Animal models of Parkinson's disease: a source of novel treatments and clues to the cause of the disease (original) (raw)
Abstract
Animal models of Parkinson's disease (PD) have proved highly effective in the discovery of novel treatments for motor symptoms of PD and in the search for clues to the underlying cause of the illness. Models based on specific pathogenic mechanisms may subsequently lead to the development of neuroprotective agents for PD that stop or slow disease progression. The array of available rodent models is large and ranges from acute pharmacological models, such as the reserpine- or haloperidol-treated rats that display one or more parkinsonian signs, to models exhibiting destruction of the dopaminergic nigro-striatal pathway, such as the classical 6-hydroxydopamine (6-OHDA) rat and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse models. All of these have provided test beds in which new molecules for treating the motor symptoms of PD can be assessed. In addition, the emergence of abnormal involuntary movements (AIMs) with repeated treatment of 6-OHDA-lesioned rats with L-DOPA has allowed for examination of the mechanisms responsible for treatment-related dyskinesia in PD, and the detection of molecules able to prevent or reverse their appearance. Other toxin-based models of nigro-striatal tract degeneration include the systemic administration of the pesticides rotenone and paraquat, but whilst providing clues to disease pathogenesis, these are not so commonly used for drug development. The MPTP-treated primate model of PD, which closely mimics the clinical features of PD and in which all currently used anti-parkinsonian medications have been shown to be effective, is undoubtedly the most clinically-relevant of all available models. The MPTP-treated primate develops clear dyskinesia when repeatedly exposed to L-DOPA, and these parkinsonian animals have shown responses to novel dopaminergic agents that are highly predictive of their effect in man. Whether non-dopaminergic drugs show the same degree of predictability of response is a matter of debate. As our understanding of the pathogenesis of PD has improved, so new rodent models produced by agents mimicking these mechanisms, including proteasome inhibitors such as PSI, lactacystin and epoximycin or inflammogens like lipopolysaccharide (LPS) have been developed. A further generation of models aimed at mimicking the genetic causes of PD has also sprung up. Whilst these newer models have provided further clues to the disease pathology, they have so far been less commonly used for drug development. There is little doubt that the availability of experimental animal models of PD has dramatically altered dopaminergic drug treatment of the illness and the prevention and reversal of drug-related side effects that emerge with disease progression and chronic medication. However, so far, we have made little progress in moving into other pharmacological areas for the treatment of PD, and we have not developed models that reflect the progressive nature of the illness and its complexity in terms of the extent of pathology and biochemical change. Only when this occurs are we likely to make progress in developing agents to stop or slow the disease progression. The overarching question that draws all of these models together in the quest for better drug treatments for PD is how well do they recapitulate the human condition and how predictive are they of successful translation of drugs into the clinic? This article aims to clarify the current position and highlight the strengths and weaknesses of available models.
LINKED ARTICLES
This article is part of a themed issue on Translational Neuropharmacology. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.164.issue-4
Keywords: Parkinson's disease, dopamine, dyskinesia, 6-hydroxydopamine, genetics, MPTP, neurodegeneration, nigro-striatal pathway, symptomatic treatment, toxin
Introduction
The classical motor symptoms of Parkinson's disease (PD) (akinesia, bradykinesia, rigidity, tremor and postural abnormalities) are associated with the loss of nigral dopaminergic cells and a decline in caudate-putamen dopamine content that led to the introduction of dopamine replacement therapy. As a consequence, there has been a key role for animal models of PD in devising novel pharmacological approaches to therapy, in developing new treatment strategies and in understanding the nature of the pathogenic processes involved in neuronal loss. The discovery that administration of reserpine or haloperidol to rodents and rabbits led to a transient parkinsonian-like state was rapidly followed by the key discovery that these symptoms were reversed by the administration of L-DOPA (Carlsson et al., 1957). This opened the door to an era where animal models of PD were used to investigate the basis of symptomatic treatment. More success followed when it was discovered that the unilateral stereotaxic injection of 6-hydroxydopamine (6-OHDA) in to the substantia nigra or the medial forebrain bundle caused the destruction of the nigro-striatal pathway and so loss of dopaminergic input to the striatum. This led to the introduction of the ‘circling’ rat model of PD that dominated research for many years and started the era of toxin use for producing animal models of PD (Ungerstedt, 1968). Through these advances came novel approaches to treatment such as the introduction of peripherally acting decarboxylase inhibitors, carbidopa and benserazide, that limited the peripheral side effects of L-DOPA and allowed a lowering of dose as more drug entered the brain (see, e.g., Pinder et al., 1976). More recently came the introduction of selective monoamine oxidase-B (MAO-B) inhibitors, selegiline and rasagiline, that slow the degradation of dopamine formed from L-DOPA and prolong its duration of effect and latterly catechol-_O_-methyl-transferase (COMT) inhibitors, entacapone and tolcapone, that stop either the peripheral or central metabolism of L-DOPA to 3-_O_-methyldopa so again prolonging its duration of effect and further increasing brain penetration of the drug (Fernandez and Chen, 2007; Lees, 2008).
Critically, the chemical and toxin animal-based models of PD ushered in the era of the use of synthetic dopamine agonists with early interest in producing anti-parkinsonian activity through post-synaptic dopamine receptor stimulation in the striatum. Large numbers of molecules were screened through the available models with, of course, many failures at both the preclinical and clinical level on route to success. Apomorphine was the first compound used experimentally that was eventually employed in the clinical treatment of PD (see, for review, Lees, 1993). An early dopamine agonist was piribedil, which was highly effective but, like many ground-breaking molecules, its clinical application in PD was not properly understood and rapid dose escalation caused high levels of nausea, vomiting and gastrointestinal disturbance that tainted its use (Rondot and Ziegler, 1992). However, there followed the introduction of ergot derivatives with bromocriptine, pergolide and cabergoline providing effective control of the motor symptoms of PD (Montastruc et al., 1993). The ergots have now been phased out due to valvular effects in the heart that may reflect the broad pharmacology of ergot derivatives and an action on 5-HT2B receptors (Elangbam, 2010). However, non-ergot drugs were already in use in PD, having been developed by employing animal models of PD, and dopaminergic therapy in PD is now centred on pramipexole, ropinirole and rotigotine as oral and transdermal medications (Bonuccelli et al., 2009). As much of the development of dopamine agonists was occurring, the cloning of dopamine receptor subtypes took place, and the animal models of PD were the test bed for examining their role in controlling motor function and specifically at examining the interaction between D1-like and D2-like receptors and the relationship to anti-parkinsonian activity and side effect profile (Jenner, 1995; 2002; 2003a).
What followed set the stage for a major advance in the development of animal models of PD and increased understanding of the processes connected to nigral dopaminergic cell loss. The discovery of the selective nigral toxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), produced through mitochondrial inhibition caused by its metabolite 1-methyl-4-phenylpyridinium (MPP+), brought a new impetus to animals models of PD (see Langston, 1987). While MPTP was toxic to nigral dopaminergic neurons in some mouse strains, it was its ability to destroy these cells in primate brain and to induce a motor syndrome closely resembling that occurring in man that allowed the first effective primate model of PD to be developed. Previously, only electrolytic or radiofrequency lesioning of the basal ganglia had taken place in primate species (Sourkes and Poirier, 1966; Poirier et al., 1975), so the discovery of MPTP was a toxin-based revolution. Very quickly it was realized that the MPTP-treated primate not only responded to all known anti-parkinsonian medication, but that it was highly predictive of the effects of dopaminergic drugs subsequently examined in clinical trial (Jenner, 2003b; 2008). The MPTP-treated primate remains a model of PD through which drugs must almost inevitably pass during the process of selection for clinical trial programmes in PD.
Very soon after the introduction of L-DOPA for the treatment of PD, it was realized that on chronic drug treatment and with disease progression, significant motor fluctuations (‘on-off’, ‘wearing off’ and freezing) and motor complications (chorea, dystonia, athetosis – collectively called dyskinesia) were common side effects (Jankovic, 2005). Attention turned to the animal models of PD to determine the cause of these side effects and to devise strategies for their prevention and treatment. Some success was achieved with reports of ‘wearing off’ in 6-OHDA-lesioned rats treated with L-DOPA (Papa et al., 1994), but in reality, it was only when the MPTP-treated primate model of PD was devised that dyskinesia as it occurs in man was seen after repeated L-DOPA treatment (Bedard et al., 1986; Clarke et al., 1987). This opened a gateway for looking at treatments that would lessen the induction of dyskinesia, such as longer-acting dopamine agonists, and as a test bed for examining novel drug molecules that might suppress established involuntary movements. More recently, similar success has been achieved in the rat with the recognition of abnormal involuntary movements (AIMs) as a rodent manifestation of dyskinesia and the use of this model to detect anti-dyskinetic strategies (Lundblad et al., 2002).
Finally, some introduction needs to be given to the role of experimental animal models of PD in understanding the pathogenic processes that occur and how these might be prevented. This is an on-going story as, so far, there has been virtually no translation from the animal model to clinical neuroprotection in PD (Jenner, 2008). However, since 6-OHDA acts through oxidative stress, MPTP/MPP+ and rotenone thorough mitochondrial complex I inhibition, PSI and epoximycin through proteasomal inhibition and lipopolysaccharide (LPS) through glial cell activation, all of these toxins have been used to test neuroprotective agents based on the knowledge that the processes involved are those known to contribute to pathogenesis in PD. In addition, the discovery of gene defects in familial PD (Hardy, 2010) and the identification of the resultant mutant proteins/enzymes have raised the hope that transgenic mice models might provide realistic models of PD as it occurs in man. Several autosomal-dominant transgenic models carrying α-synuclein or leucine-rich repeat kinase-2 (LRRK2) mutations have been described, but whilst many of these express inclusions, they fail to display robust neurodegeneration. Autosomal-recessive models with knockout of PTEN-induced putative kinase (PINK 1), Parkin or DJ-1 similarly fail to exhibit nigro-striatal pathology, so the development of more realistic transgenic models is still some way off.
So a variety of animal models of PD exist for a variety of uses. This review will undertake a detailed analysis of each model, describing its induction, key features, measureable end-points and impact on research in the PD field in terms of advancing our understanding of the disease itself and the role each has played in drug discovery programmes. The ideal model for PD would show a high degree of construct validity – that is similar pathogenesis to the disease (e.g. underlying oxidative stress, inflammation, complex I inhibition or proteasome inhibition), of face validity – that is similarity in symptoms (e.g. akinesia, rigidity), biochemistry (e.g. reduced striatal dopamine and altered downstream neurochemistry) and pathology (nigro-striatal tract degeneration and Lewy body deposition) to the human condition as well as predictive validity – that is the ability to positively identify agents that are clinically effective. Where known, these similarities will be brought out in the discussion of each model. Whilst much emphasis will inevitably be placed on the validity of models to assess drugs that can combat the motor symptoms and treatment-related complications occurring in PD, the ability of current models to assess the efficacy of neuroprotective agents will also be explored. Importantly, the review will highlight the positives and negatives of using the available animal models and look at the types of models that will be required in the future and their characteristics that will lead to the introduction of a new generation of molecules that will treat both the symptoms of PD and the disease process.
Pharmacological models
Reserpine model
The reserpine-treated rodent was one of the earliest animal models employed in PD research. Although quite a crude pharmacological mimic of the neurochemistry of PD, this model was instrumental in first demonstrating the therapeutic efficacy of what still remains the gold-standard treatment for PD, L-DOPA. It was in the late 1950s, that Carlsson et al. (1957) first demonstrated the ability of L-DOPA, the endogenous dopamine precursor to reverse the then-described ‘tranquillizing’ effects of reserpine pretreatment in mice (Carlsson et al., 1957). This effect was soon recapitulated in humans (Degkwitz et al., 1960), and the reserpine-treated mouse or more commonly rat became established as a robust screen for potential symptomatic efficacy of new drugs in PD. From a disease perspective, the reserpine model has also made important contributions to our understanding of the link between monoamine depletion and parkinsonian symptoms.
Reserpine (usual dose 4–5 mg·kg−1 s.c.) works by inhibiting the vesicular monoamine transporter, VMAT2. This leads to loss of storage capacity and hence depletion of brain (and peripheral) monoamines including noradrenaline and 5-HT as well as dopamine. Although this lack of selectivity for dopamine was once considered a failure of the reserpine model to accurately reflect the biochemistry of PD, the subsequent realization that noradrenergic and serotonergic systems are also affected in PD (Jellinger, 1991) argues in favour of the reserpine model being a relatively good mimic of the disease biochemistry. Most attention has nevertheless been paid to the dopaminergic deficit, and it is known that reserpine produces ∼85% loss of dopamine in the SNpc and >95% dopamine depletion in the striatum within 2 h of injection (Heeringa and Abercrombie, 1995). Although dopamine content in the SNpc returns to ∼30% by 24 h post injection, striatal dopamine depletion persists at >95% for at least 24 h (Heeringa and Abercrombie, 1995). Reserpine may also be given in combination with AMPT (α-methyl-_p_-tyrosine), which inhibits synthesis of dopamine and noradrenaline, to potentially prolong the neurochemical deficits, although our experience is that such combined treatment is not necessary. Reserpine also induces changes in other basal ganglia nuclei. For example, firing of the subthalamic nucleus (STN) is increased approximately 50% (Robledo and Feger, 1991), an increase that occurs in PD (Hutchison et al., 1998; Benazzouz et al., 2002), and extracellular glutamate levels are elevated in the basal ganglia output regions, specifically the entopeduncular nucleus (the rat homologue of internal globus pallidus or GPi) (Biggs et al., 1997). Behaviourally, reserpine induces features of akinesia and hind limb rigidity in rats that are representative of symptoms associated with PD. Therefore, whilst showing little in the way of construct validity, the reserpine model does, on balance, have sound face validity. Reversal of akinesia or rigidity is used as a predictive indicator of likely symptomatic efficacy of new agents. Reserpine-treated rats remain fully akinetic for up to 24 h, mirroring the sustained deficits in striatal dopamine, during which time reversal of akinesia following acute drug administration can be monitored. Beyond this time, the behaviour of rats starts to return, in line with their striatal dopamine replenishment (Betts and Duty, unpubl. obs.), so the model cannot be used to monitor efficacy produced by drugs on repeated administration.
In the early stages of target validation, direct intracerebral injection may be required as either the available tools do not adequately cross the blood–brain barrier or because specific anatomical targeting is desirable. Under these circumstances, a simple measure of contraversive circling behaviour following direct unilateral injection of agents in to reserpine-treated rodents may be taken as an index of anti-akinetic efficacy (Maneuf et al., 1996; Dawson et al., 2000; Johnston and Duty, 2003; MacInnes et al., 2004). Bilateral systemic or intracerebroventricular administration of anti-parkinsonian agents, on the other hand, produces an overall increase in locomotor activity that can be measured using any one of a number of automated or less sophisticated systems (e.g. Nash et al., 1999; MacInnes et al., 2004). Although less commonly used, reversal of hind limb rigidity, measured as muscle resistance in response to passive flexion and extension of the rat's hind limb, also reflects an anti-parkinsonian effect. In line with striatal dopamine depletion, this rigidity peaks within 1–2 h of reserpine and is maintained for up to 24 h (Goldstein et al., 1975; Lorenc-Koci and Wolfarth, 1999), again allowing only short-term drug testing.
Although the reserpine model mimics major components of the biochemistry of PD and induces akinesia and rigidity that reflect clinical features of the disease, there is no nigral dopaminergic cell degeneration, so the model is restricted to assessing novel approaches to symptomatic treatment. However, within this framework, the reserpine-treated rat has proven very useful at predicting the efficacy of both dopaminergic and non-dopaminergic drugs that are then progressed through to examination in more complex animal models. Indeed, all of the dopaminergic drugs in current clinical use to manage PD symptoms, including apomorphine, pramipexole, ropinirole, pergolide, bromocriptine and cabergoline, have, like L-DOPA, displayed efficacy in the reserpine-treated rat, supporting the predictive validity of this model (Goldstein et al., 1975; Johnson et al., 1976; Johnels, 1982; Colpaert, 1987; Miyagi et al., 1996; Maj et al., 1997; Fukuzaki et al., 2000a). Other clinically utilized agents, for example muscarinic antagonists such as benztropine and trihexyphenidyl (Goldstein et al., 1975), MAO-B or COMT inhibitors such as selegiline, rasagiline or tolcapone (Colpaert, 1987; Maj et al., 1990; Skuza et al., 1994; Finberg and Youdim, 2002) and amantadine (Goldstein et al., 1975; Colpaert, 1987; Skuza et al., 1994) show efficacy either alone or in combination with a subthreshold dose of L-DOPA in reserpine-treated rats (summarized in Table 1). These findings highlight the strong predictive validity of the reserpine-treated rat and justify its maintained position as a key model of choice for early preclinical stages of drug discovery programmes. This position is confirmed by its continued use today to assess anti-parkinsonian efficacy of both dopaminergic agents, for example D3 receptor agonists (Ghosh et al., 2010) and non-dopaminergic agents, including group III metabotropic glutamate (mGlu) receptor agonists or positive allosteric modulators (Niswender et al., 2008; Austin et al., 2010) and mixed adenosine A2A/A1 antagonists (Shook et al., 2010).
