The Tumor Inhibitor and Antiangiogenic Agent Withaferin A Targets the Intermediate Filament Protein Vimentin (original) (raw)
. Author manuscript; available in PMC: 2011 Dec 1.
SUMMARY
The natural product withaferin A (WFA) exhibits antitumor and antiangiogenesis activity in vivo, which results from this drug’s potent growth inhibitory activities. Here, we show that WFA binds to the intermediate filament (IF) protein, vimentin, by covalently modifying its cysteine residue, which is present in the highly conserved α-helical coiled coil 2B domain. WFA induces vimentin filaments to aggregate in vitro, an activity manifested in vivo as punctate cytoplasmic aggregates that colocalize vimentin and F-actin. WFA’s potent dominant-negative effect on F-actin requires vimentin expression and induces apoptosis. Finally, we show that WFA-induced inhibition of capillary growth in a mouse model of corneal neovascularization is compromised in vimentin-deficient mice. These findings identify WFA as a chemical genetic probe of IF functions, and illuminate a potential molecular target for withanolide-based therapeutics for treating angioproliferative and malignant diseases.
INTRODUCTION
In the postgenomic era, natural products are gaining importance in cell biological applications as molecular probes of protein function [1], expanding from their more traditional role as chemical scaffolds for drug development. Particularly interesting to molecular medicine are the pharmacologically active small molecules, the newly discovered binding targets of which can offer critical molecular insight into the drugable chemical space of the human proteome [2]. Such bioactive small-molecule tools can also become molecular beacons of target functions, a feature that has applications in target-based disease diagnosis and patient responses to drug therapies.
Angiogenesis, which is the growth of new blood vessels from preexisting vasculature, occurs widely in numerous human pathologies, including cancers, arthritis, endometriosis, age-related macular degeneration, diabetic retinopathy, etc. [3]. As new developments in antiangiogenesis research are providing crucial knowledge of which protein targets are sensitive to drug treatment, there is still the increasing need to discover novel drugable targets [4]. To probe the chemical diversity of plants for new classes of angiogenesis inhibitors, we recently developed a high-density, cell-based screening assay, and focused on the medicinal plant Withania somnifera [5], because of the elaboration of alkaloids and steroidal lactones (withanolides) present in this plant [6]. W. somnifera has also rich ethnopharmacological uses in traditional East Indian medicine for treatment of arthritis, immunological disorders, and bleeding conditions in women [7], which suggested to us the presence of inhibitors that target an underlying angiogenic mechanism. The rational screening strategy led to the successful isolation of withaferin A (WFA), an inhibitor known for its potent anticancer activities [8]. We demonstrated that WFA potently blocks angiogenesis in vivo [5], and investigations on the mode of action of WFA and structurally related congers has revealed that this class of withanolides target the ubiquitin-proteosome pathway (UPP) and cause rapid increases in polyubiquitinated proteins in vascular endothelial cells [5, 9].
To understand the mode of action of WFA, we exploited the chemical genetic approach to discover the in vivo binding targets of small molecules [10, 11]. To this end, we synthesized a novel WFA-biotin analog, in which the biotin affinity tag was linked to WFA via a long hydrocarbon chain residue [12], a strategy we and others have employed extensively to afford the successful characterization of small-molecule targets [13-16]. Using the novel WFA-biotin probe, which maintains the UPP-targeting activity of WFA, we previously showed that a 56 kDa protein is irreversibly targeted by WFA in human umbilical vein endothelial cells (HUVECs) [12]. Here, we have isolated this in vivo binding protein of WFA, and show that it is the type III intermediate filament (IF) protein vimentin. As vimentin is abundantly expressed by mesenchymal cells and plays a critical role in wound healing, angiogenesis, and cancer growth [17-19], here we demonstrate that WFA can be used as a chemical genetic probe of vimentin, identifying the dominant-negative role of WFA-modified vimentin as a promoter of cellular apoptosis.
RESULTS AND DISCUSSION
WFA Targets the IF Protein Vimentin
To afford the isolation of the WFA (Figure 1A) target, we performed scale-up ligand binding studies employing bovine aortic endothelial cells (BAECs). The WFA-biotin analog (WFA-B; see Figure S1 in the Supplemental Data available with this article online) was incubated with BAECs in the presence and absence of unconjugated WFA, and cell lysates containing newly biotinylated proteins were affinity-purified over NeutrAvidin columns and fractionated by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE). Gels stained with Coomassie blue dye confirmed the isolation of this 56 kDa protein from BAECs, and revealed that in vivo competition with unconjugated WFA dramatically reduced the levels of this protein (Figure 1B). LC-MS/MS characterization of this protein identified the 56 kDa protein as vimentin (27% protein coverage; Figure S2), an IF protein that is abundant in mesenchymal cells. To further support that vimentin is bound by WFA-B in vivo, HUVECs were treated with WFA-B in the presence and absence of unconjugated WFA, and total cellular lysates were fractionated by SDS-PAGE and protein blots probed with streptavidin-horseradish peroxidase (HRP). We found that the biotin label is incorporated in this 56 kDa protein in a WFA-competitive manner (Figure 1C). Additionally, when such biotinylated proteins from HUVECs were affinity purified over NeutrAvidin columns, WFA treatment competed with the binding of vimentin to the NeutrAvidin column in a dose-responsive manner, as shown by western blot experiments (Figure 1D). As prior studies have revealed that the tetrameric form of vimentin is found in vivo and exists in dynamic equilibrium with filament polymers [20], we therefore investigated whether WFA binds to purified tetrameric vimentin in vitro. We performed ligand binding assays employing unconjugated WFA as a specific competitor and 12-deoxowithastramonolide (12-D WS) as a nonspecific competitor [12], and show that vimentin is bound by WFA-B in a WFA-competitive manner (Figure 1E). Interestingly, the WFA-B-affinity chromatographic approach has also led to coisolation of two other proteins of sizes 51 kDa and 43 kDa that were simultaneously reduced in intensity by WFA treatment in BAECs (Figure 1B). The characterization of these proteins by LC-MS/MS revealed that they are 51 kDa β-tubulin and 43 kDa β-actin (Figure S3). Importantly, these NeutrAvidin-bound proteins, when subjected to 6 M urea treatment, caused disruption of β-actin and β-tubulin binding, whereas that of vimentin was not affected (Figure S4), suggesting that the in vivo binding target of WFA-B is vimentin. One possibility is that vimentin, through its interaction with plakins [21], could bind β-actin and β-tubulin and provide a cargo for microtubule-dependent protein transport [22], or it may possibly also bind to β-actin via direct interaction [23]. In this respect, we previously found a 180 kDa protein that copurifies with 56 kDa vimentin upon NeutrAvidin chromatography [12], a species which is possibly the plakin, bullous pemphigoid antigen 1. Clearly, the coisolation of vimentin-associated proteins with WFA-B unveils a novel use for this reagent in chemical proteomics.
Figure 1. Affinity Identification of Binding Target of WFA from Endothelial Cells.
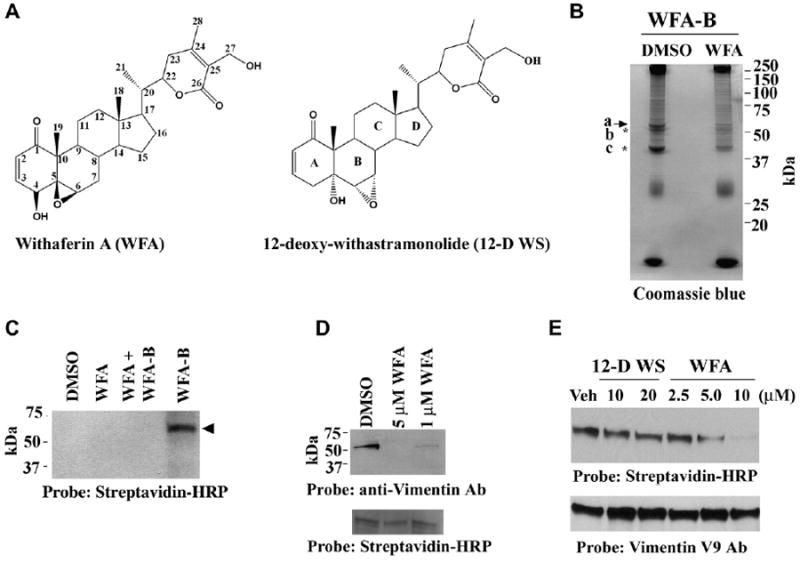
(A) Chemical structures of WFA and 12-D WS.
