Defining and redefining the nephron progenitor population (original) (raw)
. Author manuscript; available in PMC: 2012 Sep 1.
Published in final edited form as: Pediatr Nephrol. 2011 Jan 14;26(9):1395–1406. doi: 10.1007/s00467-010-1750-4
Abstract
It has long been appreciated that the mammalian kidney arises via reciprocal interactions between an epithelial ureteric epithelium and the surrounding metanephric mesenchyme. More recently, lineage tracing has confirmed that the portion of the metanephric mesenchyme closest to the advancing ureteric tips, the cap mesenchyme, represents the progenitor population for the nephron epithelia. This Six2+Cited1+ population undergoes self-renewal throughout nephrogenesis while retaining the potential to epithelialize. In contrast, the Foxd1+ portion of the metanephric mesenchyme shows no epithelial potential, developing instead into the interstitial, perivascular, and possibly endothelial elements of the kidney. The cap mesenchyme rests within a nephrogenic niche, surrounded by the stroma and the ureteric tip. While the role of Wnt signaling in nephron induction is known, there remains a lack of clarity over the intrinsic and extrinsic regulation of cap mesenchyme specification, self-renewal, and nephron potential. It is also not known what regulates cessation of nephrogenesis, but there is no nephron generation in response to injury during the postnatal period. In this review, we will examine what is and is not known about this nephron progenitor population and discuss how an increased understanding of the regulation of this population may better explain the observed variation in final nephron number and potentially facilitate the reinitiation or prolongation of nephron formation.
Keywords: Kidney development, Nephron progenitor, Cap mesenchyme, Mesenchyme to epithelial transition, Specification, Self-renewal
Introduction
The role of the nephron progenitors in mammalian kidney development
The mammalian kidney arises via reciprocal signaling between two distinct mesodermal derivatives; the ureteric bud (UB) and metanephric mesenchyme (MM) [1]. As the UB invades the MM, it produces morphogens that induce aggregation of the mesenchyme around the UB tips to form what is variously referred to as committed, capping, or cap mesenchyme. Throughout the rest of kidney development, this cap mesenchyme (CM) population exists in apposition with each ureteric tip (Fig. 1). A subset of cells within each CM forms a pre-tubular aggregate (PA) before undergoing a mesenchyme-to-epithelial transition (MET), thereby forming a single epithelial renal vesicle (RV) (Fig. 1). The formation of an RV represents the ‘birth’ of a single nephron. While described as distinct entities, the transition from CM through PA to RV is a continuum, as has become evident in the lack of definitive non-overlapping markers of each stage. PA is histologically recognized by the compaction of cells in a distinct spatial region with respect to the adjacent branching tip prior to the presence of apical-basal patterning or lumen formation. As such, PA cells continue to express some CM markers (e.g., Six2) while already expressing factors involved in epithelialization (e.g., Wnt4, cadherin 4) [2] (Fig. 1). Cells of the RV express cadherin 6, lose Six2 expression and develop a central lumen, indicating the acquisition of an epithelial phenotype [2, 3]. The functional epithelia of the final nephron then forms via a process of elongation, segmentation and patterning of this initial epithelial structure to move it from RV to comma, then S-shaped body, after which distinct convoluted proximal and distal tubules arise separated by an elongating loop of Henle. The far proximal end of this tubule becomes vascularized to form the glomerulus while the other end fuses with the ureteric epithelium to form a patent uriniferous tubule [1–6]. Via continued ureteric epithelial branching and RV induction, the full complement of nephrons is formed before cessation of nephrogenesis just before or at birth, depending upon the species [7]. The ability to form the 3,000–5,000 nephrons per kidney in a mouse, or the 200,000 to 2 million nephrons per kidney in the human [8] relies upon the survival and turnover of the CM throughout development.
Fig. 1.
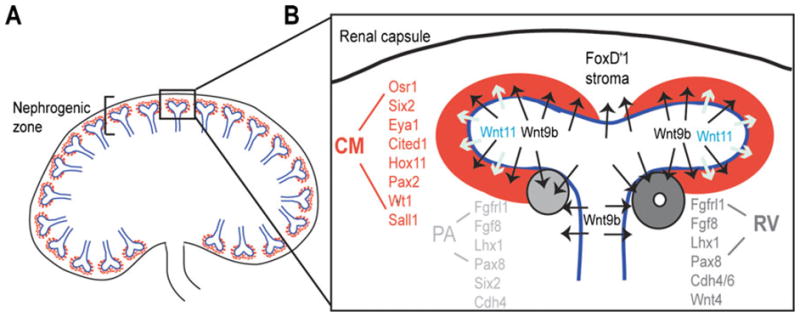
The progenitor niche within the nephrogenic zone of the developing mammalian kidney. A Diagrammatic representation of a section through a developing kidney showing the peripheral nephrogenic zone containing terminal tips of the branching ureteric tree (blue) surrounded by cap mesenchyme (red). B Progenitor niche comprising one branching tip (blue) surrounded by cap mesenchyme (CM; red) and Foxd1+ stroma. The location of Wnt9b and Wnt11 expression within the ureteric tip is indicated. Differentiation occurs in the ‘armpit’ of the branching ureteric tip, forming a pretublar aggregate and the epithelializing to form a renal vesicle. PA pre-tubular aggregate; RV renal vesicle. Key genes expressed in CM, PA, and RV are listed. Note the persistence of Six2 in the pretubular aggregate but not the RV. The first evidence of epithelialization is the expression of cadherins 4 and then 6. E-cadherin is not expressed in the RV (3)
The distinction between the role of the MM and the CM in formation of the nephrons arose via seminal studies investigating the role of the transcription factor Six2. This gene is expressed in the CM and mice lacking this gene show premature epithelialization of this region, leading to a loss of the compartment for subsequent nephron formation [9]. The resulting kidneys are small and show limited nephrons or branching and no remaining CM. Driven by the same promoter, elegant lineage tracing experiments proved that it is this specific region of the MM that gives rise to all the epithelial elements of the nephron, excluding the collecting duct [6] (Fig. 2). Via the use of a tamoxifen-inducible construct, Kobayashi et al. [6] were also able to show that the CM becomes specified as the ureteric tip enters the MM (embryonic day (E) 11–11.5 in mouse), but that after that time, no additional cells enter the CM from the MM, stroma or the tip. Via quantification of the number of CM cells present at E11.5 (approximately 6,000) and at E19.5 (approximately 120,000) it was concluded that this is a self-renewing nephron progenitor population [6]. A similar conclusion was reached via lineage tracing using the Cited1 promoter, another gene expressed in CM [10]. As such, the CM must undergo specification, self-renewal, and remain able to undergo nephron induction. Many questions remain as to how this is achieved. How is it that all renal vesicles arise in the same position with respect to the adjacent ureteric tip? How does a subset of the CM self-renew while other CM cells are undergoing differentiation? Is this a homogeneous population of cells with proliferative and differentitive equivalence or are there sub-compartments within the CM either playing different roles or at different stages of progression from one state to the next? Finally, what perturbs these interactions such that nephrogenesis ultimately ceases and the CM is lost?
Fig. 2.

