Structural evidence for the rare tautomer hypothesis of spontaneous mutagenesis (original) (raw)
Abstract
Even though high-fidelity polymerases copy DNA with remarkable accuracy, some base-pair mismatches are incorporated at low frequency, leading to spontaneous mutagenesis. Using high-resolution X-ray crystallographic analysis of a DNA polymerase that catalyzes replication in crystals, we observe that a C•A mismatch can mimic the shape of cognate base pairs at the site of incorporation. This shape mimicry enables the mismatch to evade the error detection mechanisms of the polymerase, which would normally either prevent mismatch incorporation or promote its nucleolytic excision. Movement of a single proton on one of the mismatched bases alters the hydrogen-bonding pattern such that a base pair forms with an overall shape that is virtually indistinguishable from a canonical, Watson-Crick base pair in double-stranded DNA. These observations provide structural evidence for the rare tautomer hypothesis of spontaneous mutagenesis, a long-standing concept that has been difficult to demonstrate directly.
Keywords: crystal structure, replication fidelity, mispair , polymerase structure
High-fidelity polymerases replicate double-stranded DNA with remarkable accuracy (1). Fidelity is achieved by a successive series of conformational changes and molecular recognition events encoded at different sites on the polymerase surface such that mismatches are either prevented from incorporating or are excised within a few nucleotides past their incorporation point (2–5). At the site of covalent incorporation, shape complementarity between the polymerase surface and the edges of correctly paired bases is the dominant mechanism that determines specificity (6, 7). Here, mismatched base pairs or lesions that do not conform to this stereochemical constraint misalign their incoming triphosphate moiety relative to the 3′ OH of the growing primer terminus, leading to rejection of the incorrect or damaged nucleotides (2–4). However, modified bases that maintain the stereochemistry of cognate base-pair edges are readily incorporated (6–8). Nevertheless, polymerases do incorporate mismatched nucleotide base pairs at low frequency, leading to spontaneous mutagenesis (1).
The mechanism by which spontaneous replication errors occur has long been the subject of intense speculation. In their second paper on the structure of DNA, Watson and Crick recognized that tautomerization alters the hydrogen-bonding patterns and therefore could enable mismatches to assume the structure of canonical base pairs (9). This notion was elaborated in the rare tautomer hypothesis of spontaneous mutagenesis, which states that mutations arise through the formation of high-energy tautomers at low frequency (8, 10). However, it has been challenging to obtain direct structural evidence for this mechanism. In the absence of polymerase, mismatches do not adopt a canonical base-pair structure in DNA (5, 11). Recently, a T•G mismatch has been observed to adopt a canonical base-pair structure in a polymerase, due to an ionization event, demonstrating that noncanonical hydrogen-bonding pattern can arise in a polymerase (12). Here we present the structure of an C•A mismatch in the active site of a high-fidelity DNA polymerase, the Bacillus stearothermophilus DNA polymerase I large fragment (Bacillus fragment, BF), an enzyme that has been used extensively to study the structural enzymology of nucleotide incorporation (13–16).
The C•A mismatch has the advantage that only tautomers give rise to cognate base-pair mimicry, whereas ionization leads to “wobble” base paring (Fig. 1). We show that under conditions which stabilize an enzyme conformation that places a nucleotide at the site of incorporation, the C•A mismatch adopts a tautomeric cognate base-pair shape, whereas otherwise it forms an ionized wobble that cannot be incorporated. Wobble base pairs have been observed in isolated DNA by X-ray crystallography (17) or NMR (18). We observe the C•A mismatch within the double helix past the position of incorporation, where it adopts a cognate base-pair conformation or a wobble, depending on site location on the polymerase surface. These structures unambiguously demonstrate that tautomeric base pairs can form in the polymerase active site, providing strong support of the rare tautomer hypothesis through direct structural evidence.
Fig. 1.

Inferred protonation states of C•A base pairs observed in the structures. In their canonical tautomeric state C and A do not pair (middle), because the two extracyclic amines clash. If either C or A tautomerizes (asterisk), a hydrogen-bonded base pair that mimicks a cognate shape can form (top). If A ionizes, a wobble base pair can form (bottom) (17, 18). Hydrogen bond donors and acceptors are colored blue and pink respectively.