(B) Affinity isolation of WFA-B-binding proteins. BAECs were preincubated with DMSO (vehicle) or with 5 μM WFA for 30 min and subsequently with 5 μM WFA-B for 2 hr. Cell lysates prepared in 1% Triton X-100 buffer were purified over NeutrAvidin affinity columns and subjected to SDS-PAGE. The gel was stained with Coomassie blue. The arrow points to the 56 kDa protein band and asterisks mark the coeluted 51 and 43 kDa proteins.
(C) WFA-B binds to the 56 kDa protein in HUVECs. Cells were preincubated with DMSO or WFA for 30 min and subsequently with WFA-B for 2 hr. Soluble proteins extracted in 1% Triton X-100 were fractionated by SDS-PAGE and blotted. Blots were developed with Streptavidin-HRP.
(D) WFA-B binds vimentin in HUVECs. Cell cultures were preincubated with DMSO (vehicle) or with 5 μM WFA or 1 μM for 30 min and subsequently with 5 μM WFA-B for 2 hr. Cell lysates prepared in 1% Triton X-100 buffer were purified over NeutrAvidin affinity columns and subjected to SDS-PAGE and western blotted with anti-vimentin V9 antibody. The bottom panel represents streptavidin-HRP detection of endogenous biotinylated species used as a loading control.
(E) Tetrameric soluble hamster vimentin was incubated with different doses of WFA or inactive congener 12-D WS for 1 hr and subsequently with 0.3 μM WFA-B for 1 hr at 37°C. The proteins were fractionated by SDS-PAGE, blotted and probed with vimentin V9 antibody for loading control, stripped, and reprobed with streptavidin-HRP to detect biotinylated adducts.
WFA Covalently Modifies the Unique Cysteine Residue in Tetrameric Vimentin
Next, to identify the amino acid residue(s) of vimentin modified by WFA, we incubated purified hamster tetrameric vimentin with WFA for 1 hr at 37°C and subjected the protein-ligand complexes to tryptic digestion and LC-MS/MS analysis. A search for the position of adduct formation (a molecular mass shift of 470) in the tryptic fragments of vimentin revealed that the sole cysteine residue at position 327 (position 328 in human vimentin) in the α-helical coil coiled 2B rod domain of vimentin is uniquely modified by WFA (Figure 2). This finding is consistent with prior reports that WFA is known to react with protein thiol-nucleophiles and can undergo Michael addition through its reactive A-ring (Figure 1A) pharmacophores [24].
Figure 2. WFA Modifies the Unique Cysteine Residue of Vimentin.
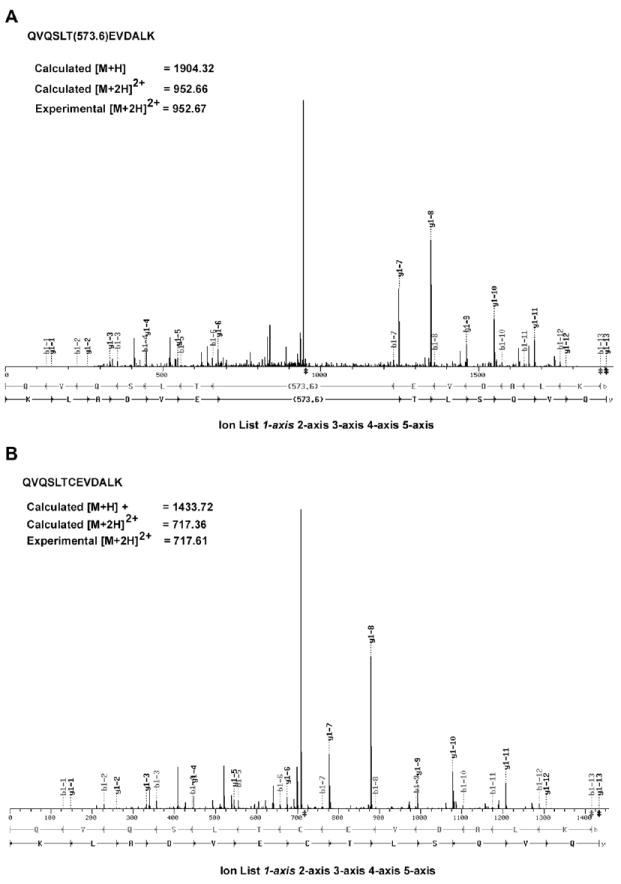
Tetrameric vimentin was incubated with WFA or DMSO and small molecule-protein complex was subjected to trypsin digestion. The tryptic fragments were subjected to LC-MS/MS analysis. The assignment of fragment ions in the MS/MS of the modified and unmodified peptides were obtained with the SEQUEST software tool “FuzzyIons” (distributed by ThermoFinnigan) to annotate the spectra (A) from WFA-treated sample and (B) from DMSO-treated sample. This analysis verifies both the peptide amino acid sequence and the location and mass of the modification (+470 Daltons on cysteine).
Molecular Model of Tetrameric Vimentin Reveals a Novel WFA-Binding Pocket
We then developed a three-dimensional model of the WFA-vimentin complex by using X-ray crystal structures of vimentin [25] and WFA [26]. Molecular modeling studies revealed a stable binding mode for WFA in the surface binding pocket of tetrameric vimentin between the pair of head-to-tail α-helical dimers (Figure 3A). In this simulated model, the C3 and C6 carbons of WFA lie in close proximity to the cysteine residue in the vimentin A helix (Figure 3B), permitting a nucleophilic attack by this thiol group on the electrophilic carbon centers (Figure 3C). Remarkably, the amino acid residues of vimentin (Gln324, Cys328, and Asp 331) that make contact with WFA (Figure 3C) are identical from mammals down to ancient fish, such as sharks [27] (Figure S5). Another feature illustrated by this model is that the orientation of C27 hydroxyl group places this moiety outside the binding cleft; thus, biotinylated WFA (Figure S1) is able to retain the flexibility to bind immobilized NeutrAvidin after modifying tetrameric vimentin in vivo. On the other hand, the simulated model of vimentin/the inactive congener 12-D WS (Figure 3D) reveals that the C5 alpha-hydroxyl and the C6-C7 epoxide of WFA are positioned to prevent rear side nucleophilic attack by the reactive thiol group on 12-D WS. This distinction between these two withanolides in their binding modes with vimentin is further corroborated by comparing the molecular docking-simulated internuclear distances between WFA and 12-D WS with vimentin (Figure S6), and these data are consistent with lack of in vitro binding activity of 12-D WS to vimentin (Figure 1D).
Figure 3. Molecular Model of WFA Binding Site in Tetrameric Vimentin.
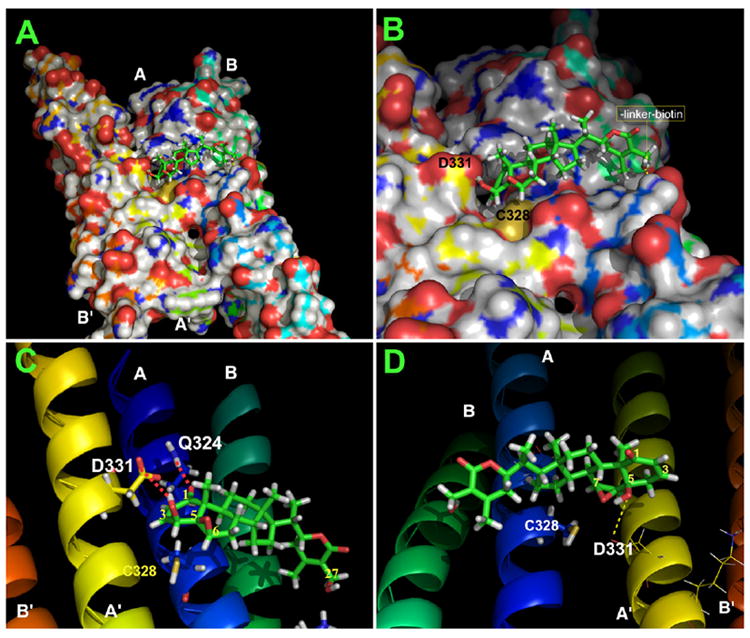
Molecular modeling of the WFA binding site in the vimentin tetramer model was derived with the crystal structures of the protein fragments associated with the PDB accession code 1gk4. Based on this model, MD-simulated annealing was performed to dock WFA to the vimentin tetramer fragment. The final protein-ligand binding structure was based on MD simulation (with the AMBER 8 program) in a water bath for 1.7 ns. The binding structure is stable in water. Images of the models were generated with PyMol software.
(A) Depicted in the figure is a snapshot of the MD-simulated solvent-accessible surface area binding structure showing WFA binding in the cleft between the A and A′ α helices of the vimentin tetramer.
