The PI3K regulatory subunit p85α can exert tumor suppressor properties through negative regulation of growth factor signalling (original) (raw)
. Author manuscript; available in PMC: 2011 Oct 30.
Abstract
PI3K plays a critical role in tumorigenesis and the PI3K p85 regulatory subunit exerts both positive and negative effects on signaling. Expression of Pik3r1, the gene encoding p85, is decreased in human prostate, lung, ovarian, bladder and liver cancers consistent with the possibility that p85 has tumor suppressor properties. We tested this hypothesis by studying mice with a liver-specific deletion of the Pik3r1 gene. These mice exhibited enhanced insulin and growth factor signaling and progressive changes in hepatic pathology, leading to the development of aggressive hepatocellular carcinomas with pulmonary metastases. Liver tumors that arose exhibited a markedly elevated level of phosphatidylinositol-3,4,5-trisphosphate (PIP3), along with Akt activation and and decreased PTEN expression, at both the mRNA and protein levels. Together, these results substantiate the concept that the p85 subunit of PI3K has a tumor suppressive role in the liver and possibly other tissues.
Keywords: liver, hepatocellular carcinoma, phosphoinositide 3-kinase, p85 subunit, tumor suppressor, PTEN, conditional gene inactivation
Introduction
The Class IA phosphoinositide 3-kinase (PI3K) pathway plays a central role in growth factor signaling and oncogenesis. Upon activation of a receptor tyrosine kinase, PI3K is activated and generates the second messenger phosphatidylinositol(3,4,5)-trisphosphate (PIP3), which activates critical downstream targets such as Akt and mTOR. When dysregulated, the PI3K pathway has a causal role in many forms of cancer, including those of the breast (1), colon (2) and liver (3).
The phosphoinositide 3-kinase enzyme is an obligate heterodimer with an SH2-containing regulatory subunit (p85) and a p110 catalytic subunit (4). The primary function of the p85 subunit is to bind, stabilize and inhibit the p110 catalytic subunit until RTK activation (5). Oncogenic mutations of PI3K enzyme can affect the kinase domain, but more commonly disable the ability of p85 to inhibit p110, thus leading to unchecked constitutive activity (6). Such cancer-causing mutations in p85 have been documented in a murine lymphoma model (7), as well as in human ovarian cancer, colon cancer (8), and glioblastoma (9).
Although p85 can function as an oncogene, knockouts of various isoforms of p85 subunits have revealed that p85 may endogenously function as a tumor suppressor (10). For instance, mice with heterozygous deletion of Pik3rl (11), the gene encoding p85α and its shorter isoforms p55α and p50α, display increased hepatic Akt activation and improved insulin sensitivity despite an overall decrease in PI3K activity (12). In a similar vein, mice with a liver-specific knockout of Pik3r1 (L-Pik3r1KO) have augmented hepatic PI3K activity and improved metabolism. In addition, mice that are heterozygous for both Pik3r1 and Pten display increased Akt and S6 activation, and a two-fold increase of intestinal neoplasia compared to Pten heterozygotes alone (13). The mechanisms of this negative regulation by the p85 monomer in these situations are multifaceted. We and others have previously shown that in most cells there is a stiochiometric imbalance between p85 and p110, with the former being in excess of the latter (14, 15). This imbalance results in a net inhibition of binding of the p85-p110 heterodimer to receptor phosphotyrosines (14). The p85 monomer also has significant effects independent of its regulation of the p110 catalytic subunit, including the sequestration of insulin receptor substrate (IRS) proteins and positive regulation of PTEN function (reviewed in (16)). Despite these significant data on the negative regulation of growth factor signaling, no studies have directly addressed the extent to which Pik3r1 alone can function to modify tumor growth and development in vivo.
Hepatocellular carcinoma (HCC) is one of the most lethal of human cancers, with a five-year survival of less than 5% (17). Worldwide, HCC is the fifth most common cancer and the third most common cause of cancer death (18). At the molecular level, there is ample literature suggesting that hepatic tumorigenesis is promoted by dysregulated growth factor signaling through receptor tyrosine kinases (RTKs) such as the insulin and IGF-1 receptors, c-MET, or epidermal growth factor receptor (EGFR), which strongly promote tumor growth, anti-apoptosis and chemotherapy resistance, primarily via the PI3K pathway (19).
In the present study, we have sought to define the role of the p85 regulatory subunit of PI3K in carcinogenesis by examining PIK3R1 expression in human cancers and determining effects of liver specific deletion of Pik3r1 (L-Pik3r1KO) on formation of liver tumors. Using the Oncomine™ database, we find that p85α expression is significantly reduced in many human cancers, including hepatocellular carcinoma. More importantly we find that mice lacking Pik3rl in the liver develop hepatocellular carcinoma by about 14 months of age, often with aggressive characteristics including lung metastases. The formation of these liver tumors correlated with increased levels of PIP3, increased Akt activation, increased PTEN phosphorylation and decreased PTEN protein and mRNA expression. Thus, PIK3R1 may function as an important tumor suppressor gene in liver and may play a similar role in other tissues.
Materials and Methods
Animals and breeding strategy
All animals were housed on a 12-h light-dark cycle and fed a standard rodent chow. All protocols for animal use and euthanasia were approved by the Animal Care Use Committee of the Joslin Diabetes Center and Harvard Medical School in accordance with National Institutes of Health guidelines. All mice in this study were on a mixed genetic background of 129Sv-C57BL/6-FVB.