Lineage relationships during kidney development. The mammalian kidney is derived from the intermediate mesoderm (IM), which gives rise to the nephric duct (ND), and the metanephric mesenchyme (MM). The ND gives rise to the collecting duct system, which is composed of two key cell types, principal cells, and intercalated cells. The MM specifies the cap mesenchyme (CM) and also gives rise to the stroma. The CM is the nephron progenitor population and differentiates in the renal vesicle via a mesenchyme-to-epithelial transition. This structure immediately becomes patterned into distal and proximal RV, with the distal RV forming the connecting cells between nephron and collecting duct and the distal cell types of the nephron (distal tubules) and the proximal RV giving rise to the glomeruli and proximal tubules. The stroma is thought to give rise to the interstitial cells of the kidney, including the pericytes and possibly also the endothelial cells. However, there is also likely to be angiogenic ingrowth of endothelial cells. Neurons and macrophages are thought to migrate into the kidney from around embryonic day 12 in the mouse [90]
Defining the nephrogenic niche
One way to envisage the CM is as a progenitor population residing within a supportive niche. The site of all nephron formation and ureteric tip branching is the periphery of the kidney in a region referred to as the nephrogenic zone (Fig. 1). This region can be viewed as a series of individual progenitor populations, with each niche containing a CM nephron progenitor population, the accompanying ureteric tip progenitors, and the surrounding non-cap or non-committed stromal progenitors. As in other stem cell niches, maintenance of these progenitors will depend upon complex cell-cell and growth factor-ligand interactions between all partners. Reciprocal interactions between the CM and the branching ureteric tip have long been appreciated. These are complex and dynamic, and involve many different growth signaling pathways, including Wnt, BMP, FGF, TGF, GDNF, and SCF signaling (reviewed in [11], [12]). Growth factors including FGF8 and bFGF/BMP7 act to maintain the CM [13], while others such as Wnt4, LIF, and TGFβ2 are involved in epithelial differentiation to RV [14–17] (note that studies on LIF have been performed in rat, not mouse). Each of these is expressed from a specific location within the niche. RA is present in the stroma [18] and in combination with Activin A has been shown to induce the formation of a pronephric field from animal cap ectoderm in Xenopus [19] as well as inducing the expression of key CM transcription factors, Pax2 and Wt1, when added to embryonic stem cells [20]. The ureteric epithelium expresses both Wnt9b and Wnt11, albeit in different sub-domains (Fig. 1). Expressed throughout the branching ureteric tree, Wnt9b is required to initiate CM differentiation via FGF8 [15, 21] and Fgflr1 [22]. Subsequent induction of Wnt4 within the pre-tubular aggregates ensures a transition to epithelium, hence the loss of either Wnt protein results in a loss of nephron formation [23–25]. Wnt11 is expressed only at the very tips of the branching ureteric tree, adjacent to the CM [26] (Fig. 1) and appears to be involved in a positive feedback loop to promote GDNF expression in the CM, which in turn promotes the continued survival and branching of the ureteric tree [27]. A role for this Wnt protein in nephron formation has not been demonstrated.
The spatial segregation of growth factors and the differential pattern of expression of a large number of CM-enriched transcription factors would predict that the CM is not a homogeneous compartment. Spatially, cells within the CM progress from self-renewing progenitor to PA to RV as you move from the most distal CM, through lateral CM to the armpit of the adjacent branching ureteric tree (Fig. 3A). This coincides with changes in relative Wnt9b and Wnt11 concentrations, both in the distal to proximal axis and from the basement membrane of the ureteric compartment to the outer edge of the CM field (Fig. 3B). This will also be the case for other growth factors secreted both from the tip and the surrounding stromal cells. The fact that the ureteric tip is not static, but continues to elongate and eventually branch again, means that this complex nephrogenic niche is temporospatially dynamic. Analyses of gene expression also provide evidence that the CM compartment is not homogeneous. Mugford et al. [2] spatially defined sub-compartments based upon gene expression within the Six2+ CM, thereby distinguishing between ‘capping mesenchyme’ (Cited1+−Six2+Wnt4−) and ‘induced mesenchyme’ (PA and RV) marked by Wnt4, Pea3, and Lef1. Of note, Six2 expression is seen to extend into the PA, overlapping with Wnt4 expression. Such populations of Six2+Wnt4+Cited1− cells were proposed to have already transitioned towards epithelial differentiation. This distinction between sites of Six2 and Cited1 expression during kidney development is seen in Fig. 3C. Of note was the observation of overlap in this region between Wnt4 and Six2 expression, suggesting Six2 expression is not confined to the self-renewal phase. Hence, the true nephron progenitor compartment was redefined as the Six2+Cited1+ cells.
Fig. 3.
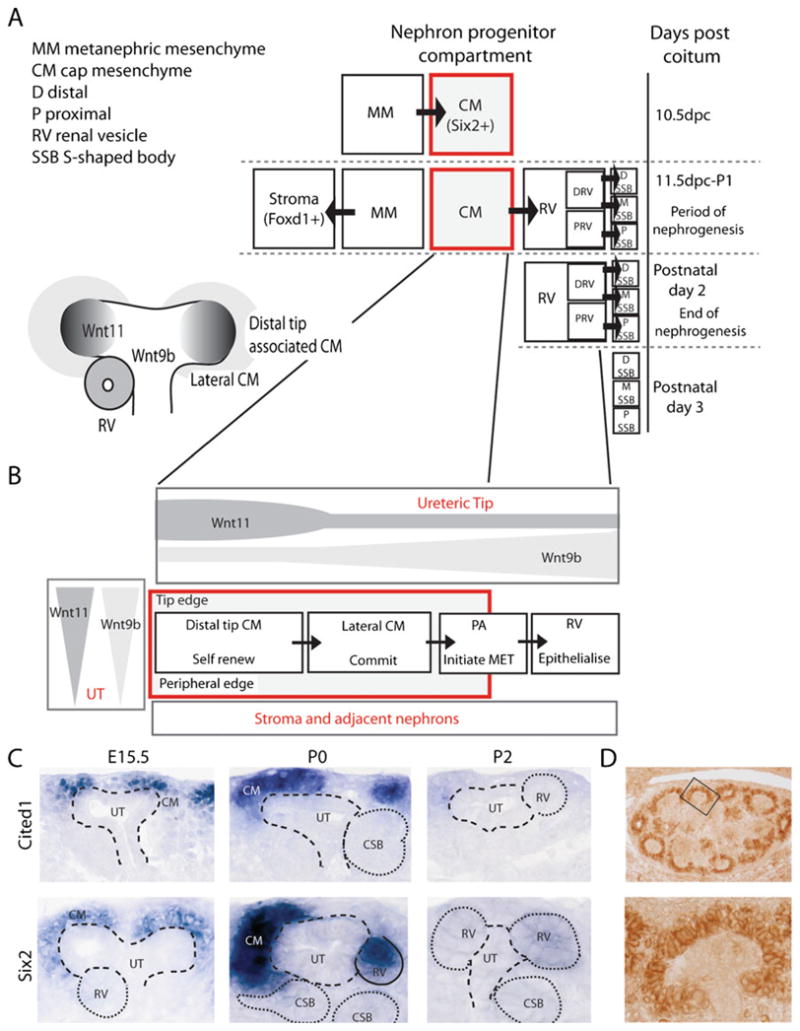
Temporospatial representations of cap mesenchyme specification, differentiation and the effect of niche architecture on growth factor gradients and gene expression. A Temporal representation of commitment of MM to CM and stroma, followed by CM differentiation and exhaustion of the CM at the end of nephrogenesis. B Diagram of the cap mesenchyme nephron progenitor population as it is aligned within the nephrogenic zone (left) and then linearized from distal tip CM to renal vesicle to illustrate the differences in concentration of two secreted morphogens, Wnt9b and Wnt11. C Section in situ hybridization of the developing mouse kidney at E15.5 and postnatal day 2 showing the heterogeneity of gene expression within the cap mesenchyme for Six2 and Cited1. D Immunohistochemistry for Bcl2 in the kidney shows specific protein staining within the cap mesenchyme (images courtesy of E. Wainwright)
Specification of the cap mesenchyme
Given that the CM is not homogeneous but represents a cell population in transition, molecular regulation of four distinct processes is required: specification, self-renewal, survival and nephron potential. In other mesodermally derived hierarchies, such as blood, lineage-instructive transcription factors drive binary cell fate choices that can be mapped from the multipotent stem cells through to their varied progeny [28]. This dichotomous specification can be sustained by cross-antagonism between transcription factors that specify the alternative fates. Fate-mapping studies conducted by Mugford et al. [29] demonstrated that within the Osr1+ mesenchyme between E9.5 and E10.5 there are already at least two separate populations; FoxD1+ and Six2+ cells. These appear to represent stroma and CM. While there is evidence that Foxd1 expression persists in the absence of Osr1, while CM specification is lost, the loss of Six2 expression in these mutant mice does not prove a role for Six2 in specification [29]. Other transcription factors upstream of Six2, such as Pax2, are more likely to be critical at this binary decision point.
