyoshi M., Motoyoshi K., Kimura F., Okada S. IL-34 and M-CSF share the receptor Fms but are not identical in biological activity and signal activation. Cell Death Differ. 2010;17:1917–1927. doi: 10.1038/cdd.2010.60. [DOI] [PubMed] [Google Scholar]
- 36.Wei S., Chitu V., Yeung Y.G., Yu W., Huang M., Williams L.T., Lin H., Stanley E.R. Functional overlap but differential expression of CSF-1 and IL-34 in their CSF-1 receptor-mediated regulation of myeloid cells. J Leukoc Biol. 2010;88:495–505. doi: 10.1189/jlb.1209822. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1
IL-34 protein expression in neurons and effect of siRNA knock-down. Expression of IL-34 in nontreated neurons (NT) was decreased by siRNA knock-down (siRNA). Intensity of IL-34 by siRNA knock-down was significantly decreased (n = 6). *P < 0.05 as compared with NT.
Supplemental Figure S2
Effect of continuous intraventricular infusion of IL-34 on associative learning in the cued and contextual conditioning tests in APP/PS1 transgenic mice. Mice were anesthetized using pentobarbital, and were placed in a stereotaxic apparatus. The infusion cannula (brain infusion kit 1; Alza Corp., Palo Alto, CA) was connected to a micro-osmotic pump (model 1002; total capacity, 90 μL), which was filled with vehicle (PBS) or IL-34 and implanted in the right ventricle [anteroposterior (AP) −0.3, mediolateral (ML) 1.0 from bregma, dorsoventral (DV) −2.5 from the skull, according to the method of Franklin and Paxinos16]. IL-34 was continuously infused into the cerebral ventricle at a dose of 6 ng/6 μL per day. Seven days after the start of infusion, the mice were subjected to the behavioral experiment. The retention session was performed at 24 hours after training. Context-dependent (A) and tone-dependent (B) freezing times were measured, and percentage of freezing behavior was calculated. Each column indicates the mean ± SEM. [WT mice, n = 10; vehicle (PBS)–treated APP/PS1 transgenic mice, n = 8; IL-34–treated APP/PS1 transgenic mice, n = 8]. *P < 0.05 as compared with WT mice; **P < 0.05 as compared with vehicle-treated APP/PS1 transgenic mice.