Table 1.
Predictive validity of the main animal models of Parkinson's disease
| Animal model | Reserpine | Haloperidol | 6-OHDA | MPTP mouse | Rotenone | MPTP primate |
|---|---|---|---|---|---|---|
| L-DOPA ± carbidopa | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Dopamine agonists | ||||||
| Apomorphine | ✓ | ✓ and X | ✓ | X | ✓ | ✓ |
| Bromocriptine | ✓ | ✓ | ✓ | ✓ | – | ✓ |
| Cabergoline | ✓ | – | ✓ | ✓ | – | ✓ |
| Pramipexole | ✓ | ✓ | ✓ | ✓ | – | ✓ |
| Pergolide | ✓ | – | ✓ | – | – | ✓ |
| Ropinirole | ✓ | – | ✓ | – | – | ✓ |
| Rotigotine | – | – | ✓ | – | – | ✓ |
| MAO-B inhibitors | ||||||
| Selegiline | ✓a | ✓a | ✓a | ✓a | – | ✓a |
| Rasagiline | ✓a | ✓ | ✓a | – | – | ✓a |
| COMT inhibitors | ||||||
| Entacapone | – | – | ✓a | – | – | ✓a |
| Tolcapone | ✓a,b | ✓a | ✓a | ✓a | – | ✓a |
| Anticholinergics | ||||||
| Trihexyphenidyl | ✓c | ✓ | – | – | – | ✓ |
| Benztropine | ✓c | ✓a | – | – | – | ✓ |
| Orphenadrine | – | – | – | – | – | – |
| Procyclidine | – | – | – | – | – | – |
| Miscellaneous | ||||||
| Amantadine | ✓ | ✓ | ✓ | ✓a | – | ✓ |
Haloperidol model
The other pharmacological model of PD is the haloperidol-treated rat, which again shows little construct validity. Haloperidol works by antagonizing dopamine D2 and, to a lesser extent, D1 receptors in medium spiny neurons that comprise the indirect and direct pathways of the motor circuit respectively. The resultant block of striatal dopamine transmission results in abnormal downstream firing within the basal ganglia circuits that is manifest as symptoms of muscle rigidity and catalepsy within 60 min of haloperidol (0.5–5 mg·kg−1, i.p.) injection (see, e.g., Sanberg, 1980). Although rigidity is a feature of PD, providing this model with some face validity, catalepsy, which is expressed as the inability of an animal to correct itself from an abnormally imposed posture, is not directly associated with PD. However, catalepsy may be likened to the inability of patients to initiate movements and so could be considered a worthwhile measure. As a biochemical mimic of PD, the haloperidol model was considered weak, but the recent demonstration that acute administration of haloperidol reduces striatal content of dopamine, noradrenaline and 5-HT (Kulkarni et al., 2009) may reverse this notion. Downstream of the striatum, the changes exhibited by haloperidol also support face validity of this model since elevated levels of extracellular glutamate (which may result from increased STN activity noted in PD) have been reported in the entopeduncular nucleus following haloperidol injection (Biggs et al., 1997).
The anti-parkinsonian efficacy of novel agents is assessed in the haloperidol model as the reversal of rigidity (as above) or catalepsy. In order to facilitate better comparison of data between laboratories, a common ‘bar test’ is utilized whereby catalepsy is measured as the time taken for an animal to remove its forepaws from a bar, so-called descent latency, although variations in bar height (set at 6–10 cm), cut-off time (60–300 s), dose of haloperidol (0.5–10 mg·kg−1) and animal sensitivities still render cross-lab comparisons difficult (Sanberg et al., 1988). However, as summarized in Table 1, a number of the drugs in current clinical use for PD have shown efficacy in the haloperidol model, including L-DOPA, bromocriptine, pramipexole, trihexyphenidyl and amantadine (Zetler, 1970; Zebrowska-Lupina et al., 1985; Kobayashi et al., 1997; Maj et al., 1997). Other drugs including benztropine, tolcapone, selegiline and rasagiline have also been shown to enhance the effects of L-DOPA (Erzin-Waters et al., 1976; Nuutila et al., 1987; Maj et al., 1990; Speiser et al., 1998), supporting the predictive validity of this model, though the effects of apomorphine are unpredictable (Erzin-Waters et al., 1976; Elliott et al., 1990). In common with the reserpine model, the haloperidol model fails to display any of the characteristic pathology associated with PD, so its use is again limited. Nevertheless, it remains a popular model of choice for assessing the potential symptomatic efficacy of novel non-dopaminergic agents including mGlu4-positive allosteric modulators, Adenosine A2A/A1 antagonists and mGlu7 agonists in PD (Niswender et al., 2008; Neustadt et al., 2009; Greco et al., 2010; Shook et al., 2010).
Although the pharmacological models have a valuable place in the discovery of symptomatic drugs for PD, they have serious limitations. First, they are only transient, and this limits their long-term usefulness. In a condition like PD where drugs will be administered chronically, the need to assess the long-term symptom relief in animal models amenable to chronic dosing regimens is paramount. Second, as these pharmacological models do not display any pathology, they are of no use when investigating novel strategies aimed at providing neuroprotection or neurorepair. Fortunately, there are other animal models of PD available in which some of these limitations are partly addressed, and it is towards these that we now turn our attention.
Classical toxin-induced rodent models of PD
The two most widely used rodent models of PD are the classical 6-OHDA-treated rat and the MPTP-treated mouse. Of these, the 6-OHDA model has been extensively used as a test bed for novel symptomatic agents as well as providing a means for assessing neuroprotective and neurorepair strategies. Although unlikely to be the first model of choice for testing symptomatic agents, since its behavioural phenotype is less robust than the 6-OHDA rat, the MPTP-treated mouse provides a useful secondary screening model and has the added advantage of being relatively easy to construct compared with the 6-OHDA rat.
6-OHDA model
The characterization of the hydroxylated analogue of dopamine, 6-OHDA, as a toxin-inducing degeneration of dopaminergic neurons in the nigro-striatal tract (Ungerstedt, 1968) has led to it being a widely used tool to induce Parkinsonism in rodents. Unlike MPTP (below), 6-OHDA does not efficiently cross the blood–brain barrier and so requires direct injection into the brain. This is undoubtedly one of its main drawbacks as specialized stereotaxic surgical instruments and training are required. Unilateral lesions of the nigro-striatal tract are almost invariably employed since bilateral lesions result in marked adispsia and aphagia, rendering tube feeding necessary for the maintained welfare and survival of the animals (see, e.g., Sakai and Gash, 1994), although some studies have utilized bilateral partial lesions (Amalric et al., 1995; Paillé_et al_., 2007). 6-OHDA is injected into the nigro-striatal tract at one of three locations: into the substantia nigra pars compacta (SNpc) where the A9 dopaminergic cell bodies are located; into the median forebrain bundle (mfb), through which the dopaminergic nigro-striatal tract ascends; or into the terminal region, the striatum. Often, the site of injection is dictated by other needs – for example, where direct mechanical damage to the SNpc is best avoided, the mfb or striatal injection models would be favoured. However, if agents being used for early target validation studies require direct supranigral infusion, then SNpc injection would be preferred since a single indwelling cannula can be used for both toxin and drug administration (see, e.g., Vernon et al., 2008; Austin et al., 2010; Iczkiewicz et al., 2010).
Following its injection, 6-OHDA is taken up into the dopaminergic neurons via the dopamine transporter, DAT (Figure 1). Given that 6-OHDA also shows high affinity for the noradrenaline transporter, NET (Luthman et al., 1989), systemic injection of the NET inhibitor, despiramine, given 30–60 min before 6-OHDA, ensures improved specificity of the toxin for dopaminergic neurons. Pargyline may also be given as a pretreatment in order to reduce any potential breakdown of 6-OHDA by MAO-B, thereby lessening the effective dose of toxin required. Although the exact mechanism behind 6-OHDA toxicity is still subject to investigation, current understanding is that, once inside dopaminergic neurons, 6-OHDA initiates degeneration through a combination of oxidative stress and mitochondrial respiratory dysfunction. Certainly, 6-OHDA readily oxidizes to form reactive oxygen species (ROS) such as H2O2 (Mazzio et al., 2004), to reduce striatal levels of antioxidant enzymes [total glutathione (GSH) or superoxide dismutase] (Perumal et al., 1992; Kunikowska and Jenner, 2001), to elevate levels of iron in the SN (Oestreicher et al., 1994) and to interact directly with complexes I and IV of the mitochondrial respiratory chain, leading to subsequent respiratory inhibition and further oxidative stress (Glinka et al., 1997). Many of these effects are thought to mirror events occurring in PD brain (Jenner, 1989), thereby supporting a high degree of construct validity for the 6-OHDA model. In addition, ongoing inflammation is also implicated in the pathogenesis and progression of PD (Whitton, 2007; Tansey and Goldberg, 2010) with microglial activation apparent in brain in PD at post-mortem using PET imaging with the ligand PK11195 (Gerhard et al., 2006). Use of PK11195 has also shown microglial activation in the striatum and SN of rats following 6-OHDA lesioning (Cicchetti et al., 2002), whilst the presence of elevated striatal inflammatory markers, for example, TNF-α (Mogi et al., 2000), further replicates what is seen in PD brain at post-mortem (Mogi et al., 1994). Thus, construct validity of these models is high. A pathogenic link between 6-OHDA and PD has been suggested following reports of the detection of 6-OHDA in the striatum and urine of L-DOPA-treated PD patients (Curtius et al., 1974; Andrew et al., 1993). Whether these findings indicate that 6-OHDA represents an endogenous component of PD pathogenesis, or that it may play a role in the enhanced oxidative stress and accelerated degeneration of residual nigral cells in patients receiving L-DOPA, remains debatable (Müller et al., 2004; Fahn and The Parkinson Study Group, 2005).
Figure 1.
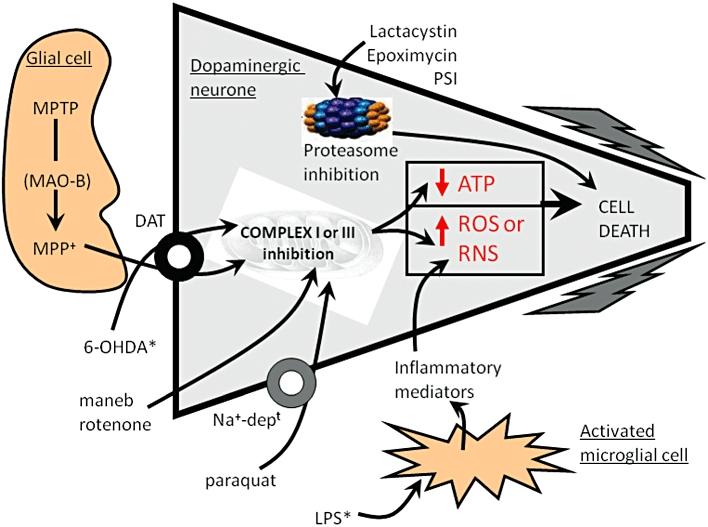
Schematic representation of a dopaminergic SNpc neuron showing the molecular targets for the various agents used to induce animal models of PD that exhibit nigro-striatal tract degeneration. *Indicates those agents that are administered directly into the brain; all other agents are delivered systemically. Maneb is believed to inhibit complex III of the mitochondrial respiratory chain, whilst the other mitochondrial toxins mainly inhibit complex I. This activity leads to the generation of ROS or reduced ATP production, which lead to apoptosis and the cells demise. 6-OHDA and MPP+ may also induce the production of ROS directly within the cytoplasm. LPS-activates microglial cells to stimulate the release of inflammatory mediators, which in turn produce reactive nitrogen species (RNS). Inhibition of proteasome activity allows a build up damaged proteins that through DNA damage (not shown for clarity), and other processes can lead to cell death. Cell death is most likely apoptotic in nature, though this remains controversial for some agents. Further details are given in the accompanying text.
The 6-OHDA model also mimics many of the biochemical features of PD, including reduced levels of striatal dopamine and tyrosine hydroxylase (TH; rate-limiting step of DA biosynthesis). Further similarities with PD include increased firing of the STN (Hassani et al., 1996; Hutchison et al., 1998; Benazzouz et al., 2002; Breit et al., 2006) and a parallel increase in glutamate levels and firing within the basal ganglia output regions (entopeduncular nucleus and substantia nigra pars reticulata) post 6-OHDA lesion (You et al., 1996; Biggs et al., 1997; Breit et al., 2006), recapitulating the increased firing in the GPi in PD patients (Hutchison et al., 1994). Though less robust, other neurochemical features of PD, such as elevated striatal enkephalin levels (Goto et al., 1990; Nisbet et al., 1995; Henry et al., 2003) or depressed striatal substance P and dynorphin levels (Fernandez et al., 1992), are also found in 6-OHDA-treated rats (Young et al., 1986; Li et al., 1990), strongly supporting its face validity. However, there is no pathology in other brain regions that are affected in PD.
The 6-OHDA model shares a common failing with many other animal models of PD as it does not lead to the formation of the pathological hallmark of PD, the Lewy body. Lewy bodies are eosinophilic inclusions that contain ubiquitinated proteins such as α-synuclein (Spillantini et al., 1997; Baba et al., 1998) and are associated with lipofuscin-containing lysosomes that have also been shown to accumulate α-synuclein in PD brain stem (Braak et al., 2001). The exact role of Lewy bodies remains to be established, but drugs to reduce aggregate formation are considered a potential future strategy for treating PD. A recent report of parkin-containing aggregate formation in 6-OHDA-lesioned rat (Um et al., 2010) is therefore an exciting advance but requires confirmation.
The one pathological feature of PD robustly displayed by the 6-OHDA model is degeneration of the nigro-striatal tract. The extent of degeneration can be established post-mortem by assessing the reduction in various parameters in the lesion (ipsilateral) hemisphere, compared with the intact (contralateral) hemisphere including number of nissl-stained cells or TH-positive neurons in the SNpc; levels of TH or DAT immunoreactivity in the striatum and levels of [3H]mazindol binding to DAT in the striatum. Behavioural indices can also be taken as a potential pre-screen for predicted lesion size. In practice, animals bearing either a near-complete (>90%) or partial lesion are produced for use in preclinical studies.
6-OHDA models with a FULL lesion
Since 6-OHDA produces a dose-dependent degeneration of the nigro-striatal tract, animals bearing a full or marked (>90%) lesion can be produced following administration of high amounts of 6-OHDA into each of the three sites, though in practice, injections into either the mfb or SNpc are most often used to produce ‘full’ lesions. In support of the face validity of these models, the pattern of dopaminergic cell loss produced by 6-OHDA mirrors, to some extent, that seen in the PD brain at post-mortem, whereby loss of A9 cells in the SNPc is more extensive than loss of neighbouring A10 cells in the ventral tegmental area (VTA) (German et al., 1989). For example, injection of 8 µg 6-OHDA into either the SNpc or mfb reliably produces over 90% degeneration of cells in the SNpc, yet only 40% loss of cells in the VTA (Carman et al., 1991). In contrast, the rapid nature of the degeneration is far removed from the slowly progressive nature of PD since nigral cell death commences within 12 h of 6-OHDA injection, before the onset of striatal terminal damage then, in line with striatal dopamine depletion, is maximal around 6 days and remains stable for at least 4 weeks post lesion (Ungerstedt, 1968; Jeon et al., 1995; Zuch et al., 2000).
Accompanying this marked degeneration is a number of robust spontaneous and drug-induced behavioural phenotypes. So while the rapid nature of cell death means this model is open to criticism when investigating novel neuroprotective agents, it provides a stable baseline against which to monitor the efficacy of symptomatic agents. The most widely used locomotor assessments of unilaterally lesioned rats are the circling responses induced by the systemic injection of either the mixed D1/D2 agonist apomorphine or amphetamine, which can be monitored using automated rotometer systems. Through its ability to induce dopamine release, amphetamine (1–5 mg·kg−1) creates an imbalance in dopamine transmission favouring the intact, contralateral (i.e. opposite to the lesion side) striatum, resulting in ipsiversive (i.e. towards the lesion side) rotation. Such an imbalance can be detected with as little as a 50% loss of dopaminergic neurons (Hefti et al., 1980; Hudson et al., 1993; Barneoud et al., 1995), so, although evident in fully lesioned animals, amphetamine-induced rotation cannot be used as a screen for a marked (>90%) lesion. Apomorphine (0.01–1 mg·kg−1) on the other hand creates an imbalance in striatal dopaminergic transmission favouring excess stimulation of the ipsilateral (lesioned) side where denervation-induced dopamine receptor supersensitivity results following nigral cell destruction, producing contraversive (i.e. away from the lesion) rotation. Contraversive rotation to apomorphine is only consistently seen when >90% of dopaminergic neurons have been lost, the point at which receptor supersensitivity occurs (Hefti et al., 1980; Hudson et al., 1993; Barneoud et al., 1995). It can therefore be confidently used as a pre-screen to detect those animals where 6-OHDA injection has resulted in a ‘full’ lesion.