Oncomine and gene expression studies
The Oncomine™ 3.0 database was interrogated for expression levels of PIK3R1 using the online interface http://www.oncomine.org/ (discussed and reviewed in (20)). The data from Oncomine™ was converted to raw expression levels by taking the inverse log(2) of the raw values. Expression levels of PIK3R1 were then normalized to the corresponding control tissue in each experiment. Gene expression from liver tissue in lox/lox and L-Pik3r1KO mice was assessed by quantitative reverse transcription (RT)-PCR, as described previously (21). Primers can be found in Supplemental table 1.
Histology and Immunohistochemistry
Tissues were harvested immediately and stained in hematoxylin and eosin using standard techniques. Phospho-Akt and PIP3 immunofluroescence were performed as described previously (22).
Western blotting
Tissue homogenates prepared in a tissue homogenization buffer from livers in the random fed state, except when indicated (21). Rabbit polyclonal pan-p85α antibody was generated as described previously (23), otherwise all other antibodies were purchased from Cell Signaling Technology (Beverly, MA). Protein expression was quantified by densitometry using NIH Image and presented as ±SEM.
Primary hepatocyte isolation and culture
Hepatocytes were isolated by a collagenase digestion technique as described previously (24). For experiments involving growth factor stimulation, hepatocytes were serum starved for 10 hours in DMEM-H plus glutamine and antibiotics, then stimulated for 15 minutes with either saline, human insulin 100 nM (Novo Nordisk, Copenhagen, Denmark), EGF 20 ng/mL (R&D systems) or PDGF-AB 1 ng/mL (R&D systems).
Statistics
Data are presented as ± s.e.m. Student’s t-test was used for statistical analysis between two groups. Statistical significance between multiple treatment groups was determined by analysis of variance (ANOVA) and Tukey’s _t_-test
Results
PIK3R1 expression is reduced several human cancers, including hepatocellular carcinoma
Changes in expression of the PI3K p85 regulatory subunit modifies insulin/IGF-1 action and PI3K activity (12) and enhances tumorigenic effects of Pten deficiency (13). To determine the potential role of the p85 regulatory subunit of PI3K in human cancer, we analyzed expression levels of PIK3R1, the gene that encodes p85, in Oncomine™, an on-line database of over 18,000 gene array studies (available on-line http://www.oncomine.org/, or see (20)). The data collection was interrogated for differences in PIK3R1 mRNA expression between cancerous tissue and corresponding normal tissue, primarily from human tissue samples. PIK3R1 was significantly decreased in samples from human cancer tissues compared to control tissues (p<0.05) in almost thirty different cancer studies (Supplemental Table 2). These data included over 1600 tissue specimens representing cancers of the prostate, liver, lung, breast, kidney, and others (Supplemental Table 2). For instance, PIK3R1 expression was significantly decreased by 17-75% in prostate cancer and by 19-46% in lung cancer (25-28), and reduced by 18% (p<0.001) in bladder cancer (29), 22% (p<0.01) in ovarian cancer (30), and 18% (p<0.05) in breast cancer ((31) Figures 1a and 1b). These data demonstrate a pattern of reduced expression in human cancer and suggest a possible role of p85 as a tumor suppressor in a number of situations. In the case of hepatocellular carcinoma, the expression patterns of p85 demonstrated a correlation with grade of malignancy (Figure 1c). Thus, p85 mRNA levels were inversely correlated with stage of hepatocellular carcinoma, both at Stage IIIA (32) and metastatic disease within the liver (Figure 1d, (33)). These data suggest a potential negative regulatory effect of p85 on tumorigenesis. To directly test this hypothesis in vivo, we created a mouse model with a tissue-specific knockout of Pik3r1 in liver.
Figure 1. Pik3r1 expression is decreased in many human cancers, including hepatocellular carcinoma.

Expression data from thousands of microarray experiments were analyzed for decreased Pik3r1 levels. For a complete list, see Supplemental Table 1. (a) PIK3R1 expression in normal and cancer samples from two representative studies on prostate cancer (n=189 (total), p<0.0001) and lung cancer (n=231(total), p<1×10×−14). Data were normalized to the controls from each individual study. (b) Representative PIK3R1 levels from normal tissue and cancer from the indicated tissues and studies. (c) PIK3R1 levels as a function of increased stage of hepatocellular carcinoma in the indicated study (n=68, p<0.05 nl vs stage IIIa). (d) Expression of Pik3r1 as a function the presence of HCC or hepatic metastases (mets) from the indicated study (n=161 (total), p<0.01 normal vs metastases). Bars represent mean±SEM, *p<0.05, **p<0.01, ***p<1×10−4; ****p<1×10−14. For specific n values, please see cited studies or Supplemental Table 1.