(B) The close-up image shows the A-ring twist-boat and B-ring half-chair conformation of WFA [26] (see Figure 1A) is accommodated deep within the binding cleft of the vimentin tetramer, allowing for proper orientation with Cys328 (yellow) to form a covalent bond with the C3 or C6 electrophilic carbon centers of WFA. Note that the exocyclic C27 hydroxyl group (arrow) of WFA to which the linker-biotin was conjugated is orientated toward the solvent-side of the binding cleft that makes it possible for WFA-B to bind tetrameric vimentin.
(C) The model shows hydrogen bonding between Gln324 of the vimentin A-helix and the C1 position oxygen atom (2.3 Å), and Asp331 of the vimentin A′ helix and the C4 hydroxyl group (1.7 Å).
(D) The alpha orientations of the C5 (OH) and C6-C7 (epoxide) of the inactive withanolide congener 12-D WS appose Cys328 of vimentin, thus preventing nucleophilic attack by the thiol group on the electrophilic carbon centers.
Covalent Modification of Tetrameric Vimentin by WFA Causes Formation of Filament Aggregates In Vitro and In Vivo
The clinical importance of the cysteine residue in vimentin lies in its propensity for being preferentially oxidized in vimentin compared to other cytoskeletal proteins from rheumatoid arthritis patients [28]. Because this cysteine residue under oxidizing conditions can participate in disulfide cross-linking between a pair of vimentin dimers, leading to disruption to the filament structure in vitro [29], we next investigated how chemical modification of cysteine by WFA affects the vimentin IF structure. Employing soluble tetrameric vimentin in filament polymerization assays in vitro, we show that, although WFA at high doses does not block filament assembly per se, the drug induces formation of filamentous aggregates, many of which display amorphous condensed structures, as revealed in negatively stained transmission electron micrographs (Figure 4). This phenotype is not observed with lower doses of WFA or equivalent high doses of the inactive congener, 12-D WS. To further corroborate that vimentin aggregation by WFA treatment is associated with perturbation of the cytoskeleton structure, we investigated drug effects in BAECs by immunostaining. Cells treated with 3 μM WFA showed condensation of vimentin filaments around the perinuclear region and the presence of numerous, vimentin-positive staining particles in the cytoplasm (Figure 4; compare Figures 4E and 4F). Importantly, these vimentin particulates strongly costain for actin (Figure 4; compare Figures 4H and 4J), exemplifying the role of the WFA-binding domain in control of cytoskeletal structure through actin-perturbing activity of vimentin [21-23].
Figure 4. WFA Causes Aggregation of Vimentin Filaments In Vitro and In Vivo.
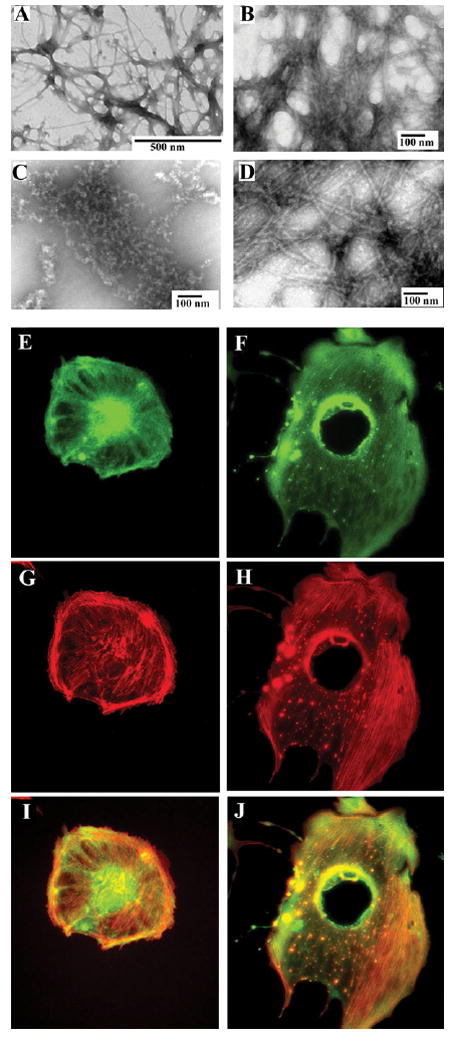
(A) Tetrameric soluble hamster vimentin was polymerized in presence of 170 mM NaCl by incubation at 37°C for 1 hr. The protein was fixed with 0.5% glutaraldehyde, stained with uranyl acetate, and observed by transmission EM. The presence of vehicle solvent does not interfere with filament formation. (B) Polymerization of tetrameric vimentin in the presence of 25 μM WFA produces extensive filamentous aggregates and (C) many irregular fragmented aggregated structures. (D) 12-D WS (25 μM) does not disrupt vimentin polymerization. BAECs treated with DMSO (E, G, I) or 3 μM WFA (F, H, J) for 18 hr were stained for vimentin with a monoclonal anti-vimentin antibody (green) and cos-tained with phalloidin-rhodamine (red). The vimentin (E) and (F) and phalloidin-stained images (G) and (H) in fluorescence overlap reveal the presence of numerous cytoplasmic particulate granules that costain for vimentin and disrupted F-actin in WFA-treated cells compared to controls (I) and (J).
WFA Causes Vimentin Fragmentation In Vivo
As it became apparent that vimentin targeting by WFA may initiate signaling events leading to cellular apoptosis, we wanted to know whether soluble tetrameric vimentin, which is difficult to differentiate from the abundant filaments by staining, is also affected by WFA. Through western blot analysis of soluble proteins from HUVECs treated with WFA, we show dose-dependent (above 2 μM) increases in vimentin cleavage products (Figure 5A), which became more pronounced by 2 hr, paralleling the decreased expression of the 56 kDa form after drug treatment (Figure 5B). Since several posttranslationally modified isoforms of vimentin exist in cells, we performed two-dimensional western blot analysis to better characterize the vimentin-targeting activity of WFA. As compared with the vehicle-treated cells (Figures 5C and 5D), WFA treatment was found to cause reductions in the levels of several ~53–56 kDa isoforms (Figure 5E), with WFA-B treatment also inducing similar reduction in levels of vimentin isoforms at cyctotoxic doses of this WFA analog (Figure 5F). These data further support the in vivo drug-mimetic effect of WFA-B.
Figure 5. WFA Induces Vimentin Fragmentation in Endothelial Cells.
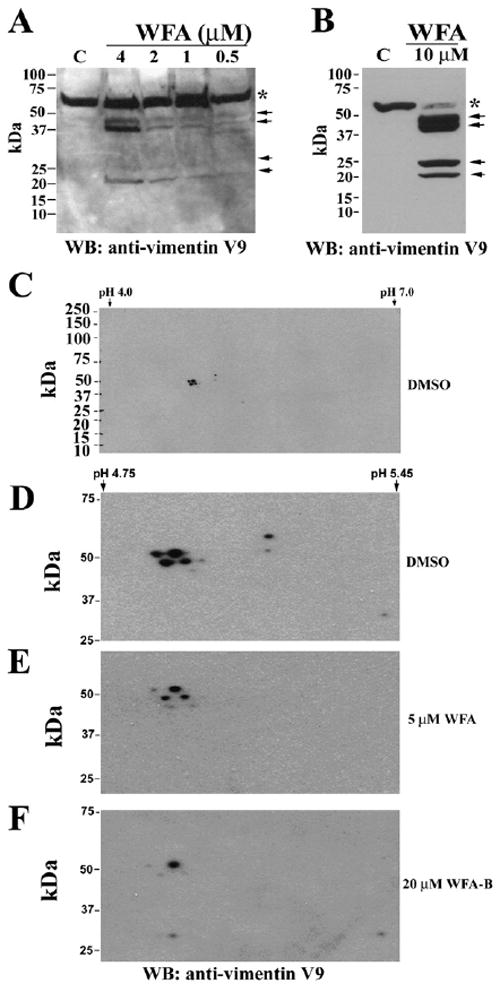
(A) Western blot analysis of HUVECs shows dose-responsive increases in vimentin cleavage products (arrows) with WFA treatment after 2 hr as detected with the monoclonal anti-vimentin V9 antibody.
(B) Higher concentrations and longer periods of exposure to WFA (4 hr) cause reduction in levels of the 56 kDa protein (asterisk) and increased abundance of cleavage products of vimentin (arrows).
(C) Two-dimensional western blotting analysis of WFA-treated HU-VECs. Cells were extracted with buffer A and soluble lysates were subjected to isoelectric focusing with IPG strips (Biorad). The second dimension was performed with 4%–20% polyacrylamide gels and proteins transferred on membranes for western blot analysis. Protein blots were probed with anti-vimentin monoclonal antibody (V9). IPG strips in the range of pH 4–7 were employed to allow for fine resolution of closely migrating vimentin isoforms.
