Recurrent DNMT3A Mutations in Patients with Myelodysplastic Syndromes (original) (raw)
. Author manuscript; available in PMC: 2012 Jan 1.
Published in final edited form as: Leukemia. 2011 Mar 18;25(7):1153–1158. doi: 10.1038/leu.2011.44
Abstract
Alterations in DNA methylation have been implicated in the pathogenesis of myelodysplastic syndromes (MDS), although the underlying mechanism remains largely unknown. Methylation of CpG dinucleotides is mediated by DNA methyltransferases, including DNMT1, DNMT3A, and DNMT3B. DNMT3A mutations have recently been reported in patients with de novo acute myeloid leukemia (AML), providing a rationale for examining the status of DNMT3A in MDS samples. Here, we report the frequency of DNMT3A mutations in patients with de novo MDS, and their association with secondary AML. We sequenced all coding exons of DNMT3A using DNA from bone marrow and paired normal cells from 150 patients with MDS and identified 13 heterozygous mutations with predicted translational consequences in 12/150 patients (8.0%). Amino acid R882, located in the methyltransferase domain of DNMT3A, was the most common mutation site, accounting for 4/13 mutations. DNMT3A mutations were expressed in the majority of cells in all tested mutant samples regardless of blast counts, suggesting that DNMT3A mutations occur early in the course of MDS. Patients with DNMT3A mutations had worse overall survival compared to patients without DNMT3A mutations (p=0.005) and more rapid progression to AML (p=0.007), suggesting that DNMT3A mutation status may have prognostic value in de novo MDS.
Keywords: myelodysplastic syndrome, DNMT3A, mutation
INTRODUCTION
Cancer initiation and progression is caused by genetic and epigenetic alterations in DNA. Cancer genomes are characterized by global DNA hypomethylation with concomitant hypermethylation of gene promoter regions. The skewing of cytosine methylation may contribute to tumor development due to decreased expression of critical tumor suppressor genes that are hypermethylated in cancer cells.1-3 Currently, the underlying mechanism of altered DNA methylation in cancer genomes and the critical target genes affected by methylation remain largely unknown.2
Both acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) genomes contain different global DNA methylation patterns compared to normal bone marrow cells and compared to each other, suggesting that there may be methylation-specific gene alterations that contribute to these diseases.4-6 Indirect evidence supporting the hypothesis that DNA methylation changes may be important in AML and MDS genomes comes from the observation that cytosine analog drugs that interfere with methylation (5-azacytidine and decitabine (5-aza-2’-deoxycytidine)) have clinical activity in these diseases.7-10 However, inhibition of DNA methyltransferases (DNMTs), which are responsible for the covalent linking of methyl groups to the CpG dinucleotide, is only one potential mechanism of action of 5-azacytidine and decitabine.
DNMTs are critical for establishing and maintaining CpG methylation. The major DNMTs with methyltransferase activity in humans are DNMT1, DNMT3A, and DNMT3B. DNMT3A and DNMT3B are the dominant DNA methyltransferases involved in de novo DNA methylation and act independent of replication, while DNMT1 acts predominantly during replication to maintain hemimethylated DNA, although their precise roles in cancer cells are less well defined.11 The link between DNMTs and cancer comes from observations that elevated levels of DNMT proteins and activities occur in many types of cancers, and overexpression of DNMT1 in cells can lead to transformation (reviewed in3). In mice, altered DNMTs have variable effects on cancer. The number of colonic polyps that occur in compound Dnmt1 heterozygous knock-out/Apcmin mice is reduced, while T-cell lymphomas are increased in mice carrying a Dnmt1 hypomorphic allele (~10% normal levels).12, 13 Inherited mutations in DNMT3B are the most common cause of the ICF (immunodeficiency, centrosome instability, and facial anomalies) syndrome in humans.14 ICF patients have loss of methylated pericentromeric sequences, resulting in chromosomal instability in lymphocytes and a severe immunodeficiency. ICF patients are not prone to cancer, although they tend to die at an early age from infections.14 Collectively, the data suggest that alterations in DNMTs are associated with cancer, but direct evidence in humans has been lacking.
Our group has recently found that DNMT3A mutations are common (22% frequency) in de novo AML and are associated with poor survival,15 providing a rationale for screening patients with de novo MDS and secondary AML for mutations in DNMT3A. In this study, we performed total exonic resequencing of DNMT3A using DNA from 150 de novo MDS patients, of which 46 developed secondary AML, and identified 13 somatic mutations in 12 patients (8.0% of cases).