Hepatocytes lacking p85 display enhanced sensitivity to multiple growth factors
Mice lacking Pik3r1 in the liver (L-Pik3r1KO) were generated by crossing mice homozygous for a floxed allele of Pik3r1 with those heterozygous for the albumin-Cre transgene, as described previously (22). Although p85α plays a critical role in hepatic insulin signaling (16) we wondered if this molecule also negatively regulated other signaling pathways such as the epidermal growth factor (EGF) receptor and platelet-derived growth (PDGF) factor receptor since they have both been shown to play critical roles in HCC progression (34, 35). We isolated primary hepatocytes from L-Pik3r1KO mice and stimulated them with insulin, PDGF or EGF and assessed the response of downstream mediators Akt and MAPK (Figure 2). As observed in other cell types that lack p85α (11), L-Pik3r1KO primary hepatocytes showed a 1.5-fold increase in insulin-stimulated Akt phosphorylation, but no differences in insulin-stimulated MAPK phosphorylation (Figure 2 and Supplemental Figure 1). Primary hepatocytes from L-Pik3r1KO mice also displayed augmented Akt phosphorylation in response to EGF or PDGF stimulation by 1.6 and 1.8 fold, respectively (Figure 2), while there were no differences between control and knockout hepatocytes in EGF- or PDGF-stimulated MAPK phosphorylaton (Supplemental Figure 1). Thus, deletion of p85α selectively enhances insulin and growth factor action through the Akt pathway with no change in their effects on the MAPK pathway.
Figure 2. Loss of Pik3r1 enhances insulin sensitivity and Akt activation to multiple growth factors.
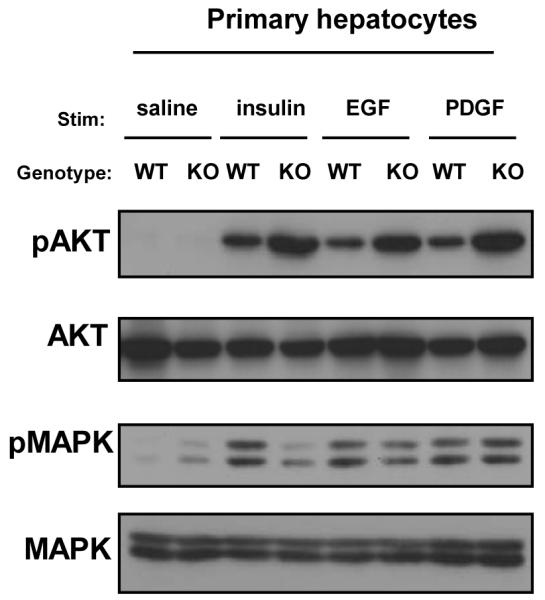
Western blots of total lysates from primary hepatocytes of indicated genotype using antibodies against phospho-Akt or phospho-MAPK. Cells were treated with indicated growth factor for 15 minutes following a 12-hour serum starvation.
HCC is preceded by dysplastic nodules in the setting of chronic aseptic hepatitis
Since HCC rarely occurs without preceding liver damage, we analyzed livers from younger L-Pik3r1KO and lox/lox controls to determine the presence of pre-malignant pathology. A tabular summary of these findings is presented as Supplemental Table 3. At six months of age, there were no detectable abnormalities in lox/lox control livers while 4/10 six month-old knockout mice examined showed evidence of hepatitis and necrosis (Figure 3). On gross inspection, small necrotic lesions were readily visible on the surface of the mouse livers (Figure 3a, left panel) and histology of these same livers showed significant neutrophilic infiltrate throughout the liver and periportal foci of necrotic hepatocytes with monocytic and neutrophilic infiltration (representative section in Figure 3a, right panel). Seven of twelve young L-Pik3r1KO mice and ten of fourteen older knockout mice showed evidence of this chronic hepatitis by liver histology. Gram stain and culture of these lesions revealed no evidence of bacterial infection (Taniguchi, Bronson and Kahn, unpublished data).
Figure 3. Hepatitis and dysplastic nodules precede the formation of hepatocellular carcinoma in L-Pik3r1KO mice.

(a, left) Gross dissection of a six month old L-Pik3r1KO mouse. The liver is shown with a small sterile abscess (black arrow) (a, right) Section from a six month-old L-Pik3r1KO mouse showing periportal inflammation and patches of hepatocyte necrosis. (b) PAS-stained sections of livers from of six-month old lox/lox (left) and knockout (right) livers. The deeper purple color denotes increased glycogen deposition. (c) L-Pik3r1KO livers at twelve months exhibit nodularity with focal areas of abnormal growth (black arrows). Histology of this same liver showed extensive dysplastic nodules (artificially highlighted with dashed lines) of both low (denoted by “L”) and high-grade (“denoted by “H”).
These L-Pik3r1KO mice also exhibited significant hepatic glycogen accumulation beginning at 2 months of age as determined by periodic acid-Schiff (PAS) staining, which selectively binds to glycogen within cells (Figure 3b). This abnormal glycogen deposition is likely a consequence of increased insulin sensitivity. Indeed, 6 month-old L-Pik3r1KO mice have lower levels of fasting blood glucose and insulin (Supplemental Figure 2a and b) as well as improved intraperitoneal glucose tolerance tests (Supplemental Figure 2c). In addition, expression of the essential gluconeogenic enzymes phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6 phosphatase (G6Pase) were attenuated by 50% and 70%, respectively, in L-Pik3r1KO livers (Supplemental Figure 2d). Both PEPCK and G6Pase are key hepatic enzymes that break down glycogen during fasting to maintain blood glucose levels. Thus, livers of L-Pik3r1KO mice accumulate abnormally high amounts of glycogen, most likely due to enhanced insulin sensitivity and decreased gluconeogenesis. These glycogen deposits then create a source of inflammation and associated hepatosteatitis, which is similar to the pathobiology of humans glycogen storage diseases (36).