Results
Five sites at which fidelity filters are encoded (19–22) have been identified for BF (15) (Fig. 2): the preinsertion site where the incoming DNA template strand resides; the insertion site where the incoming nucleotide triphosphate pairs with the template; the catalytic site where the metals and catalytic groups are aligned; the postinsertion site where the 3′ end of the nascent duplex strand resides; and a four-base-pair DNA duplex-binding region. Recognition events at the insertion site are the most critical for replication fidelity (1, 7), and results from the binding energy arising from shape complementarity between the enzyme and cognate base pairing of the incoming nucleotide with the template strand (23), and precise alignment of the catalytic groups on the enzyme with the reactive groups on the substrates (24, 25). This process involves concerted motions of the polymerase O helix (4, 5), base pairing of the incoming nucleotide with the template strand, and positioning of the triphosphate moiety next to the binuclear metal center in the polymerase catalytic site and the 3′ hydroxyl of the nascent strand (23, 24).
Fig. 2.

DNA polymerase replication fidelity filters. Shaded areas correspond to fidelity filters: preinsertion site (n, orange), insertion site (n, blue), catalytic site (magenta), postinsertion site (_n_-1, pink), and DNA duplex-binding region (_n_-2 to _n_-5, gray). DNA primer (copper) and template strands (orange) are also shown. The O helix transitions from an open (magenta) through an ajar (gray) to a closed (blue) conformation. Cognate-shaped base pairs (blue) are positioned for catalysis in the closed state. Noncanonical shapes (gray) tend to be selected against in the ajar conformation. The polymerase makes hydrogen bonds with the minor groove of base pairs positioned at sites _n_-1 to _n_-5 in the duplex-binding region following incorporation. This figure combines information derived from four structures: open (1L3U) (15), ajar (3HP6) (29), and closed (2HVI and 3EZ5) (34, 42).
The C•A Mismatches Can Form a Cognate Base-Pair Shape at the Insertion Site.
Substitution of Mg2+ with the mutagenic Mn2+ ion in the active site significantly enhances the misincorporation of C•A mismatches (26, 27). Here we compare high-resolution structures of C•A mismatches positioned at the insertion site in the presence of Mg2+ or Mn2+ (Fig. 3, Tables S1 and S2, and Fig. S1). In the 1.58 Å resolution Mg2+ structure, we find that the C•A mismatch forms a noncognate wobble (Fig. 1) which does not match the shape of a T•A base pair placed in the same position (Fig. 3 A_–_C). Furthermore, the polymerase O helix does not adopt the fully closed conformation associated with productive catalysis (15, 21, 28), but remains in an “ajar” conformation that tends to prevent noncognate shapes from moving into the closed conformation necessary for the chemical incorporation step (29). The structure of the triphosphate moiety also is distorted from that observed in a cognate base pair. Taken together, these differences between cognate and mismatch recognition events are expected to interfere with catalysis, preventing mismatch incorporation.
Fig. 3.

Comparison of C•A mismatch and T•A cognate base pairs placed at the polymerase insertion site. (A), (B) The C•A wobble (green) and T•A base (gray) pair obtained in the presence of Mg2+. For the C•A wobble pair, the O helix adopts the ajar conformation (A), the triphosphate is distorted, and the catalytic site is incompletely assembled (B). (D), (E) The C•A cognate shape (magenta) obtained in the presence of Mn2+. Comparison with a T•A base pair shows that the O helix is closed (D), the triphosphate is undistorted, and the active site fully assembled (E). (C), (F) Two views of composite omit maps of the C•A base pair (contoured at 1.2_σ_, C•A wobble; contoured at 2_σ_, C•A cognate) (green, Mg2+; purple, Mn2+). The presence of Mn2+ is confirmed by anomalous difference map (red, contoured at 4_σ_). (G), (H) Superposition of C•A wobble and C•A cognate at two different views showing the structural differences between the wobble and cognate conformations of this mismatch. (I) Variations of minor groove angles of C•A mismatch structures (wb, wobble; m, cognate mimic) or average cognate, Watson-Crick base-pair structures (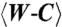 ) captured at five different positions. _λ_primer and _λ_template are defined as the angle between the glycosidic bond of primer or template nucleotide and a line between the C1′ atoms of the base pair. Complete tables of all nine base-pair parameters are included (Table S3). Analysis shown here is based on molecule 1 of the two molecules in the asymmetric unit; molecule 2 is described in Table S3. The capture of a nucleotide at the insertion site involves the use of dideoxy analogs (34). Additional structures were determined with a 2′-deoxycytidine triphosphate which confirms the results described here (Fig. S1).