(D–F) Expanded images (range, pH 4–6) of western blots from control (D), WFA- (E), and WFA-B-treated (F) HUVECs show the dramatic reduction in spot intensity of many vimentin isoforms in drug-treated cells.
The Ubiquitin-Proteasome Pathway-Targeting Activity of WFA is Mediated by Vimentin
Since IF protein aggregation has been shown to negatively impact the UPP, resulting in proteasome inhibition [30], we queried whether the UPP-targeting activity of WFA [5, 9] is also regulated in a vimentin-dependent manner. Employing the widely used MCF-7 vimentin-deficient cell culture model [31], we found that WFA causes increased levels of polyubiquitinated proteins in vimentin-transfected MCF-7 cells, but only moderately in the vector controls (Figure 6A). We addressed the importance of the 2-3 unsaturated position of the A-ring on UPP activity of WFA by structure-activity relationship studies. We found that the semisynthetic analogs, 3β-methoxy-dihydrowithaferin A or 3β-thiophenoxy-dihydrowithaferin A, like the inactive congener, 12-D WS [12], also failed to increase cellular levels of ubiquitinated species (Figure S7A). Similarly, vimentin-transfected MCF-7 cells treated with 12-D WS also failed to increase levels of ubiquitinated proteins (Figure S7B). These results are consistent with the binding mode of WFA for vimentin (Figure 3). Next, to corroborate that vimentin targeting, and not the possible direct inhibition of the 20S proteasome by WFA, mediates its UPP-targeting function [9], we tested WFA in proteasome kinetic assays. We show that, whereas the highly selective 20S proteasome inhibitor, epoxomicin [13], significantly inhibits the 20S proteasome’s major catalytic function at 10 nM, WFA minimally inhibits this catalytic activity, even at cytotoxic concentrations of 10 μM (Figure 6B). The Kobs/[I] (M−1S−1), a measure of the efficiency of inactivation [13], was calculated to be 340 ± 80 (0.5–10 μM) for WFA, while that of epoxomicin is 44,510 ± 7,000 (10–75 nM). Contrary to a recent report [32], our data suggest that WFA-modified vimentin may mediate sequestration of ubiquitinated proteins and result in proteasome inhibition in vivo. Thus, employing cell lines derived from vimentin-deficient and isogenic wild-type mouse strains [33], we show that vimentin deficiency confers enhanced resistance to WFA-induced apoptosis with wild-type cells having a 7-fold higher rate of apoptosis, whereas 12-D WS does not induce apoptosis in wild-type cells (Figure 6C). Furthermore, exploiting a protein transduction methodology to introduce the WFA-modified, dominant-negative vimentin tetramers into cells, we found by immunostaining that endogenous vimentin IFs and F-actin are found to aggregate, whereas cells transduced with vehicle-treated vimentin do not produce this phenotype (Figure 6; compare Figures 6D and 6F). Thus, vimentin targeting by WFA has a dominant-negative affect on the cystosketon architecture, as seen by staining for F-actin (Figures 6E and 6G). Collectively, these findings identify a potent cytoskeleton-perturbing activity of WFA, which is mediated by vimentin.
Figure 6. WFA-Induced Upregulation of Protein Ubiquitination is Mediated by Vimentin.
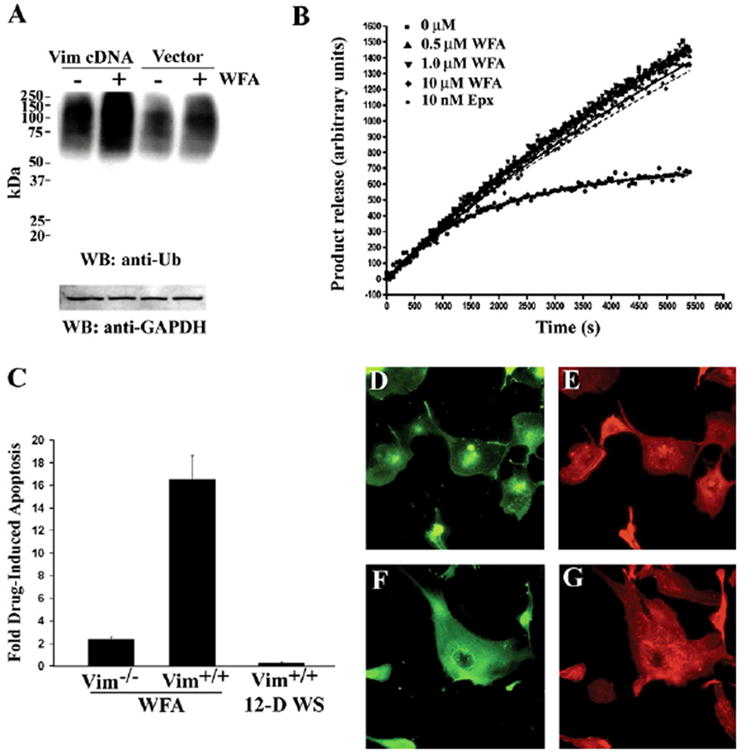
(A) MCF-7 cells that lack endogenous IF proteins were transfected with human vimentin cDNA or a vector control and, after 24 hr, cells were treated with either vehicle or 2 μM WFA for 1 hr. Cell lysates were prepared and equal amounts of protein subjected to SDS-PAGE and protein blots probed with anti-ubiquitin antibody. Blots were reprobed with anti-GAPDH antibody.
(B) 20S proteasome (bovine) preparation was incubated with vehicle (control), 0.5 μM, 1.0 μM and 10 μM WFA or with 10 nM Epoxomicin (Epx) in presence of LLVY-AMC substrate at room temperature. Fluorescence readings from triplicate samples were obtained at different time intervals from the 96-well plate with excitation at 355 nm and emission at 430 nm. The release of product was plotted against time for each concentration of inhibitor.
(C) Embryonic fibroblast cell lines derived from vimentin-deficient mice and wild-type litter-mates [33] were treated with vehicle, 5 μM WFA, or 5 μM 12-D WS for 24 hr. Cells were harvested and stained with annexin V-FITC and pro-pidium iodide, and extent of apoptosis assessed by flow cytometry. The fold-apoptosis for drug over vehicle-treated samples for each cell line was plotted (n = 2 experiments); error bars represent SD.
(D) BAECs were transduced with WFA-modified vimentin and cells were stained after 18 hr.
(E) Cells transduced with WFA-modified vimentin show both condensed vimentin filaments localized largely in and around the nucleus and the presence of vimentin-staining particulates.
(F and G) Cells transduced with vehicle-treated vimentin show well-distributed orchestration of vimentin IFs (F) and actin cytoskeleton (G). Results representative of three experiments are shown.
The In Vivo Antiangiogenic Activity of WFA is Mediated by Vimentin
We previously reported that WFA exhibits potent angiogenesis inhibitory activity in vivo [5]. Thus, here we have investigated whether the pharmacological activity of WFA on de novo capillary growth is altered by vimentin deficiency. Through the use of the mouse model of injury-induced corneal neovascularization, we found that WFA markedly suppressed neovascularization in wild-type mice (73% inhibition; n = 8; p = 0.002), whereas it only marginally attenuated neovascularization in vimentin-null mice (29% inhibition; n = 10; p = 0.005) (Figure 7), revealing that inhibition of capillary growth by WFA is largely mediated by vimentin. It is likely that vimentin in corneoscleral capillaries and fibroblastic cells of the injured corneal stroma [34] is targeted by WFA in this angiogenesis model. Furthermore, in accord with our in vitro studies, we find that the inactive congener, 12-D WS, does not alter corneal neovascularization in wild-type Svev mice (<1% activation; n = 6; p = 0.1724). Interestingly, the vascularization response of vimentin-deficient mice is not as extensive as in wild-type mice, which is consistent with previous reports on impaired angiogenesis in vimentin deficiency [19] and the likely impairment of vimentin-dependent me-chanosignaling events in the vasculature [35].
Figure 7. Vimentin Mediates the Antiangiogenic Activity of WFA.