At 10-12 months of age, this inflammation and glycogen deposition progressed to the formation of pre-cancerous dysplastic nodules. In humans, hepatocellular carcinoma is often preceded by the development of small dysplastic nodules seen on biopsy or MRI (37, 38)). Five of twelve L-Pik3r1KO mice aged 10-12 months exhibited diffuse low and high-grade dysplastic nodules within the liver parenchyma were readily apparent on gross dissection (Figure 3c, black arrows, left panel) and at the microscopic level (Figure 3c, right panel). In addition, we performed micro-MRI on several L-Pik3r1KO mice prior to sacrifice and found that these lesions appeared radiologically similar to dysplastic nodules in humans, as exhibited by contrast enhancement characteristics on T1-weighted imaging (Supplemental Figure 3).
L-Pik3r1KO mice develop an aggressive, high-grade hepatocellular carcinoma
The heightened sensitivity to multiple growth factors in L-Pik3r1KO hepatocytes led us to posit that the chronic loss of Pik3r1 could be hepatocarcinogenic in older mice. Indeed, 86% (12/14) of the mice aged 14-20 months spontaneously formed hepatocellular carcinoma, while none of the control lox/lox mice had any evidence of cancer (see Supplemental Table 3). One lox/lox mouse developed a benign hepatic adenoma at age 23 months, which is an entirely normal finding in male BL/6 or 129 mice of this advanced age (39). Upon gross dissection, these older L-Pik3r1KO mice displayed large, irregular livers that were infiltrated with tumors (Figure 4a). Several of the livers exhibited cystic lesions that were filled with bile and necrotic debris (Figure 4a, green arrows). Other mice exhibited massive livers that filled the entire peritoneal cavity, accompanied with intracapsular hemorrhage (Figure 4a, red arrows). The livers of L-Pik3r1KO mice over 14 months of age were also larger than age matched controls, with an average liver weight of L-Pik3r1KO mice of 2.0±0.3 g (n=11) compared to age-matched wild-type controls with average liver weights of 1.3±0.1 g (n=6, p<0.05 compared to KO), primarily due to tumor weight. When adjusted for body weight, livers lacking Pik3r1 were nearly double the relative size of control livers (6.9% vs. 3.4% KO vs. WT, p<0.01).
Figure 4. L-Pik3r1KO mice develop an aggressive hepatocellular carcinoma at 14-20 months of age.

(a) Gross dissection of an L-Pik3r1KO mouse at 16 months (left) and 20 months (right) of age. Note the nodules spread diffusely over the liver (yellow arrows) and a bile-filled cyst (green arrow). Focal intracapsular hemorrhage is present (red arrows) (b, left) High-powered view of HCC. Note the high nuclear/cytoplasmic ratio, thickened trabeculae and microsteatosis and mitotic figures (black arrows). (b, right) Bile duct hyperplasia with neutrophilic and monophilic infiltrates. (c) Lung metastases from the mouse in (c). (d) Quantitative RT-PCR analysis of mRNA levels of CD68, tumor necrosis factor-alpha (TNF–α) and interleukin-6 (IL-6)) in 16 -18 month-old mice of the indicated genotypes. (*p<0.05 compared to lox/lox, error bars represent SEM)
Histological analysis of these mice from 14-24 months of age often revealed significant infiltration of the normal liver parenchyma with HCC and dysplastic nodules. The histological features of the HCC in L-Pik3r1KO mice were consistent with a high-grade cancer, with a high nuclear/cytoplasm ratio, prominent nucleoli, coarsened trabeculae with layers 3-6 cells wide, and multiple mitoses in nearly every high-powered field (Figure 4b, left). In addition many livers displayed prominent bile duct hyperplasia and periportal inflammation (Figure 4b, right). These high-grade lesions were strongly correlated with the presence of pulmonary metastases, which occurred in nearly 60% (7/12) of the knockout mice with HCC (Figure 4c). Of note, metastases were detected exclusively in the lung and were only observed in mice older than 16 months of age.
To confirm that inflammation may have played a role in the pathophysiology of these tumors, we performed quantitative RT-PCR analysis on HCC samples from L-Pik3r1KO mice (14-18 months old) and compared them to age-matched controls (Figure 4d). HCC samples demonstrated a 3.0±0.4 fold increase in the expression of CD68, a specific macrophage marker that is elevated in human HCC (40). In addition, this increase was associated with a 3.1 ± 0.98 fold increase in the expression of TNF-alpha, however, there were no statistically significant difference in the expression interleukin 6 (IL-6).
Upregulated Akt signaling of hepatocellular carcinoma in L-Pik3r1KO mice
To determine the molecular mechanisms by which Pik3r1 deletion promotes hepatocarcinogenesis, we performed western blots on liver lysates from knockout and control mice at ages between 6 and 18 months of age. These liver extracts were prepared from mice in a random fed state without exogenous hormone stimulation. Compared to controls, whole liver extracts of L-Pik3r1KO mice showed an 80-90% decrease in p85α expression and a complete abrogation of p55α and p50α expression (Figure 5a). Since the p85 subunit of PI3K is known to stabilize the p110α subunit (5), the loss of the p85 expression also resulted in a 75-85% decrease in levels of the catalytic p110α subunit of PI3K.
Figure 5. HCC in L-Pik3r1KO mice exhibits enhanced activation of the PI3K-Akt axis.

(a) Random-fed liver lysates from the indicated genotype and age were blotted with specified antibodies. (b) Immunofluorescent staining for phospho-Akt of frozen sections from the corresponding livers from mice of the indicated genotype and age (see Methods). (c) Quantification of immunofluorescence in the images in (b). (d) As in (a), but the lysates are blotted for phosphorylated TSC2 and S6 proteins.