) captured at five different positions. _λ_primer and _λ_template are defined as the angle between the glycosidic bond of primer or template nucleotide and a line between the C1′ atoms of the base pair. Complete tables of all nine base-pair parameters are included (Table S3). Analysis shown here is based on molecule 1 of the two molecules in the asymmetric unit; molecule 2 is described in Table S3. The capture of a nucleotide at the insertion site involves the use of dideoxy analogs (34). Additional structures were determined with a 2′-deoxycytidine triphosphate which confirms the results described here (Fig. S1).
By contrast, in the presence of Mn2+ (observed at 1.59 Å resolution), the C•A mismatch exhibits all the hallmarks of cognate base-pair recognition and incorporation (Fig. 3 D_–_F): shape matching, triphosphate alignment, and O helix closure. Furthermore, in this cognate conformation the cytosine O2 atom forms a hydrogen bond with a water molecule that is anchored by three polymerase side chains (Fig. 4A). This hydrogen bond is absent in the wobble base pairs (Fig. 4B), but its equivalent is found in all four cognate base pairs (Fig. 4 C_–_F), indicating that it represents a critical feature of correctly formed base-pair edge recognition at the insertion site.
Fig. 4.

A water mediated hydrogen bond encodes edge recognition of cognate base-pair shapes. (A) C•A cognate shape mimic (Mn2+, magenta); (B) C•A wobble (Mg2+, green); (C) T•A; (D) A•T; (E) G•C; (F) C•G (from previously published structure; PDB code, 2HVI) (34). Composite omit maps (gray) at 1.5_σ_ (A), (C_–_E) and 1.2_σ_ (B) are shown around the base pairs and the anchored water molecule. Dashed lines (black) indicate hydrogen bonds.
The Cognate Shape of the Mismatch Corresponds to a Tautomer.
Only tautomerization of C or A results in a hydrogen-bonding pattern that enables formation of a base pair mimicking the cognate A•T shape (10) (Fig. 1). However, a similar shape also could arise from deamination of the cytosine to form uracil, which is rare, but can occur (in the range of 10-10 sec-1 by in vitro measurement) (30). We carried out mass spectrometric analysis of the crystallization drops and determined that cytosine, not uracil, is present (Fig. S2). The mimicry of the T•A shape by a C•A mismatch is therefore the consequence of stabilizing a noncanonical tautomerized state, consistent with the original proposal of the rare tautomer hypothesis (9, 10).
Structures of the C•A Mismatch Placed at the DNA Duplex Region.
Noncanonical protonation states of the C•A base pair also are observed at the other fidelity filters. At the postinsertion site and duplex DNA-binding region (Fig. 5), in addition to steric shape complementarity, readout of hydrogen bonds in the minor groove contributes to edge recognition (1, 23). Using successive rounds of nucleotide incorporation in the crystal (14, 15), we have placed the C•A mismatch at the postinsertion site, as well as the _n_-3 and _n_-4 sites of the DNA duplex-binding region. At all three locations the two nucleotides form base pairs, indicating formation of noncanonical protonation states. At the _n_-3 position the C•A mismatch forms a near-cognate interaction and accordingly is tautomerized (Fig. 5D), whereas at the postinsertion (Fig. 5 A_–_C) and _n_-4 (Fig. 5E) sites a wobble is adopted corresponding to ionization events. At the postinsertion site, the wobble results in a 0.5 Å displacement of the 3′ hydroxyl. Otherwise, distortions are moderate compared to other mismatches captured at this position (16). The effects of the wobble at the _n_-4 position are also mild and do not induce a “memory effect” by transmitting distortions back to the active site, as has been observed in several mismatches (16). At the _n_-6 position the C•A mismatch forms a wobble base pair (Fig. 5F) similar to the structure obtained in free DNA dodecamer (17) and is no longer interacting with the DNA polymerase. These observations indicate that the altered hydrogen bonds between C and A at the filter sites arise from local interactions between the mismatch and the polymerase.