6-OHDA-lesioned rats with ‘full’ lesions also display forelimb akinesia that can be measured using a range of non-invasive tests such as the adjusted stepping test, cylinder test and forelimb placement test. The stepping test picks out impairment in the ability of a 6-OHDA-lesioned rat to initiate stepping movements with the contralateral paw when moved steadily in forehand and backhand directions (Olsson et al., 1995; Kirik et al., 1998; Grealish et al., 2008; Austin et al., 2010). In the cylinder or rearing test, animals bearing an approximate 90% lesion display a preference for using their non-impaired (ipsilateral) forelimb for vertical exploration of a 20 cm diameter, 30 cm height perspex cylinder (Schallert et al., 2000; Austin et al., 2010). Deficits in use of the contralateral forelimb (reflective of akinesia) are also found post lesion in the vibrissae-elicited forelimb placement test, which assesses the ability of an animal to place its forelimb on a bench edge in response to detection of vibrissae movements caused by contact with said bench edge (Schallert et al., 2000; Grealish et al., 2008). These spontaneous behaviours are, like the lesions themselves, stable for at least 30 days post lesion (Schallert et al., 2000) and are reversed by L-DOPA administration (Olsson et al., 1995; Lundblad et al., 2002; Monville et al., 2005; Marin et al., 2007), supporting their use as valid measures of parkinsonian motor features against which to assess the efficacy of new symptomatic drug treatments. However, given that treatment with symptomatic drugs occurs when PD is first diagnosed, these models, which most likely reflect end-stage illness, are subject to criticism.
6-OHDA models with a PARTIAL lesion
Motor symptoms of PD appear when around 60–70% of the nigro-striatal tract has degenerated; hence, this is the size of lesion aimed for in partial lesion models that attempt to mimic the earlier-stage PD. Partial lesions have been produced in some cases by reducing the dose of 6-OHDA injected into the SNpc or mfb. Although the degree of degeneration is more variable with reduced doses of 6-OHDA, in general, a dose of around 6 µg 6-OHDA injected into either the mfb or SNpc is sufficient to produce around 70% loss of nigral cells and striatal dopamine depletion within 2 weeks (see, e.g., Costa et al., 2001a; Visanji et al., 2006b). Unfortunately, the behavioural readouts obtained from animals bearing a partial lesion of this nature have turned out to be less reliable. Thus, whilst animals bearing a partial lesion do produce ipsiversive rotations in response to amphetamine, this response may be seen in as few as half the animals tested (Hefti et al., 1980). In addition, animals bearing lesions of less than 80% do not show robust deficits in the cylinder reaching test (Hefti et al., 1980), although other measures of forelimb akinesia have not been reported. Therefore, this model can only be reliably used for assessing the histological and neurochemical benefits of potential neuroprotective agents, although the rapid progression of cell death is still a limitation for such studies.
A more slowly developing partial lesion of the nigro-striatal pathway has been achieved by administering 6-OHDA into the striatum, which produces striatal terminal damage within 1 day of injection, whilst nigral cell loss is minimal at 1 week, reaching a maximum within 2–3 weeks (Sauer and Oertel, 1994; Przedborski et al., 1995; Blandini et al., 2007). The pattern of cell loss again mirrors that in PD, with the SNpc showing around 20% more cell loss compared with the VTA (Przedborski et al., 1995). Unfortunately, the overall extent of nigral cell loss achieved varies between laboratories, with the intrastriatal injection of 20–28 µg 6-OHDA producing between 20% and 85% loss of cells in the SNpc and 60–90% reductions in striatal DA levels or terminals by around 2 weeks following treatment (Sauer and Oertel, 1994; Przedborski et al., 1995; Lee et al., 1996; Kirik et al., 1998; Blandini et al., 2007).
Behavioural readouts are also less than ideal for assessing the efficacy of neuroprotective drugs. Animals with a partial nigro-striatal lesion (50–70%) produced by intra-striatal 6-OHDA injection may display deficits in the adjusted stepping test, which are maximal around 2 weeks post lesion and stable for up to 12 weeks (Winkler et al., 1996; Kirik et al., 1998, 2001). However, other investigations failed to detect deficits in animals with <80% lesion (Barneoud et al., 2000). In contrast, amphetamine-induced rotation is consistently observed, but the intensity of rotation does not change significantly across a wide range of nigral cell loss (between 50% and 90%) (Lee et al., 1996; Kirik et al., 1998; Barneoud et al., 2000), so, although of use in pre-screening for a partial lesion, amphetamine-induced rotation will not reliably detect functional improvements even in animals where a marked degree of neuroprotection has been achieved. Interestingly, apomorphine, which is generally considered to only produce rotation when 90% or more of striatal DA content is lost, may produce rotations in animals with partial lesions (Cadet and Zhu, 1992; Przedborski et al., 1995; Lee et al., 1996; Kirik et al., 2001; Blandini et al., 2007). This unexpected effect may reflect that whilst the overall _average_ DA loss is below the necessary threshold for evoking receptor supersensitivity, a localized loss of >90% that is sufficient to evoke receptor supersensitivity may occur in some striatal areas, thus producing sufficient imbalance in firing to facilitate rotations (Kirik et al., 1998). On balance, although not perfect, the adjusted stepping test may provide the most consistent behavioural readout in rats with a partial lesion induced by intra-striatal 6-OHDA. However, spontaneous recovery in the form of sprouting of dopaminergic fibres and TH recovery in the striatum that has been observed by 4–6 months post lesion (Blanchard et al., 1995; Stanic et al., 2003) needs to be taken into account when planning long-term studies with neuroprotectve agents.
With the exception of the antimuscarinic drugs, all the drugs in clinical use today have shown efficacy in the 6-OHDA lesion models (Table 1), supporting their predictive validity. For example, L-DOPA and the clinically utilized dopamine agonists (bromocroptine, cabergoline, pramipexole, pergolide, ropinirole and rotigotine) produce contraversive rotations akin to those described above with apomorphine (Johnson et al., 1976; Broekkamp et al., 1990; Eden et al., 1991; Mierau and Schingnitz, 1992; Fukuzaki et al., 2000a; Prikhojan et al., 2000; Schmidt et al., 2008). Amantadine also produces rotation – this time in an ipsiversive manner, reflecting its pre-synaptic action to enhance dopaminergic transmission on the intact side, as seen with amphetamine (Reavill et al., 1983). Finally, the MAO-B inhibitors selegiline and rasagiline and the COMT inhibitors tolcapone and entacapone have been shown to potentiate the actions of L-DOPA in producing contraversive rotation (Heikkila et al., 1981; Nuutila et al., 1987; Tornwall and Mannisto, 1993; Moses et al., 2004).
In conclusion, the 6-OHDA lesion model resembles PD in a number of key areas. It has construct validity, combining mitochondrial dysfunction, oxidative stress and inflammation and face validity, combining biochemistry (dopamine depletion, neuropeptide changes, etc.), nigro-striatal pathology (SNpc cells lost > VTA) and forelimb akinesia. However, the model does not capture all features of the illness. In PD, pathological change occurs in many brain areas outside of the basal ganglia such as the locus coeruleus and raphe nuclei, and this is not recapitulated in the 6-OHDA model. In most forms of the 6-OHDA model, cell death occurs far more rapidly than in PD, and, although this is less so following intra-striatal 6-OHDA administration, this model shows marked variability in the size of lesion and behavioural readouts produced. Finally, the presence of intracellular proteinous aggregates resembling Lewy bodies remains to be established, and the unilateral nature of the symptoms does not mimic that of PD. Nevertheless, as long as these limitations are understood and accounted for when interpreting effects, the 6-OHDA model will remain at the forefront of preclinical drug discovery for PD.
Indeed, the 6-OHDA-lesioned rat appears to be a good predictor of subsequent efficacy in the primate model of PD (see later) and perhaps beyond in to man. A number of new dopaminergic drugs entering phase II/III clinical trials show efficacy in the 6-OHDA-lesioned rat, including the D2 partial agonist, aplindore (Heinrich et al., 2006) and the D2, D3 and 5-HT1A partial agonist, pardoprunox (Jones et al., 2010). Other agents showing positive outcomes in the MPTP-treated primate, such as the non-selective monoamine uptake inhibitor BTS 74–398, have also shown efficacy in the 6-OHDA-lesioned rat (Lane et al., 2005). Similarly, non-dopaminergic approaches to improving motor symptoms in PD, such as the adenosine A2A antagonist istradefylline show efficacy in the 6-OHDA-lesioned rat (Lundblad et al., 2003) and subsequently in the primate model and in man. This suggests a significant role for the 6-OHDA-lesioned rodent in predicting symptomatic approaches outside of the dopaminergic arena. In contrast, the predictive validity of the model for neuroprotective strategies remains uncertain at this stage since strategies that reduced nigral dopaminergic cell loss in the rat have not so far translated to the clinic. However, adeno-associated virus (AAV)-mediated intra-striatal delivery of the neurotrophic factor neurturin that restores nigral cell numbers in MPTP-treated primates (Kordower et al., 2006a) was shown to protect against 6-OHDA lesions in the rat (Gasmi et al., 2007). Unfortunately, this preclinical success has not been recapitulated in clinical trials where intraputaminal delivery of AAV-mediated neurturin failed to show efficacy (Marks et al., 2010). This and other failures of translation through to the clinic will be considered in greater detail towards the end of the review.
Modelling dyskinesia in the 6-OHDA-lesioned rat model
One of the key challenges in providing adequate long-term drug treatment for PD is in avoiding the occurrence of the treatment-related motor complications such as dyskinesia that affect the long-term efficacy of L-DOPA. For many years, the only animal model available for assessing the likelihood of new agents to provoke dyskinesia or for investigating the efficacy of potential anti-dyskinetic treatments, was the MPTP-treated primate, detailed discussion of which appears later in this review. Since few laboratories have expertise in using primates, and there are substantial costs involved, the emergence of a cheaper, more accessible rodent model of dyskinesia based on the 6-OHDA rat model is a welcome addition. In 6-OHDA-lesioned rats, repeated administration of either L-DOPA or dopamine agonists such as apomorphine produces a gradually increasing number of contraversive rotations (Bevan, 1983; Deshaies et al., 1984). This exaggerated or hyperkinetic response correlates with changes in expression of certain neuropeptides (see, e.g., Duty and Brotchie, 1997), which are believed to play a role in the emergence of dyskinesias in PD and in MPTP-treated primates (see Bezard et al., 2001a; Henry et al., 2003). Moreover, classes of drug, which are known to reduce L-DOPA-induced dyskinesia in man, such as α2-adrenergic antagonists and 5-HT1A agonists (Durif et al., 1995; Rascol et al., 2001), similarly reduce this hyperkinetic circling response to repeated L-DOPA administration in the 6-OHDA-lesioned rat (Henry et al., 1998). However, the relevance of the circling response alone to the complex dyskinesia expressed by patients (and MPTP-treated primates) is questionable, and a more detailed examination of this phenomenon has led to the description of additional dyskinetic-like features in these animals, termed abnormal involuntary movements, or AIMs. AIMs, which were first described at length by Bjorklund's group (Cenci et al., 1998; Lee et al., 2000), are believed to mirror more closely dyskinesia as it appears in primates. The AIMs rating scale (Cenci et al., 1998) combines measures of a contraversive rotational response (so-called locomotive dyskinesia), with measures of stereotypic behaviour, classified into three categories: forelimb dyskinesia, expressed as repetitive rhythmic jerks or dystonic posturing of the contralateral forelimb; axial dystonia, manifest as contralateral twisted posture of the neck and upper body; orolingual dyskinesia, characterized by stereotyped jaw movements and contralateral tongue protrusion. The AIMs model has since been validated by a number of groups that have shown again that α2-adrenergic antagonists and 5-HT1A agonists, classes of drug known to have anti-dyskinetic efficacy in PD or MPTP-treated primates, reduce AIMs (Lundblad et al., 2002) and, equally importantly, agents like bromocriptine and ropinirole, which do not provoke significant dyskinesia when given de novo to MPTP-treated primates also fail to evoke AIMs in the 6-OHDA-lesioned rat (Lundblad et al., 2002; Stockwell et al., 2008). As with contraversive rotation, the occurrence of AIMs also correlates with changes in neuropeptide gene expression (Cenci et al., 1998). It also appears possible to distinguish between anti-dyskinetic and anti-akinetic effects by combining AIMs measurement with that of forelimb placing test (Schallert et al., 2000; Monville et al., 2005), but not with performance in the cylinder test, which is affected by the development of dyskinesia (Lundblad et al., 2002). So, although the MPTP-treated primate model will remain as the gold standard for assessing dyskinesia and new potential treatments, we are now armed with a useful rodent model for early-stage preclinical evaluation. Indeed, translation from this model through to primate and clinical trials has already gained support. Thus, in line with demonstrations that mGlu5 antagonists reduce L-DOPA-induced dyskinesia in rodents (Mela et al., 2007) and primates (Rylander et al., 2010), the mGlu5 antagonist, AFQ-056 has been shown to lessen L-DOPA-induced dyskinesia in two small-scale phase II trials (Berg et al., 2011). Finally, it should be acknowledged that in addition to facilitating the discovery of new drug treatments for PD, the 6-OHDA lesion and AIMs models are providing a wealth of information on mechanisms underlying nigral cell degeneration and the cause of these treatment-related side effects, such as dyskinesia.
The MPTP-treated mouse model
MPTP is a commonly used toxin for inducing both rodent and primate models of PD based on its ability to induce persistent Parkinsonism in man (Davis et al., 1979; Langston et al., 1983). Subsequent investigations in non-human primates identified that selective destruction of dopaminergic neurons of the nigro-striatal tract was the pathological basis behind the motor deficits observed (Burns et al., 1983; Jenner et al., 1984; Langston et al., 1984), and out of this came the most relevant animal model of PD that persists today. The impact of the MPTP-treated primate model in the PD field is second to none, but first we will focus attention on the use of MPTP in non-primate species. Many species, including rats, are insensitive to the toxic effects of MPTP, possibly due to the relatively rapid clearance of MPP+, the toxic metabolite of MPTP (Johannessen et al., 1985). However, specific strains of mice, notably black C57, and Swiss Webster are sensitive to MPTP (Sonsalla and Heikkila, 1988) and have enabled development of the MPTP mouse model of PD.
The mechanism behind the neurotoxic action of MPTP has been the subject of intense investigation and is relatively well understood (Figure 1). MPTP is a lipophilic protoxin that, following systemic injection (usually i.p. or s.c.), rapidly crosses the blood–brain barrier (Riachi et al., 1989). Once inside the brain, MPTP is converted by MAO-B (principally in glia and serotonergic neurons) into the intermediary, 1-methyl-4-phenyl-2,3,dihydropyridinium (MPDP+) before its rapid and spontaneous oxidation to the toxic moiety, 1-methyl-4-phenylpyridinium (MPP+) (Chiba et al., 1984). Following its release into the extracellular space, MPP+ is taken up via DAT into dopaminergic neurons where cytoplasmic MPP+ can trigger the production of ROS, which may contribute to its overall neurotoxicity (Javitch et al., 1985). However, the majority of MPP+ is eventually accumulated within mitochondria where the key toxic mechanism occurs. Once inside mitochondria, MPP+ impairs mitochondrial respiration via inhibition of complex I of the electron transport chain (Nicklas et al., 1987). This action impairs the flow of electrons along the respiratory chain, leading to reduced ATP production and the generation of ROS, such as superoxide radicals. The combined effects of lowered cellular ATP and elevated ROS production are most likely responsible for initiation of cell death-related signalling pathways such as p38 mitogen-activated kinase (Karunakaran et al., 2008), c-jun N-terminal kinase (JNK) (Saporito et al., 2000) and bax (Hassouna et al., 1996; Vila et al., 2001), all of which have been demonstrated in vivo following MPTP treatment and may contribute to apoptotic cell death (Jackson-Lewis et al., 1995; Tatton and Kish, 1997). Given that many of these mechanisms are also features of pathogenesis in PD, this model shows a high degree of construct validity.
The MPTP-treated mouse has some clear advantages over the 6-OHDA lesion model, not least of all economical benefits in terms of the cheaper costs associated with purchasing and housing mice. Being systemically active, MPTP administration does not require the type of skilled stereotaxic surgery that production of a 6-OHDA lesion requires. The systemic injection also produces a bilateral degeneration of the nigro-striatal tract, more reflective of that seen in PD. The MPTP model also mimics many of the known biochemical features of PD. For example, in addition to the well-known reductions in striatal dopamine and TH, there are also elevated levels of both striatal PPE-A (Gudehithlu et al., 1991) and ACh (Hadjiconstantinou et al., 1985). Further downstream in the basal ganglia, extracellular glutamate levels have been shown to be elevated in the SN of MPTP-treated mice, a rise associated with the induction of programmed cell death (Meredith et al., 2009), whilst glutathione (GSH) levels are significantly reduced (Ferraro et al., 1986) as in PD itself. Finally, in further support of the face validity of this model, inflammatory markers are elevated in the striatum and SN of MPTP-treated mice (Kurkowska-Jastrzebska et al., 1999; Hebert et al., 2003), which occurs as a result of reactive microgliosis in PD.