MATERIALS AND METHODS
Patients
Adults diagnosed with myelodysplastic syndrome and no prior history of exposure to chemotherapy and/or radiotherapy were included in the study. A punch biopsy of normal skin and a bone marrow aspirate were obtained with informed consent in accordance with a tissue acquisition protocol approved by the Human Research Protection Office at the Washington University School of Medicine.
DNA Samples
Genomic DNA samples were extracted from the skin and unfractionated bone marrow specimens and then subjected to whole genome amplification, as previously described.16
Sequencing
Primers were designed to amplify the coding sequences and splice sites of DNMT3A (provided in15). Amplicons were sequenced using BigDye chemistry and analyzed on an ABI 3730 sequencer. Sequence variants, including single nucleotide variants (SNVs) and insertion/deletion events (indels) were called by The Genome Center’s mutational profiling pipeline and manually reviewed. Potential somatic mutations (present in tumor, but not in the matched normal sample) were confirmed by independent PCR and sequencing.
Expression profiling and cDNA analysis
CD34+ cell purification, RNA preparation, hybridization to the Affymetrix U133plus2 arrays, and analysis of data were performed as previously described for these samples.16 cDNA was prepared according to the manufacturer’s recommendations (Invitrogen, Carlsbad, CA). Primers for cDNA amplification and sequencing crossed exon boundaries in all cases.
Statistical Analysis
Differences in clinical characteristics of patients with or without DNMT3A mutations were assessed using the Fisher’s exact or Mann-Whitney tests. Survival data are current to 01/06/2011. Survival curves were generated using the Kaplan-Meier method and differences were assessed by Log-Rank analysis. The analyses were generated using SAS/STAT software (Version 9.2 of SAS System for Windows, SAS Institute Inc., Cary, NC). Association testing of germline polymorphisms was performed using the Fisher’s exact or Chi-square tests. The predicted severity of DNMT3A mutations with translational consequences was evaluated using the SIFT and Polyphen2 algorithms.17, 18
RESULTS
Patient Characteristics
We identified a total of 150 patients with de novo MDS who had paired bone marrow (tumor) and skin (normal) DNA samples available. Cases were classified according to the French-American-British (FAB) system upon banking of their bone marrow specimens. The diagnoses included refractory anemia (RA; n=67), RA with ringed sideroblasts (RARS; n=5), RA with excess blasts (RAEB; n=72), and RA with excess blasts in transformation (RAEB-T; n=6) (Table 1). The median International Prognostic Scoring System (IPSS) score was 1 (range 0-3), and the median myeloblast count was 5 (range 0-28%).
Table 1.
Patient Characteristics
| DNMT3A wildtype no. (%) | DNMT3A mutant no. (%) | P-value | |
|---|---|---|---|
| Total (n=150) | 138 (92.0) | 12 (8.0) | |
| Age at diagnosis | 60 | 69 | 0.03† |
| range (median) | 20-87 (62) | 39-86 (71) | |
| Median Survival | 965 days | 433 days | 0.02* |
| 0.005** | |||
| Gender | |||
| Male | 83 (60) | 9 (75) | 0.31 |
| Female | 55 (40) | 3 (25) | |
| FAB subtype | |||
| RA | 63 (45) | 4 (33) | 0.30 |
| RARS | 4 (3) | 1 (8) | |
| RAEB | 66 (48) | 6 (50) | |
| RAEB-T | 5 (4) | 1 (8) | |
| CMML | 0 (0) | 0 (0) | |
| Blood count mean | |||
| Hb (g/dL) | 10.1 | 9.7 | 0.50† |
| ANC (K/mcL) | 3.0 | 2.2 | 0.90† |
| Plt (K/mcL) | 97.0 | 68.3 | 0.85† |
| Cytogenetics | |||
| normal | 62 | 7 | 0.39 |
| -Y only | 2 | 0 | 1.00 |
| -5, del(5q) | 28 | 2 | 1.00 |
| -7, del(7q) | 16 | 1 | 1.00 |
| -17, del(17q) | 4 | 1 | 0.34 |
| -20, del(20q) | 11 | 1 | 1.00 |
| +8 | 17 | 1 | 1.00 |
| complex (≥ 3) | 34 | 2 | 0.74 |
| other | 17 | 1 | 1.00 |
| not available | 4 | 0 | 1.00 |
| IPSS | |||
| low | 22 (17) | 1 (8) | 0.82 |