Fig. 5.

Comparison of C•A mismatch and T•A cognate base-pair structures in the duplex region. (A_–_C) The C•A wobble base pair captured at the postinsertion site (1.53 Å resolution) showing overall structure (A), minor-groove interactions (B), and composite omit map (contoured at 1.8_σ_) around the mismatch (C). The next template base is disordered. (D), (E) C•A adopts a near-cognate shape at the _n_-3 (D) position (1.65 Å resolution) and a wobble shape at the _n_-4 (E) position (1.65 Å resolution). At both positions, minor groove interactions are maintained. (F) The C•A wobble observed at the _n_-6 position (1.60 Å resolution) where there are no contacts between the duplex DNA and the polymerase.
Discussion
The tautomeric form of the C•A mismatch mimics the shape of a cognate base pair in the insertion site prior to incorporation. Although this tautomeric form is of higher energy than the canonical protonation state (31), the local environment of the DNA polymerase can contribute to its stabilization in two ways: through binding interactions with features present in the tautomeric cognate stereochemistry but absent in the ground or ionized states, and by electrostatically altering the intrinsic equilibrium between the tautomers (32, 33). The first effect is evident in the structure of the C•A mismatch (Fig. 4). In its cognate shape, the cytosine O2 atom makes a hydrogen bond with a tightly bound water, whereas the wobble cannot present this acceptor in the appropriate geometry. This interaction is present in all four cognate base pairs placed at this site, and is equivalent to the minor groove readout mechanism that recognizes cognate base pairs at the postinsertion site and beyond (1). This critical water is tightly bound by three residues that are highly conserved in the A-family polymerases (to which BF belongs), replaced by other hydrogen-bonding groups in the C-, X-, and Y-family polymerases, but absent in the B-family polymerases (Fig. 6). This conservation pattern suggests that mismatch incorporation by polymerase-mediated perturbation of tautomeric equilibria also is present in polymerases other than BF, and varies with family, perhaps influencing their degree of fidelity in accordance with biological function.
Fig. 6.

Insertion sites of representative members of five DNA polymerase families. (A) Superposition of three members of the A-family DNA polymerases: BF (yellow; PDB code, 2HVI) (34), Thermus aquaticus DNA polymerase I large fragment (cyan; 3KTQ) (21), and T7 bacteriophage DNA polymerase (pink;1T7P) (28). The interactions between the water molecule and the base, and the three anchoring protein residues are conserved in all three complexes. (B) A member of the C-family DNA polymerase, Geobacillus kaustophilus DNA polymerase PolC (3F2B) (43). A water molecule makes similar interaction with the incoming nucleotide base which is coordinated by a single histidine instead of the anchoring side chains. (C) A member of the X-family DNA polymerase, human DNA polymerase beta (2FMP) (44). The incoming nucleotide is hydrogen-bonded directly to an asparagine side chain instead of a water. Similar interactions are also present in another member of the family, human DNA polymerase lambda (1XSN) (45). (D) A member of the Y-family DNA polymerase, Sulfolobus solfataricus DNA polymerase IV (Dpo4) (2AGQ) (46). The water molecule contacting the nucleotide base is coordinated by a tyrosine instead of three anchoring residues. Similar interactions are also present in another member of the family, human DNA polymerase iota (1ZET) (47) (reviewed in ref. 48). (E) A member of the B-family DNA polymerase, Enterobacteria phage RB69 DNA polymerase (3NCI) (49). The base-pair edges are read out by Van der Waals interactions only, perhaps augmented by weak electrostatic interactions mediated by the glycine and the two ring protons of tyrosine (49). Similar interactions are also present in another member of the family, Bacillus phage phi29 DNA polymerase (2PYJ) (22). The selection of the structure for a representative polymerase family was based on resolution of the ternary complex.