The Pax2 transcription factor is expressed in the intermediate mesoderm well before the commencement of kidney development. Pax2 is required for the formation of the nephric duct and continues to be expressed in the branching ureteric tree. While not expressed in the MM prior to the arrival of the UB, Pax2 is one of the first transcription factors to define the CM as distinct from the remaining mesenchyme [30] and has been assumed to play a role in specification [31]. However, there is debate about the onset of Pax2 expression. A loss of mesenchymal Pax2 expression in the developing kidneys of Danforth’s short tail mutant mice results from the absence of induction of the outgrowth of the ureteric bud, and hence the loss of CM specification [32]. In mice null for Pax2, the mesenchyme is described as not competent to undergo MET [32–34], again suggesting a loss of CM specification. However, it has also been reported that in the c-Ret knockout mouse, where once again no ureteric bud forms, Pax2 expression is still initiated within the MM [34]. The promiscuous involvement of Pax2 at so many stages and within so many different cellular compartments of the developing kidney begs the question of whether it is playing the same role or different roles at different times and places. It transpires that this DNA binding protein, via interaction with an adaptor protein, PTIP, becomes linked to the histone methyltransferase complex, thereby mediating alterations to chromatin domains [31, 35]. Hence, the resulting transcriptional profile will depend upon what part of the chromatin Pax2 is influencing at a given time. This is likely to be influenced by what other interacting transcription factors are present at the time and suggests a context-dependant role for Pax2.
There are many other key transcription factors expressed in the MM whose expression persists in the CM. These include Osr1, Sall1, Eya1, Hox11 paralogs, and WT1. Like Pax2, Osr1 and Eya1 have key roles in specifying the intermediate mesoderm and all play roles in nephric duct formation. All three genes are also expressed later in the CM. Eya1 has been proposed to be genetically upstream of Six1 and Pax2, which are thought to work together to induce Gdnf, thereby initiating the outgrowth of the nephric duct [36]. Other key players here are the Hox11 paralogs, Hoxa11, Hoxc11, and Hoxd11. These proteins are thought to complex with Pax2 and Eya1 to activate Six2 and Gdnf [37].
Osr1, which initially specifies intermediate mesoderm from paraxial and lateral plate fates [38], is nonetheless critical for CM specification and survival [29, 39]. Osr1 expression becomes restricted to the CM and Osr1 null mice fail to show expression of key CM genes (Pax2, Six2, GDNF, Eya1, and Sall1) [39]. In contrast, Osr1 is not required for the formation of the Foxd1+ stromal compartment despite being expressed in MM prior to the separation of these lineages.
Another homolog of the Drosophila homeobox gene “sinus oculis” [40, 41], Six1, is expressed in the developing mesonephros and in the CM and is required for kidney development [42]. Loss of Six1 also disrupts mesonephric tubule formation. Six1 forms a transcriptional complex with Pax2 and Eya1 [36, 42] and a loss of Six1 does affect the expression of Pax2 and Six2 expression but not Eya1. The dual knockout of Six1 and Six4, both expressed within the CM, results in a loss of Pax2, Pax8 and GDNF expression [43].
Sall1 expression is not restricted to CM, with expression seen also in the uncommitted mesenchyme and the early epithelial structures [44]. However, it is the Sall1hi fraction of the developing kidney that responds to Wnt4 by undergoing an epithelial transition. This suggests that the highest Sall1 expression in CM. The Sall1 promoter shows multiple binding sites for both WT1 and Six1 and transient assays suggest that Six1 synergizes with Eya1 to upregulate Sall1 expression [45]. However, Sall1 expression is far broader that Eya1 or Six1 and persists after MET. Over-expression of Sall1 in the Six2+ CM had no effect on the survival or function of this subcompartment [46]. However, in both the Hox11 triple mutant and the Sall1 mutant mice the metanephric mesenchyme forms but the ureteric bud fails to invade and branch, leading to renal agenesis in both cases [44, 47]. In humans, mutations in Sall1 result in the Townes-Brocks syndrome of renal agenesis/dysgenesis. In mice, while the MM still forms in the absence of Sall1, there is no initiation of Six2 expression. Hence, Sall1 may be required for CM specification or play a lesser role in cap turnover. Additionally, ectopic anterior activation of Hoxd11 can activate Six2 expression, imposing a metanephric development pattern on the mesonephros [48]. This also suggests a role for Hoxd11 in CM specification.
Self-renewal and survival within the cap mesenchyme: two separable parameters
While self-renewal of cells within the CM must occur in order to generate the vast number of nephrons present at the end of kidney development, how this is controlled is unclear. The gene most clearly associated with CM self-renewal is Six2. Inactivation of Six2 in the developing kidney results in ectopic epithelialization and thus depletion of the CM [9]. This in turn results in the loss of a source of GDNF for the ureteric epithelium, resulting in a stunting of branching [6, 9]. Hapolinsufficiency for Six2 results in a reduction in nephron number, hypoplasia, and glomerulopathy [49]. Regulation of Six2 is not fully understood, however Six2 is known to be downstream of Hox11 genes during developing of both kidney and branchial arch mesenchyme. Here, Six2 regulation is mediated by a single enhancer element to which Hox11 binds [50]. Six2 expression is also likely to be downstream of Six1 and Sall1. It might be presumed that given only the Cited1+ fraction of the Six2-positive cells represents progenitors that Cited1 may also have a role in self-renewal. In contrast, a knockout of this gene did not show any apparent effect on nephron production [51].
A recent study by Coulliard and Trudel [52] demonstrated that expression of c-myc is essential for normal kidney development. c-myc is expressed in the MM, CM, and pre-tubular aggregates, but is down-regulated upon epithelialization [53]. In mice, conditional inactivation of c-myc in the CM at E11.5 resulted in decreased cellular proliferation and a subsequent 35% reduction in CM cells by E18.5, however nephrogenesis still occurred. This indicates c-myc is not lineage-instructive but likely to regulate proliferation/self-renewal of the CM. While deletion of c-myc in early kidney development resulted in hypoplasia, c-myc deletion after E17.5 had no effect on self-renewal, suggesting a temporal variation in the relative importance of CM proliferation [52].
While self-renewal is required to generate sufficient cells for nephron formation, survival of these cells is likely to be a separable attribute. Indeed, it has long been known that the survival of the mesenchymal portion of the kidney depended upon the arrival and continued presence of the ureteric epithelium. Hence, culture of the mesenchyme in the absence of UB resulted in apoptosis. Equally, the MM was shown to undergo apoptosis in the absence of the key MM-specifying genes, including WT1, Eya1, and Hox11. While an MM is present in WT1 knockout mice, loss of this gene results in a failure of the ureteric bud to arise. Consequently, the MM undergoes apoptosis [54]. The ureteric factor determining MM survival is not known. However, there is strong expression of Bcl2 confined to the CM (Fig. 3D), suggesting that there is active suppression of apoptosis. This is likely to be downstream of signals from the ureteric tip or surrounding stroma but is not necessarily driving self-renewal.
Maintaining a population with renal epithelial potential
The final requirement for the CM is that it remains able to respond to the appropriate signals to form renal vesicles. Regulating the balance between self-renewal and differentiation into nephron epithelia is critical for determining nephron number. Early co-culture studies demonstrated that the ureteric bud could be replaced by a number of inducer tissues, including the spinal cord, and that in the presence of such inducing tissues, the mesenchyme was able to undergo epithelial transition. Subsequent studies identified Wnt proteins as the inducing signals [23, 25]. While loss of induction results in apoptosis, the CM can be rescued in vitro by the presence of a number of growth factors, including TGFβ2 [55]. However, such cultured CM loses its nephron-forming potential. Isolated mesenchyme can also be maintained as a long-term self-renewing mesenchy-mal population in the presence of bFGF and thrombin [56]. We defined such cultures as ‘nephrospheres’ as they represented clonal, long-term self-renewing multipotent stem cells able to be serially passaged from floating spheres of cells. Here again, while a broad pan-mesodermal multi-potentiality remained, the mesenchyme loses its nephron epithelial potential. Culture with TGFβ2 results in the formation of a blastemal and chondrogenic phenotypes, as is frequently observed in Wilms’ tumors [57], suggesting a misdirection of differentiation or loss of specification. As nephrospheres can be derived from Sall1- or Osr1-positive populations that will include CM, the loss of renal potential by these cultures may simply indicate that the CM component itself is selectively lost, leaving only the stromal progenitor population. However, the cells within these cultures are not Foxd1-positive. What these two studies do show is an apparent separation between survival and renal potential.