| INT-1 | 55 (41) | 5 (42) | |
| INT-2 | 36 (27) | 3 (25) | |
| high | 20 (15) | 3 (25) | |
| not available | 5 | 0 |
DNMT3A mutations in MDS
We designed and validated 28 primer pairs covering the coding sequences and splice sites of all 23 exons for DNMT3A. To ensure comprehensive coverage of DNMT3A we performed bidirectional sequencing of all amplicons in the 150 paired DNA samples, producing 16,890 reads. At least one sequencing read (forward or reverse) was obtained for 83% of the 460.1 kbp of target sequence. Only two amplicons failed to produce adequate sequence coverage (amplicon 0001788_083, covering 5’ UTR in exon 1;amplicon 0001788_055, covering exon 14). The Genome Center’s analysis pipeline16 was used to identify sequence variants and we restricted our analysis to changes with predicted translational consequences (i.e., nonsynonymous substitutions, insertion/deletions, and splice site nucleotide changes) and identified 13 mutations in 12 bone marrow samples from 150 MDS samples (8.0% of cases) (Table 2). All 13 mutations were confirmed as somatic by repeat sequencing of the paired tumor/normal DNA samples (Supplemental Figure 1). All 13 mutations were heterozygous and 7 mutations have not been previously described.15, 19 One patient (UPN 379929) had two mutations in different exons. Four of the 13 mutations occurred at amino acid R882, which was also the most common mutation site in AML.15, 19 Another mutation (at amino acid P904) is also recurrent in MDS and was detected in one de novo AML patient.15 To identify tumor samples that might contain a somatic deletion or copy number neutral loss of heterozygosity (LOH) involving the DNMT3A gene, we used the Affymetrix SNP array 6.0 to analyze three samples containing two or more consecutive SNPs that appeared to be heterozygous in the germline (skin) and homozygous in the paired tumor sample. No deletions or LOH events were detected in these three samples (suggesting that the apparent LOH at these isolated SNPs were sequencing artifacts).
Table 2.
DNMT3A Mutations in MDS Patients.
| UPN | Genome coordinate | Nucleotide change | Mutation in cDNA? | Zygosity | Exon | Protein change | SIFT |
|---|---|---|---|---|---|---|---|
| 176267 | 2:25,310,746 | G>A | Yes | Het | 23 | R882H | Damaging |
| 317598 | 2:25,310,746 | G>A | Yes | Het | 23 | R882H | Damaging |
| 457721 | 2:25,310,746 | G>A | NE | Het | 23 | R882H | Damaging |
| 958595 | 2:25,310,746 | G>A | NE | Het | 23 | R882H | Damaging |
| 319179 | 2:25,310,680 | C>T | Yes | Het | 23 | P904L | Damaging |
| 379929 | 2:25,310,680 | C>T | NE | Het | 23 | P904L | Damaging |
| 379929 | 2:25,316,787 | T>G | NE | Het | 19 | L737R | Damaging |
| 988428 | 2:25,316,685 | G>T | Yes | Het | 19 | R771L | Damaging |
| 989739 | 2:25,316,688 | C>G | Yes | Het | 19 | S770W | Damaging |
| 207282 | 2:25,317,045 | C>G | Yes | Het | 18 | S714C | Damaging |
| 917011 | 2:25,320,304 | C>T | Yes | Het | 16 | R635W | Damaging |
| 690100 | 2:25,323,007 | INS [GGCCCTTAGGGCCAGAAGGCTG] | NE | Het | 10 | L422 [PAFWP*] | NE |
| 975079 | 2:25,324,556 | C>T | Yes | Het | 7 | Q237* | NE |
There was no association between mutation detection and the myeloblast count of the banked bone marrow specimen (median blast count was 5% in patients without DNMT3A mutations, vs. 7% in patients with DNMT3A mutations, p=0.31), suggesting that mutations were not missed due to the cellular heterogeneity in the samples. In fact, the mutant allele trace peak heights were consistently equal to the normal allele trace peak heights for each sample and independent of the myeloblast count (p=0.40) (Supplemental Figure 1). This supports our previous observation that the myeloblast count underestimates the size of the mutant clone in MDS16 and further suggests that hematopoiesis is clonal in early stage MDS and that the DNMT3A mutations are acquired early in MDS pathogenesis.