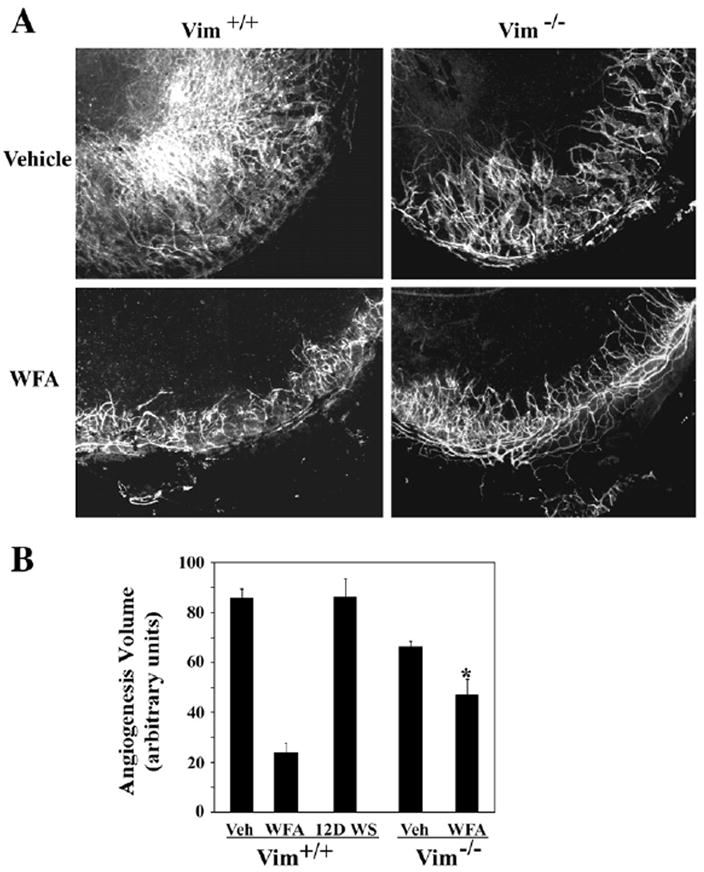
Wild-type mice and vimentin-deficient mice were subjected to corneal chemical injury with limbal and corneal epithelial cell debridement. Mice were treated by daily intraperiteoneal injection with vehicle, 2 mg/kg WFA, or 2 mg/kg 12-D WS for 10 days. Neovascularization responses in corneas were assessed by flat-mount staining with anti-CD31-flourescent antibodies.
(A) Flat-mount stained corneas were photographed and representative images of CD31+ (fluorescent) capillaries are shown.
(B) Quantification of neovascularization from each group of mice (n = 14, Vim+/+; n = 10, Vim−/−) was performed as previously described [45]. Error bars represent SD; * p < 0.05 by Mann-Whitney U test.
WFA has recently been reported to bind annexin II in cancer cell lines [36]. However, this 36 kDa protein is not detected in affinity-purified proteins from bovine (Figure 1B) or human (Figure 1C) [12] endothelial cells, nor was it detected by western blotting of NeutrAvidin-purified proteins in our studies (data not shown). Thus, we are drawn to the interpretation that differences in the construction of the biotinylated WFA analogs (e.g., inclusion of a long, linear hydrocarbon linker between the natural product and biotin) [10, 37] or their applications for target isolation (in vitro versus in vivo labeling) could account for different targets being bound to WFA. It is also possible that WFA has binding specificities for different targets in vivo, which may also be context related. Since vimentin deficiency does not abrogate WFA’s biological activities totally, the existence of other relevant targets of this compound must be considered. The related IF protein, desmin, could be a relevant target, because the WFA-binding motif is conserved in desmin (Figure S5) [27]. Thus, the modest WFA inhibitory activity on corneal neovascularization in vimentin-deficient mice may be due to the targeting of desmin in mural cells of invasive capillaries [34]. While this theory remains to be tested, our findings underscore the importance of multiple target activity of WFA that could be exploited for drug development.
In conclusion, this study illustrates the cell biological and pharmacological use of a small molecule that targets type III IF proteins via modification of the conserved rod 2B domain. Intriguingly, a recent report has shown that the antiangiogenic and chemopreventive agent, epigallocatechin gallate, also binds to vimentin, although via the N-terminal region of this protein [38]. The revelation that vimentin is a critical target of such chemically diverse natural products (withanolides and flavanoids) provides new opportunities to investigate potential synergistic effects of these inhibitors on cell signaling networks evoked under malignant, angiofibrotic, and inflammatory conditions [39].
SIGNIFICANCE
Withaferin A (WFA) is a prototype of the withanolide class of natural products that exhibit diverse pharmacological activities, including antitumor, antiangiogenic, cardioprotective, anti-inflammatory, and immunomodulatory effects [7]. Our findings showing that site-specific modification of vimentin by WFA in vivo causes endothelial cell apoptosis illuminates the chemical genetic approach of perturbing the conserved rod 2B domain to produce a dominant-negative effect on intermediate filament (IF) protein function. Use of such a small molecule to alter IF function can serve as a complementary approach to classical genetic studies of disorders of IFs [40], and, potentially, WFA-B can also be exploited as a chemical proteomic reagent for investigating the IF proteome. Thus, WFA and derivative steroidal lactones represent a class of useful chemical genetic tools for studies of the type III IF proteins. As vimentin modulates the immune response [41], and is overexpressed in prostate and other cancers [42], WFA holds great promise as a potential drug lead for the development of small-molecule therapeutics.
EXPERIMENTAL PROCEDURES
All cell culture supplies were purchased from GIBCO, unless otherwise specified. Antibodies were from Santa Cruz Biotechnology, unless otherwise specified. WFA and 12-D WS were obtained from Chromadex, and stock solutions were freshly prepared in DMSO. The synthesis of WFA analogs, 3β-methoxy-dihydrowithaferin A and 3β-thiophenoxy-dihydrowithaferin A, is described in Supplemental Experimental Procedures provided in the Supplemental Data. All cell reagents were purchased from Invitrogen.
Cell Cultures and Reagents
HUVECs and BAECs were obtained from Cascade Biologicals and cultured according to the vendor’s protocols. The MCF-7 breast cancer cell line was obtained from American Type Culture Collection and cultured in RPMI 1640 medium containing 10% fetal bovine serum (FBS). MFT-16 cells from embryo fibroblasts of vimentin homozygous-deficient mice (Vim−/−) and MFT-6 cells from embryo fibroblasts of wild-type (Vim+/+) mice were obtained from Robert Evans (University of Colorado, Denver, CO) and cultured in F12:DMEM (1:1) medium supplemented with 5% FBS. All cells were cultured in humidified incubators at 37°C-5% CO2 conditions.
Isolation of WFA-B-Binding Proteins by Affinity Chromatography
The synthesis and chemical characterization of WFA-B and its use to identify biotinylated proteins from HUVECs was previously reported [12]. For scale-up studies, BAECs were preincubated with DMSO or 5 μM WFA for 1 hr and subsequently treated with 5 μM WFA-B for 2 hr. Cells were washed in ice-cold phosphate-buffered saline (PBS) and cytoplasmic extracts were prepared in buffer A (5 mM Tris, pH 7.6, 50 mM NaF, 1% Triton X-100, 5 mM EGTA) supplemented with a proteinase inhibitor cocktail (Roche). After centrifugation, equal amounts of protein were precleared on agarose beads (Sigma) to remove nonspecific agarose-binding proteins. The beads were centrifuged and precleared cell lysates were repeatedly loaded three times on columns containing NeutrAvidin-agarose beads (Pierce) to maximize immobilization of biotinylated proteins. After extensive washing with ice-cold buffer A, bound biotinylated proteins were eluted in Lae-meli gel loading buffer containing β-mercaptoethanol, and fractioned by SDS-PAGE on 15 × 15 cm gels. Gels were stained with Coomassie blue dye, and bands corresponding to 56 kDa, 51 kDa, and 43 kDa protein were excised for mass spectrometric analysis.
Identification of Affinity-Purified Proteins by LC-MS/MS
All mass spectra reported in this study were acquired by the University of Kentucky Mass Spectrometry Facility. Gel pieces were digested with trypsin, and LC-MS/MS spectra were acquired on a ThermoFinnigan LCQ “Classic”; quadrupole ion trap mass spectrometer (Finnigan Co., San Jose, CA). Separations were performed with an HP 1100 high-performance liquid chromatograph modified with a custom splitter to deliver 4 μl/min to a custom C18 capillary column (300 μm inner diameter × 15 cm). Gradient separations consisted of a 2 min isocratic step at 95% water and 5% acetonitrile (both phases containing 0.1% formic acid). The organic phase was increased to 20% acetonitrile over 8 min, and then increased to 90% acetonitrile over 25 min, held at 90% acetonitrile for 8 min, and then increased to 95% in 2 min; finally, they were returned to the initial conditions in 10 min (total acquisition time, 45 min with a 10 min recycle time). Tandem mass spectra were acquired in a data-dependent manner. Three microscans were averaged to generate the data-dependent full-scan spectrum. The most intense ion was subjected to tandem mass spectrometry, and three microscans were averaged to produce the MS/MS spectrum. Masses subjected to the MS/MS scan were placed on an exclusion list for 2 min. Resulting MS/MS spectra were searched against mammalian proteins in the Swiss-Prot database with the Mascot search engine (Matrix Science).