The architecture of any cellular niche is critical for the maintenance of somatic stem cells, including skin, intestine, and blood [58–60]. As with the CM, when removed from their specific niche environment and cultured in vitro, many stem and progenitor cells undergo cell death or spontaneously differentiate. During development, some progenitor cell populations are able to establish a temporary niche that is eventually lost, leading to the exhaustion of that embryonic progenitor population. This has recently been shown to be the case in the Drosophila larval midgut, where establishment of a temporary niche during development maintains the progenitors until the permanent intestinal stem cells of the adult are established [61]. It would appear that during kidney development, the CM also resides in a transient niche defined by the surrounding ureteric tip and stoma and that removal from this niche results in the loss of progenitor potential.
Cessation of nephrogenesis and nephron progenitor exhaustion
In mammals, nephron formation is an embryological phenomenon and does not occur in adults. Cessation of nephrogenesis occurs around gestational week 36 in humans, after which time no new nephrons are formed. In the mouse, the last CM markers disappear around postnatal day 3 (P3) [7]. During this period of time, the tips lose their normal ampullary shape and renal vesicles begin to form around the end of the tip [7]. Six2 expression at this point in time is present in structures that are clearly in the process of forming nephrons and fusing with the adjacent tips (Fig. 3C). Studies in mouse have shown that the exhaustion of the CM does not occur via selective apoptosis, nor does it involve the differentiation of CM into FoxD1+ stromal precursors. What does appear to happen is a global commitment of remaining CM to epithelial differentiation [7]. Cessation of nephrogenesis may be viewed as an active process or passive exhaustion. Active cessation of nephrogenesis suggests a threshold or a binary decision such that at a given point in time the signals from around the CM either trigger differentiation and/or the signal to ‘self-renew’ ends. Such an active process may be triggered by signals from the tip or surrounding stroma. As there is no obvious physiological association with the timing of cessation (varies from species to species), such a trigger may simply arise due to alterations in growth factor concentrations within the niche as a result of changes in spatial arrangement over time. As birth approaches, the number of stromal cells between the CM and the renal capsule reduces. The resulting changes in growth factor signaling may be enough to eliminate self-renewal or trigger differentiation of the CM.
Another way to envisage cessation of nephrogenesis is as passive exhaustion. In this model, self-renewing stem cells represent a subpopulation of the CM, with the remainder of the CM population representing a transit amplifying population without the self-renewal properties of the stem cell fraction of the CM. If the capacity for any given cap to survive rests on the presence of such a cell, nephrogenesis would cease when the size of any given cap population became too small to contain a self-renewing (stem) cell. In the mouse, existing data does suggest that the overall number of CM cells per tip is actually declining as development proceeds, with approximately 6,000 cap cells per tip at E11.5 and 120 per tip at E19.5 [6]. This view predicts that if there was an increase in either the number of such stem cells per cap or their rate of self-renewal, nephrogenesis would be prolonged. This would not be the case if cessation of nephrogenesis is actively triggered. The argument against this model is the paucity of evidence for conditions under which nephrogenesis can be prolonged, although this may have simply been overlooked.
Redefining the renal stem cell of importance
There has been growing interest over the last decade in the possibility of identifying a renal stem cell in the postnatal kidney so as to develop novel regenerative approaches to the treatment of kidney disease. From the discussion here, and from the pathological observations over decades, it is evident that no new nephrons form after birth and that the embryonic nephron progenitors that give rise to the functional units of the postnatal kidney do not persist in the adult kidney. This is supported by the fact that the culture conditions able to sustain the embryonic mesenchymal progenitors failed to support the growth of any postnatal renal population [56]. In addition, Humphreys et al. [62] have shown that renal turnover in response to acute damage does not involve the recruitment or epithelialization of any non-epithelial cell type. All cellular turnover in response to damage occurred within cells of the nephron [62]. While these epithelial cells are all derived from a common embryonic nephron progenitor population, the fact that that population no longer exists raises the question of the pertinence of understanding the origin of the nephron itself. However, it does remain possible that a hierarchy of epithelial progenitors, acting in normal tubular repair or able to be facultatively recruited under specific circumstances, also exists in the adult kidney.
There is tremendous variability in the total number of nephrons present in an adult human kidney (200,000 to 2 million per kidney) [8, 63] and it has been shown that both environmental and genetic factors can affect how many nephrons are formed [64]. In turn, low nephron number is a contributing factor in the development of hypertension and renal disease [65–67]. In numerous models in which the fetus is exposed to a suboptimal uterine environment, including maternal undernutrition [68], uteroplacental insufficiency [69], or glucocorticoid exposure [70], nephron number is reduced, which is often associated with hypertension in the adult. This leads to hyperfiltration of remaining glomeruli, which in turn leads to glomerular damage and sclerosis [64, 67]. In humans, low birth weight, as a result of intrauterine growth retardation (placental insufficiency, cord compression, maternal anemia, or drug abuse) and premature birth, correlate strongly with reduced nephron endowment [8, 71, 72]. These individuals are at particular risk.
Were it possible to prolong nephrogenesis in such fetuses, this could have profound effects on long-term health for the individual and the health burden of hypertension and renal failure of the population as a whole. Evidence that this may be feasible comes from the sheep, in which it has been reported that the removal of one kidney (uninephrectomy) from sheep fetuses at 100 days of gestation (term=150 days) resulted in a significant degree of compensatory growth and nephrogenesis in the remaining kidney by birth [73]. Of particular importance was the observation that involved a prolongation of nephrogenesis in utero resulting in an almost doubling of nephron number (530,000 glomeruli) in comparison to a normal sheep kidney (365,000 glomeruli) [73]. This suggests that prolongation of nephrogenesis is possible but does not explain how this has occurred.
While no longer present in the postnatal kidney, analyses of human fetal kidneys both confirms the conservation of expression of CM markers between human and mouse as well as demonstrating the capacity to isolate such cells from human fetal material based upon the cell surface markers Ncam and Fzd7 [74]. Such material may form the basis for bioengineering. In addition, with the progress being made in the stem cell field, regeneration of CM from exogenous sources, such as embryonic stem cells or via the reprogramming of differentiated cells, is potentially feasible. Directed differentiation of human embryonic stem cells to specific cells types of all three lineages has been reported. This has included the generation of neurons, oligodendrocytes, beating cardiac myocytes, hematopoietic and pancreatic progenitors [75, 76]. In the majority of cases, defining the protocol for directing such differentiation has drawn on an intimate understanding of the embryological development of that cell type in vivo. Kidney is a mesodermal derivative, sharing with other mesodermal tissues including blood, a mesendodermal origin. Mileposts between mesendoderm and nephron progenitor include definitive mesoderm, intermediate mesoderm and metanephric mesenchyme with each step requiring unique growth factor combinations to induce appropriate specification (Fig. 4). Fully defining the genetic regulation of CM specification and how this is induced during kidney morphogenesis may allow the directed differentiation of hES cell in culture to this tissue type. Fully defining the environment then required for this population to self-renew and remain committed to a nephron epithelial fate would also be needed. Another approach would be the reprogramming of an adult cell type to a CM phenotype. This would require the enforced expression of lineage-instructive transcription factors, including those required for specification of the CM in vivo (Fig. 4). Reprogramming from adult fibroblast to induced pluripotent stem cell is now feasible via the reintroduction of as few as four key transcription factors [77]. The enforced expression of key transcriptional regulators has also been shown to facilitate reprogramming from one differentiated cell type to another [78–80]. If a given progenitor phenotype has a stable enough phenotype and can be provided with the extrinsic requirements to support this phenotype, it should also be possible to reprogram any differentiated cell into a specific progenitor. Again, for reprogramming to a CM phenotype, a clear understanding of the niche requirements and transcriptional state that defines the nephron progenitors will be required.