The mutagenic effect of substituting Mn2+ for Mg2+ is probably also the consequence of enhancing binding to cognate base-pair shapes. Mn2+ stabilizes the formation of the closed state relative to the ajar state (Fig. 3) thereby enhancing the binding energy of the insertion site.
The intrinsic equilibrium of the tautomeric states is a function of the p_K_ a values of the two groups that change protonation states. These values are affected by the relative stabilities of dipoles within the base heterocycle, and therefore are strongly dependent on the local electrostatic environment (33). Accordingly, the DNA polymerase could affect the spontaneous mutagenesis rate by shaping the local electrostatic field.
The results obtained here demonstrate that at least in one case spontaneous mutagenesis (prior to subsequent DNA repair processes) arises as a consequence of base tautomerization that enables a mismatch to assume the shape of a cognate base pair, consistent with the original rare tautomer hypothesis (9, 10). Such tautomers can be stabilized by binding interactions that favor cognate stereochemical shapes, conformational equilibria that affect such binding energies, the chemical character of the residues encoding the readout, local electrostatics that alter the intrinsic tautomerization equilibria, and the chemical character of the bases themselves. These effects provide a general framework within which the observed differences in spontaneous mutagenesis frequencies of DNA polymerases, their mutants, and individual base pairs can be rationalized.
Methods
Preparation of Protein.
Wild-type and D598A/ F710Y mutant proteins were purified as described (13). The D598A/F710Y double mutant (34) was used to capture ternary complexes. D598A destabilizes a crystal contact thereby favoring the closed state in the crystal (15). F710Y facilitates incorporation of a 2′,3′-dideoxynucleotide chain terminator (35) which prevents further incorporation and therefore traps ternary complexes before chemistry (some other members of the A family DNA polymerases have a wild-type Tyr at the equivalent position for example, T7 DNA polymerase) (36). Wild-type protein was used to capture C•A at the postinsertion site, _n_-3, _n_-4, and _n_-6 positions.
BF Primer-Template Complexes with Nucleotides Placed at the Insertion Site (Crystal Form II).
Unincorporated 2′,3′-dideoxynucleoside triphosphates were trapped at the insertion site of complexes ddCTP•dA, ddTTP•dA, ddGTP•dC, and ddATP•dT by incubation of protein, primer-template duplexes (protein:DNA in a 1∶3 molar ratio), dideoxynucleotides (10 mM), and Mg2+ or Mn2+ sulphate (20 mM) for 1 h at room temperature. Template sequences were designed such that a single nucleotide is incorporated to form dideoxy primer terminus thereby trapping the next nucleotide at the insertion site (Table S2). These reactions were used to set up crystallization as described previously (15) to obtain Crystal Form II crystals. dCTP was trapped at the insertion site by exchanging ddCTP, first soaking the crystals of ddCTP•dA (grown in the presence of both Mg2+ and Mn2+) in a stabilization solution in the absence of nucleotides (60% saturated (NH4)2SO4, 2.5% 2-methyl-2,4-pentanediol, 100 mM MES 2-(N-Morpholino)ethanesulfonic acid pH 5.8, 30 mM MnSO4 and 30 mM MgSO4) at 17 °C for 2 d to remove the ddCTP, followed by soaking in the stabilization solution with 21.5 mM dCTP at 17 °C for 36 h to add dCTP (Fig. S1).
BF Complexes with Mismatches Incorporated into the Primer-Template Duplex (Crystal Form I).
The C•A mismatch positioned at the postinsertion site were obtained either by catalysis of primer-template in the crystal or by crystallization of a primer-template complex with a mismatch at the 3′ primer terminus (Table S2). BF-DNA binary complexes were formed by incubating wild-type protein with the respective primer-template duplex (protein:DNA in a 1∶3 molar ratio) in 20 mM MgSO4 on ice for 1 h, followed by setting up the crystallization as described to obtain Crystal Form I. To incorporate a C•A mismatch by catalysis, crystals were soaked in a stabilization solution (51.5% saturated (NH4)2SO4, 2.5% 2-methyl-2,4-pentanediol, 100 mM MES pH 5.8) supplemented with 30 mM dCTP and 60 mM MnSO4 at 17 °C for 24 h. There were no discernible structural differences between C•A mismatch positioned at the postinsertion site using either method (We present data obtained from the catalysis experiment, because it was collected to higher resolution.). C•A mismatches were positioned at various sites in the DNA duplex region by adding nucleotides in various combinations and incubating in stabilization solution at 17 °C for 24 h to stimulate catalysis in the crystal of the C•A mismatch placed at the postinsertion site (obtained by either method): _n_-3, 15 mM dATP and dCTP; _n_-4, 10 mM dATP, dCTP, and dGTP; _n_-6, 7.5 mM dATP, dCTP, dGTP, and dTTP (Table S2).