WFA Binding Site Identification in Vimentin
Hamster tetrameric vimentin (0.5 mg/ml) was incubated with 10 μM WFA or an equivalent amount of DMSO for 1 hr at 37°C. Protein-ligand complexes were subjected to tryptic digestion and LC-MS/MS analysis. The assignment of fragment ions in the MS/MS of the modified and unmodified peptides was obtained with the SEQUEST software tool “FuzzyIons” (distributed by ThermoFinnigan) to annotate the spectra from WFA-treated and DMSO-treated samples. This analysis verifies both the peptide amino acid sequence and the location and mass of the modification (+470 daltons on cysteine).
3D Model of the Human Vimentin Fragment
The initial coordinates of 2B human vimentin fragment used in our computational studies came from the X-ray crystal structure (Protein Data Bank [PDB] code: 1gk4) [25] deposited in the PDB. To encompass the structure of the protein environment surrounding the active residue Cys328, the missing residues of the 2A fragment (i.e., residues 313–327 in A and B helices) were built with the α-helical template structure and the automated homology modeling tool, Modeler/InsightII software (Accelrys, Inc.). Then, the best 3D model was solvated in water and refined by performing a long-time molecular dynamics (MD) simulation in water (see Supplemental Experimental Procedures).
Molecular Docking
To explore the possible vimentin-ligand binding mode, the first step was to dock the ligand (i.e., WFA) or 12-D WS to vimentin tetramer fragment by virtue of their geometric complementarity. We aimed to find where the ligand could be inserted most comfortably. The molecular docking for each vimentin-ligand binding was carried out in the same way as we recently did for studying other protein-ligand binding systems [43]. Briefly, a ligand-binding site was defined as that consisting of the residues within a sphere (with a radius of 20 Å) centered at Cys328 residue. The ligand was initially positioned at ~10 Å in front of Cys328 of the binding site. The initial docking calculations were performed on the ligand with the vimentin fragment binding site by the “automatic docking” Affinity module of the InsightII package (Accelrys, Inc.). The Affinity methodology uses a combination of Monte Carlo-type and simulated annealing procedures to dock the guest molecule (the ligand) to the host (the receptor). The vimentin-ligand binding structure obtained from the initial docking was further refined by performing an MD simulation in a water bath (see Supplemental Experimental Procedures).
Western Blotting Experiments
After treatments, cells were washed in PBS and extracts having equal amount of proteins were subjected to SDS-PAGE on 4%–20% Tris-glycine gels (BioEpress) and transferred to Immun-Blot PVDF membrane (Bio-Rad) by standard techniques. Primary antibodies were diluted in 5% nonfat dry milk Tris-buffered saline, 0.02% Tween-20 (NFDM-TBST) at the concentration of 1:500, and secondary antibodies were used at 1:1000 dilution. Blots were extensively washed in TBST buffer and developed by enhanced chemiluminescence method (Amersham) and exposed to X-ray film.
Analysis of Vimentin Isoforms by 2D Gel Electrophoresis
HUVECs cultured in 60 mm dishes were treated with vehicle or 5 μM WFA or 20 μM WFA-B for 2 hr. Cells were washed in ice-cold PBS and collected in buffer A. After centrifugation at 14,000 × g for 10 min, equal amounts of protein from supernatants were suspended in 8 M urea, 50 mM dithiothreitol (DTT), and 0.2% Bio-lite ampholytes pH 3–5 and pH 5–8 (2:1 ratio; Biorad) with 0.01% bromophenol blue. The protein lysate was centrifuged at 14,000 × g for 5 min to precipitate insoluble material before incubation with 11 cm IPG strips (pH 4–7; Biorad). Gels were actively rehydrated at 50 mA for 12 hr with a Protean IEF cell system (Biorad). Isoelectric focusing was subsequently conducted for 5.5 hr (250 V for 20 min, 8,000 V for 2.5 hr, and 8,000 V for 25,000 V hr). Strips were dipped in equilibration buffer 1 containing 6 M urea, 2% SDS, 0.375 M Tris-HCl (pH 8.8), 20% glycerol, and 2% (w/v) DTT for 10 min, followed by an incubation in equilibration buffer 2 containing 6 M urea, 2% SDS, 0.375 M Tris-HCl (pH 8.8), 20% glycerol, and 2.5% (w/v) iodoacetamide for 10 min. Strips were laid on second dimension 4%–20% gradient gels (Biorad) and run at 200 V for 1 hr. Proteins were transferred at 100 V for 45 min for western blotting.
Transfection Studies
MCF-7 cells were transfected with a pCMV6-XL5 vector containing the human vimentin cDNA under CMV promoter, according to vendor instructions (Origene). Control samples were transfected with an empty vector (PCMV6-XL4). Transfected cells were allowed to recover for 12 hr and subsequently treated with vehicle or 2 μM WFA or 12-D WS for 18 hr. An equal amount of protein lysates were subjected to western blotting and probed with an antiubiquitin mouse monoclonal antibody.
Protein Transduction Studies
Tetrameric vimentin (0.5 μg) (Cytoskeleton) was incubated with 10 μM WFA or an equivalent amount of vehicle (DMSO) for 1 hr at 37°C to form protein-WFA adducts. Vimentin-WFA or vimentin alone (0.5 μg) was mixed with the Chariot protein transduction reagent (Active Motif) and incubated for 30 min at 24°C to form complex according to the manufacturer’s instructions. The protein-Chariot complex was subsequently added to BAECs in serum-free medium and incubated for 1 hr in 37°C-5% CO2 conditions. Fresh complete medium was then added and cells were incubated for an additional 18 hr under normal culture condition. Cells were processed for immunohistochemistry analysis as described previously here.
Apoptosis by Flow Cytometry
To assess the apoptosis activity of WFA and 12-D WS, embryonic fibroblast vimentin-deficient cell lines (Vim−/−; MFT-16) and wild-type (Vim+/+; MFT-6) cells [32] were treated with 5 μM WFA, 5 μM 12-D WS, or an equivalent amount of vehicle (DMSO) for 24 hr in 37°C-5% CO2 conditions in complete medium. Apoptotic cells were measured with the Vybrant Apoptosis Assay Kit (Molecular Probes) according to the manufacturer’s instructions. Flow cytometric analysis was conducted at the University of Kentucky Core Flow Cytometry Center.
Cell Staining Procedures and Fluorescence Imaging
After treatments, BAECs were washed with PBS and fixed with 4% paraformaldehyde for 5 min. Cells were permeabilized with 0.1% Triton-X in PBS for 20 min at 4°C and blocked for 30 min in 3% BSA. Rabbit polyclonal vimentin antibody (Vim) or mouse monoclonal vimentin antibody (V9) was applied to cells for 1 hr at 24°C at 1:400 dilution in PBS. After washing with PBS, cells were incubated with anti-rabbit FITC-conjugated secondary antibody (1:500) or anti-mouse Texas Red-conjugated secondary antibody (1:500) for 30 min. Cells were washed, and those labeled with Vim-antibody were incubated with phalloidin-Rhodamine (1:200) for 20 min. After washes for 2 hr, staining was visualized with a Nikon TE2000 microscope.
Transmission Electron Microscopy of Vimentin Filaments
Tetrameric hamster vimentin was subjected to in vitro filament formation assays with vendor-supplied reagents and instructions (Vimentin Filament Biochemistry Kit, Denver, CO). Vimentin (0.5 mg/ml) was mixed with WFA (5 or 25 μM), DMSO, or 12D-WS (25 μM) in filament polymerization buffer (170 mM NaCl final concentration) and incubated for 1 hr at 37°C. Protein was immediately fixed in 0.5% glutaraldehyde, stained with uranyl acetate, and applied to copper grids for EM staining (University of Kentucky Core Microscopy and Imaging Facilities). Over 100 grids for each treatment were viewed at 80 kV on an FEI Biotwin 12 transmission electron microscope, and 25 representative images were collected. The entire experiment was then repeated.