Fig. 4.
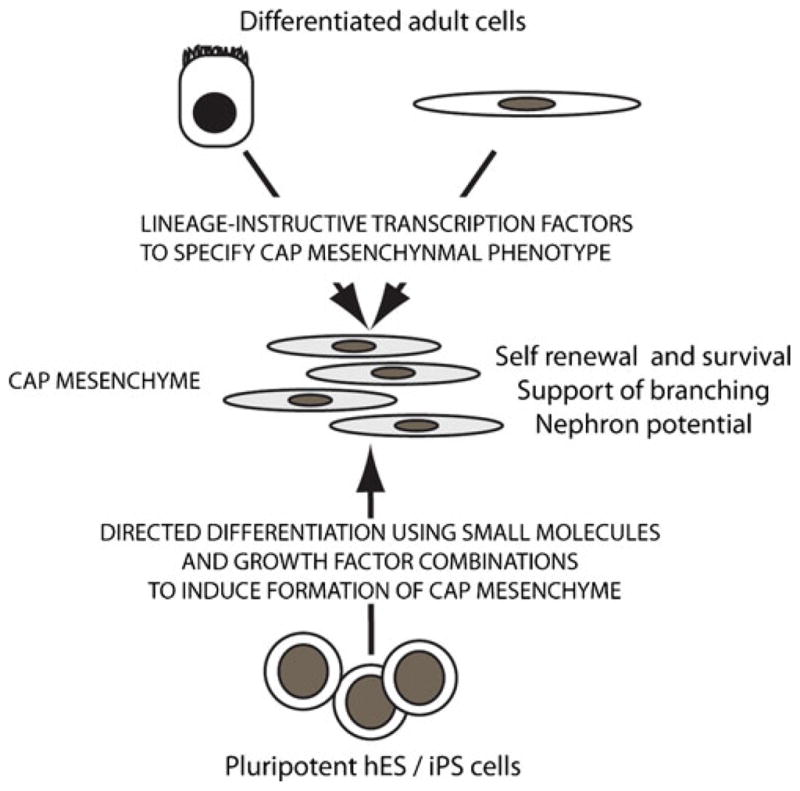
Model of approaches for the generation of nephron progenitors. This may include the directed differentiation of human pluripotent stem cells (embryonic stem cells or induced pluripotent cells) so as to recapitulate the differentiative steps normally taken to reach cap mesenchyme. Alternatively, reprogramming of differentiated adult cells may be achieved via the enforced re-expression of lineage-instructive transcription factors
The in vitro generation of CM is not likely to assist in cases of reduced nephron endowment. However, a greater understanding of the regulation of the niche itself may allow us to modify the process of nephrogenesis in situ such that cessation of nephrogenesis is delayed.
Reinitiation of nephrogenesis
One major challenge to the regeneration of nephron progenitors includes identifying a successful endpoint. For any population to be regarded as CM, it should be specified, able to self renew, able to support tip branching and able to differentiate appropriately. As culture of isolated CM in vitro has not been achieved without a loss of phenotype, knowing when CM has been generated and maintaining it in that state is difficult. In the absence of such conditions, an in vivo or ex vivo assay of renal progenitor potential is required. The renal field lags considerably behind other stem cell fields in the lack of definitive progenitor assays. The most convincing of these to date has involved the incorporation of a test cell population into a developing organ, either via the microinjection into kidney explants [81] or developing embryos [82]. In this way, Yokoo et al. [82] demonstrated the capacity of GDNF-transfected MSC populations to replace MM. More recently, we have used a recombination approach to show the successful integration of freshly isolated CM into a developing mouse kidney ex vivo. What would be preferable for a screen would be the incorporation of our knowledge of the nephrogenic niche into an in vitro environment able to support specification, self-renewal, survival, and renal differentiative potential. This may be feasible in the form of a micro-bioreactor.
If a nephron progenitor population could be generated and cultivated, could nephrogenesis be reinitiated in situ for the treatment of renal disease in adult life? This is the scenario that exists in the mesonephroi of some lower vertebrates. After nephrectomy in the cartilaginous skate Leucoraja erinacea, both the injured and the contralateral kidneys undergo de novo nephrogenesis via mesenchymal condensation followed by MET [83]. This process does not result from dedifferentiation of epithelial cells, but rather from the maintenance throughout adulthood of a persistent nephrogenic mesenchymal blastema located at the ventro-lateral portion of the skate kidney. Reports in other teleost fish, such as goldfish [84, 85], catfish [86], zebrafish [87], and Medaka [88] describe similar regeneration in response to various nephrotoxins. This response is biphasic, with an initial phase proliferation of existing epithelial cells (as in mammals), followed by a second phase of repair that involves de novo nephrogenesis very similar to normal nephron development [89]. Watanabe et al. [88] reported a significant increase in the number of Wt1+ clusters in Medaka mesonephros at 14 days post-injury, which corresponded to an increase in the number of developing nephrons observed histologically [88]. Such de novo nephrogenic responses appear robust. Salice et al. [85] challenged individual goldfish with repeated gentamicin exposure and observed de novo nephrogenesis after every injury. Hence, this response is not a last resort but is a bonafide repair mechanism amongst fish utilizing a self-renewing progenitor field that appears to have been lost in higher vertebrates. In mammals, the postnatal kidney is the metanephros and not the mesonephros. However, Hartman et al. [7], in their study of cessation of nephrogenesis, did suggest that while the CM has exhausted, the remaining ureteric tips could still respond to exogenous embryonic CM. Hence, while no such nephrogenic zone exists in postnatal mammalian kidneys, it is not known whether reintroduced CM introduced post-injury could act autonomously to reinitiate nephrogenesis in a similar way. In situ reprogramming within the postnatal pancreas has already demonstrated a capacity to re-specify exocrine pancreatic cells into insulin-secreting, glucose responsive beta-islet cells [80]. Such an in vivo re-specification would overcome the need to recreate the environment of the nephrogenic niche, but would continue to require a responsive ureteric compartment, without which any neo-nephrons would have no patent connection to the collecting duct system.
In conclusion, our understanding of the nephron progenitor population within the developing kidney has advanced considerably. However, the intricate interplay between this compartment and its surrounding niche, and its interrelationship with the ureteric and stromal progenitor populations, has much left to reveal. With greater understanding, the possibilities for reinitiation or prolongation of nephrogenesis will expand, potentially changing forever our approach to renal disease prevention and treatment.
Acknowledgments
We acknowledge Eleanor Wainwright and Dagmar Wilhelm for the images of Bcl2 protein localization within the developing kidney. We also acknowledge Kylie Georgas for technical assistance. CH is a Rosamond Siemon Postdoctoral Scholar. KM is a Senior Research Fellow and ML is a Principal Research Fellow with the National Health and Medical Research Council of Australia. The research of the authors is supported by the National Health and Medical Research Council of Australia (ID631362) and the National institutes of Health (DK070136).
Contributor Information
Caroline Hendry, Institute for Molecular Bioscience, The University of Queensland, St. Lucia 4072, Australia.
Bree Rumballe, Institute for Molecular Bioscience, The University of Queensland, St. Lucia 4072, Australia.
Karen Moritz, Email: k.moritz1@uq.edu.au, School of Biomedical Sciences, The University of Queensland, St. Lucia 4072, Australia.