Diffraction Data Collection and Structure Determination.
Crystals were flash frozen in liquid nitrogen either directly out of the crystallization drop (Crystal Form II) or after soaking in a cryoprotectant solution (60% saturated (NH4)2SO4, 100 mM MES pH 5.8, 24% sucrose) (Crystal Form I). Data were collected at SIBYLS and SER-CAT beamlines and processed with XDS (X-ray Detector Software) (37). Structures were determined and refined using starting model Crystal Form II (closed conformation, 2HVI) or Crystal Form I [open conformation, 1L3T, 1L5U, 1L5U, and 1L3V for C•A (_n_-1), C•A (_n_-3), C•A (_n_-4), and C•A (_n_-6) respectively] in REFMAC5 (38) and PHENIX (39). Model building was carried out in COOT (40). Data and refinement statistics are listed in Table S1. Composite omit maps were generated in CNS (41). All figures and superpositions were prepared in PyMOL.
Mass Spectrometry Experiments.
Crystallization drops of ddCTP•dA cognate (Mn2+) and ddCTP•dA wobble (Mg2+) crystals, or soaking solution of dCTP•dA cognate (Mn2+, Mg2+) crystals, or a positive control of dUTP added to the dCTP∙dA soaking solution were analyzed using Mass Spectrometry to determine the amination state of the nucleotide (Fig. S2). In all samples, only the expected nucleotide was detected. Therefore the observed cognate structure of the C•A mismatch is not due to cytosine deamination.
Supplementary Material
Supporting Information
Acknowledgments.
We thank G.R. Dubay for assistance with the Mass Spectrometry experiments, E.Y. Wu for discussions, and S.M. Armstrong for assistance with protein expression and purification. This work was supported by National Institutes of Health (NIH) grants P01 CA092584 and R01 GM091487 to L.S.B.
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3PX6, 3PX4, 3TAN, 3TAP, 3TAQ, 3TAR, 3PX0, 3PV8, 3THV, 3TI0).
References
- 1.Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 2.Rothwell PJ, Waksman G. Structure and mechanism of DNA polymerases. Adv Protein Chem. 2005;71:401–440. doi: 10.1016/S0065-3233(04)71011-6. [DOI] [PubMed] [Google Scholar]
- 3.Joyce CM, Benkovic SJ. DNA polymerase fidelity: kinetics, structure, and checkpoints. Biochemistry. 2004;43:14317–14324. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- 4.Steitz TA, Yin YW. Accuracy, lesion bypass, strand displacement and translocation by DNA polymerases. Philos T Roy Soc B. 2004;359:17–23. doi: 10.1098/rstb.2003.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson KA. Conformational coupling in DNA polymerase fidelity. Annu Rev Biochem. 1993;62:685–713. doi: 10.1146/annurev.bi.62.070193.003345. [DOI] [PubMed] [Google Scholar]
- 6.Kool ET. Active site tightness and substrate fit in DNA replication. Annu Rev Biochem. 2002;71:191–219. doi: 10.1146/annurev.biochem.71.110601.135453. [DOI] [PubMed] [Google Scholar]
- 7.Echols H, Goodman MF. Fidelity mechanisms in DNA replication. Annu Rev Biochem. 1991;60:477–511. doi: 10.1146/annurev.bi.60.070191.002401. [DOI] [PubMed] [Google Scholar]
- 8.Harris VH, et al. The effect of tautomeric constant on the specificity of nucleotide incorporation during DNA replication: support for the rare tautomer hypothesis of substitution mutagenesis. J Mol Biol. 2003;326:1389–1401. doi: 10.1016/s0022-2836(03)00051-2. [DOI] [PubMed] [Google Scholar]
- 9.Watson JD, Crick FHC. Genetical implications of the structure of deoxyribonucleic acid. Nature. 1953;171:964–967. doi: 10.1038/171964b0. [DOI] [PubMed] [Google Scholar]
- 10.Topal MD, Fresco JR. Complementary base pairing and the origin of substitution mutations. Nature. 1976;263:285–289. doi: 10.1038/263285a0. [DOI] [PubMed] [Google Scholar]