Consequences of DNMT3A mutations
Mutation of the R882 position in DNMT3A has previously been shown to reduce the methyltransferase activity of the protein and reduces its ability to bind DNA.19, 20 The functional consequences of the remaining mutations has not been studied, therefore we applied a computational algorithm to assess the consequences of the remaining missense mutations. The frameshift and nonsense mutations were not testable using this approach. Of the remaining 6 mutations, SIFT uniformly predicted the mutations to be damaging (Table 2), whereas PolyPhen2 predicted all but one, S714C, to be damaging.17, 18 In addition to the R882 position, only the P904 position has been previously reported to be mutated.15 The locations of the remaining missense mutations fall within the methyltransferase domain (Figure 1). DNMT3A mRNA expression measured by microarray was similar in normal CD34+ bone marrow cells and in CD34-selected cells from MDS samples with (n=3) or without DNMT3A mutations (n=20) (Supplemental Figure 2). In 8 samples from MDS patients with DNMT3A mutations, cDNA sequencing detected both the mutant and wildtype alleles, suggesting that both were expressed (Table 2, Supplemental Figure 3).
Figure 1. Location of mutations in the DNMT3A gene.
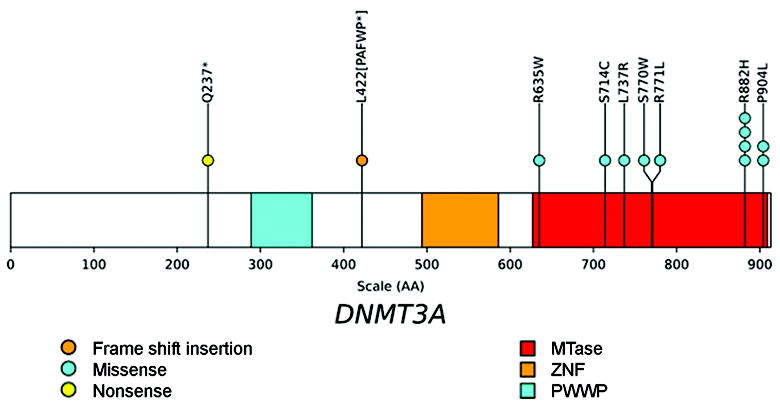
The positions and predicted translational consequences of DNMT3A mutations detected in 150 MDS samples are shown. Each circle is a mutation found in one patient. One patient has two mutations in DNMT3A. The conserved proline-tryptophan-tryptophan-proline (PWWP), zinc finger (ZNF), and methyltransferase (MTase) domains are shown.
*, stop codon.
Clinical significance of DNMT3A mutations in MDS
The clinical characteristics of the 12 patients with DNMT3A mutations were similar to that of the 138 patients without mutations (Table 1). DNMT3A mutations occurred in all FAB subtypes tested and were not associated with a specific karyotype. Six patients with mutant DNMT3A had low or INT-1 IPSS scores, and 6 patients had INT-2 or high scores, indicating that mutations occurred in patients that were low and high risk for AML progression based on their IPSS. We compared the overall survival (OS) of 12 patients with DNMT3A mutations vs. 138 patients without a mutation and observed significantly worse OS in patients with mutations (log-rank p=0.005), although our sample size is small and transplantation status was not considered (Figure 2A). There was also worse event-free survival (EFS) for patients with mutations (log-rank p=0.009) (Figure 2B). 7/12 (58%) of patients with a DNMT3A mutation progressed to AML, compared to 39/138 (28%) of patients without a mutation (Figure 2C, log-rank p=0.007). The median survival of patients with a mutation was 433 days compared to 965 days for patients without a mutation (Table 1, log-rank p=0.005). A multivariate analysis for outcomes could not be performed due to the small sample size of patients with mutations, indicating that a larger cohort from a clinical trial will be needed to definitively address the effect of DNMT3A mutations on outcomes. 8 of 12 MDS patients with DNMT3A mutations received some treatment with the DNMT inhibitors, azacitidine or decitabine (Supplemental Table 1). The small sample size and non-uniform treatment precludes an assessment of the impact of DNMT3A mutations on response to DNMT inhibitors in this cohort.
Figure 2. Survival analysis of MDS patients with DNMT3A mutations.