Melissa H. Little, Email: M.Little@imb.uq.edu.au, Institute for Molecular Bioscience, The University of Queensland, St. Lucia 4072, Australia
References
- 1.Saxen L, Sariola H. Early organogenesis of the kidney. Pediatr Nephrol. 1987;1:385–392. doi: 10.1007/BF00849241. [DOI] [PubMed] [Google Scholar]
- 2.Mugford JW, Yu J, Kobayashi A, McMahon AP. High-resolution gene expression analysis of the developing mouse kidney defines novel cellular compartments within the nephron progenitor population. Dev Biol. 2009;333(2):312–323. doi: 10.1016/j.ydbio.2009.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Georgas KM, Rumballe BA, Valerius MT, Chiu HS, Thiagarajan RD, Lesieur E, Aronow BJ, Brunskill EW, Combes AN, Tang D, Taylor D, Grimmond SM, Potter SS, McMahon AP, Little MH. Analysis of early nephron patterning reveals a role for distal RV proliferation in fusion to the ureteric tip via a cap mesenchyme-derived connecting segment. Dev Biol. 2009;332(2):273–286. doi: 10.1016/j.ydbio.2009.05.578. [DOI] [PubMed] [Google Scholar]
- 4.Davies JA, Bard JB. The development of the kidney. Curr Top Dev Biol. 1998;39:245–301. doi: 10.1016/S0070-2153(08)60458-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Little MH. Regrow or repair – potential regenerative therapies for the kidney. J Am Soc Nephrol. 2006;17(9):2390–2401. doi: 10.1681/ASN.2006030218. [DOI] [PubMed] [Google Scholar]
- 6.Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, McMahon AP. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell. 2008;3(2):169–181. doi: 10.1016/j.stem.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hartman HA, Lai HL, Patterson LT. Cessation of renal morphogenesis in mice. Dev Biol. 2007;310:379–387. doi: 10.1016/j.ydbio.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hughson M, Farris AB, 3rd, Douglas-Denton R, Hoy WE, Bertram JF. Glomerular number and size in autopsy kidneys: the relationship to birth weight. Kidney Int. 2003;63:2113–2122. doi: 10.1046/j.1523-1755.2003.00018.x. [DOI] [PubMed] [Google Scholar]
- 9.Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler GR, Oliver G. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 2006;25:5214–5228. doi: 10.1038/sj.emboj.7601381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyle S, Misfeldt A, Chandler KJ, Deal KK, Southard-Smith EM, Mortlock DP, Baldwin HS, de Caestecker M. Fate mapping using Cited1-CreERT2 mice demonstrates that the cap mesenchyme contains self-renewing progenitor cells and gives rise exclusively to nephronic epithelia. Dev Biol. 2008;313:234–245. doi: 10.1016/j.ydbio.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dressler GR. The cellular basis of kidney development. Ann Rev Cell Dev Biol. 2006;22:509–529. doi: 10.1146/annurev.cellbio.22.010305.104340. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt-Ott KM, Barasch J. WNT/beta-catenin signaling in nephron progenitors and their epithelial progeny. Kidney Int. 2008;74:1004–1008. doi: 10.1038/ki.2008.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dudley AT, Godin RE, Robertson EJ. Interaction between FGF and BMP signaling pathways regulates development of metanephric mesenchyme. Genes Dev. 1999;13:1601–1613. doi: 10.1101/gad.13.12.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osafune K, Takasato M, Kispert A, Asashima M, Nishinakamura R. Identification of multipotent progenitors in the embryonic mouse kidney by a novel colony-forming assay. Development. 2006;133:151–161. doi: 10.1242/dev.02174. [DOI] [PubMed] [Google Scholar]
- 15.Perantoni AO, Timofeeva O, Naillat F, Richman C, Pajni-Underwood S, Wilson C, Vainio S, Dove LF, Lewandoski M. Inactivation of FGF8 in early mesoderm reveals an essential role in kidney development. Development. 2005;132:3859–3871. doi: 10.1242/dev.01945. [DOI] [PubMed] [Google Scholar]
- 16.Barasch J, Yang J, Ware CB, Taga T, Yoshida K, Erdjument-Bromage H, Tempst P, Parravicini E, Malach S, Aranoff T, Oliver JA. Mesenchymal to epithelial conversion in rat metanephros is induced by LIF. Cell. 1999;99:377–386. doi: 10.1016/s0092-8674(00)81524-x. [DOI] [PubMed] [Google Scholar]
- 17.Plisov SY, Yoshino K, Dove LF, Higinbotham KG, Rubin JS, Perantoni AO. TGF beta2, LIF and FGF2 cooperate to induce nephrogenesis. Development. 2001;128:1045–1057. doi: 10.1242/dev.128.7.1045. [DOI] [PubMed] [Google Scholar]
- 18.Marlier A, Gilbert T. Expression of retinoic acid-synthesizing and -metabolizing enzymes during nephrogenesis in the rat. Gene Expr Patterns. 2004;5:179–185. doi: 10.1016/j.modgep.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Osafune K, Nishinakamura R, Komazaki S, Asashima M. In vitro induction of the pronephric duct in Xenopus explants. Dev Growth Differ. 2002;44:161–167. doi: 10.1046/j.1440-169x.2002.00631.x. [DOI] [PubMed] [Google Scholar]
- 20.Kim D, Dressler GR. Nephrogenic factors promote differentiation of mouse embryonic stem cells into renal epithelia. J Am Soc Nephrol. 2005;16:3527–3534. doi: 10.1681/ASN.2005050544. [DOI] [PubMed] [Google Scholar]
- 21.Grieshammer U, Cebrian C, Ilagan R, Meyers E, Herzlinger D, Martin GR. FGF8 is required for cell survival at distinct stages of nephrogenesis and for regulation of gene expression in nascent nephrons. Development. 2005;132:3847–3857. doi: 10.1242/dev.01944. [DOI] [PubMed] [Google Scholar]
- 22.Gerber SD, Steinberg F, Beyeler M, Villiger PM, Trueb B. The murine Fgfrl1 receptor is essential for the development of the metanephric kidney. Dev Biology. 2009;335:106–119. doi: 10.1016/j.ydbio.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 23.Stark K, Vainio S, Vassileva G, McMahon AP. Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature. 1994;372:679–683. doi: 10.1038/372679a0. [DOI] [PubMed] [Google Scholar]
- 24.Kispert A, Vainio S, McMahon AP. Wnt-4 is a mesenchymal signal for epithelial transformation of metanephric mesenchyme in the developing kidney. Development. 1998;125:4225–4234. doi: 10.1242/dev.125.21.4225. [DOI] [PubMed] [Google Scholar]
- 25.Carroll TJ, Park JS, Hayashi S, Majumdar A, McMahon AP. Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev Cell. 2005;9:283–292. doi: 10.1016/j.devcel.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 26.Kispert A, Vainio S, Shen L, Rowitch DH, McMahon AP. Proteoglycans are required for maintenance of Wnt-11 expression in the ureter tips. Development. 1996;122:3627–3637. doi: 10.1242/dev.122.11.3627. [DOI] [PubMed] [Google Scholar]
- 27.Majumdar A, Vainio S, Kispert A, McMahon J, McMahon AP. Wnt11 and Ret/Gdnf pathways cooperate in regulating ureteric branching during metanephric kidney development. Development. 2003;130:3175–3185. doi: 10.1242/dev.00520. [DOI] [PubMed] [Google Scholar]
- 28.Graf T, Busslinger M. B young again. Immunity. 2008;28:606–608. doi: 10.1016/j.immuni.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Mugford JW, Sipila P, McMahon JA, McMahon AP. Osr1 expression demarcates a multi-potent population of intermediate mesoderm that undergoes progressive restriction to an Osr1-dependent nephron progenitor compartment within the mammalian kidney. Dev Biol. 2008;324:88–98. doi: 10.1016/j.ydbio.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dressler GR, Deutsch U, Chowdhury K, Nornes HO, Gruss P. Pax2, a new murine paired-box-containing gene and its expression in the developing excretory system. Development. 1990;109:787–795. doi: 10.1242/dev.109.4.787. [DOI] [PubMed] [Google Scholar]