- 11.Echols H, Goodman MF. Fidelity mechanisms in DNA replication. Annu Rev Biochem. 1991;60:477–511. doi: 10.1146/annurev.bi.60.070191.002401. [DOI] [PubMed] [Google Scholar]
- 12.Bebenek K, Pedersen LC, Kunkel TA. Replication infidelity via a mismatch with Watson-Crick geometry. Proc Natl Acad Sci USA. 2011;108:1862–1867. doi: 10.1073/pnas.1012825108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiefer JR, et al. Crystal structure of a thermostable Bacillus DNA polymerase I large fragment at 2.1 A resolution. Structure. 1997;5:95–108. doi: 10.1016/s0969-2126(97)00169-x. [DOI] [PubMed] [Google Scholar]
- 14.Kiefer JR, Mao C, Braman JC, Beese LS. Visualizing DNA replication in a catalytically active Bacillus DNA polymerase crystal. Nature. 1998;391:304–307. doi: 10.1038/34693. [DOI] [PubMed] [Google Scholar]
- 15.Johnson SJ, Taylor JS, Beese LS. Processive DNA synthesis observed in a polymerase crystal suggests a mechanism for the prevention of frameshift mutations. Proc Natl Acad Sci USA. 2003;100:3895–3900. doi: 10.1073/pnas.0630532100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson SJ, Beese LS. Structures of mismatch replication errors observed in a DNA polymerase. Cell. 2004;116:803–816. doi: 10.1016/s0092-8674(04)00252-1. [DOI] [PubMed] [Google Scholar]
- 17.Hunter WN, Brown T, Anand NN, Kennard O. Structure of an adenine-cytosine base pair in DNA and its implications for mismatch repair. Nature. 1986;320:552–555. doi: 10.1038/320552a0. [DOI] [PubMed] [Google Scholar]
- 18.Boulard Y, et al. The pH dependent configurations of the C.A mispair in DNA. Nucleic Acids Res. 1992;20:1933–1941. doi: 10.1093/nar/20.8.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doublie S, Ellenberger T. The mechanism of action of T7 DNA polymerase. Curr Opin Struct Biol. 1998;8:704–712. doi: 10.1016/s0959-440x(98)80089-4. [DOI] [PubMed] [Google Scholar]
- 20.Brautigam CA, Steitz TA. Structural and functional insights provided by crystal structures of DNA polymerases and their substrate complexes. Curr Opin Struct Biol. 1998;8:54–63. doi: 10.1016/s0959-440x(98)80010-9. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Korolev S, Waksman G. Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J. 1998;17:7514–7525. doi: 10.1093/emboj/17.24.7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berman AJ, et al. Structures of phi29 DNA polymerase complexed with substrate: the mechanism of translocation in B-family polymerases. EMBO J. 2007;26:3494–3505. doi: 10.1038/sj.emboj.7601780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kunkel TA. DNA replication fidelity. J Biol Chem. 2004;279:16895–16898. doi: 10.1074/jbc.R400006200. [DOI] [PubMed] [Google Scholar]
- 24.Steitz TA. DNA polymerases: structural diversity and common mechanisms. J Biol Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- 25.Steitz TA. DNA- and RNA-dependent DNA polymerases. Curr Opin Struct Biol. 1993;3:31–38. [Google Scholar]
- 26.Sirover MA, Loeb LA. Infidelity of DNA synthesis in vitro: screening for potential metal mutagens or carcinogens. Science. 1976;194:1434–1436. doi: 10.1126/science.1006310. [DOI] [PubMed] [Google Scholar]
- 27.Weymouth LA, Loeb LA. Mutagenesis during in vitro DNA synthesis. Proc Natl Acad Sci USA. 1978;75:1924–1928. doi: 10.1073/pnas.75.4.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doublié S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 29.Wu EY, Beese LS. The structure of a high fidelity DNA polymerase bound to a mismatched nucleotide reveals an “ajar” intermediate conformation in the nucleotide selection mechanism. J Biol Chem. 2011;286:19758–19767. doi: 10.1074/jbc.M110.191130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frederico LA, Kunkel TA, Shaw BR. Cytosine deamination in mismatched base pairs. Biochemistry. 1993;32:6523–6530. doi: 10.1021/bi00077a005. [DOI] [PubMed] [Google Scholar]