The MPTP model does, however, have some clear _dis_advantages over the 6-OHDA model, particularly in terms of reproducibility and the range of behavioural readouts that can be obtained. Mice are far less sensitive to MPTP than primates, and the higher doses required can be acutely lethal as a result of the peripheral neuro- or cardiotoxicity induced (see Jackson-Lewis and Przedborski, 2007). Given that the risk of mortality usually occurs within 24 h of the first dose of MPTP and is dose-dependent, the high rates (up to 50%) seen following acute bolus dosing in the earlier studies (see, e.g., Ferger et al., 2000) can be reduced to acceptable levels (<20%) with reductions in the dose administered and can be almost completely avoided using alternative protocols in which the same or even higher total dose is given in multiple doses (e.g. Gibrat et al., 2009). Clearly from both an ethical and practical perspective, these more chronic protocols are favoured. The handling of large doses of MPTP also represents a risk to researchers, and therefore, care must be taken to reduce exposure when handling both the toxin and biological waste products from the treated animals, a detailed account of which is found in Przedborski et al. (2001). Many factors are known to influence the reproducibility of the lesion, including strain of mice (and even supplier and line), age, gender and weight (Miller et al., 1998), and together with the myriad of published dosing paradigms that contribute further to the variability in lesion size, this makes for a rather complicated backdrop against which to design drug discovery studies.
The various dosing regimens used to generate the MPTP mouse model have been extensively reviewed elsewhere (Jackson-Lewis and Przedborski, 2007). In many cases, MPTP is given with probenecid (250 mg·kg−1), a uricosuric agent that reduces the renal clearance of MPTP, thereby prolonging its action. The most common protocols and the degree of nigro-striatal tract denervation produced by these can be summarized as follows: acute bolus, 1 × 30–40 mg·kg−1 giving 80–90% striatal DA depletion; acute multiple, 2 × 40 mg·kg−1 or 4 × 12.5–25 mg·kg−1 given at 2 h intervals producing variable 60–90% striatal DA loss; sub-acute, 25–40 mg·kg−1/day for 5 days ± probenecid giving 76% loss striatal DA and 60% SNpc cell loss; chronic intermittent, 25 mg·kg−1 twice weekly for 5 weeks ± probenecid, giving 95% loss around 1 week, but reducing to a stable 70–80% loss by 12 weeks post treatment (Pothakos et al., 2009); chronic infusion, 20–40 mg·kg−1/day for up to 28 days given via osmotic minipumps, giving most variable degree of cell loss so far ranging from 25% to 80% loss of cells in the SNpc and 28–90% loss of striatal dopamine (Fornai et al., 2005; Alvarez-Fischer et al., 2008; Gibrat et al., 2009). The pattern of cell death produced is similar to that seen in humans, with the SNpc affected more than the VTA (German et al., 1989; Sundstrom et al., 1990; Hung and Lee, 1996), and chronic infusion may also induce loss of noradrenergic cells in the locus coeruleus, further resembling the clinical picture (Fornai et al., 2005). However, in all cases, the cell death is rapid in onset, with first signs appearing within 12–72 h, and is maintained for up to 28 d (Jackson-Lewis et al., 1995; Tatton and Kish, 1997; Novikova et al., 2006), although striatal dopamine depletion may show signs of recovery when using acute or sub-acute MPTP dosing paradigms (Lau and Meredith, 2003). As noted for the 6-OHDA model, this rapidity of cell death is not reflective of the disease itself and is an obvious weakness of this model. Care should also be taken in studies where assessment of the effects of MPTP are limited to measurement of striatal dopamine content, as MPTP can exert a reserpine-like effect and deplete catecholamines with recovery over the following 2 months (Hallman et al., 1985). This may occur in the absence of nigral cell death, and indeed, not all laboratories have been able to reproducibly observe loss of dopaminergic neurons with MPTP in mice.
Controversy still surrounds the issue of whether MPTP-treated mice exhibit Lewy body-like inclusions. In one of the earliest studies examining this phenomenon, whilst very few inclusions were noted 3 weeks post-chronic MPTP/probenecid treatment, by 24 weeks, several of the remaining TH-positive SN neurons contained α-synuclein- and ubiquitin-immunoreactive inclusions, though these did not resemble classical Lewy bodies found in the disease (Meredith et al., 2002). Later studies failed to find inclusions using the same treatment regimen or following multiple-acute or sub-chronic paradigms (Fornai et al., 2005; Shimoji et al., 2005), and a similarly conflicting picture has emerged in animals receiving chronic infusion of MPTP, with α-synuclein- and ubiquitin-positive inclusion bodies noted in some studies with 14 or 28d infusion, but with others failing to pick out any (Fornai et al., 2005; Alvarez-Fischer et al., 2008; Gibrat et al., 2009). Although more positive than the 6-OHDA model in this respect, further work is needed before these models can be reliably used for assessing agents that may prevent aggregate formation.
Because MPTP is administered systemically, it results in a bilateral degeneration of the nigro-striatal tract, so observations of lateralized differences in motor behaviour used with the 6-OHDA model cannot be used here. However, MPTP-treated mice display signs of akinesia and catalepsy, which have been monitored using for example the pole test, beam walking test, overall rotarod performance and locomotor activity and rearing behaviour in the open-field arena (Sedelis et al., 2001). Of these, measures of locomotor activity and rearing in open field arenas are most often used as readouts of parkinsonian-like behaviour in MPTP mice, but the phenotypes encountered differ greatly depending on the dosing schedule adopted. Mice treated with MPTP via acute bolus or acute multiple dosing paradigms display only a transient reduction in locomotor activity and rearing behaviour, which is lost or even reverts to a hyperactive state within 2–3 days (Colotla et al., 1990). These acute models are therefore of little use when assessing the symptomatic efficacy of drugs. In contrast, sub-acute dosing with MPTP produces a more persistent hypoactivity, evident within 3 h post-treatment and lasting for at least 10 days, whilst chronic intermittent dosing also produces a long-lasting hypoactivity and impaired rotarod performance evident from 2 weeks post-MPTP treatment and lasting for up to 6-months (Petroske et al., 2001; Luchtman et al., 2009; Pothakos et al., 2009). Possibly reflecting the wide variation in nigro-striatal lesion size, chronic infusion of MPTP produces a varied behavioural phenotype ranging from a reduction in locomotor activity and rearing behaviour in the open-field arena that is reversed by apomorphine (Fornai et al., 2005) through to no behavioural deficits at all (Alvarez-Fischer et al., 2008; Gibrat et al., 2009).
Where behavioural deficits are displayed, they have been shown to be reversed by some of the drugs in clinical use today, confirming a certain degree of predictive validity of some MPTP models for assessing symptomatic agents. For example, L-DOPA and the dopamine agonists bromocriptine, cabergoline and pramipexole reverse these behavioural deficits (Fredriksson et al., 1990; Rozas et al., 1998; Archer et al., 2003; Viaro et al., 2010), whilst the MAO-B inhibitor selegiline, the COMT inhibitor tolcapone, and amantadine have been shown to potentiate the effects of L-DOPA in these mice (Fredriksson and Archer, 1995; Fredriksson et al., 2001). The effects of apomorphine are again more varied, with some studies showing no effect (Rozas et al., 1998; Archer et al., 2003), yet others showing reversal of hypokinesia (Fornai et al., 2005). The MPTP mouse model has also been able to predict the efficacy of non-dopaminergic agents, such as the A2A antagonist, istradefylline (Shiozaki et al., 1999). The model is also expected to predict the ability of agents to provide protection or repair against degeneration in the MPTP-treated primate, especially given they share a common inducer. This is certainly borne out by some studies, such as that showing the ability of 5-HT1A agonists to protect against MPTP-induced degeneration in both mice and primates (Bezard et al., 2006). However, the ability to predict agents with clinical neuroprotective efficacy is another story; if the model was predictive, the myriad of compounds shown to protect against MPTP toxicity would surely have led to disease modification in PD by now.
Pesticide-induced models
Rotenone model
The realization that MPTP produced nigro-striatal tract degeneration through the targeting of mitochondrial complex I led to the search for other mitochondrial toxins that might be used to model PD. The best known of the models to emerge from this is the rotenone model of PD, but since its first introduction (Betarbet et al., 2000), it has continued to provoke much debate (e.g. Cicchetti et al., 2009; Greenamyre et al., 2010). Like MPTP, the insecticide rotenone is highly lipophilic, so it readily crosses the blood–brain barrier and diffuses into neurons where, in a manner similar to MPTP, it accumulates within mitochondria and inhibits complex I (Figure 1). The ensuing reductions in ATP are not, however, considered a cause of the toxicity; rather the production of ROS, subsequent to glutathione depletion, is thought to induce oxidative stress (Sherer et al., 2003a). Oxidative damage, in the form of protein carbonyl formation, has certainly been found in the midbrain, olfactory bulb, striatum and cortex of rats treated with rotenone (Sherer et al., 2003a), just as is reported in the PD brain at post-mortem (Alam et al., 1997). The extensive microglial activation seen in both the SNpc and striatum following rotenone infusion (Sherer et al., 2003b) is consistent with the inflammatory features found in idiopathic PD (Gerhard et al., 2006; Whitton, 2007; Tansey and Goldberg, 2010), lending support to the construct validity of this model. Further support is offered by the recent finding that rotenone inhibits proteasomal activity (Wang et al., 2006b), which, as will be described below, is also implicated in PD.
Unfortunately, in addition to its central toxicity, rotenone shows a high degree of systemic (primarily cardiovascular) toxicity that produces high mortality rates (∼30% of animals) regardless of administration route (see, e.g., Betarbet et al., 2000). There also appears to be an intrinsic resistance of some rats to rotenone, with as few as 50% of treated animals displaying neurodegeneration (Betarbet et al., 2000). All of these factors combined, result in the necessity for using a larger numbers of animals at the start of any study to ensure relevant numbers are available for biochemical and histological analysis.
The first report on the rotenone model of PD offered the promise of a realistic model of PD. Betarbet et al. (2000) used osmotic mini-pumps to drive intravenous infusion of low doses of rotenone (2–3 mg·kg−1/day) for between 1 and 5 weeks and produced variable degrees of nigro-striatal degeneration in around half the animals. The pattern of cell death mirrored that seen in idiopathic PD, with greater cell loss evident in the ventral tier of the SNc, relative sparing of the VTA and some degeneration of noradrenergic neurons in the locus coeruleus. This study also reported α-synuclein- and ubiquitin-positive Lewy body-like cytoplasmic inclusions or aggregates with a dense core and fibrillar surround within the SNpc and the rats that showed dopaminergic deficits all developed motor and postural abnormalities. Others have since challenged the idea that rotenone produces selective toxicity of the nigro-striatal system but rather reveal a multisystem degeneration incorporating 20–30% losses in the striatum of 5-HT fibres,DARPP-32 projection neurons and cholinergic interneurons as well as ∼30% loss of noradrenergic neurons in the locus coeruleus and SNpc cells (Hoglinger et al., 2003). The presence of α-synuclein inclusions and reduced spontaneous locomotor activity did, however, corroborate the earlier findings. The outcome appears just as variable when rotenone is given by subcutaneous, rather than intravenous infusion (Sherer et al., 2003c; Lapointe et al., 2004), casting doubt upon the reproducibility of these rotenone models for use in drug discovery programmes. Interestingly, a recent report has provided evidence of peripheral enteric NS pathology (in the form of α-synuclein inclusions) following s.c. rotenone infusion (Drolet et al., 2009), indicating that it might, however, be useful for examining the less well studied peripheral pathology and Lewy body formation that occurs in PD.
Administration of rotenone using intermittent i.p. dosing schedules has proven more effective to date. When low doses (1.5–2.5 mg·kg−1 i.p) are administered daily for up to 2 months, a dose-dependent reduction in striatal TH and dopamine levels is found (Alam and Schmidt, 2002), and animals exhibit reduced locomotor activity in the open-field test and marked catalepsy that are reversed by L-DOPA (Alam and Schmidt, 2002; 2004). Use of a novel fatty acid-based vehicle has also helped reduce mortality rates considerably and, when given i.p. in this vehicle daily for up to 30 days, rotenone produces loss of striatal dopamine terminals, nigral pathology (α-synuclein- and ubiquitin-positive intracellular inclusions and ∼45% cell loss) and a clear parkinsonian phenotype characterized by postural instability and reduced paw reaching in the cylinder test (Cannon et al., 2009). Although this mode of rotenone administration shows much promise, until these findings are replicated by other laboratories, doubt will remain regarding the reproducibility and hence robustness of the model (Cicchetti et al., 2009).
In an attempt to mimic the most likely route of rotenone exposure to humans, recent studies have begun to investigate the effects of intra-gastric administration of rotenone to C57Bl/6J mice (Pan-Montojo et al., 2010). Although in early days, it appears that intragastric administration (5 mg·kg−1 5 days a week for 1.5 or 3 months) causes by 3 months a modest degeneration of SNpc cells, accompanied by α-synuclein inclusions and reduced rotarod performance. Interestingly, inclusions were found in the enteric nervous system and the dorsal motor nucleus of the vagus at an earlier time point (1.5 months), leading the authors to suggest a possible transynaptic mechanism by which PD might spread throughout the nervous system, from a peripheral starting point, as suggested by Braak et al. (2006).
Because of the variable nature of rotenone's effects, whilst being useful to examine aspects related to the pathophysiology of PD, the current rotenone models do not offer robust test beds for assessing the effects of symptomatic drugs. Indeed, as Table 1 summarizes, only L-DOPA and apomorphine among the drugs in clinical use today have been shown to reverse locomotor deficits in the rotenone model and then only in those models utilizing repeated i.p. injections (Alam and Schmidt, 2004; Cannon et al., 2009). However, it is worth noting that both pramipexole and selegiline, drugs that were examined for disease-modifying potential in clinical trials but then failed to show clear efficacy, did protect against rotenone-induced degeneration (Saravanan et al., 2006; Inden et al., 2009), suggesting the rotenone model may show promise for selecting agents with potential neuroprotective efficacy.
Paraquat and Maneb model
Given that exposure to the herbicide paraquat (1,1′-dimethyl-4,4′-bipyridinium) or the fungicide Maneb (manganese ethylene-bis-dithiocarbamate) has been associated with an increased incidence of PD (Ascherio et al., 2006; Costello et al., 2009), it is not surprising that attempts have been made to model PD using these agents.
Paraquat enters the brain via the neutral amino acid transporter (Shimizu et al., 2001) before Na+-dependent uptake into cells occurs (Figure 1). Once inside cells, paraquat leads both to indirect mitochondrial toxicity via redox cycling and also direct inhibition of complex I (at higher doses) (Miller, 2007). Maneb, on the other hand, preferentially inhibits complex III of the mitochondrial respiratory chain following entry into the brain (Zhang et al., 2003). Paraquat and Maneb have been shown to produce enhanced toxicity when combined (Thiruchelvam et al., 2000), possibly as a result of Maneb increasing the brain concentration and reducing clearance of paraquat (Barlow et al., 2003). Coupled with the fact that human exposure to one of these pesticides alone is unlikely as they are used in the same geographical regions, this provides a clear rationale for combining their administration in order to produce an animal model of PD.
The combined administration of paraquat (10 mg·kg−1 i.p.) and Maneb (30 mg·kg−1 i.p.) twice weekly for up to 6 weeks in either C57bl/6 mice or Wistar rats produces only a modest but fairly consistent level of nigro-striatal degeneration (20–35%), with relative sparing of the VTA (Thiruchelvam et al., 2000; Cicchetti et al., 2005). These changes are surprisingly accompanied in most cases (Thiruchelvam et al., 2000; Cicchetti et al., 2005) but not all (Saint-Pierre et al., 2006) by motor deficits manifest as hunched posture and a decline in locomotor activity of the mice. Co-administration of L-DOPA to mice prevents both the motor deficits and nigral cell losses induced by paraquat and Maneb (Li et al., 2005), probably reflective of L-DOPA competing with paraquat for access into the brain. Significant microglial activation and lipid peroxidation is found in the striatum, indicating an inflammatory component and the involvement of oxidative stress in the toxicity of rotenone (Thiruchelvam et al., 2000; Cicchetti et al., 2005; Gupta et al., 2010). Some treated animals suffer from progressive weight loss and respiratory pathology, leading to quite high mortality rates that need to be taken into account (Saint-Pierre et al., 2006), and the model has also received criticism because of its minimal degree of cell death and variable loss (if at all) of striatal dopamine content (Miller, 2007). There has been no mention of inclusions in this model either, so at this stage the model has a limited use in drug discovery programmes. Indeed, none of the currently used drugs to treat PD have been examined in the model, hence its exclusion from Table 1. The exception being selegiline, which protects against paraquat toxicity (Liou et al., 2001), implying a potential role for this model in assessing neuroprotective agents.