- 31.Dressler GR. Advances in early kidney specification, development and patterning. Development. 2009;136:3863–3874. doi: 10.1242/dev.034876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Torres M, Gomez-Pardo E, Dressler GR, Gruss P. Pax-2 controls multiple steps of urogenital development. Development. 1995;121:4057–4065. doi: 10.1242/dev.121.12.4057. [DOI] [PubMed] [Google Scholar]
- 33.Brophy PD, Ostrom L, Lang KM, Dressler GR. Regulation of ureteric bud outgrowth by Pax2-dependent activation of the glial derived neurotrophic factor gene. Development. 2001;128:4747–4756. doi: 10.1242/dev.128.23.4747. [DOI] [PubMed] [Google Scholar]
- 34.Phelps DE, Dressler GR. Aberrant expression of Pax-2 in Danforth’s short tail (Sd) mice. Dev Biol. 1993;157:251–258. doi: 10.1006/dbio.1993.1129. [DOI] [PubMed] [Google Scholar]
- 35.Patel SR, Kim D, Levitan I, Dressler GR. The BRCT-domain containing protein PTIP links PAX2 to a histone H3, lysine 4 methyltransferase complex. Dev Cell. 2007;13:580–592. doi: 10.1016/j.devcel.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sajithlal G, Zou D, Silvius D, Xu PX. Eya1 acts as a critical regulator for specifying the metanephric mesenchyme. Dev Biol. 2005;284:323–336. doi: 10.1016/j.ydbio.2005.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gong KQ, Yallowitz AR, Sun H, Dressler GR, Wellik DM. A Hox-Eya-Pax complex regulates early kidney developmental gene expression. Mol Cell Biol. 2007;27(21):7661–7668. doi: 10.1128/MCB.00465-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.James RG, Schultheiss TM. Bmp signaling promotes intermediate mesoderm gene expression in a dose-dependent, cell-autonomous and translation-dependent manner. Dev Biol. 2005;288:113–125. doi: 10.1016/j.ydbio.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 39.James RG, Kamei CN, Wang QR, Jiang R, Schultheiss TM. Odd-skipped related 1 is required for development of the metanephric kidney and regulates formation and differentiation of kidney precursor cells. Development. 2006;133:2995–3004. doi: 10.1242/dev.02442. [DOI] [PubMed] [Google Scholar]
- 40.Ohto H, Takizawa T, Saito T, Kobayashi M, Ikeda K, Kawakami K. Tissue and developmental distribution of Six family gene products. Int J Dev Biol. 1998;42:141–148. [PubMed] [Google Scholar]
- 41.Boucher CA, Winchester CL, Hamilton GM, Winter AD, Johnson KJ, Bailey ME. Structure, mapping and expression of the human gene encoding the homeodomain protein, SIX2. Gene. 2000;247:145–151. doi: 10.1016/s0378-1119(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 42.Xu PX, Zheng W, Huang L, Maire P, Laclef C, Silvius D. Six1 is required for the early organogenesis of mammalian kidney. Development. 2003;130:3085–3094. doi: 10.1242/dev.00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kobayashi H, Kawakami K, Asashima M, Nishinakamura R. Six1 and Six4 are essential for Gdnf expression in metanephric mesenchyme and ureteric bud formation, while Six1 deficiency alone causes mesonephric-tubule defects. Mech Dev. 2007;124:290–303. doi: 10.1016/j.mod.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 44.Nishinakamura R, Matsumoto Y, Nakao K, Nakamura K, Sato A, Copeland NG, Gilbert DJ, Jenkins NA, Scully S, Lacey DL, Katsuki M, Asashima M, Yokota T. Murine homolog of SALL1 is essential for ureteric bud invasion in kidney development. Development. 2001;128:3105–3115. doi: 10.1242/dev.128.16.3105. [DOI] [PubMed] [Google Scholar]
- 45.Chai L, Yang J, Di C, Cui W, Kawakami K, Lai R, Ma Y. Transcriptional activation of the Sall1 by the human Six1 homeo-domain during kidney development. J Biol Chem. 2006;281:18918–18926. doi: 10.1074/jbc.M600180200. [DOI] [PubMed] [Google Scholar]
- 46.Jiang Q, Fujimura S, Kobayashi C, Nishinakamura R. Overexpression of Sall1 in vivo leads to reduced body weight without affecting kidney development. J Biochem. 2010;147:445–450. doi: 10.1093/jb/mvp214. [DOI] [PubMed] [Google Scholar]
- 47.Wellik DM, Hawkes PJ, Capecchi MR. Hox11 paralogous genes are essential for metanephric kidney induction. Genes Dev. 2002;16:1423–1432. doi: 10.1101/gad.993302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mugford JW, Sipila P, Kobayashi A, Behringer RR, McMahon AP. Hoxd11 specifies a program of metanephric kidney development within the intermediate mesoderm of the mouse embryo. Dev Biol. 2008;319:396–405. doi: 10.1016/j.ydbio.2008.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fogelgren B, Yang S, Sharp IC, Huckstep OJ, Ma W, Somponpun SJ, Carlson EC, Uyehara CF, Lozanoff S. Deficiency in Six2 during prenatal development is associated with reduced nephron number, chronic renal failure and hypertension in Br/+ adult mice. Am J Physiol Renal Physiol. 2009;296:F1166–F1178. doi: 10.1152/ajprenal.90550.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yallowitz AR, Gong KQ, Swinehart IT, Nelson LT, Wellik DM. Non-homeodomain regions of Hox proteins mediate activation versus repression of Six2 via a single enhancer site in vivo. Dev Biol. 2009;335:156–165. doi: 10.1016/j.ydbio.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boyle S, Shioda T, Perantoni AO, de Caestecker M. Cited1 and Cited2 are differentially expressed in the developing kidney but are not required for nephrogenesis. Dev Dyn. 2007;236:2321–2330. doi: 10.1002/dvdy.21242. [DOI] [PubMed] [Google Scholar]
- 52.Couillard M, Trudel M. C-myc as a modulator of renal stem/progenitor cell population. Dev Dyn. 2009;238:405–414. doi: 10.1002/dvdy.21841. [DOI] [PubMed] [Google Scholar]
- 53.Mugrauer G, Ekblom P. Contrasting expression patterns of three members of the myc family of protooncogenes in the developing and adult mouse kidney. J Cell Biol. 1991;112:13–25. doi: 10.1083/jcb.112.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, Jaenisch R. WT-1 is required for early kidney development. Cell. 1993;74:679–691. doi: 10.1016/0092-8674(93)90515-r. [DOI] [PubMed] [Google Scholar]
- 55.Sims-Lucas S, Young R, Martinez G, Taylor D, Grimmond SM, Teasdale R, Little MH, Bertram J, Caruana G. Redirection of renal mesenchyme to stromal and chondrocytic fates in the presence of TGF-β2. Differentiation. 2010;79:272–284. doi: 10.1016/j.diff.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 56.Lusis M, Li J, Ineson J, Li J, Little MH. Isolation and culture of metanephric mesenchyme-derived nephrospheres reinforces evidence that embryonic renal progenitors are multipotent and exhaust during cessation of nephron formation. Stem Cell Research. 2010;5:23–39. [Google Scholar]
- 57.Zhuang Z, Merino MJ, Vortmeyer AO, Bryant B, Lash AE, Wang C, Deavers MT, Shelton WF, Kapur S, Chandra RS. Identical genetic changes in different histologic components of Wilms’ tumors. J Natl Cancer Inst. 1997;89:1148–1152. doi: 10.1093/jnci/89.15.1148. [DOI] [PubMed] [Google Scholar]
- 58.Fuchs E. Finding one’s niche in the skin. Cell Stem Cel. 2009;4:499–502. doi: 10.1016/j.stem.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walker MR, Patel KK, Stappenbeck TS. The stem cell niche. J Pathol. 2009;217(2):169–180. doi: 10.1002/path.2474. [DOI] [PubMed] [Google Scholar]
- 60.Eliasson P, Jönsson JI. The hematopoietic stem cell niche: low in oxygen but a nice place to be. J Cell Physiol. 2010;222:17–22. doi: 10.1002/jcp.21908. [DOI] [PubMed] [Google Scholar]