- 31.Fonseca Guerra C, Bickelhaupt FM, Saha S, Wang F. Adenine tautomers: relative stabilities, ionization energies, and mismatch with cytosine. J Phys Chem A. 2006;110:4012–4020. doi: 10.1021/jp057275r. [DOI] [PubMed] [Google Scholar]
- 32.Fogarasi G. Water-mediated tautomerization of cytosine to the rare imino form: an ab initio dynamics study. Chem Phys. 2008;349:204–209. [Google Scholar]
- 33.Hobza P, Sponer J. Structure, energetics, and dynamics of the nucleic acid base pairs: nonempirical ab initio calculations. Chem Rev. 1999;99:3247–3276. doi: 10.1021/cr9800255. [DOI] [PubMed] [Google Scholar]
- 34.Warren JJ, Forsberg LJ, Beese LS. The structural basis for the mutagenicity of O(6)-methyl-guanine lesions. Proc Natl Acad Sci USA. 2006;103:19701–19706. doi: 10.1073/pnas.0609580103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tabor S, Richardson CC. A single residue in DNA polymerases of the Escherichia coli DNA polymerase I family is critical for distinguishing between deoxy- and dideoxyribonucleotides. Proc Natl Acad Sci USA. 1995;92:6339–6343. doi: 10.1073/pnas.92.14.6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Minnick DT, et al. Side chains that influence fidelity at the polymerase active site of Escherichia coli DNA polymerase I (Klenow fragment) J Biol Chem. 1999;274:3067–3075. doi: 10.1074/jbc.274.5.3067. [DOI] [PubMed] [Google Scholar]
- 37.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell dimensions. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- 38.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 39.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Emsley P, Cowtan K. COOT: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 41.Brünger AT, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 42.Golosov AA, Warren JJ, Beese LS, Karplus M. The mechanism of the translocation step in DNA replication by DNA polymerase I: a computer simulation analysis. Structure. 2010;18:83–93. doi: 10.1016/j.str.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Evans RJ, et al. Structure of PolC reveals unique DNA binding and fidelity determinants. Proc Natl Acad Sci USA. 2008;105:20695–20700. doi: 10.1073/pnas.0809989106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Batra VK, et al. Magnesium-induced assembly of a complete DNA polymerase catalytic complex. Structure. 2006;14:757–766. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia-Diaz M, Bebenek K, Krahn JM, Kunkel TA, Pedersen LC. A closed conformation for the Pol lambda catalytic cycle. Nat Struct Mol Biol. 2005;12:97–98. doi: 10.1038/nsmb876. [DOI] [PubMed] [Google Scholar]
- 46.Vaisman A, Ling H, Woodgate R, Yang W. Fidelity of Dpo4: effect of metal ions, nucleotide selection and pyrophosphorolysis. EMBO J. 2005;24:2957–2967. doi: 10.1038/sj.emboj.7600786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang J. DNA polymerases: Hoogsteen base-pairing in DNA replication? Nature. 2005;437:E6–7. doi: 10.1038/nature04199. [DOI] [PubMed] [Google Scholar]
- 48.Pata JD. Structural diversity of the Y-family DNA polymerases. Biochim Biophys Acta. 2010;1804:1124–1135. doi: 10.1016/j.bbapap.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang M, et al. Insights into base selectivity from the 1.8 A resolution structure of an RB69 DNA polymerase ternary complex. Biochemistry. 2011;50:581–590. doi: 10.1021/bi101192f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information