MPTP primate model
The discovery of the ability of MPTP to induce Parkinsonism in man led to an opportunity to use systemic toxin administration to produce a model of PD in primates with a high degree of construct validity (Davis et al., 1979; Burns et al., 1983; Langston et al., 1983; 1984; Jenner et al., 1984). This had never been achieved before and those models previously available involved surgical approaches to destroy the nigro-striatal pathway or the use of manganese toxicity, which causes pallidal rather that nigral neuronal loss (see, e.g., Olanow et al., 1996). Indeed, MPTP has a particular efficacy in destroying dopaminergic neurons in the primate substantia nigra that is not seen in lower species, with the exception of some mouse strains (see earlier). The reason for this peculiar sensitivity is not well understood, but it may relate to the persistence of its metabolite MPP+ in the brain for long periods of time compared with the rapid clearance that occurs in other species (Herkenham et al., 1991). It may also reflect a higher sensitivity of primate nigral dopaminergic neurons to toxins such as MPTP since PD appears to be a syndrome that specifically affects man. All species of primates in which MPTP has been tested appear to be sensitive to the toxin, and these include macaques, vervet monkeys, squirrel monkeys, baboons and marmosets.
The repeated systemic administration of MPTP by intraperitoneal, subcutaneous and intravenous administration in doses that vary with species and route over 3–5 days leads to the onset of a parkinsonian syndrome almost immediately and certainly within a few days of commencing treatment. It consists of akinesia, bradykinesia, rigidity of the limb and trunk and postural abnormalities which form cardinal symptoms of PD as they occur in man. Without a shadow of doubt, this model therefore has the strongest face validity of all the animal models of PD. However, classical rest tremor is not commonly observed; rather postural tremor is present. Those primates that exhibit rest tremor may have a differential loss of dopaminergic neurons in the A8 and A9 region of substantia nigra (SN). Overall, the physiological basis of tremor in PD is itself poorly understood, and it is rare for tremor to be produced by selective lesion of the SN. In most studies, a very marked parkinsonian state is initially induced from which the animals partially recover over a period of weeks to then exhibit stable motor deficits. The recovery may relate to the adaptation of remaining dopaminergic neurons, but it may also reflect a reserpine-like action of MPTP that initially depletes vescicular stores of dopamine, noradrenaline and 5-HT but which subsequently recover (Rose et al., 1989a,b). Some variants of the model exist in which MPTP can be given by intracarotid injection to initiate asymmetric motor deficits (Przedborski et al., 1991; Schneider and Dacko, 1991). However, although PD itself is invariably unilateral in presentation, this model is more difficult to assess for drug effect and for the onset of motor complications, although it induces less severe motor deficits in the early stages of MPTP treatment. The motor deficits in MPTP-treated primates can be readily assessed through the automated measurement of locomotor activity and the assessment of motor disability using semi-quantitative rating scales that assess many features of motor function. In hemi-parkinsonian animals, asymmetric changes in motor ability occur and rotation occurs on treatment with dopaminergic drugs. On occasions, MPTP has been given in small repeated doses or over longer periods of time to induce partial lesions of the nigro-striatal pathway or to try and produce a model of PD that is more ‘progressive’ in nature than occurs with acute toxin treatment (Blanchet et al., 1998b; Bezard et al., 2001b; Meissner et al., 2003). Interestingly, the effects of MPTP might manifest themselves in non-motor symptoms associated with PD. For example, in common marmosets, there is clearly constipation, bladder hyper-reflexia, excessive salivation and sleep disturbance (Albanese et al., 1988; Yoshimura et al., 1993; 1998; Barraud et al., 2009). Rhesus monkeys on the other hand, when treated chronically (>3 months) with low doses of MPTP (0.025–0.1 mg·kg−1, given i.v. 2–3 times per week) that do not evoke motor deficits, display cognitive deficits similar to those that accompany PD (Taylor et al., 1986; Schneider and Kovelowski, 1990). Although the latter model is starting to be used in the serach for agents able to treat this cognitive impairment (Decamp and Schneider, 2009), it is surprising that there has never been a full examination of the standard MPTP-treated primate for non-motor symptoms, given their clinical importance and the need for animal models of non-motor signs in which pharmacological approaches to treatment could be assessed.
The pathology and biochemistry of the MPTP-treated primate shows that this is a model of selective nigro-striatal degeneration that is only similar in some respects to that seen in PD. There is extensive loss of nigral dopaminergic neurons (>70%) with some of the regional differences in the extent of cell loss and subsequent reductions in dopamine content in the caudate nucleus and in the putamen (>90%) that occur in man although this has been a matter of debate (Schneider et al., 1987; German et al., 1988; 1992; Pifl et al., 1988; Gibb and Lees, 1991; Perez-Otano et al., 1994; Varastet et al., 1994; Elsworth et al., 2000). Other major differences are that the loss of dopaminergic neurons is not progressive and the pathological hallmark of PD, the Lewy body, does not appear (Halliday et al., 2009), although accumulations of α-synuclein may be present (Kowall et al., 2000). Pathology does not affect the VTA or other dopaminergic nuclei to the same extent, but there are reports of cell loss in the hypothalamus and the noradrenergic cells of locus coeruleus (Mitchell et al., 1985; Forno et al., 1986; Gibb et al., 1986; 1989; Miyoshi et al., 1988). Otherwise, cell death does not seem to occur in a range of other brain regions that are known to be affected in PD, for example the raphe nuclei, substantia innominata, dorsal motor nucleus of the vagus (see, e.g., Garvey et al., 1986). Transient changes in mesolimbic dopamine content, cardiac noradrenaline content (largely in rodents) and adrenal dopamine levels have been reported, but these are probably a reflection of the reserpine like actions of MPTP (Fuller et al., 1984; Fine et al., 1985; Rose et al., 1989a). Despite these distinctions between the MPTP primate model and the disease itself, its face validity is supported in other ways. For example, as in PD, the loss of dopaminergic neurons leads to a reactive microgliosis that can persist long after toxin administration and that has been suggested to reflect an on-going loss of dopaminergic neurons that also occurred in MPTP exposed drug addicts (Langston et al., 1999; McGeer et al., 2003; Barcia et al., 2004; McGeer and McGeer, 2008). The loss of striatal dopaminergic input leads to alterations in the density of D1, D2 and D3 dopamine receptors and mRNA, but the nature of these is not consistent between studies (Bedard et al., 1986; Joyce et al., 1986; Falardeau et al., 1988; Alexander et al., 1991; Przedborski et al., 1991; Gnanalingham et al., 1993; Hurley et al., 1996; Morissette et al., 1996; Herrero et al., 1996a; Decamp et al., 1999; Quik et al., 2000). Changes in the activity of striatal output pathways are known to occur with altered levels of pre-proenkephalin-A mRNA, pre-proenkephalin-B mRNA, dynorphin mRNA and glutamic acid decarboxylase mRNA and changes in the expression or levels of neuropeptides, GABA receptors, adenosine A2A receptors and opioid receptors that translate into altered activity of the indirect (strio-GPe) pathway and direct (strio-GPi) pathway (including Taquet et al., 1988; Lavoie et al., 1991; Taylor et al., 1991; Herrero et al., 1995; 1996a,b,c; Vila et al., 1996a,b; 1997; Morissette et al., 1999; Obeso et al., 2000c; Calon et al., 2001; Tel et al., 2002).
It is from the pharmacological perspective that the MPTP-treated primate model has proved so useful. Almost immediately, the ability of L-DOPA to reverse the MPTP-induced motor deficits was recognized (Burns et al., 1983). Subsequently, every dopaminergic drug used in the treatment of PD was shown to be effective – bromocriptine, pergolide, cabergoline, apomorphine, ropinirole, pramipexole and piribedil as were those antimuscarinic agents tested such as trihexyphenidyl and benztropine (Close et al., 1990; Fukuzaki et al., 2000b; Jenner, 2003b; 2008). The MAO B inhibitors selegiline and rasagiline produced mild motor improvement and potentiated the effects of L-DOPA. Similarly, the COMT inhibitors, entacapone and tolcapone, were shown to potentiate the actions of L-DOPA (see, e.g., Smith et al., 1997). So here was a model with strong predictive validity for therapeutic effect in PD (see Table 1) that is now an almost essential step between preclinical and clinical investigations. New dopaminergic approaches also showed effectiveness in the model. New drug delivery systems were developed such as the rotigotine transdermal patch (Loschmann et al., 1989; Stockwell et al., 2009). Partial dopamine agonists, such as aplindore and pardoprunox, reversed motor deficits and are now in phase II/III clinical evaluation (Jackson et al., 2010; Johnston et al., 2010; Tayarani-Binazir et al., 2010). A range of D1 agonists, including some that were active in early clinical evaluation such as ABT-431, CY 208–243 and a range of benzazepine derivatives, all showed effectiveness in the model although no D1 agonist has so far been introduced in to general clinical practice (Nomoto et al., 1988; Temlett et al., 1988; 1989; Kebabian et al., 1992; Loschmann et al., 1992; Gnanalingham et al., 1995a,b; Shiosaki et al., 1996). More recently, drugs acting on D3 receptors have also shown promise, but none has so far entered clinical evaluation (Bezard et al., 2003; Millan et al., 2004; Silverdale et al., 2004). Moreover, there has been a notable failure that urges caution in interpreting everything coming from MPTP-treated primates. A series of studies in MPTP-treated common marmosets showed drugs that were non-specific inhibitors of monoamine reuptake, such as brasofensine, tesofensine and BTS 74–398, were highly effective in reversing motor disability (Hansard et al., 2002a,b; 2004; Pearce et al., 2002). However, early clinical evaluation in PD has failed to reveal any obvious anti-parkinsonian activity (Bara-Jimenez et al., 2004; Hauser et al., 2007; Rascol et al., 2008). The reasons for the discrepancy are not clear but may relate to differences between the model and PD in relation to loss of noradrenergic and serotoninergic neurons. In MPTP-treated primates, these could act as substrates for drug action, whereas in PD, their loss would remove any such action. Also, it is surprising that monoamine reuptake inhibitors do have effects in MPTP-treated primates as the majority of striatal dopaminergic terminals have degenerated, so the substrate for their ability to increase synaptic dopamine levels is missing. Indeed, it appears that the effects of monoamine reuptake blockers is not striatally mediated and perhaps, a limbic or cortical arousal effect, explains the increased motor activity observed.
Another major use of the MPTP-treated primate lies in the study of the major motor complication occurring in PD, namely dyskinesia. As in MPTP-exposed drug addicts, it became clear that MPTP-treated monkeys repeatedly exposed to L-DOPA showed dyskinesia rapidly after drug treatment was started (Langston and Ballard, 1984; Bedard et al., 1986; Clarke et al., 1987; Langston et al., 2000). The rapidity of onset differs from events in PD where dyskinesia may take years to emerge, but it reflects the high degree of nigral denervation in these animals that lowers the extent and duration of L-DOPA exposure required for involuntary movements to appear (Smith et al., 2003; Kuoppamaki et al., 2007). However, the nature of the dyskinesia is almost indistinguishable from that occurring in man and consists of chorea, dystonia and athetosis, and these can be assessed using semi-quantitative rating scales akin to those used in man (Langston et al., 2000). The intensity of dyskinesia is reduced by the only drug known to be effective in the control of these involuntary movements in PD, namely the weak NMDA antagonist amantadine (Blanchet et al., 1998a). MPTP-treated primates also show other motor complications and fluctuations associated with the treatment of PD, such as ‘wearing off’, ‘on-off’ and freezing, but surprisingly these have received scant attention considering their importance to the treatment of PD in man (Clarke et al., 1987; Kuoppamaki et al., 2002; Jenner, 2009). More recently, non-motor complications of L-DOPA treatment have been described in MPTP-treated primates. In both marmoset and macaque PD models, L-DOPA treatment has been shown to evoke neuropsychiatric-like behaviours including agitation, ‘halucinationory-like’ behaviour and obsessive grooming, which in some cases were responsive to antipsychotic drugs (Visanji et al., 2006a; 2009; Fox et al., 2010). These demonstrations open up new avenues for exploring the mechanisms behind these L-DOPA complications and pharmacological ways in which to treat them. However, to date, the MPTP model has been the most instrumental in the search for the cause of treatment-related dyskinesia by allowing electrophysiological, anatomical and biochemical analysis of the alterations occurring in the basal ganglia (see as examples Crossman, 1987; 1989; 1990; 2000; Obeso et al., 2000a,b,c; 2002; 2008). Whilst these have led to concepts on the roles of altered activity in the direct and indirect output pathways and overactivity of the subthalamic nucleus, it is the pharmacological evaluation of dyskinesia in the MPTP-treated primate that has led to the most significant advances.
The repeated administration of longer-acting dopamine agonists, such as bromocriptine and ropinirole, to MPTP-treated primates caused only mild dyskinesia compared with the intense dyskinesia produced by repeated L-DOPA administration (Bedard et al., 1986; Pearce et al., 1998). The same was observed in clinical trials where compared with L-DOPA monotherapy in early PD, ropinirole, pramipexole, cabergoline and pergolide produced a lower incidence of dyskinesia in the first 4–5 years of treatment (Parkinson Study Group, 2000; Rascol et al., 2000; Hubble, 2002; Oertel et al., 2006). This gave rise to the concept of continuous dopaminergic stimulation (CDS), with the argument being that dopamine is normally released in a continuous manner causing a tonic stimulation of dopamine receptors that is lost in PD (Olanow and Obeso, 2000). Replacing dopaminergic tone using short-acting drugs such as L-DOPA results in a non-physiological stimulation that leads to the onset of abnormal basal ganglia function and the expression of dyskinesia. Consequently, the longer-acting dopamine agonists would cause less dyskinesia and so should be used in the early treatment of PD. However, there has never been a head-to-head comparison of dopamine agonists of differing duration of effect in PD, and the literature from the MPTP-treated primate now shows that there is no difference in the intensity of dyskinesia produced by a range of dopamine agonists with differing half-lives (Jenner, 2009). In fact, dopamine agonists seem to prime the basal ganglia for dyskineisa but do not lead to its expression until L-DOPA treatment is introduced. This can be seen experimentally in the MPTP-treated primate where repeated administration of ropinirole or piribedil causes little or no dyskinesia but on first exposure to L-DOPA intense dyskinesia appears (Smith et al., 2006; Jackson et al., 2007). The same may be true in PD where in the long term, there seems to be no difference in troublesome dyskinesia prevalence in patients who were started on a dopamine agonist compared to those receiving L-DOPA treatment as inevitably L-DOPA had to be introduced to supplement the lower efficacy of dopamine agonists in controlling motor symptoms (Parkinson Study Group, 2000; Katzenschlager et al., 2008). This raises the question of why there is a difference in the ability of L-DOPA to control motor function in PD and to induce dyskinesia compared with the agonist drugs. This may simply be a question of pharmacology and have nothing to do with the duration of drug effect. Dopamine agonists, such as ropinirole and pramipexole, are highly focused on stimulating the post-synaptic D2/D3 receptors and nothing else. In contrast, L-DOPA is rich in its pharmacology and forms dopamine that stimulates D1, D2, D3, D4 and D5 receptors. Some dopamine is converted to noradrenaline, and L-DOPA can displace 5-HT from serotoninergic neurons as well as altering glutamate release and potentially acting as an amino acid neuromodulator in its own right. So it may be incorrect to think of L-DOPA and dopamine agonists as similar pharmacological agents.
One component of the concept of CDS does appear to be valid and that relates to the effects of continuous drug delivery compared with discontinuous administration, and again the MPTP-treated primate model has proved formative. The continuous delivery of apomorphine, ropinirole or rotigotine from osmotic mini pumps or subcutaneous depots resulted in less dyskinesia induction than occurred on oral administration or by repeated subcutaneous injection (Bibbiani et al., 2005; Stockwell et al., 2008; 2009). Similarly, delivering L-DOPA more continuously by combining it with the peripheral COMT inhibitor entacapone resulted in less dyskinesia than with L-DOPA alone when a four times daily treatment regimen was employed (Smith et al., 1997). All of this has suggested that more continuous drug delivery should be used in the treatment of PD, and this mantra is now widely accepted and utilized. Indeed, continuous delivery of apomorphine by subcutaneous infusion and L-DOPA by intraduodenal infusion in late stage PD has been shown to improve motor function over oral therapy and to reduce the intensity of dyskinesia (Manson et al., 2002; Stocchi et al., 2005).