- 61.Mathur D, Bost A, Driver I, Ohlstein B. A transient niche regulates the specification of Drosophila intestinal stem cells. Science. 2010;8327(5962):210–213. doi: 10.1126/science.1181958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Humphreys BD, Valerius MT, Kobayashi A, Mugford JW, Soeung S, Duffield JS, McMahon AP, Bonventre JV. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell. 2008;2:284–291. doi: 10.1016/j.stem.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 63.Hoy WE, Bertram JF, Denton RD, Zimanyi M, Samuel T, Hughson MD. Nephron number, glomerular volume, renal disease and hypertension. Curr Opin Nephrol Hypertens. 2008;17:258–265. doi: 10.1097/MNH.0b013e3282f9b1a5. [DOI] [PubMed] [Google Scholar]
- 64.Moritz KM, Cullen-McEwen LA. Kidney Development and Fetal Programming. In: Wintour-Coghlan EM, Owens JA, editors. Early Life Origins of Health and Adult Disease. Landes Bioscience; 2006. pp. 130–144. [Google Scholar]
- 65.Brenner BM, Garcia DL, Anderson S. Glomeruli and blood pressure. Less of one, more the other? Am J Hypertens. 1988;1:335–347. doi: 10.1093/ajh/1.4.335. [DOI] [PubMed] [Google Scholar]
- 66.Moritz KM, Wintour EM, Dodic M. Renal development and programming. In: Gluckman PD, Hanson MA, editors. Developmental Origins of Health and Disease – A Biomedical Perspective. 23. University Press; Cambridge: 2006. pp. 310–322. [Google Scholar]
- 67.Brenner BM, Anderson S. The interrelationships among filtration surface area, blood pressure, and chronic renal disease. J Cardiovasc Pharmacol. 1992;19(Suppl 6):S1–S7. doi: 10.1097/00005344-199219006-00002. [DOI] [PubMed] [Google Scholar]
- 68.Hoppe CC, Evans RG, Bertram JF, Moritz KM. The effects of dietary protein restriction on nephron number in the mouse. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1768–R1774. doi: 10.1152/ajpregu.00442.2006. [DOI] [PubMed] [Google Scholar]
- 69.Wlodek ME, Mibus A, Tan A, Siebel AL, Owens JA, Moritz KM. Normal lactational environment restores nephron endowment and prevents hypertension after placental restriction in the rat. J Am Soc Nephrol. 2007;18:1688–1696. doi: 10.1681/ASN.2007010015. [DOI] [PubMed] [Google Scholar]
- 70.Singh R, Cullen-McEwen LA, Kett KM, Boon WM, Dowling J, Bertram JF, Moritz KM. Prenatal corticosterone exposure results in altered AT1/AT2, nephron deficit and hypertension in the rat offspring. J Physiol. 2007;579:503–513. doi: 10.1113/jphysiol.2006.125773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rodríguez MM, Gómez AH, Abitbol CL, Chandar JJ, Duara S, Zilleruelo GE. Histomorphometric analysis of postnatal glomerulogenesis in extremely preterm infants. Pediatr Dev Pathol. 2004;7:17–25. doi: 10.1007/s10024-003-3029-2. [DOI] [PubMed] [Google Scholar]
- 72.Rodriguez MM, Gomez A, Abitbol C, Chandar J, Montané B, Zilleruelo G. Comparative renal histomorphometry: a case study of oligonephropathy of prematurity. Pediatr Nephrol. 2005;20:945–949. doi: 10.1007/s00467-004-1800-x. [DOI] [PubMed] [Google Scholar]
- 73.Douglas-Denton R, Moritz KM, Bertram JF, Wintour EM. Compensatory renal growth after unilateral nephrectomy in the ovine fetus. J Am Soc Nephrol. 2002;13:406–410. doi: 10.1681/ASN.V132406. [DOI] [PubMed] [Google Scholar]
- 74.Metsuyanim S, Harari-Steinberg O, Buzhor E, Omer D, Pode-Shakked N, Ben-Hur H, Halperin R, Schneider D, Dekel B. Expression of stem cell markers in the human fetal kidney. PLoS ONE. 2009;4:e6709. doi: 10.1371/journal.pone.0006709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Irion S, Nostro MC, Kattman SJ, Keller GM. Directed differentiation of pluripotent stem cells: from developmental biology to therapeutic applications. Cold Spring Harb Symp Quant Biol. 2008;73:101–110. doi: 10.1101/sqb.2008.73.065. [DOI] [PubMed] [Google Scholar]
- 76.Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132:661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 77.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 78.Graf T. Differentiation plasticity of hematopoietic cells. Blood. 2002;99:3089–3101. doi: 10.1182/blood.v99.9.3089. [DOI] [PubMed] [Google Scholar]
- 79.Bussmann LH, Schubert A, Vu Manh TP, De Andres L, Desbordes SC, Parra M, Zimmermann T, Rapino F, Rodriguez-Ubreva J, Ballestar E, Graf T. A robust and highly efficient immune cell reprogramming system. Cell Stem Cell. 2009;5:554–566. doi: 10.1016/j.stem.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 80.Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to β-cells. Nature. 2008;455:627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Challen GA, Ivan Bertoncello I, Deane J, Ricardo S, Little MH. Kidney side population cells represent a non-haematopoietic but heterogeneous population with multilineage and renal potential. J Amer Society Nephrol. 2006;17:1896–1912. doi: 10.1681/ASN.2005111228. [DOI] [PubMed] [Google Scholar]
- 82.Yokoo T, Ohashi T, Shen JS, Sakurai K, Miyazaki Y, Utsunomiya Y, Takahashi M, Terada Y, Eto Y, Kawamura T, Osumi N, Hosoya T. Human mesenchymal stem cells in rodent whole-embryo culture are reprogrammed to contribute to kidney tissues. Proc Natl Acad Sci USA. 2005;102:3296–3300. doi: 10.1073/pnas.0406878102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Elger M, Hentschel H, Litteral J, Wellner M, Kirsch T, Luft FC, Haller H. Nephrogenesis is induced by partial nephrectomy in the elasmobranch Leucoraja erinacea. J Am Soc Nephrol. 2003;14:1506–1518. doi: 10.1097/01.asn.0000067645.49562.09. [DOI] [PubMed] [Google Scholar]
- 84.Reimschuessel R, Bennett RO, May EB, Lipsky MM. Development of newly formed nephrons in the goldfish kidney following hexachlorobutadiene-induced nephrotoxicity. Toxicol Pathol. 1990;18:32–38. doi: 10.1177/019262339001800105. [DOI] [PubMed] [Google Scholar]
- 85.Salice CJ, Rokous JS, Kane AS, Reimschuessel R. New nephron development in goldfish (Carassius auratus) kidneys following repeated gentamicin-induced nephrotoxicosis. Comp Med. 2001;51:56–59. [PubMed] [Google Scholar]
- 86.Cormier SM, Neiheisel TW, Racine RN, Reimschuessel R. New nephron development in fish from polluted waters: A possible biomarker. Ecotoxicology. 1995;4:157–168. doi: 10.1007/BF00116479. [DOI] [PubMed] [Google Scholar]
- 87.Zhou W, Boucher RC, Bollig F, Englert C, Hildebrandt F. Characterization of mesonephric development and regenaration using transgenic zebrafish. Am J Physiol Renal Physiol. 2010;229(5):F1040–F1047. doi: 10.1152/ajprenal.00394.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Watanabe N, Kato M, Suzuki N, Inoue C, Fedorova S, Hashimoto H, Maruyama S, Matsuo S, Wakamatsu Y. Kidney regeneration through nephron neogenesis in medaka. Dev Growth Differ. 2009;51:135–143. doi: 10.1111/j.1440-169X.2009.01090.x. [DOI] [PubMed] [Google Scholar]
- 89.Reimschuessel R. A fish model of renal regeneration and development. ILAR J. 2001;42:285–291. doi: 10.1093/ilar.42.4.285. [DOI] [PubMed] [Google Scholar]
- 90.Rae F, Woods K, Sasmono T, Campanale N, Taylor D, Ovchinnikov D, Grimmond S, Hume DA, Ricardo S, Little MH. Characterisation and trophic functions of murine embryonic macrophages based upon the use of a CSF-1R-EGFP transgene reporter. Dev Biol. 2007;308:232–246. doi: 10.1016/j.ydbio.2007.05.027. [DOI] [PubMed] [Google Scholar]