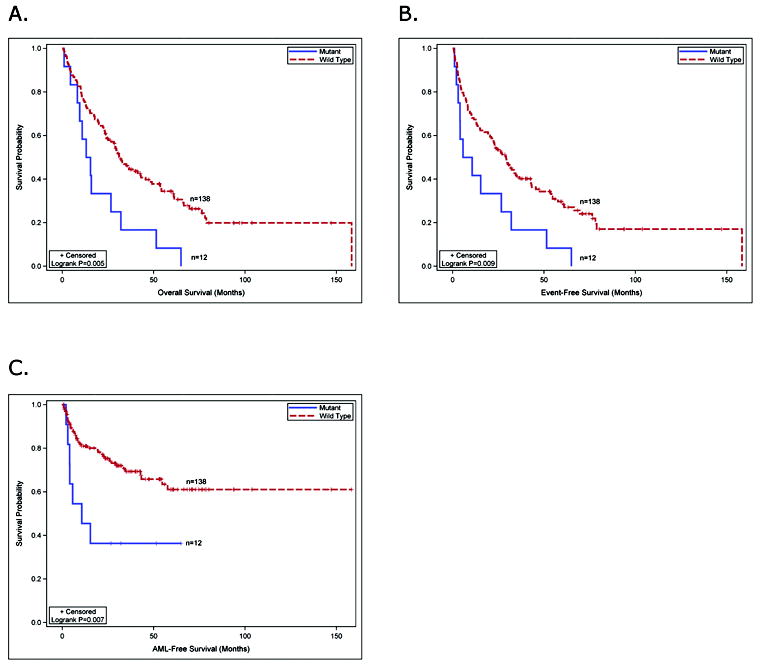
(A) the overall survival of MDS patients with DNMT3A mutations compared to MDS patients without DNMT3A mutations. (B) the event-free survival of MDS patients with or without DNMT3A mutations. (C) progression to AML in MDS patients with or without DNMT3A mutations.
No novel coding SNPs were detected in DNMT3A. We examined the allele and genotype frequencies of two known synonymous SNPs in our cohort (rs2276598, rs41284843) and found no significant differences, comparing either MDS patients to race-matched control subjects (Supplemental Table 2) or comparing MDS patients with DNMT3A mutations to MDS patients without DNMT3A mutations (Supplemental Table 3).
DISCUSSION
In this study, we identified 13 somatic mutations in the DNA methyltransferase gene DNMT3A in 12/150 (8.0%) de novo MDS patients. The mutations were heterozygous and were computationally predicted to alter the protein function. A mutation hot spot at amino acid R882 occurred in 4 MDS samples and is located in the methyltransferase domain of the protein. The mutations occurred in all MDS FAB subtypes (excluding CMML, which was not examined) as well as IPSS scores ranging from 0-3. MDS patients harboring a mutation in DNMT3A appear to have worse overall survival and more rapid progression to AML, suggesting that this mutation may have prognostic value in MDS.
DNMT3A, in conjunction with DNMT3B, is critical for proper de novo DNA methylation and development of mammals. Mice that are homozygous null for Dnmt3a-/- are born runted, die of aganglionic megacolon, and males lack germ cells.21 Conditional knockout of Dnmt3a in the germ cells showed lack of methylation of normally imprinted genes H19 and Dlk1-Gtl2.22 Dnmt3b-/- mice die at 9.5 days post coitus (dpc) and display demethylation of minor satellite repeats, and the double mutant Dnmt3a-/-/Dnmt3b-/- mice are embryonic lethal at day 8.5 dpc with global demethylation.21 Conditional deletion of Dnmt3a-/- or Dnmt3b-/- individually in murine Kit+/Lineage-/Sca+ (KLS) bone marrow cells did not affect myeloid colony formation, multilineage differentiation, or self-renewal, but did alter methylation patterns in differentiating hematopoietic cells.23 In contrast, Dnmt3a-/-/Dnmt3b-/- KLS cells were unable to contribute to long-term hematopoiesis in a murine transplantation model, suggesting that either Dnmt3a-/- or Dnmt3b-/- is necessary to maintain hematopoietic stem cell self-renewal.23 It remains unclear how acquired mutations in DNMT3A may alter hematopoiesis, but murine knockout models suggest that simple loss of function may not be the predominant mechanism. In fact, overexpression of a Dnmt3a isoform (Dnmt3a1) was recently shown to increase penetrance and reduce latency of _PML/RARA_-initiated myeloid leukemia in a murine transplantation model.24
All the DNMT3A mutations in MDS are heterozygous and it is not known whether these nucleotide mutations result in a loss of function, gain of function, or have a dominant negative property. The missense mutations involve highly conserved residues within the methyltransferase domain of DNMT3A, suggesting that they may not be simple loss of function mutations but may confer a novel protein function. In contrast, the frameshift and nonsense mutations occur upstream of the methyltransferase domain and are likely to be loss of function mutations. Although previous reports suggest that the R882 mutation has loss of function properties,19, 20 the R882 mutant was not tested in the presence of the wild-type allele, which may alter the mutant phenotype. The DNA and cDNA sequencing data revealed that the mutant DNMT3A allele was present and expressed in nearly all cells in the MDS samples, even though the myeloblast count was far less than <30% for most samples. This suggests that DNMT3A mutations are very early genetic events in MDS, and may confer a clonal advantage to cells bearing a mutation in this gene. Ultimately, modeling these mutations in an organism will be required to understand their transforming potential.