Enzyme Kinetic Studies
KA values were determined as follows. Inhibitors were mixed with a fluorogenic peptide substrate and assay buffer (20 mM Tris [pH 8.0], 0.5 mM EDTA, and 0.035% SDS) in a 96-well plate. The chymotrypsin-like activity was assayed with the fluorogenic peptide substrates, Suc-Leu-Leu-Val-Tyr-AMC (Sigma-Aldrich). Hydrolysis was initiated by the addition of bovine 20S proteasome, and the reaction was followed by fluorescence (360 nm excitation/460 nm detection) with a Micro-plate Fluorescence Reader (FL600; Bio-Tek Instruments, Inc., Win-noski, VT) employing the software KC4 v.2.5 (Bio-Tek Instruments, Inc.). Reactions were allowed to proceed for 60–90 min, and fluorescence data were collected every 1 min. Fluorescence was quantified as arbitrary units, and progression curves were plotted for each reaction as a function of time. Kobs/[I] values were obtained with the PRISM program by nonlinear least-squares fit of the data to the following equation: fluorescence = _v_st + ([_v_0 − _v_s]/Kobs)(1 − exp[−Kobs t]), where _v_0 and _v_s are the initial and final velocities, respectively, and Kobs is the reaction rate constant. The range of inhibitor concentrations tested was chosen so that several half-lives could be observed during the course of the measurement. Reactions were performed with inhibitor concentrations that were < 100-fold of those of the proteasome assayed.
Corneal Neovascularization Assays in Mouse
All animal experiments were conducted in accordance with the Declaration of Helsinki, and procedures approved by IACUC committee of the University of Kentucky. Mice were housed in specific pathogen-free cages in designated lab animal housing facilities. Vimentin homozygous-deficient mice (Vim−/−) and mice that were vimentin-heterozygous deficient (Vim+/−) in the 129/Svev background were obtained from David Markovitz (University of Michigan Medical Center) and breeding colonies established at the University of Kentucky. Age-matched littermates were genotyped by polymerase chain reaction, as previously described [44], and Vim−/− and Vim+/+ mice were employed for corneal vascularization experiments [45]. In brief, mice between 4 and 6 wk of age were anesthetized by intraperiteoneal (i.p.) injection of ketamine and xylazine. Corneas were topically anesthetized by application of proparacain eye drop, and 1 μl drop of dilute 0.15 M sodium hydroxide was applied for 1 min. The cornea was immediately washed extensively in saline solution, and corneal and limbal epithelium gently removed by scraping with a blunt Tooke corneal knife. The cornea was topically treated with atropine eye drop and covered with tobramycin and erythromycin antibiotic eye ointment. WFA or 12-D WS (2 mg/kg solubilized in DMSO) or vehicle (DMSO) was injected i.p. in respective drug or control groups of mice after their recovery from corneal injury, and subsequently every day thereafter for a period of 10 days. Mice were humanely killed and eyes enucleated. The anterior segment half of eyes were dissected and corneal buttons were prepared. Corneal tissues were fixed in 100% acetone for 20 min, washed in PBS for 1 hr, and blocked for 18 hr in 1% BSA-PBS at 4°C. Cornea whole-mount staining was performed by incubating tissues in FITC-conjugated rat anti-mouse CD31 antibody (BD PharMingen; 1:333 dilution in 1% BSA-PBS) for 12 hr, washed away for 24 hr at 4°C in 1% BSA-PBS, and affixed to glass slides with a coverslip. Fluorescent staining was visualized on a Nikon TE2000 microscope, and quantified by importing digital images to NIH ImageJ [45].
Supplementary Material
Supplementary Files
Acknowledgments
We thank Jack Goodman from the UK Mass Spectrometry Core Facility, Greg Bauman and Jennifer Strange from Flow Cytometry Core Facility, and Mary Engle from Imaging and Histology Core Center for their expert technical assistance, and J. Ambati and A. Pearson for scientific discussions. R.M. is supported by a Fight for Sight Foundation grant-in-aid, Kentucky Science and Engineering Foundation award, and a Research to Prevent Blindness Challenge Award to the Department of Ophthalmology; C.-G.Z. by NIDA/NIH; K.B.K. by the Kentucky Lung Cancer Research Program; D.M.M is the recipient of a Burroughs Wellcome Fund Clinical Scientist Award in Translational Research, and is supported by the Arthritis Foundation and NHLB/NIH and NIAID/NIH.
Footnotes
The authors declare no conflicts of interest.
Supplemental Data Supplemental Data, including Supplemental Experimental Procedures used in this work, Supplemental References, and seven figures are available online at http://www.chembiol.com/cgi/content/full/14/6/623/DC1/.
References
- 1.Zhang Y, Yeh JR, Mara A, Ju R, Hines JF, Cirone P, Griesbach HL, Schneider I, Slusarski DC, Holley SA, Crews CM. A chemical and genetic approach to the mode of action of fumagillin. Chem Biol. 2006;13:1001–1009. doi: 10.1016/j.chembiol.2006.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shen J, Xu X, Cheng F, Liu H, Luo X, Shen J, Chen K, Zhao W, Shen X, Jiang H. Virtual screening on natural products for discovering active compounds and target information. Curr Med Chem. 2003;10:2327–2342. doi: 10.2174/0929867033456729. [DOI] [PubMed] [Google Scholar]
- 3.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 4.Phung MW, Dass CR. In-vitro and in-vivo assays for angiogenesis-modulating drug discovery and development. J Pharm Pharmacol. 2006;58:153–160. doi: 10.1211/jpp.58.2.0001. [DOI] [PubMed] [Google Scholar]
- 5.Mohan R, Hammers HJ, Bargagna-Mohan P, Zhan XH, Herbstritt CJ, Ruiz A, Zhang L, Hanson AD, Conner BP, Rougas J, Pribluda VS. Withaferin A is a potent inhibitor of angiogenesis. Angiogenesis. 2004;7:115–122. doi: 10.1007/s10456-004-1026-3. [DOI] [PubMed] [Google Scholar]
- 6.Upton R. Ashwagandha Root. Scotts Valley, CA: American Herbal Pharmacopoeia; 2000. [Google Scholar]
- 7.Mishra LC, Singh BB, Dagenais S. Scientific basis for the therapeutic use of Withania somnifera (ashwagandha): a review. Altern Med Rev. 2000;5:334–346. [PubMed] [Google Scholar]
- 8.Shohat B, Gitter S, Abraham A, Lavie D. Antitumor activity of withaferin A (NSC-101088) Cancer Chemother Rep. 1967;51:271–276. [PubMed] [Google Scholar]
- 9.Bargagna-Mohan P, Ravindranath PP, Mohan R. Small molecule anti-angiogenic probes of the ubiquitin proteasome pathway: potential application to choroidal neovascularization. Invest Ophthalmol Vis Sci. 2006;47:4138–4145. doi: 10.1167/iovs.05-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crews CM, Splittgerber U. Chemical genetics: exploring and controlling cellular processes with chemical probes. Trends Biochem Sci. 1999;24:317–320. doi: 10.1016/s0968-0004(99)01425-5. [DOI] [PubMed] [Google Scholar]
- 11.Schreiber SL. Chemical genetics resulting from a passion for synthetic organic chemistry. Bioorg Med Chem. 1998;6:1127–1152. doi: 10.1016/s0968-0896(98)00126-6. [DOI] [PubMed] [Google Scholar]
- 12.Yokota Y, Bargagna-Mohan P, Ravindranath PP, Kim KB, Mohan R. Development of withaferin A analogs as probes of angiogenesis. Bioorg Med Chem Lett. 2006;16:2603–2607. doi: 10.1016/j.bmcl.2006.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc Natl Acad Sci USA. 1999;96:10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim KB, Myung J, Sin N, Crews CM. Proteasome inhibition by the natural products epoxomicin and dihydroepone-mycin: insights into specificity and potency. Bioorg Med Chem Lett. 1999;9:3335–3340. doi: 10.1016/s0960-894x(99)00612-5. [DOI] [PubMed] [Google Scholar]