As expected, there was little activation of the PI3K or MAPK pathways in lox/lox mice at 6 months or 16-18 months, and little activation in 6-month old L-Pik3r1KO livers since these mice were not stimulated with exogenous growth factors before harvesting their livers (Figure 5a). On the other hand, older L-Pik3r1KO livers that contained hepatocellular carcinoma demonstrated a striking upregulation of Akt phosphorylation, by 3.84 ±0.35 fold (Supplemental Figure 4a, p<0.01) compared to 6 month-old lox/lox controls. Likewise, MAPK phosphorylation/activity was significantly enhanced in knockout mice with HCC by 2.37 ± 0.22 fold as compared to young lox/lox mice (Supplemental Figure 4b, p<0.001). The significantly increased Akt phosphorylation in tumor samples was confirmed by direct visualization with immunofluorescent staining. Phosphorylated Akt detected in situ was increased by 4-fold in tumor samples compared to lox/lox and KO controls (Figure 5b and quantitated in 5c). These signaling changes are likely cell autonomous, and not a result of systemic endocrine signals, since blood glucose and serum levels of insulin did not differ between 18 month-old control and L-Pik3r1KO mice (Supplemental Figures 5a and b).
The mammalian target of rapamycin (mTOR) pathway is critical for the activating protein synthesis and cell growth, and is regulated by PI3K/Akt via the phosphorylation and inhibition of TSC1 and TSC2. The increased activity of Akt in the L-Pik3r1KO mice with HCC resulted in a significant increase in the phosphorylation of TSC2, which lies upstream of mTOR (Figure 5d). In addition, the phosphorylation of ribosomal S6 protein, which is the ultimate downstream target of mTOR, was increased eight-fold (Figure 5d).
L-Pik3r1KO mice exhibit increased PIP3 levels and diminished PTEN expression
The activation of Akt requires the binding of the enzyme to the second messenger phosphatidylinositol(3,4,5)-trisphosphate (PIP3) via its pleckstrin homology (PH) domain. To determine levels of PIP3 in the tumors, we used an in situ immunofluorescence technique that allowed us to semi-quantitatively measure PIP3 levels in hepatocytes (Figure 6a, (22)). This technique showed that very little PIP3 accumulated in the control lox/lox and young L-Pik3r1KO mice, which was expected since these livers were not exposed to stimulation before their collection. Tumor samples from L-Pik3r1KO mice, however, displayed significant PIP3 accumulation. Quantitation of immunofluorescence levels showed that the hepatocellular carcinoma from L-Pik3r1KO mice had 4.92±0.3 fold more PIP3 than age-matched control lox/lox mice (1.0±0.06) or younger L-Pik3r1KO mice (0.96±0.05).
Figure 6. Elevated levels of PIP3 and Decrease PTEN expression in HCC from L-Pik3r1KO mice.

(a) Immunofluorescent staining of the frozen sections of the liver from mice of the indicated age and genotype with a primary anti-PIP3 antibody (IgM) and an anti-mouse secondary antibody conjugated to Alexafluor Red. The sections were counterstained with DAPI. (n=4 for each genotype, 16 slides analyzed per mouse) (b) Western blots of liver lysates from mice of the indicated genotype and age were blotted with antibodies against PTEN and phospho-PTEN proteins (Ser 380/Thr382/383). (c) RT-PCR analysis of PTEN levels in HCC tumor samples and normal liver from age-matched lox/lox controls. *p<0.05 (d) A scheme of HCC progression in L-Pik3r1KO mice
This enhancement of PIP3 levels suggested that degradation by PTEN or some other lipid phosphatase might be dysregulated. We have previously shown that the loss of p85 partially abrogates PTEN activity and decreases PIP3 turnover (12). Western blots on liver lysates from lox/lox and L-Pik3r1KO mice found a 53±5.8% decrease in PTEN protein expression in 18 month-old KO mice compared to age-matched lox/lox controls and younger KO animals (Figure 6b). This correlated with a nearly 60% decrease in Pten mRNA as determined by quantitative PCR analysis of RNA transcripts from tumor samples compared to age-matched control livers (Figure 6c).
Phosphorylation of PTEN on its C-terminal tail has also been shown to negatively regulate PTEN activity at the protein levels (41). Western blotting with an antibody that recognizes multiple phosphorylation sites in the C-terminus of PTEN revealed a modest decrease in overall levels of phospho-PTEN, but after adjusting for overall decreased PTEN expression, this represented a 2.1±0.4 fold increase in level of PTEN phosphorylation at the protein level (Figure 6b) Taken together, these data demonstrate that PTEN protein and mRNA expression may be altered in the setting of chronic p85alpha deletion, and that PTEN activity may also be negatively regulated by a state of relative hyperphosphorylation.
Discussion
Over the past two decades, a number of studies have demonstrated an integral role of PI3K in tumorigenesis (reviewed in (4)). This can occur through direct activation of PI3K by oncoproteins, such as polyoma middle T and receptor tyrosine kinases, deletion of the PIP3 phosphatase PTEN, and genetic mutation of proteins in the PI3K signaling pathway, including p85, p110, and AKT. In the present study, we provide evidence that decreased expression of the PI3K p85 regulatory subunit can also directly lead to tumor formation in a novel murine model of hepatocellular carcinoma.