This report and the recent identification of mutations and deletions in histone modifying genes (UTX, ASXL1, TET2, EZH2) in MDS patients provides compelling evidence that epigenetic alterations contribute to MDS pathogenesis.25-32 UTX is a histone demethylase, EZH2 is a histone methyltransferase, ASXL1 helps recruit polycomb and trithorax complexes to chromatin, and TET2 converts methylcytosine to hydroxymethylcytosine.26, 33-37 Many, but not all, of the somatic variants in TET2, ASXL1, and EZH2 result in frameshifts and nonsense mutations, suggesting that they result in loss of function. In addition, ATRX is a member of the SWI/SNF group of proteins that associate with chromatin and ATRX is mutated in a rare form of MDS that is associated with alpha-thalassemia (ATMDS).38 Given that these genes affect epigenetic pathways, it will be important to know whether mutations in these genes are mutually exclusive, or whether the genes could have nonoverlapping functional consequences. Likewise, it is unknown whether mutations in different genes will impact the expression of a common set of genes, or whether each mutation results in a unique gene expression signature. These questions will need to be addressed using larger cohorts of patients that are comprehensively genotyped for mutations in all the genes.
Collectively, the growing list of somatic mutations in MDS that affect histone modifying proteins, and now the DNA methyltransferase gene DNMT3A, may provide a potential mechanism for the clinical activity observed in some MDS patients treated with histone deacetylase inhibitors and DNA methyltransferase inhibitors. The response to hypomethylating agents will be an important association to explore in large retrospective studies, and prospectively in future clinical trials. The potential utility of DNMT3A mutation status for determining prognosis in de novo MDS is compelling, but will need to be validated in larger clinical studies. If reproduced, DNMT3A mutation status could help risk stratify de novo MDS patients for more aggressive treatment early in their disease course, such as allogeneic transplant for eligible candidates.
Supplementary Material
1. Supplemental Figure 1. Sequence traces.
(A) Chromatograms from capillary sequencing of paired normal (skin biopsy) and tumor (bone marrow aspirate) samples at 13 positions in DNMT3A that are somatically mutated in MDS samples. (B) The peak heights of mutant:wildtype DNMT3A alleles detected by sequencing is plotted as a function of the bone marrow blast count for each sample. There is no correlation between mutant allele proportion and blast count. The mutant:wildtype allele ratios are ~50% in all cases, implying that there is a single dominant clone containing a heterozygous DNMT3A mutation.
2. Supplemental Figure 2. DNMT3A expression.
Expression of DNMT3A RNA in CD34+ selected bone marrow cells from normal subjects (n=6), MDS patients with wild type DNMT3A (n=20), or MDS patients with mutant DNMT3A (n=3) was measured on Affymetrix U133plus2 arrays. Results are shown for three probesets targeting DNMT3A. The gene is expressed at low levels in most samples with no significant differences between the groups.
3. Supplemental Figure 3. Expression of mutant DNMT3A cDNA.
Sequencing results from pooled tumor cDNA generated from six of the samples shown in Supplemental Figure 1. The mutant:wildtype allele ratios are ~50% in all cases, suggesting that the mutant and wildtype alleles are expressed at similar levels.
4. Supplemental Table 1. Clinical characteristics of MDS patients with DNMT3A mutations.
5. Supplemental Table 2. Association between germline DNMT3A polymorphisms and somatic DNMT3A mutation status in MDS patients.
6. Supplemental Table 3. Association between germline DNMT3A polymorphisms in MDS cases vs. normal controls.
Acknowledgments
This work was supported by NIH grants R01HL082973 (Graubert), RC2HL102927 (Graubert), U54HG003079 (Wilson), P01CA101937 (Ley), and a Howard Hughes Medical Institute Physician-Scientist Early Career Award (Walter).
Footnotes
AUTHOR CONTRIBUTIONS MW, LD, TL, and TG designed the study. DS, JS, MG, MM, RF, HS, JKV, and MO performed research and generated data. MW, LD, DS, JS, JB, PW, JD, EM, RW, TL, and TG analyzed data. MW, LD, DS, and TG wrote the paper.