- 15.Sin N, Meng L, Wang MQ, Wen JJ, Bornmann WG, Crews CM. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proc Natl Acad Sci USA. 1997;94:6099–6103. doi: 10.1073/pnas.94.12.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griffith EC, Su Z, Turk BE, Chen S, Chang YH, Wu Z, Biemann K, Liu JO. Methionine aminopeptidase (type 2) is the common target for angiogenesis inhibitors AGM-1470 and ovalicin. Chem Biol. 1997;4:461–471. doi: 10.1016/s1074-5521(97)90198-8. [DOI] [PubMed] [Google Scholar]
- 17.Eckes B, Colucci-Guyon E, Smola H, Nodder S, Babinet C, Krieg T, Martin P. Impaired wound healing in embryonic and adult mice lacking vimentin. J Cell Sci. 2000;113:2455–2462. doi: 10.1242/jcs.113.13.2455. [DOI] [PubMed] [Google Scholar]
- 18.van Beijnum JR, Dings RP, van der Linden E, Zwaans BM, Ramaekers FC, Mayo KH, Griffioen AW. Gene expression of tumor angiogenesis dissected: specific targeting of colon cancer angiogenic vasculature. Blood. 2006;108:2339–2348. doi: 10.1182/blood-2006-02-004291. [DOI] [PubMed] [Google Scholar]
- 19.Lundkvist A, Reichenbach A, Betsholtz C, Carmeliet P, Wolburg H, Pekny M. Under stress, the absence of intermediate filaments from Muller cells in the retina has structural and functional consequences. J Cell Sci. 2004;117:3481–3488. doi: 10.1242/jcs.01221. [DOI] [PubMed] [Google Scholar]
- 20.Soellner P, Quinlan RA, Franke WW. Identification of a distinct soluble subunit of an intermediate filament protein: tetrameric vimentin from living cells. Proc Natl Acad Sci USA. 1985;82:7929–7933. doi: 10.1073/pnas.82.23.7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leung CL, Green KJ, Liem RK. Plakins: a family of versatile cytolinker proteins. Trends Cell Biol. 2002;12:37–45. doi: 10.1016/s0962-8924(01)02180-8. [DOI] [PubMed] [Google Scholar]
- 22.Clarke EJ, Allan V. Intermediate filaments: vimentin moves in. Curr Biol. 2002;12:R596–R598. doi: 10.1016/s0960-9822(02)01102-8. [DOI] [PubMed] [Google Scholar]
- 23.Esue O, Carson AA, Tseng Y, Wirtz D. A direct interaction between actin and vimentin filaments mediated by the tail domain of vimentin. J Biol Chem. 2006;281:30393–30399. doi: 10.1074/jbc.M605452200. [DOI] [PubMed] [Google Scholar]
- 24.Fuska J, Fuskova A, Rosazza JP, Nicholas AW. Novel cytotoxic and antitumor agents. IV. Withaferin A: relation of its structure to the in vitro cytotoxic effects on P388 cells. Neoplasma. 1984;31:31–36. [PubMed] [Google Scholar]
- 25.Strelkov SV, Herrmann H, Geisler N, Wedig T, Zimbelmann R, Aebi U, Burkhard P. Conserved segments 1A and 2B of the intermediate filament dimer: their atomic structures and role in filament assembly. EMBO J. 2002;21:1255–1266. doi: 10.1093/emboj/21.6.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bandhoria PG, Vivek K, Kumar P, Satti NK, Dutt P, Suri KA. Crystal structure of 5beta, 6beta-epoxy-4beta, 27-dihydroxy-1-oxo-22R-witha-2,24-dienolide isolated from Withania somnifera leaves. Anal Sci. 2006;22:x91. Published online April 20, 2006 10.2116/analscix.22.x89. [Google Scholar]
- 27.Schaffeld M, Herrmann H, Schultess J, Markl J. Vimentin and desmin of a cartilaginous fish, the shark Scyliorhinus stellaris: sequence, expression patterns and in vitro assembly. Eur J Cell Biol. 2001;80:692–702. doi: 10.1078/0171-9335-00206. [DOI] [PubMed] [Google Scholar]
- 28.Rogers KR, Morris CJ, Blake DR. Cytoskeletal rearrangement by oxidative stress. Int J Tissue React. 1989;11:309–314. [PubMed] [Google Scholar]
- 29.Rogers KR, Herrmann H, Franke WW. Characterization of disulfide crosslink formation of human vimentin at the dimer, tetramer, and intermediate filament levels. J Struct Biol. 1996;117:55–69. doi: 10.1006/jsbi.1996.0069. [DOI] [PubMed] [Google Scholar]
- 30.Liu J, Chen Q, Huang W, Horak KM, Zheng H, Mestril R, Wang X. Impairment of the ubiquitin-proteasome system in desminopathy mouse hearts. FASEB J. 2006;20:362–364. doi: 10.1096/fj.05-4869fje. [DOI] [PubMed] [Google Scholar]
- 31.Bar H, Kostareva A, Sjoberg G, Sejersen T, Katus HA, Herrmann H. Forced expression of desmin and desmin mutants in cultured cells: impact of myopathic missense mutations in the central coiled-coil domain on network formation. Exp Cell Res. 2006;312:1554–1565. doi: 10.1016/j.yexcr.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 32.Yang H, Shi G, Dou QP. The tumor proteasome is a primary target for the natural anticancer compound withaferin A isolated from “Indian Winter Cherry”. Mol Pharmacol. 2007;71:426–437. doi: 10.1124/mol.106.030015. [DOI] [PubMed] [Google Scholar]
- 33.Holwell TA, Schweitzer SC, Evans RM. Tetracycline regulated expression of vimentin in fibroblasts derived from vimentin null mice. J Cell Sci. 1997;110:1947–1956. doi: 10.1242/jcs.110.16.1947. [DOI] [PubMed] [Google Scholar]
- 34.Ishizaki M, Wakamatsu K, Matsunami T, Yamanaka N, Saiga T, Shimizu Y, Zhu G, Kao WW. Dynamics of the expression of cytoskeleton components and adherens molecules by fibroblastic cells in alkali-burned and lacerated corneas. Exp Eye Res. 1994;59:537–549. doi: 10.1006/exer.1994.1139. [DOI] [PubMed] [Google Scholar]
- 35.Terzi F, Henrion D, Colucci-Guyon E, Federici P, Babinet C, Levy BI, Briand P, Friedlander G. Reduction of renal mass is lethal in mice lacking vimentin: role of endothelin-nitric oxide imbalance. J Clin Invest. 1997;100:1520–1528. doi: 10.1172/JCI119675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Falsey RR, Marron MT, Gunaherath GM, Shirahatti N, Mahadevan D, Gunatilaka AA, Whitesell L. Actin microfilament aggregation induced by withaferin A is mediated by annexin II. Nat Chem Biol. 2006;2:33–38. doi: 10.1038/nchembio755. [DOI] [PubMed] [Google Scholar]
- 37.Berger AB, Vitorino PM, Bogyo M. Activity-based protein profiling: applications to biomarker discovery, in vivo imaging and drug discovery. Am J Pharmacogenomics. 2004;4:371–381. doi: 10.2165/00129785-200404060-00004. [DOI] [PubMed] [Google Scholar]
- 38.Ermakova S, Choi BY, Choi HS, Kang BS, Bode AM, Dong Z. The intermediate filament protein vimentin is a new target for epigallocatechin gallate. J Biol Chem. 2005;280:16882–16890. doi: 10.1074/jbc.M414185200. [DOI] [PubMed] [Google Scholar]
- 39.Hordijk PL. Endothelial signalling events during leukocyte transmigration. FEBS J. 2006;273:4408–4415. doi: 10.1111/j.1742-4658.2006.05440.x. [DOI] [PubMed] [Google Scholar]
- 40.Bar H, Strelkov SV, Sjoberg G, Aebi U, Herrmann H. The biology of desmin filaments: how do mutations affect their structure, assembly, and organisation? J Struct Biol. 2004;148:137–152. doi: 10.1016/j.jsb.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 41.Mor-Vaknin N, Punturieri A, Sitwala K, Markovitz DM. Vimentin is secreted by activated macrophages. Nat Cell Biol. 2003;5:59–63. doi: 10.1038/ncb898. [DOI] [PubMed] [Google Scholar]
- 42.Singh S, Sadacharan S, Su S, Belldegrun A, Persad S, Singh G. Overexpression of vimentin: role in the invasive phenotype in an androgen-independent model of prostate cancer. Cancer Res. 2003;63:2306–2311. [PubMed] [Google Scholar]
- 43.Hamza A, Zhan C-G. How Can (−)-epigallocatechin gallate from green tea prevent HIV-1 infection? mechanistic insights from computational modeling and the implication for rational design of anti-HIV-1 entry inhibitors. J Phys Chem B. 2006;110:2910–2917. doi: 10.1021/jp0550762. [DOI] [PubMed] [Google Scholar]
- 44.Colucci-Guyon E, Portier MM, Dunia I, Paulin D, Pournin S, Babinet C. Mice lacking vimentin develop and reproduce without an obvious phenotype. Cell. 1994;79:679–694. doi: 10.1016/0092-8674(94)90553-3. [DOI] [PubMed] [Google Scholar]
- 45.Ambati BK, Anand A, Joussen AM, Kuziel WA, Adamis AP, Ambati J. Sustained inhibition of corneal neovascularization by genetic ablation of CCR5. Invest Ophthalmol Vis Sci. 2003;44:590–593. doi: 10.1167/iovs.02-0685. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Files