An initial clue of the role of expression of the regulatory subunit of PI3K in cancer comes from the finding that PIK3R1 expression is decreased in many types of human cancers, including prostate, lung, breast and hepatocellular carcinoma (Figure 1a-d). Other human studies have also suggested the possible importance of p85 in human carcinogenesis. For instance, a functional missense mutation in PIK3R1 resulting in reduced p85 expression has been strongly linked with colon cancer (42). Likewise, the PIK3R1 gene is located on human chromosome 5q13, a region that is commonly deleted in cancers that utilize the PI3K pathways for growth, including hepatocellular carcinoma (43). Since other putative tumor suppressors involved in DNA damage repair are contained within the 5q13 chromosomal region, including XRCC4 and hRad17 (44), further studies must be performed to determine the extent to which PIK3R1 haploinsufficiency might account for the association of the 5q13 chromosomal deletion and cancer. However, in the case of hepatocellular carcinoma, the data from this study indicate that PIK3R1 may indeed be a meaningful tumor suppressor gene.
In our mouse model of a selective deletion of Pik3r1 in liver, a complex combination of cellular and molecular events contribute to progressive pathological events that ultimately causeHCC, as illustrated schematically in Figure 6d. Many features of this murine HCC model resemble human HCC. Human HCC is preceded by chronic inflammation from infectious or chemical etiologies (18), while this mouse model develops chronic hepatitis from abnormal glycogen deposition that arises from an intrinsic enhancement of hepatic insulin sensitivity (Figure 3 and (12)). Inflammation is readily observed at the cellular level via histology and at the molecular levels with increased levels of a macrophage marker and TNF–α (Figure 4d). This chronic hepatitis and HCC is also described in mouse models of glycogen storage disease, as well as in humans with this disorder (36).
By 14 months of age, almost all L-Pik3r1KO mice develop an aggressive hepatocellular carcinoma as demonstrated by the presence of pulmonary metastases in 60% of the mice with HCC, which is an uncommon feature in murine cancer models that exhibit spontaneous tumor growth. These results agree with our analysis of clinical human data that show a correlation of decreased Pik3r1 expression with Stage IIIA or metastatic hepatocellular carcinoma, supporting the notion that the p85 can serve as an endogenous tumor suppressor and that reduced levels of Pik3r1 may enable the formation of hepatocellular carcinoma.
The molecular mechanisms of HCC formation in L-Pik3r1KO mice are similar to those human HCC in several respects. Notably, human HCC is correlated with dysregulated growth factor signaling, particularly through receptor tyrosine kinases (RTKs) such as IGF-1, PDGF and EGF receptors. L-Pik3r1KO mice have exquisite growth factor sensitivity, particularly through the PI3K/Akt axis, as shown by enhanced Akt signaling through insulin/IGF-1 and other RTKs, including the PDGF-R and EGFR/ErbB3, which have all been implicated in human cancers (Figure 2).
This enhancement of Akt signaling is the net effect of several contributing factors. We have previously described that p85 has a positive regulatory effect on PTEN, where the loss of p85 leads to decreased PTEN function and decreased breakdown of PIP3, leading to accumulation of this second messenger and resulting in enhanced downstream signaling, most notably by PH-domain containing proteins like Akt. In this study, we also find that despite a significant defect in the enzyme capacity of PI3K, tumor specimens from L-Pik3r1KO mice displayed five times more hepatic PIP3 than control mice and a resultant four-fold increase in Akt activation.
The mechanism of PTEN regulation by p85 in the context of HCC appears to be due to a diminished expression of PTEN protein levels that is a result of both decreased PTEN transcript production and increased phosphorylation of the PTEN protein. Phosphorylation of the C-terminal tail of PTEN is known to play a crucial role in regulating both its expression and activity in vitro, but it is unclear how much this contributes to this human cancer or this murine model (41, 45). Nonetheless, the loss of PTEN expression that occurs in the older L-Pik3r1KO mice likely accelerates the formation of HCC, as it has been suggested to do in humans (46). The mechanism of how p85 might modulate PTEN mRNA expression or PTEN phosphorylation is unclear and further studies must be conducted to determine the regulation of PTEN phosphorylation in terms of membrane recruitment of the enzyme, epigenetic events regulating PTEN (47), as well as the potential effects of p85 deletion on regulation of other PIP3 phosphatases.
Despite many similarities with human HCC, this murine model differs in several key respects. First, the gross morphology of this murine cancer is different from the human form, marked by the presence of bilious cysts and bile duct proliferation. These histological findings may reflect the role of p85 in bile canalicular transport (48). Secondly, despite the correlation between abrogated hepatic p85 and reduced PTEN expression in the murine HCC, many of the human cancer studies with decreased PIK3R1 expression did not also show a concomitant decrease in PTEN expression (Supplemental Figure 6). The reason for this discrepancy is unknown, but likely stems from the fact that the gene expression data may not adequately capture the multiple different ways that PTEN function can be compromised, including functional mutations that do not alter RNA transcript production (49), promoter methylation (47), or post-translational regulation (50). Moreover, it may be possible that the HCC in different individuals or different populations results from different inciting events, such as infection (such as hepatitis) versus toxin (ethanol or aflatoxin B), and these could have completely different mechanisms that are masked in the gene array data.
In summary, we have demonstrated that the p85 regulatory subunit of PI3K functions as an inhibitor of growth factor signaling and a novel tumor suppressor in the liver and most likely in other cancers as well. Increasing or restoring expression of p85 may thus represent an exciting potential therapeutic possibility against cancer.