CONFLICT OF INTEREST DISCLOSURES The authors declare no competing financial interests.
References
- 1.Esteller M. Epigenetics in cancer. N Engl J Med. 2008 Mar 13;358(11):1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 2.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2009 Jan;31(1):27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ting AH, McGarvey KM, Baylin SB. The cancer epigenome--components and functional correlates. Genes Dev. 2006 Dec 1;20(23):3215–3231. doi: 10.1101/gad.1464906. [DOI] [PubMed] [Google Scholar]
- 4.Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010 Jan 19;17(1):13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Figueroa ME, Skrabanek L, Li Y, Jiemjit A, Fandy TE, Paietta E, et al. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood. 2009 Oct 15;114(16):3448–3458. doi: 10.1182/blood-2009-01-200519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang Y, Dunbar A, Gondek LP, Mohan S, Rataul M, O’Keefe C, et al. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood. 2009 Feb 5;113(6):1315–1325. doi: 10.1182/blood-2008-06-163246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cashen AF, Schiller GJ, O’Donnell MR, DiPersio JF. Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J Clin Oncol. 2009 Feb 1;28(4):556–561. doi: 10.1200/JCO.2009.23.9178. [DOI] [PubMed] [Google Scholar]
- 8.Chitambar CR, Libnoch JA, Matthaeus WG, Ash RC, Ritch PS, Anderson T. Evaluation of continuous infusion low-dose 5-azacytidine in the treatment of myelodysplastic syndromes. Am J Hematol. 1991 Jun;37(2):100–104. doi: 10.1002/ajh.2830370207. [DOI] [PubMed] [Google Scholar]
- 9.Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006 Apr 15;106(8):1794–1803. doi: 10.1002/cncr.21792. [DOI] [PubMed] [Google Scholar]
- 10.Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002 May 15;20(10):2429–2440. doi: 10.1200/JCO.2002.04.117. [DOI] [PubMed] [Google Scholar]
- 11.Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 12.Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, et al. Induction of tumors in mice by genomic hypomethylation. Science. 2003 Apr 18;300(5618):489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 13.Laird PW, Jackson-Grusby L, Fazeli A, Dickinson SL, Jung WE, Li E, et al. Suppression of intestinal neoplasia by DNA hypomethylation. Cell. 1995 Apr 21;81(2):197–205. doi: 10.1016/0092-8674(95)90329-1. [DOI] [PubMed] [Google Scholar]
- 14.Xu GL, Bestor TH, Bourc’his D, Hsieh CL, Tommerup N, Bugge M, et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999 Nov 11;402(6758):187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- 15.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010 Dec 16;363(25):2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graubert TA, Payton MA, Shao J, Walgren RA, Monahan RS, Frater JL, et al. Integrated genomic analysis implicates haploinsufficiency of multiple chromosome 5q31.2 genes in de novo myelodysplastic syndromes pathogenesis. PLoS One. 2009;4(2):e4583. doi: 10.1371/journal.pone.0004583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002 Mar;12(3):436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002 Sep 1;30(17):3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamashita Y, Yuan J, Suetake I, Suzuki H, Ishikawa Y, Choi YL, et al. Array-based genomic resequencing of human leukemia. Oncogene. 2010 Jun 24;29(25):3723–3731. doi: 10.1038/onc.2010.117. [DOI] [PubMed] [Google Scholar]
- 20.Gowher H, Loutchanwoot P, Vorobjeva O, Handa V, Jurkowska RZ, Jurkowski TP, et al. Mutational analysis of the catalytic domain of the murine Dnmt3a DNA-(cytosine C5)-methyltransferase. J Mol Biol. 2006 Mar 31;357(3):928–941. doi: 10.1016/j.jmb.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 21.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999 Oct 29;99(3):247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 22.Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, et al. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004 Jun 24;429(6994):900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- 23.Tadokoro Y, Ema H, Okano M, Li E, Nakauchi H. De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J Exp Med. 2007 Apr 16;204(4):715–722. doi: 10.1084/jem.20060750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subramanyam D, Belair CD, Barry-Holson KQ, Lin H, Kogan SC, Passegue E, et al. PML-RAR{alpha} and Dnmt3a1 cooperate in vivo to promote acute promyelocytic leukemia. Cancer research. Nov 1;70(21):8792–8801. doi: 10.1158/0008-5472.CAN-08-4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009 May 28;360(22):2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 26.Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010 Aug;42(8):722–726. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- 27.Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009 Jun 18;113(25):6403–6410. doi: 10.1182/blood-2009-02-205690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nature genetics. 2009 Jul;41(7):838–842. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 29.Mohamedali AM, Smith AE, Gaken J, Lea NC, Mian SA, Westwood NB, et al. Novel TET2 mutations associated with UPD4q24 in myelodysplastic syndrome. J Clin Oncol. 2009 Aug 20;27(24):4002–4006. doi: 10.1200/JCO.2009.22.6985. [DOI] [PubMed] [Google Scholar]
- 30.Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nature genetics. 2010 Aug;42(8):665–667. doi: 10.1038/ng.620. [DOI] [PubMed] [Google Scholar]
- 31.Tefferi A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Patnaik MM, et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia. 2009 Jul;23(7):1343–1345. doi: 10.1038/leu.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viguie F, Aboura A, Bouscary D, Ramond S, Delmer A, Tachdjian G, et al. Common 4q24 deletion in four cases of hematopoietic malignancy: early stem cell involvement? Leukemia. 2005 Aug;19(8):1411–1415. doi: 10.1038/sj.leu.2403818. [DOI] [PubMed] [Google Scholar]
- 33.Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, et al. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science (New York, NY) 2007 Oct 19;318(5849):447–450. doi: 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- 34.Laible G, Wolf A, Dorn R, Reuter G, Nislow C, Lebersorger A, et al. Mammalian homologues of the Polycomb-group gene Enhancer of zeste mediate gene silencing in Drosophila heterochromatin and at S. cerevisiae telomeres. Embo J. 1997 Jun 2;16(11):3219–3232. doi: 10.1093/emboj/16.11.3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010 Dec 9;468(7325):839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Acquaviva C, Gelsi-Boyer V, Birnbaum D. Myelodysplastic syndromes: lost between two states? Leukemia. 2010 Jan;24(1):1–5. doi: 10.1038/leu.2009.157. [DOI] [PubMed] [Google Scholar]
- 37.Sinclair DA, Milne TA, Hodgson JW, Shellard J, Salinas CA, Kyba M, et al. The Additional sex combs gene of Drosophila encodes a chromatin protein that binds to shared and unique Polycomb group sites on polytene chromosomes. Development. 1998 Apr;125(7):1207–1216. doi: 10.1242/dev.125.7.1207. [DOI] [PubMed] [Google Scholar]
- 38.Steensma DP, Higgs DR, Fisher CA, Gibbons RJ. Acquired somatic ATRX mutations in myelodysplastic syndrome associated with alpha thalassemia (ATMDS) convey a more severe hematologic phenotype than germline ATRX mutations. Blood. 2004 Mar 15;103(6):2019–2026. doi: 10.1182/blood-2003-09-3360. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1. Supplemental Figure 1. Sequence traces.
(A) Chromatograms from capillary sequencing of paired normal (skin biopsy) and tumor (bone marrow aspirate) samples at 13 positions in DNMT3A that are somatically mutated in MDS samples. (B) The peak heights of mutant:wildtype DNMT3A alleles detected by sequencing is plotted as a function of the bone marrow blast count for each sample. There is no correlation between mutant allele proportion and blast count. The mutant:wildtype allele ratios are ~50% in all cases, implying that there is a single dominant clone containing a heterozygous DNMT3A mutation.
2. Supplemental Figure 2. DNMT3A expression.
Expression of DNMT3A RNA in CD34+ selected bone marrow cells from normal subjects (n=6), MDS patients with wild type DNMT3A (n=20), or MDS patients with mutant DNMT3A (n=3) was measured on Affymetrix U133plus2 arrays. Results are shown for three probesets targeting DNMT3A. The gene is expressed at low levels in most samples with no significant differences between the groups.
3. Supplemental Figure 3. Expression of mutant DNMT3A cDNA.
Sequencing results from pooled tumor cDNA generated from six of the samples shown in Supplemental Figure 1. The mutant:wildtype allele ratios are ~50% in all cases, suggesting that the mutant and wildtype alleles are expressed at similar levels.
4. Supplemental Table 1. Clinical characteristics of MDS patients with DNMT3A mutations.
5. Supplemental Table 2. Association between germline DNMT3A polymorphisms and somatic DNMT3A mutation status in MDS patients.
6. Supplemental Table 3. Association between germline DNMT3A polymorphisms in MDS cases vs. normal controls.