Recently, the MPTP-treated primate has been used to examine the potential of non-dopaminergic therapies for the motor symptoms of PD, and there have been several recent reviews of this work (see, e.g., Brotchie, 2005; Fox et al., 2006; 2008). Outside of the dopaminergic area, only muscarinic antagonists, such as benzhexol, and glutamate antagonists, such as amantadine, have been shown to exert efficacy. The recent approach is based on the multiple sites of pathology in the brain in PD that affect a variety of neurotransmitters including noradrenaline, 5-HT, acetylcholine and glutamate and on the alterations in basal ganglia input and output pathways that occur as a result of the loss of striatal dopaminergic tone. The latter offers a wealth of opportunities for manipulating motor function beyond the damaged dopaminergic system. There are alterations in GABA, acetylcholine and glutamatergic neurons, and these can be manipulated directly and through adenosine, opioid, 5-HT, noradrenaline, histamine and cannabinoid receptors among others. There have been promising effects in the MPTP-treated primate with α2-adrenergic antagonists, such as fipamezole, with 5-HT1A agonists, such as sarizotan, and with adenosine A2A antagonists, such as istradefylline (Kanda et al., 2000; Bibbiani et al., 2001; Savola et al., 2003; Fox et al., 2006; Gregoire et al., 2009). However, translation from the MPTP-treated primate has not gone well, and there are worrying examples of lack of efficacy in PD. For example, sarizotan decreased L-DOPA-induced dyskinesia in the primate without interfering with the improvement in motor function. In contrast in PD, sarizotan both suppressed dyskinesia and worsened motor function in phase II studies and did nothing in phase III investigations (Olanow et al., 2004; Goetz et al., 2007). Istradefylline had a modest symptomatic effect in the primate and did not provoke established dyskinesia, but in man, its effects on motor function were limited and increased dyskinesia was observed (Hauser et al., 2008). In this case, it may be the trial design and not the model that is at fault. The preclinical studies showed istradefylline only to improve motor function when administered with low doses of L-DOPA but not with high doses. In the phase III clinical studies, the drug was administered to patients on optimal dopaminergic medication, and only small further improvements in motor function occurred.
So while the MPTP-treated primate has value as a predictive model, there are caveats that must be considered in examining the results of evaluations in what is, after all, a relative simple model of alterations in movement. The MPTP-treated primate can also be used to evaluate neuroprotective strategies, and this will be considered later. In addition, it is an effective primate model in which to examine neurorestorative strategies such as neurotrophic factors, cell-based therapies and gene therapies (see as examples Iravani et al., 2001; Costa et al., 2001b; Kordower et al., 2006a; Jarraya et al., 2009).
Moving from animal models of symptomatic activity to neuroprotection and neurorestoration
So far, this review has largely focused on the use of animal models in the detection of novel symptomatic agents for the treatment of the motor symptoms of PD or the complications arising from its treatment. However, the major challenge in trying to advance the treatment of PD lies in dealing with the progression of the disease process and in reversing the neuronal damage that occurs as a result. As a consequence, we will now review models of PD that might have value in detecting neuroprotective and neurorestorative agents. Earlier mention has been made of the use of 6-OHDA, MPTP, rotenone and paraquat/Maneb to destroy the dopaminergic nigro-striatal pathway, but in this section, those models that mimic some known component of the disease process occurring in man will be considered. From the outset, it should be pointed out that many of these models have yet to be widely utilized for examining neuroprotective or restorative strategies, and this remains a challenge for the future.
Animal models based on hallmarks of PD
Despite the extensive knowledge of pathogenic mechanisms, such as oxidative stress, mitochondrial dysfunction and excitotoxicity, underlying neuronal loss in PD, we have not found any clinically effective means of stopping or slowing the disease process. We have also not devised animal models of PD that truly reflect the widespread and progressive pathology of the illness. For these reasons, there has been considerable interest in returning to the basic elements of PD that form the hallmarks of the disease process. These are the formation of Lewy bodies and the presence of activated microglial cells. Both are being assessed for their ability to provide novel animal models of PD with a high degree of construct validity and to understand the role these processes play in cell death.
Proteasomal inhibitor models
Interest in Lewy body formation was rekindled by the discovery of mutations in α-synuclein responsible for rare forms of familial PD (Polymeropoulos et al., 1997). The toxicity of α-synuclein was associated with misfolding of the mutated protein and altered ability to be degraded by proteasomal and lysosomal mechanisms (Cookson and van der Brug, 2008). When Lewy bodies were shown to be intensely immunoreactive for α-synuclein and also to contain nitrated forms of the protein along with a wide range of other proteinous material, the failure of protein metabolism in PD was proposed as being core to the neuronal loss (Spillantini et al., 1998; Duda et al., 2000). The subsequent discovery of two further mutations in familial PD, namely ubiquitin carboxy terminal hydrolase-1 (UCH-L1) and parkin, that affect the functioning of the ubiquitin–proteasome system, further focused attention (Kitada et al., 1998; Leroy et al., 1998). When a reduction in proteasomal catalytic activity and subunit expression in the SN in PD was reported, attempts to use this as a means of producing a new animal model of PD commenced (McNaught and Jenner, 2001; McNaught et al., 2002a). Proteasomal inhibitors, such as lactacystin, PSI and epoximycin, were shown to selectively kill dopaminergic cells in culture and subsequently on direct intranigral injection to destroy the nigro-striatal pathway, reduce striatal dopamine content and to induce motor deficits in the rat (McNaught et al., 2002b,c). Infusions or repeated injections of lactacystin or PSI were proposed as providing progressive models of PD. However, it was a report that the systemic administration of PSI/epoximycin could reproduce many components of PD as it affected man, suggestive of strong face validity, which raised intense interest in this model (McNaught et al., 2004). The peripheral administration of PSI/epoximycin for some days was shown to cause a progressive loss of nigro-striatal neurons and the progressive onset of motor disability. Motor disability was reversed by the administration of L-DOPA. In addition, proteinous inclusions related to Lewy bodies were present along with pathology in other brain regions affected in PD, namely the locus coeruleus, raphe nuclei and substantia innominata.
Unfortunately, these initial findings proved difficult to reproduce and reports of failure in mice, rats and primates quickly appeared (Kordower et al., 2006b; Manning-Bog et al., 2006). However, in our hands and those of others, a partial reproduction of the original report was obtained (Zeng et al., 2005; Schapira et al., 2006; Bukhatwa et al., 2010). An approximate 40% loss of nigral dopaminergic neurons was consistently obtained on repeated subcutaneous administration of PSI at a somewhat higher dose than used before, but while this was incremental over 8 weeks, it did not progress further. A variable loss of motor function was observed, and only a small increase on administration of dopaminergic drugs presumably reflecting the low degree of dopaminergic cell death that hovered around the point at which dopamine loss resulted in decreased motor activity. In agreement with the original study, there was pathology in other brain regions and proteinous inclusions and glial activation were observed. The key question is why these changes are so difficult to reproduce.
Recent investigations in these laboratories suggest that the dose of PSI is critical with optimal dosage levels above which toxicity decreases (Bukhatwa et al., 2010). There are also differences between routes of administration with effects observed after subcutaneous and oral administration but not after intraperitoneal treatment. What underlies these differences is a mystery, as nothing is known about the absorption, distribution or metabolism of PSI and its plasma half-life has not been measured. It is also unknown whether PSI penetrates in to brain. There may also be differences in the purity of PSI from different suppliers and between batches from the same supplier. Perhaps surprisingly, and contrary to previous conjecture, PSI does not appear to be unstable in solution, and it does not seem to be rapidly metabolized in plasma.
The variability in response to PSI is frustrating as this could potentially be a valuable model of PD in which to test neuroprotective strategies, but nothing has so far appeared in the literature to this effect. It appears that some drugs that are effective in more classical models of PD as neuroprotectants are not able to stop PSI toxicity but, then again, they did not have any effect in man. There needs to be more investigation of the potential of proteasomal inhibitors to provide an animal model of PD alongside inhibitors of lysosomal function or even a combination of the two.
Glial activation models
The other key hallmark of PD is the presence of a reactive microgliosis in the SN that accompanies the loss of dopaminergic neurons (McGeer et al., 1988; McGeer and McGeer, 2008). Glial cells would normally support neuronal viability, but the activation of microglia leads to an inflammatory response that is accompanied by cytokine release and by an induction of iNOS that leads to nitrative stress as shown by the presence of 3-nitrotyrosine immunoreactivity (Hirsch et al., 1999; 2003). It is presumed that glial cell activation follows the start of neuronal cell death, and that it contributes to disease progression, although it could potentially be an initiator of cell loss. For example, increased release of TNF-α could lead to ceramide-dependent NF-κβ-mediated apoptosis.
For these reasons, lipolysaccharide (LPS) has been used to activate glial cells in experimental models of PD and to reproduce to some extent the inflammatory events that occur in man. LPS has been shown in vitro to kill dopaminergic neurons through glial cell activation, and for this to be accompanied by increased release of cytokines, iNOS induction, oxidative and nitrative stress and reduced secretion of the trophic factors, BDNF and GDNF (McNaught and Jenner, 1999; 2000a,b). Its unilateral stereotaxic injection in to the substantia nigra results also in neuronal loss and destruction of the nigro-striatal pathway that leads to asymmetric motor function when challenged with amphetamine or apomorphine (Herrera et al., 2000; Castano et al., 2002; Iravani et al., 2005). The administration of LPS causes induction of iNOS and the expression of 3-nitrotyrosine immunoreactivity, indicating peroxynitrite formation and its attack on proteins (Figure 1). The effects of LPS are partially blocked by iNOS inhibitors and can also be partially attenuated by the administration of anti-inflammatory agents.
The LPS model appears to be a good model of the inflammatory events occurring in PD, but it does not replicate the disease condition in a number of respects having, therefore limited face validity. It leads to a primary loss of dopaminergic neurons through inflammatory mechanisms, which flies in the face of the current concepts of how PD occurs. LPS may activate microglia, but it also leads to astrocytosis, which is not a major component of the glial reaction occurring in PD. On stereotaxic injection, it is not progressive, but versions of this model have been examined that involve either multiple small injections of LPS or its continuous infusion, but these do not produce effects that outlast acute toxin action. Interestingly, a single administration of LPS given to adult mice has been reported to cause progressive dopaminergic neuronal loss (Qin et al., 2007). If replicated, this may be a valuable addition to animal models of PD. The other shortcoming of the LPS model is the lack of pathology in other brain regions that are affected by PD. However, despite the limitations of the LPS model, it does seem like a good test bed for strategies aimed at limiting inflammatory change in PD and so slowing its progression.
Animal models based on gene abnormalities in familial PD
There have been major advances in determining the underlying gene defects in familial PD that have led to the identification of gene products and attempts to produce transgenic models of PD in mice. The gene products uncovered have shown commonality with mechanisms of neuronal cell death in sporadic PD, including mitochondrial dysfunction (α-synuclein, PINK1, DJ-1, LRRK2) and alterations in protein folding and metabolism (α-synuclein, parkin, UCH-L1, glucocerebrosidase) (see, e.g., recent reviews Yang et al., 2009; Cookson and Bandmann, 2010; Hardy, 2010). Initial excitement over the description of the A30P and A53T mutations in α-synuclein has so far failed to translate into truly effective transgenic models of PD (see Chesselet, 2008). Attempts to produce α-synuclein knockouts, over-expressers and transgenics led to a variety of abnormalities in the brain and spinal cord, including mitochondrial abnormalities, gliosis, loss of motor neurons, α-synuclein aggregate formation and some functional abnormalities in the nigro-striatal system, but there has not been any consistent reports of loss of nigral dopaminergic neurons (Dawson et al., 2010). LRKK2 transgenic mice similarly display dopaminergic dysfunction and some behavioural deficits that are L-DOPA responsive but no noticeable nigral cell degeneration (Lin et al., 2009). The models of autosomal-recessive PD, based on knockout of parkin (Perez and Palmiter, 2005), PINK1 (Kitada et al., 2007) or DJ-1 (Goldberg et al., 2005) genes, despite showing the expected mitochondrial dysfunction and subtle abnormalities in dopaminergic transmission such as reduced evoked striatal dopamine release, also failed to replicate nigral pathology.
The reason for the failure of the transgenic approach to produce an effective animal model of PD in mice is puzzling as is the inability to get these gene defects to reproduce the pathological changes with which they are associated in man. Perhaps the mouse is not appropriate since, for example, the wild-type α-synuclein in mice is one of the mutant forms in man associated with familial PD. Perhaps ageing is a crucial factor, or may be these gene defects do not operate in isolation, and either multiple gene defects are required to trigger neuronal loss or the gene defects operate in conjunction with environmental triggers. However, even when all three of the recessive genes are silenced together, as achieved in the recent parkin/DJ-1/PINK1 triple knockout mice, this is still not sufficient to cause dopaminergic cell loss (Kitada et al., 2009). The gene defects reported may not themselves be the cause of familial PD, but they may operate through epigenetic effects that allow, for example the expression of effects of otherwise silent genes that then initiate dopaminergic cell loss. One such area that might prove useful is in changes in ceramide metabolism linked to glucocerebrosidase mutations and to lysomal/autophagic protein metabolism. Individuals with Gaucher's disease have an 8–10-fold increased risk of developing PD because of glucocerebrosidase mutations, and this would seem highly relevant to the development of new animal models of PD (Sidransky et al., 2009; Velayati et al., 2010). Other suggestions as to why the dopaminergic neurons do not generate in these transgenic models have been highlighted in the recent review by Dawson et al. (2010). One proposal is that when genes are either knocked out or over-expressed embryonically, compensatory mechanisms may occur in the dopaminergic system to effectively mask the effects of the genetic manipulation. This implies that moving towards conditional knockouts or viral vector-mediated delivery of transgenes in the adult might yield better models, and there is already some evidence to support this. For example, viral vector-driven over-expression of α-synuclein in both adult mice and primates has produced models that more closely reproduce the pathology of the human condition in terms of exhibiting nigro-striatal tract degeneration (Kirik et al., 2002; 2003; Lo Bianco et al., 2004).
In passing, it is of interest to mention some other gene defects that may be relevant to PD or to the pathogenic processes that underlie neuronal loss. The transcription factors Nurr-1 and Pitx3 are essential for the development of a dopaminergic phenotype in nigral neurons (see, for example, Zetterstrom et al., 1997; Nunes et al., 2003). Decreased expression of the Nurr1 gene and Nurr-1 and Pitx3 polymorphisms have been associated with PD (Hwang et al., 2003; Jankovic et al., 2005; Le et al., 2008; Fuchs et al., 2009; Bergman et al., 2010). Nurr-1 knockout mice show marked loss of dopaminergic neurons, increased sensitivity to toxins such as MPTP and lactacystin and impaired locomotion compared to wild type animals (Zetterstrom et al., 1997; Le et al., 1999a,b; Jiang et al., 2005; Pan et al., 2008). Pitx3 deficient (aphakia) mice also show marked loss of nigral dopaminergic neurons and L-DOPA-sensitive motor deficits (Hwang et al., 2003; van den Munckhof et al., 2006). Despite these promising phenotypes, these models have yet to be widely employed by others. Disruption of complex I of the mitochondrial respiratory chain in the ‘Mitopark’ mouse has also been reported to produce nigral cell degeneration and motor deficits in a progressive manner, but this has not been widely available to other investigators, and its value as an animal model of PD remains to be determined (Ekstrand and Galter, 2009; Galter et al., 2010). In addition, mice with discrete deletions in proteasomal subunit proteins are also available, and these do exhibit loss of dopaminergic neurons but the life span of these animals seems extremely short (Bedford et al., 2008). Finally, deletion of VMAT-2 may also produce a model of PD but not one that reflects the pathology of the disease in man (Colebrooke et al., 2006).
Although the genetic mice models have not yet contributed to drug discovery for PD, it is possible that with further optimization, such models will one day contribute in this way. However, as things stand, the current genetic models have most utility in shedding light on how the gene mutations associated with PD might contribute to disease pathogenesis.
Emerging genetic models in multicellular model organisms
The disappointing outcomes obtained so far by modelling the genetics of PD in mice have stimulated interest in developing alternative genetic models in multicellular model organisms. The fruitfly Drosophila melanogaster, the nematode Caenorhabditis elegans (C. elegans) and the zebrafish Danio rerio offer some clear advantages over rodents in terms of the relative ease with which the genome can be manipulated to model the gene mutations associated with PD and of the much reduced costs involved in the development of genetic models of PD, but of course, their face validity is limited by the nature of the ‘symptoms’ these species present with. Given that these models are in much earlier stages of development, they have yet to play a role in drug discovery for PD; however, they may prove invaluable in the future development of disease-modifying strategies that have so far yielded little success in clinical efforts. The review would not be complete therefore without a brief mention of the promise these models hold.