Supplementary Material
1
2
Acknowledgments
We greatly appreciate the technical assistance of Michael Rourk. The authors would like to thank Jonathan Glickman for his discussion on the HCC pathology. This work was supported by National Institutes of Health Grants DK33201 and DK55545, Joslin Diabetes and Endocrinology Research Center Grant DK34834 (to C.R.K.) and a GM41890 and CA089021 for L.C.C. RW acknowledges support from SAIR grant 2U24CA092782-07. C.M.T. acknowledges support from the American Diabetes Association Medical Scholars Award (C.M.T.) and Medical Scientist Training Program scholarship (Harvard Medical School).
Abbreviations
PI3K
Phosphoinositide-3 kinase
HCC
hepatocellular carcinoma
PTEN
phosphatase and tensin homolog
L-Pik3r1KO
mice with liver specific knockout of the Pik3r1 allele
References
- 1.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084–91. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gustin JP, Karakas B, Weiss MB, et al. Knockin of mutant PIK3CA activates multiple oncogenic pathways. Proc Natl Acad Sci U S A. 2009;106:2835–40. doi: 10.1073/pnas.0813351106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tommasi S, Pinto R, Pilato B, Paradiso A. Molecular pathways and related target therapies in liver carcinoma. Curr Pharm Des. 2007;13:3279–87. doi: 10.2174/138161207782360663. [DOI] [PubMed] [Google Scholar]
- 4.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 5.Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Regulation of the p85/p110 phosphatidylinositol 3′-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol. 1998;18:1379–87. doi: 10.1128/mcb.18.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miled N, Yan Y, Hon WC, et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007;317:239–42. doi: 10.1126/science.1135394. [DOI] [PubMed] [Google Scholar]
- 7.Jimenez C, Jones DR, Rodriguez-Viciana P, et al. Identification and characterization of a new oncogene derived from the regulatory subunit of phosphoinositide 3-kinase. Embo J. 1998;17:743–53. doi: 10.1093/emboj/17.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Philp AJ, Campbell IG, Leet C, et al. The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001;61:7426–9. [PubMed] [Google Scholar]
- 9.McLendon R, Friedman A, Bigner D, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008 doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo J, Cantley LC. The negative regulation of phosphoinositide 3-kinase signaling by p85 and it’s implication in cancer. Cell Cycle. 2005;4:1309–12. doi: 10.4161/cc.4.10.2062. [DOI] [PubMed] [Google Scholar]
- 11.Mauvais-Jarvis F, Ueki K, Fruman DA, et al. Reduced expression of the murine p85alpha subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. J Clin Invest. 2002;109:141–9. doi: 10.1172/JCI13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taniguchi CM, Tran TT, Kondo T, et al. Phosphoinositide 3-kinase regulatory subunit p85alpha suppresses insulin action via positive regulation of PTEN. Proc Natl Acad Sci U S A. 2006;103:12093–7. doi: 10.1073/pnas.0604628103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo J, Sobkiw CL, Logsdon NM, et al. Modulation of epithelial neoplasia and lymphoid hyperplasia in PTEN+/− mice by the p85 regulatory subunits of phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2005;102:10238–43. doi: 10.1073/pnas.0504378102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ueki K, Fruman DA, Brachmann SM, Tseng YH, Cantley LC, Kahn CR. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol Cell Biol. 2002;22:965–77. doi: 10.1128/MCB.22.3.965-977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barbour LA, Rahman S Mizanoor, Gurevich I, et al. Increased P85alpha is a potent negative regulator of skeletal muscle insulin signaling and induces in vivo insulin resistance associated with growth hormone excess. J Biol Chem. 2005;280:37489–94. doi: 10.1074/jbc.M506967200. [DOI] [PubMed] [Google Scholar]
- 16.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 17.Ince N, Wands JR. The increasing incidence of hepatocellular carcinoma. N Engl J Med. 1999;340:798–9. doi: 10.1056/NEJM199903113401009. [DOI] [PubMed] [Google Scholar]
- 18.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–76. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 19.Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;25:3787–800. doi: 10.1038/sj.onc.1209556. [DOI] [PubMed] [Google Scholar]
- 20.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–80. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taniguchi CM, Ueki K, Kahn R. Complementary roles of IRS-1 and IRS-2 in the hepatic regulation of metabolism. J Clin Invest. 2005;115:718–27. doi: 10.1172/JCI23187. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Taniguchi CM, Kondo T, Sajan M, et al. Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKClambda/zeta. Cell Metab. 2006;3:343–53. doi: 10.1016/j.cmet.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 23.Ueki K, Yballe CM, Brachmann SM, et al. Increased insulin sensitivity in mice lacking p85beta subunit of phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2002;99:419–24. doi: 10.1073/pnas.012581799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taniguchi CM, Aleman JO, Ueki K, et al. The p85alpha regulatory subunit of phosphoinositide 3-kinase potentiates c-Jun N-terminal kinase-mediated insulin resistance. Mol Cell Biol. 2007;27:2830–40. doi: 10.1128/MCB.00079-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lapointe J, Li C, Higgins JP, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci U S A. 2004;101:811–6. doi: 10.1073/pnas.0304146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu YP, Landsittel D, Jing L, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22:2790–9. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 27.Beer DG, Kardia SL, Huang CC, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–24. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 28.Stearman RS, Dwyer-Nield L, Zerbe L, et al. Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen-induced murine model. Am J Pathol. 2005;167:1763–75. doi: 10.1016/S0002-9440(10)61257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez-Carbayo M, Socci ND, Lozano J, Saint F, Cordon-Cardo C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J Clin Oncol. 2006;24:778–89. doi: 10.1200/JCO.2005.03.2375. [DOI] [PubMed] [Google Scholar]
- 30.Lu KH, Patterson AP, Wang L, et al. Selection of potential markers for epithelial ovarian cancer with gene expression arrays and recursive descent partition analysis. Clin Cancer Res. 2004;10:3291–300. doi: 10.1158/1078-0432.CCR-03-0409. [DOI] [PubMed] [Google Scholar]
- 31.Richardson AL, Wang ZC, De Nicolo A, et al. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006;9:121–32. doi: 10.1016/j.ccr.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 32.Okada T, Iizuka N, Yamada-Okabe H, et al. Gene expression profile linked to p53 status in hepatitis C virus-related hepatocellular carcinoma. FEBS Lett. 2003;555:583–90. doi: 10.1016/s0014-5793(03)01345-0. [DOI] [PubMed] [Google Scholar]
- 33.Chen X, Cheung ST, So S, et al. Gene expression patterns in human liver cancers. Mol Biol Cell. 2002;13:1929–39. doi: 10.1091/mbc.02-02-0023.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanabe KK, Lemoine A, Finkelstein DM, et al. Epidermal growth factor gene functional polymorphism and the risk of hepatocellular carcinoma in patients with cirrhosis. JAMA. 2008;299:53–60. doi: 10.1001/jama.2007.65. [DOI] [PubMed] [Google Scholar]
- 35.Gotzmann J, Fischer AN, Zojer M, et al. A crucial function of PDGF in TGF-beta-mediated cancer progression of hepatocytes. Oncogene. 2006;25:3170–85. doi: 10.1038/sj.onc.1209083. [DOI] [PubMed] [Google Scholar]
- 36.Lee PJ. Glycogen storage disease type I: pathophysiology of liver adenomas. Eur J Pediatr. 2002;161(Suppl 1):S46–9. doi: 10.1007/s00431-002-1002-0. [DOI] [PubMed] [Google Scholar]
- 37.Sakamoto M. Early HCC: diagnosis and molecular markers. J Gastroenterol. 2009;44(Suppl 19):108–11. doi: 10.1007/s00535-008-2245-y. [DOI] [PubMed] [Google Scholar]
- 38.Robinson P. Hepatocellular carcinoma: development and early detection. Cancer Imaging. 2008;8(Spec No A):S128–31. doi: 10.1102/1470-7330.2008.9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fox JG. The Mouse in Biomedical Research. Second ed Academic Press; 2006. [Google Scholar]
- 40.Kuang DM, Peng C, Zhao Q, Wu Y, Chen MS, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma promote expansion of memory T helper 17 cells. Hepatology. 51:154–64. doi: 10.1002/hep.23291. [DOI] [PubMed] [Google Scholar]
- 41.Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20:5010–8. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L, Plummer SJ, Thompson CL, Tucker TC, Casey G. Association between phosphatidylinositol 3-kinase regulatory subunit p85alpha Met326Ile genetic polymorphism and colon cancer risk. Clin Cancer Res. 2008;14:633–7. doi: 10.1158/1078-0432.CCR-07-1211. [DOI] [PubMed] [Google Scholar]
- 43.Katoh H, Shibata T, Kokubu A, et al. Genetic profile of hepatocellular carcinoma revealed by array-based comparative genomic hybridization: identification of genetic indicators to predict patient outcome. J Hepatol. 2005;43:863–74. doi: 10.1016/j.jhep.2005.05.033. [DOI] [PubMed] [Google Scholar]
- 44.Collis SJ, DeWeese TL, Jeggo PA, Parker AR. The life and death of DNA-PK. Oncogene. 2005;24:949–61. doi: 10.1038/sj.onc.1208332. [DOI] [PubMed] [Google Scholar]
- 45.Valiente M, Andres-Pons A, Gomar B, et al. Binding of PTEN to specific PDZ domains contributes to PTEN protein stability and phosphorylation by microtubule-associated serine/threonine kinases. J Biol Chem. 2005;280:28936–43. doi: 10.1074/jbc.M504761200. [DOI] [PubMed] [Google Scholar]
- 46.Yao YJ, Ping XL, Zhang H, et al. PTEN/MMAC1 mutations in hepatocellular carcinomas. Oncogene. 1999;18:3181–5. doi: 10.1038/sj.onc.1202659. [DOI] [PubMed] [Google Scholar]
- 47.Wang L, Wang WL, Zhang Y, Guo SP, Zhang J, Li QL. Epigenetic and genetic alterations of PTEN in hepatocellular carcinoma. Hepatol Res. 2007;37:389–96. doi: 10.1111/j.1872-034X.2007.00042.x. [DOI] [PubMed] [Google Scholar]
- 48.Misra S, Ujhazy P, Gatmaitan Z, Varticovski L, Arias IM. The role of phosphoinositide 3-kinase in taurocholate-induced trafficking of ATP-dependent canalicular transporters in rat liver. J Biol Chem. 1998;273:26638–44. doi: 10.1074/jbc.273.41.26638. [DOI] [PubMed] [Google Scholar]
- 49.Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22:2954–63. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- 50.Leslie NR, Batty IH, Maccario H, Davidson L, Downes CP. Understanding PTEN regulation: PIP2, polarity and protein stability. Oncogene. 2008;27:5464–76. doi: 10.1038/onc.2008.243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
2