Drosophila model
Of the multicellular model organisms available, the Drosophila model has received most attention to date. A large number of human genes, including those implicated in PD such as parkin, UCH-L1, PINK1, DJ-1 and LRRK2 have highly conserved homologues in Drosophila (Whitworth et al., 2006). Drosophila models with mutations in a number of these genes have already been developed – but how robust are they? Although the degree and exact pattern of dopaminergic neuron loss may vary between models and between groups for a given model, in general they exhibit a good level of reproducibility. Moreover, many of the models exhibit motor deficits, manifest as a premature loss of climbing ability when the flies are permitted to ‘escape’ from a vial housing them, although often only 50% of flies will exhibit such losses indicating inter-fly variation.
Feany and Bender (2000) first reported production of the α-synuclein transgenic Drosophila model of PD. In their study, expression of either wild-type A53T or A30P mutant forms of α-synuclein resulted in a premature loss of climbing ability, from 20 days through to 40 days of age, that paralleled the time course of degeneration of dopaminergic neurons and the formation of α-synuclein-positive inclusions. Some later studies confirmed an ∼50% loss of dopaminergic neurons in 20 day-old flies (Auluck et al., 2002), although others failed to pick out any losses, possibly due to the reduced sensitivity of using whole-mount immunohistochemistry techniques (Pesah et al., 2005). Subsequent studies have further confirmed the appearance of climbing defects in ∼50% of flies (Pendleton et al., 2002; Haywood and Staveley, 2004) and, of key interest from a drug testing perspective, these climbing defects were restored following treatment with L-DOPA, dopamine agonists (pergolide, bromocriptine) or muscarinic antagonists (atropine) (Pendleton et al., 2002).
Most studies agree that loss-of function Parkin mutant or Parkin-null Drosophila also exhibit a relatively selective loss of dopaminergic neurons at both 1 and 20 days of age, accompanied by an ∼60% reduction in brain dopamine levels and the characteristic climbing deficit (Greene et al., 2003; Cha et al., 2005; Whitworth et al., 2005; Wang et al., 2007). Again, the behavioural phenotype could be rescued by L-DOPA administration (Cha et al., 2005). LRKK2 mutant Drosophila, with either a loss-of-function R1441G mutation in the ROC domain (Lee et al., 2007), or a gain-of-function G2019S mutation in the kinase domain, also show decreased climbing ability and reduced TH expression (Lee et al., 2007) or reduced numbers of dopaminergic neurons (Liu et al., 2008; Ng et al., 2009). L-DOPA can partially restore the behavioural phenotype in these flies (Liu et al., 2008). PINK1 inactivation in Drosophila also leads to reduced numbers of dopaminergic neurons and impaired climbing ability (Park et al., 2006; Wang et al., 2006a), whilst DJ-1 mutant Drosophila do not produce a consistent PD-like phenotype (Park et al., 2005).
It appears therefore that, contrary to the situation in mice, a wide range of PD-associated gene mutations do produce a PD-like phenotype in Drosophila with both behavioural deficits and dopaminergic neuron loss. Just what role these models can play in drug discovery programmes awaits clarification, but they are certainly more amenable to high throughput screening than the rodent models. In addition, the ability to undertake genome-wide genetic screens for mutations in as yet unrecognized genes that may result in the disease phenotype means that Drosophila provides a powerful tool for the investigation of both existing and new genetic links with PD. It is worth noting that rotenone has also been used to induce a PD-like phenotype in Drosophila, which is manifest as inhibition of climbing in 80% flies alongside a 30–50% loss of TH-positive neurons (Coulom and Birman, 2004). So Drosophila models may provide a useful way of looking at the combined effects of genetic and environmental toxins in the future.
C. elegans model
A well-characterized genome and multiple offspring (∼350) for rapid production of models and high-throughput screening make C. elegans another ideal platform for genetic studies. The adult wild-type worm comprises 1000 cells, about a third of which (320) are neurons, exactly eight of which are dopaminergic playing a role in mechanosensation, basal motor activity, habituation, egg laying and defaecation. Despite its simplicity, at the molecular level C. elegans harbours all the fundamental elements for dopamine transmission, including orthologues of all the enzymes involved in DA biosynthesis and metabolism (e.g. Nass et al., 2001). The dopamine neurons of C. elegans are sensitive to the neurotoxins and pesticides introduced earlier including 6-OHDA, which induces blebbing of dopaminergic neurites and rounding of cell bodies before apoptotic cell death occurs within a few hours (Nass et al., 2002), MPP+ (Braungart et al., 2004) and rotenone or paraquat (Ved et al., 2005), though the latter leads to death of the whole worm, not just dopaminergic neurons! The most common functional readout of dopaminergic dysfunction in C. elegans is the loss of basal slowing response, a characteristic dopamine-dependent reduction of bending frequency when near food (e.g. bacteria), to facilitate feeding (Sawin et al., 2000), but other behaviours such as coiling and freezing have also been noted (Braungart et al., 2004). However, the behaviours induced by toxin exposure are not clearly correlated with the loss of the dopaminergic neurons. For example, on the one hand, 6-OHDA produces marked degeneration without motor deficits, whereas on the other hand, MPP+ produces motor deficits even at doses of MPP+ below the threshold for inducing cell death (Harrington et al., 2010). Dopamine agonists were, in this case, able to rescue the behavioural phenotype (Braungart et al., 2004). As with Drosophila, many of the PD-associated genes (parkin LRRK2, DJ-1 and PINK1, but not α-synuclein) are also conserved in C. elegans, opening this worm up to exploitation as a genetic model of PD. Fewer models have been developed so far, but those that have show promise. Although orthologus of α-synuclein are not found, models produced by over-expressing wild-type or mutant human α-synuclein (A53T or A30P) display degeneration of dopaminergic neurons alongside loss of the basal slowing response (Kuwahara et al., 2006). PD-associated mutations in parkin and DJ-1 have been shown to further increase susceptibility of C. elegans to rotenone (Ved et al., 2005), whilst over-expression of wild-type LRRK2 increased survival in response to paraquat and rotenone (Saha et al., 2009), indicating LRRK2 mutations may enhance vulnerability in PD. C. elegans may therefore offer another means of assessing the effects of a combined genetic and environmental hit associated with PD. However, the power of C. elegans genetic models most likely lies in dissecting out the molecular pathways involved in PD and, in doing so, we may identify novel targets for rational drug discovery aimed at slowing or even reversing disease progression.
Zebrafish model
The 3–4 cm long vertebrate zebrafish that has been used for many years to study development and gene function is the last of the contenders as a potential model of PD amenable to high throughput in vivo drug screening. In zebrafish, dopaminergic neurons in the posterior tuberculum of the ventral diencephalon (∼14 in total; analogous to the human SNpc) ascend towards the striatum and are thus more anatomically comparable with the nigro-striatal tract in mammals. These neurons are sensitive to some of the classical PD model toxins, namely 6-OHDA and MPTP showing reduced brain levels of dopamine, noradrenaline and histamine within 2 days of systemic injection (Anichtchik et al., 2004). Although this altered neurochemistry is mirrored by reduced swimming, indicative of bradykinesia, the chemical alterations are not maintained by day 8. Parallel studies exposing zebrafish to MPTP via the tank water found a similar reduction in swimming post-treatment, which lasted over 7 days and was accompanied by an ∼20% reduction in TH-positive neurons in diencephalon, but sparing of the locus coeruleus (Bretaud et al., 2004; Wen et al., 2008). In contrast, exposure via the tank water to either rotenone or paraquat was ineffective in this model (Bretaud et al., 2004). Recent attention has focused on producing genetic models of PD in the zebrafish. To date, inactivation or knockdown of a variety of PD-related genes in the embryo using morpholino oligonucleotide approaches has produced zebrafish with a wide variety of phenotypes. For example, DJ-1 knockdown produces no dopaminergic neuron loss by itself but enhances susceptibility to oxidative stress delivered in the form of hydrogen peroxide (Bretaud et al., 2007). Parkin-deficient zebrafish have been shown to exhibit specific 20% loss of ascending dopaminergic neurons in the posterior tuberculum and impaired complex I activity, although disappointingly no alteration in swimming behaviour (Flinn et al., 2009), whilst in almost mirror image, PINK1 knockdown did not result in loss of DA neurons but altered their projection patterns and resulted in reduced swimming (Xi et al., 2010). Perhaps most promising to date, LRKK2 knockdown has been shown to reduce TH and DAT expression in the diencephalon, reduce TH-positive cells some 25–30% (though other cell types were also affected) and half the distance the zebrafish swim (Xi et al., 2010). Although less amenable to high throughput screening the LRRK2 mutant in particular looks very promising as a future vertebrate model in which to examine some aspects of the genetics of PD.
Animal models of PD and neuroprotection
Animal models of PD based on toxin-induced destruction of the nigro-striatal pathway have proved highly effective in detecting novel dopaminergic approaches to treatment and in the avoidance or reversal of motor fluctuations and motor complications that occur during therapy and as a result of disease progression. In contrast, there has been no translation from animal models of PD into a clinically proven neuroprotective or disease-modifying strategy. Many potentially neuroprotective compounds from a wide range of pharmacological classes have been identified in rodent and primate models, and it is worrysome that so far none has proved effective in man. The way to make the current state of the art clear is through a few examples.
The discovery of the toxicity of MPTP to the nigro-striatal pathway through its conversion to MPP+ by MAO-B led to the observation that this was prevented by the use of non-selective MAO inhibitors and selective MAO-B inhibitors. As a consequence, MAO-B inhibitors, such as selegiline, were thought to be potentially neuroprotective in PD. Indeed, initial clinical trials indicated a disease-modifying effect, although in the longer term, this turned out to be due to symptomatic improvements mediated by potentiation of the actions of dopamine. In retrospect, interfering with the conversion of a toxin to its active metabolite would seem a very limited approach to the general problem of treating the progression of PD. More recently, rasagiline, another MAO-B inhibitor has been shown to prevent MPTP toxicity in mice and monkeys, and again there are suggestions that it might be disease modifying, although the current clinical evidence is limited. Subsequently, the MPTP-treated mouse has been used as a test bed for a myriad of potentially neuroprotective molecules, and many positive effects have been reported. Importantly, these reports must now show that the test substance does not interfere with the conversion of MPTP to MPP+ or its uptake into dopaminergic neurons or any other kinetic component of its toxicity as a prerequisite for claiming a neuroprotective effect. Even so, the very large number of positives that have appeared in the literature without clinical translation is a matter of concern, and there are methodological issues that need to be addressed. In some studies, only striatal dopamine levels are reported, and there is no assessment of either the effects of MPTP or the test molecule on numbers of dopaminergic neurons in the SNpc. It should be re-emphasized here that the use of MPTP in susceptible mouse strains is not as reproducible from laboratory to laboratory as it was initially considered, with very different treatment regimens being used and no certainty that these will cause a loss of dopaminergic neurons as opposed to a transient fall in forebrain dopamine content. In many studies, only pretreatment with the study drug is employed, and effects subsequent to MPTP treatment are not assessed. This does not reflect events in PD where over 60% of nigral dopaminergic neurons are lost prior to the onset of motor symptoms.
The same problems of lack of translation apply to a whole variety of other potential neuroprotective agents. Dopamine agonists, such as ropinirole and pramipexole, have been reported to be neuroprotective in a range of animal models of PD, including the MPTP-treated mouse and 6-OHDA-lesioned rat, yet there is no clinical evidence to support such effects. Drugs that modify cell death cascades leading to apoptosis seemed to be highly effective in preventing the toxicity of a range of toxins in animal models of PD but were inactive in the clinical studies that were subsequently undertaken. And so the list goes on, with failure of translation for glutamate antagonists, antioxidants, neurotrophic factors and anti-inflammatory agents amongst others.
So where does the problem lie? It may be that killing dopaminergic neurons through toxin use provides excellent animal models for testing the effects of symptomatic treatments but does not reflect the pathogenic events occurring in man. Indeed, the cause or causes of PD at the molecular level remain unclear, so the target for neuroprotective therapy is unknown. The animal models of PD currently employed reflect current thinking on the pathogenesis of PD, but this thinking may be incorrect. Alternatively, it could be that the animal models are giving the correct answer, but it is the subsequent clinical trials that are at fault. The number of dose levels used in man is limited and may not result in the concentrations reached in brain in animals. More importantly, it is very likely that there are multiple pathogenic events leading to PD, and these vary between individuals. While clinical trials continue to be based on large patient groups of mixed pathology, it may be impossible to detect disease modification that affects small numbers of those receiving active treatment against the background of lack of effect in most. Urgent attention needs to be given to sub-typing patients with PD based on genetics or biomarkers.
It is difficult to see how proceeding with more of the same is going to lead to neuroprotective strategies for PD. We need new approaches to generate animal models of PD that are progressive, that reflect the disease process more closely and that reflect the widespread pathology associated with the disease in man and perhaps, the genetic models of familial PD or genes that increase the risk of developing PD will eventually provide these. However, there is continued activity in progress based on established animal models, and it will be interesting to see if the neuroprotective/neurorestorative effects of isradipine, exendin or cogane, which have all shown efficacy in the MPTP mouse or 6-OHDA-lesioned rat models (Harkavyi and Whitton, 2010; Li et al., 2009; Meredith et al., 2008; Visanji et al., 2008), are reproduced in the on-going clinical trials.
Conclusions
There is little doubt that the availability of experimental animal models of PD has dramatically altered dopaminergic drug treatment of the motor signs of the illness and in the prevention and reversal of drug related side effects that emerge with disease progression. However, so far, we have made little progress in moving in to other pharmacological areas for the treatment of PD, and we have not developed models that reflect the progressive nature of the illness and its complexity in terms of the extent of pathology and biochemical change. Only when this occurs are we likely to make progress in developing the next generation of drugs for PD that aim to stop or slow the disease progression.
Acknowledgments
The authors wish to acknowledge funding from Cure Parkinson's Trust, Medical Research Council, National Parkinson's Foundation and Parkinson's UK.
Glossary
Abbreviations
6-OHDA
6-hydroxydopamine
AAV
adeno-associated virus
AIMs
abnormal involuntary movements
AMPT
α-methyl-_p_-tyrosine
DARPP-32
dopamine and cAMP-regulated phosphoprotein 32
DAT
dopamine transporter
GPi
globus pallidus internus
LID
L-DOPA-induced dyskinesia
LRRK2
leucine-rich repeat kinase 2
MPP+
1-methyl-4-phenylpyridinium
MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
NET
noradrenaline transporter
PD
Parkinson's disease
PINK1
PTEN-induced putative kinase 1
ROS
reactive oxygen species
SNpc
substantia nigra pars compacta
UCHL-1
ubiquitin carboxy terminal hydrolase-1
VMAT2
vesicular monoamine uptake transporter 2
VTA
ventral tegmental area
Conflict of interest
The authors report no conflict of interest in the content of this manuscript.
References
- Alam M, Schmidt WJ. Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav Brain Res. 2002;136:317–324. doi: 10.1016/s0166-4328(02)00180-8. [DOI] [PubMed] [Google Scholar]
- Alam M, Schmidt WJ. L-DOPA reverses the hypokinetic behaviour and rigidity in rotenone-treated rats. Behav Brain Res. 2004;153:439–446. doi: 10.1016/j.bbr.2003.12.021. [DOI] [PubMed] [Google Scholar]
- Alam ZI, Daniel SE, Lees AJ, Marsden DC, Jenner P, Halliwell B. A generalised increase in protein carbonyls in the brain in Parkinson's but not incidental Lewy body disease. J Neurochem. 1997;69:1326–1329. doi: 10.1046/j.1471-4159.1997.69031326.x. [DOI] [PubMed] [Google Scholar]
- Albanese A, Jenner P, Marsden CD, Stephenson JD. Bladder hyperreflexia induced in marmosets by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurosci Lett. 1988;87:46–50. doi: 10.1016/0304-3940(88)90143-7. [DOI] [PubMed] [Google Scholar]
- Alexander GM, Brainard DL, Gordon SW, Hichens M, Grothusen JR, Schwartzman RJ. Dopamine receptor changes in untreated and (+)-PHNO-treated MPTP parkinsonian primates. Brain Res. 1991;547:181–189. doi: 10.1016/0006-8993(91)90960-4. [DOI] [PubMed] [Google Scholar]
- Alvarez-Fischer D, Guerreiro S, Hunot S, Saurini F, Marien M, Sokoloff P, et al. Modelling Parkinson-like neurodegeneration via osmotic minipump delivery of MPTP and probenecid. J Neurochem. 2008;107:701–711. doi: 10.1111/j.1471-4159.2008.05651.x. [DOI] [PubMed] [Google Scholar]
- Amalric M, Moukhles H, Nieoullon A, Daszuta A. Complex deficits on reaction time performance following bilateral intrastriatal 6-OHDA infusion in the rat. Eur J Neurosci. 1995;7:972–980. doi: 10.1111/j.1460-9568.1995.tb01085.x. [DOI] [PubMed] [Google Scholar]
- Andrew R, Watson DG, Best SA, Midgley JM, Wenlong H, Petty RK. The determination of hydroxydopamines and other trace amines in the urine of parkinsonian patients and normal controls. Neurochem Res. 1993;18:1175–1177. doi: 10.1007/BF00978370. [DOI] [PubMed] [Google Scholar]
- Anichtchik OV, Kaslin J, Peitsaro N, Scheinin M, Panula P. Neurochemical and behavioural changes in zebrafish Danio rerio after systemic administration of 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Neurochem. 2004;88:443–453. doi: 10.1111/j.1471-4159.2004.02190.x. [DOI] [PubMed] [Google Scholar]
- Archer T, Palomo T, McArthur R, Fredriksson A. Effects of acute administration of DA agonists on locomotor activity: MPTP versus neonatal intracerebroventricular 6-OHDA treatment. Neurotox Res. 2003;5:95–110. doi: 10.1007/BF03033375. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Chen H, Weisskopf MG, O'Reilly E, McCullough ML, Calle EE, et al. Pesticide exposure and risk for Parkinson's disease. Ann Neurol. 2006;60:197–203. doi: 10.1002/ana.20904. [DOI] [PubMed] [Google Scholar]
- Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of α-synuclein toxicity in a Drosophila model for Parkinson's disease. Science. 2002;295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- Austin PJ, Betts MJ, Broadstock M, O'Neill MJ, Mitchell SN, Duty S. Symptomatic and neuroprotective effects following activation of nigral group III metabotropic glutamate receptors in rodent models of Parkinson's disease. Br J Pharmacol. 2010;160:1741–1753. doi: 10.1111/j.1476-5381.2010.00820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, et al. Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- Bara-Jimenez W, Dimitrova T, Sherzai A, Favit A, Mouradian MM, Chase TN. Effect of monoamine reuptake inhibitor NS 2330 in advanced Parkinson's disease. Mov Disord. 2004;19:1183–1186. doi: 10.1002/mds.20124. [DOI] [PubMed] [Google Scholar]
- Barcia C, Sanchez BA, Fernandez-Villalba E, Bautista V, Poza YP, Fernandez-Barreiro A, et al. Evidence of active microglia in substantia nigra pars compacta of parkinsonian monkeys 1 year after MPTP exposure. Glia. 2004;46:402–409. doi: 10.1002/glia.20015. [DOI] [PubMed] [Google Scholar]
- Barlow BK, Thiruchelvam MJ, Bennice L, Cory-Slechta DA, Ballatori N, Richfield EK. Increased synaptosomal dopamine content and brain concentration of paraquat produced by selective dithiocarbamates. J Neurochem. 2003;85:1075–1086. doi: 10.1046/j.1471-4159.2003.01773.x. [DOI] [PubMed] [Google Scholar]
- Barneoud P, Parmentier S, Mazadier M, Miquet JM, Boireau A, Dubedat P, et al. Effects of complete and partial lesions of the dopaminergic mesotelencephalic system on skilled forelimb use in the rat. Neuroscience. 1995;67:837–848. doi: 10.1016/0306-4522(95)00112-v. [DOI] [PubMed] [Google Scholar]
- Barneoud P, Descombris E, Aubin N, Abrous DN. Evaluation of simple and complex sensorimotor behaviours in rats with a partial lesion of the dopaminergic nigrostriatal system. Eur J Neurosci. 2000;12:322–336. doi: 10.1046/j.1460-9568.2000.00896.x. [DOI] [PubMed] [Google Scholar]
- Barraud Q, Lambrecq V, Forni C, McGuire S, Hill M, Bioulac B, et al. Sleep disorders in Parkinson's disease: the contribution of the MPTP non-human primate model. Exp Neurol. 2009;219:574–582. doi: 10.1016/j.expneurol.2009.07.019. [DOI] [PubMed] [Google Scholar]
- Bedard PJ, Di Paolo T, Falardeau P, Boucher R. Chronic treatment with L-DOPA, but not bromocriptine induces dyskinesia in MPTP-parkinsonian monkeys. Correlation with [3H]spiperone binding. Brain Res. 1986;379:294–299. doi: 10.1016/0006-8993(86)90783-3. [DOI] [PubMed] [Google Scholar]
- Bedford L, Hay D, Devoy A, Paine S, Powe DG, Seth R, et al. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J Neurosci. 2008;28:8189–8198. doi: 10.1523/JNEUROSCI.2218-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benazzouz A, Breit S, Koudsie A, Pollak P, Krack P, Benabid AL. Intraoperative microrecordings of the subthalamic nucleus in Parkinson's disease. Mov Disord. 2002;17(Suppl 3):S145–S149. doi: 10.1002/mds.10156. [DOI] [PubMed] [Google Scholar]
- Berg D, Godau J, Trenkwalder C, Eggert K, Csoti I, Storch A, et al. AFQ056 treatment of levodopa-induced dyskinesia: results of 2 randomized controlled trials. Mov Disord. 2011 doi: 10.1002/mds.23616. [Epub ahead of print] doi: 10.1002/mds.23616. [DOI] [PubMed] [Google Scholar]
- Bergman O, Hakansson A, Westberg L, Nordenstrom K, Carmine Belin A, Sydow O, et al. PITX3 polymorphism is associated with early onset Parkinson's disease. Neurobiol Aging. 2010;31:114–117. doi: 10.1016/j.neurobiolaging.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Bevan P. Repeated apomorphine treatment causes behavioural supersensitivity and dopamine D2 receptor hyposensitivity. Neurosci Lett. 1983;35:185–189. doi: 10.1016/0304-3940(83)90548-7. [DOI] [PubMed] [Google Scholar]
- Bezard E, Brotchie JM, Gross CE. Pathophysiology of levodopa-induced dyskinesia: potential for new therapies. Nat Rev Neurosci. 2001a;2:577–588. doi: 10.1038/35086062. [DOI] [PubMed] [Google Scholar]
- Bezard E, Dovero S, Prunier C, Ravenscroft P, Chalon S, Guilloteau D, et al. Relationship between the appearance of symptoms and the level of nigrostriatal degeneration in a progressive 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaque model of Parkinson's disease. J Neurosci. 2001b;21:6853–6861. doi: 10.1523/JNEUROSCI.21-17-06853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezard E, Ferry S, Mach U, Stark H, Leriche L, Boraud T, et al. Attenuation of levodopa-induced dyskinesia by normalizing dopamine D3 receptor function. Nat Med. 2003;9:762–767. doi: 10.1038/nm875. [DOI] [PubMed] [Google Scholar]
- Bezard E, Gerlach I, Moratalla R, Gross CE, Jork R. 5-HT1A receptor agonist-mediated protection from MPTP toxicity in mouse and macaque models of Parkinson's disease. Neurobiol Dis. 2006;23:77–86. doi: 10.1016/j.nbd.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Bibbiani F, Oh JD, Chase TN. Serotonin 5-HT1A agonist improves motor complications in rodent and primate parkinsonian models. Neurology. 2001;57:1829–1834. doi: 10.1212/wnl.57.10.1829. [DOI] [PubMed] [Google Scholar]
- Bibbiani F, Costantini LC, Patel R, Chase TN. Continuous dopaminergic stimulation reduces risk of motor complications in parkinsonian primates. Exp Neurol. 2005;192:73–78. doi: 10.1016/j.expneurol.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Biggs CS, Fowler LJ, Whitton PS, Starr MS. Extracellular levels of glutamate and aspartate in the entopeduncular nucleus of the rat determined by microdialysis: regulation by striatal dopamine D2 receptors via the indirect striatal output pathway? Brain Res. 1997;753:163–175. doi: 10.1016/s0006-8993(97)00033-4. [DOI] [PubMed] [Google Scholar]
- Blanchard V, Chritin M, Vyas S, Savasta M, Feuerstein C, Agid Y, et al. Long-term induction of tyrosine hydroxylase expression: compensatory response to partial degeneration of the dopaminergic nigrostriatal system in the rat brain. J Neurochem. 1995;64:1669–1679. doi: 10.1046/j.1471-4159.1995.64041669.x. [DOI] [PubMed] [Google Scholar]
- Blanchet PJ, Konitsiotis S, Chase TN. Amantadine reduces levodopa-induced dyskinesias in parkinsonian monkeys. Mov Disord. 1998a;13:798–802. doi: 10.1002/mds.870130507. [DOI] [PubMed] [Google Scholar]
- Blanchet PJ, Konitsiotis S, Hyland K, Arnold LA, Pettigrew KD, Chase TN. Chronic exposure to MPTP as a primate model of progressive Parkinsonism: a pilot study with a free radical scavenger. Exp Neurol. 1998b;153:214–222. doi: 10.1006/exnr.1998.6906. [DOI] [PubMed] [Google Scholar]
- Blandini F, Levandis G, Bazzini E, Nappi G, Armentero MT. Time-course of nigrostriatal damage, basal ganglia metabolic changes and behavioural alterations following intrastriatal injection of 6-hydroxydopamine in the rat: new clues from an old model. Eur J Neurosci. 2007;25:397–405. doi: 10.1111/j.1460-9568.2006.05285.x. [DOI] [PubMed] [Google Scholar]
- Bonuccelli U, Del DP, Rascol O. Role of dopamine receptor agonists in the treatment of early Parkinson's disease. Parkinsonism Relat Disord. 2009;15(Suppl 4):S44–S53. doi: 10.1016/S1353-8020(09)70835-1. [DOI] [PubMed] [Google Scholar]
- Braak E, Sandmann-Keil D, Rub U, Gai WP, de Vos RA, Steur EN, et al. α-synuclein immunopositive Parkinson's disease-related inclusion bodies in lower brain stem nuclei. Acta Neuropathol. 2001;101:195–201. doi: 10.1007/s004010000247. [DOI] [PubMed] [Google Scholar]
- Braak H, Muller CN, Rub U, Ackermann H, Bratzke H, de Vos RA, et al. Pathology associated with sporadic Parkinson's disease – where does it end? J Neural Transm Suppl. 2006;70:89–97. doi: 10.1007/978-3-211-45295-0_15. [DOI] [PubMed] [Google Scholar]
- Braungart E, Gerlach M, Riederer P, Baumeister R, Hoener MC. Caenorhabditis elegans MPP+ model of Parkinson's disease for high-throughput drug screenings. Neurodegener Dis. 2004;1:175–183. doi: 10.1159/000080983. [DOI] [PubMed] [Google Scholar]
- Breit S, Lessmann L, Unterbrink D, Popa RC, Gasser T, Schulz JB. Lesion of the pedunculopontine nucleus reverses hyperactivity of the subthalamic nucleus and substantia nigra pars reticulata in a 6-hydroxydopamine rat model. Eur J Neurosci. 2006;24:2275–2282. doi: 10.1111/j.1460-9568.2006.05106.x. [DOI] [PubMed] [Google Scholar]
- Bretaud S, Lee S, Guo S. Sensitivity of zebrafish to environmental toxins implicated in Parkinson's disease. Neurotoxicol Teratol. 2004;26:857–864. doi: 10.1016/j.ntt.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Bretaud S, Allen C, Ingham PW, Bandmann O. p53-Dependent neuronal cell death in a DJ-1-deficient zebrafish model of Parkinson's disease. J Neurochem. 2007;100:1626–1635. doi: 10.1111/j.1471-4159.2006.04291.x. [DOI] [PubMed] [Google Scholar]
- Broekkamp CL, De Graaf JS, van Delft AM. Behavioural pharmacology of trans-5-chloro-2-methyl-2,3,3a,12b-tetrahydro- 1H-dibenz[2,3:6,7]oxepino-[4,5-c]pyrrolidine maleate, a compound interacting with dopaminergic and serotonergic receptors. Arzneimittelforschung. 1990;40:544–549. [PubMed] [Google Scholar]
- Brotchie JM. Nondopaminergic mechanisms in levodopa-induced dyskinesia. Mov Disord. 2005;20:919–931. doi: 10.1002/mds.20612. [DOI] [PubMed] [Google Scholar]
- Bukhatwa S, Zeng BY, Rose S, Jenner P. The effects of dose and route of administration of PSI on behavioural and biochemical indices of neuronal degeneration in the rat brain. Brain Res. 2010;1354:236–242. doi: 10.1016/j.brainres.2010.07.060. [DOI] [PubMed] [Google Scholar]
- Burns RS, Chiueh CC, Markey SP, Ebert MH, Jacobowitz DM, Kopin IJ. A primate model of Parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc Nat Acad Sci USA. 1983;80:4546–4550. doi: 10.1073/pnas.80.14.4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet JL, Zhu SM. The intrastriatal 6-hydroxydopamine model of hemiparkinsonism: quantitative receptor autoradiographic evidence of correlation between circling behavior and presynaptic as well as postsynaptic nigrostriatal markers in the rat. Brain Res. 1992;595:316–326. doi: 10.1016/0006-8993(92)91066-n. [DOI] [PubMed] [Google Scholar]
- Calon F, Lavertu N, Lemieux AM, Morissette M, Goulet M, Grondin R, et al. Effect of MPTP-induced denervation on basal ganglia GABA(B) receptors: correlation with dopamine concentrations and dopamine transporter. Synapse. 2001;40:225–234. doi: 10.1002/syn.1045. [DOI] [PubMed] [Google Scholar]
- Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT. A highly reproducible rotenone model of Parkinson's disease. Neurobiol Dis. 2009;34:279–290. doi: 10.1016/j.nbd.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A, Lindqvist M, Magnusson T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature. 1957;180:1200. doi: 10.1038/1801200a0. [DOI] [PubMed] [Google Scholar]
- Carman LS, Gage FH, Shults CW. Partial lesion of the substantia nigra: relation between extent of lesion and rotational behavior. Brain Res. 1991;553:275–283. doi: 10.1016/0006-8993(91)90835-j. [DOI] [PubMed] [Google Scholar]
- Castano A, Herrera AJ, Cano J, Machado A. The degenerative effect of a single intranigral injection of LPS on the dopaminergic system is prevented by dexamethasone, and not mimicked by rh-TNF-alpha, IL-1beta and IFN-gamma. J Neurochem. 2002;81:150–157. doi: 10.1046/j.1471-4159.2002.00799.x. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Lee CS, Bjorklund A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur J Neurosci. 1998;10:2694–2706. [PubMed] [Google Scholar]
- Cha GH, Kim S, Park J, Lee E, Kim M, Lee SB, et al. Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc Natl Acad Sci USA. 2005;102:10345–10350. doi: 10.1073/pnas.0500346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesselet MF. In vivo alpha-synuclein overexpression in rodents: a useful model of Parkinson's disease? Exp Neurol. 2008;209:22–27. doi: 10.1016/j.expneurol.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba K, Trevor A, Castagnoli N., Jr Metabolism of the neurotoxic tertiary amine, MPTP, by brain monoamine oxidase. Biochem Biophys Res Commun. 1984;120:574–578. doi: 10.1016/0006-291x(84)91293-2. [DOI] [PubMed] [Google Scholar]
- Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O. Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur J Neurosci. 2002;15:991–998. doi: 10.1046/j.1460-9568.2002.01938.x. [DOI] [PubMed] [Google Scholar]
- Cicchetti F, Lapointe N, Roberge-Tremblay A, Saint-Pierre M, Jimenez L, Ficke BW, et al. Systemic exposure to paraquat and maneb models early Parkinson's disease in young adult rats. Neurobiol Dis. 2005;20:360–371. doi: 10.1016/j.nbd.2005.03.018. [DOI] [PubMed] [Google Scholar]
- Cicchetti F, Drouin-Ouellet J, Gross RE. Environmental toxins and Parkinson's disease: what have we learned from pesticide-induced animal models? Trends Pharmacol Sci. 2009;30:475–483. doi: 10.1016/j.tips.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Clarke CE, Sambrook MA, Mitchell IJ, Crossman AR. Levodopa-induced dyskinesia and response fluctuations in primates rendered parkinsonian with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) J Neurol Sci. 1987;78:273–280. doi: 10.1016/0022-510x(87)90041-4. [DOI] [PubMed] [Google Scholar]
- Close SP, Elliott PJ, Hayes AG, Marriott AS. Effects of classical and novel agents in a MPTP-induced reversible model of Parkinson's disease. Psychopharmacology. 1990;102:295–300. doi: 10.1007/BF02244093. [DOI] [PubMed] [Google Scholar]
- Colebrooke RE, Humby T, Lynch PJ, McGowan DP, Xia J, Emson PC. Age-related decline in striatal dopamine content and motor performance occurs in the absence of nigral cell loss in a genetic mouse model of Parkinson's disease. Eur J Neurosci. 2006;24:2622–2630. doi: 10.1111/j.1460-9568.2006.05143.x. [DOI] [PubMed] [Google Scholar]
- Colotla VA, Flores E, Oscos A, Meneses A, Tapia R. Effects of MPTP on locomotor activity in mice. Neurotoxicol Teratol. 1990;12:405–407. doi: 10.1016/0892-0362(90)90061-g. [DOI] [PubMed] [Google Scholar]
- Colpaert FC. Pharmacological characteristics of tremor, rigidity and hypokinesia induced by reserpine in rat. Neuropharmacology. 1987;26:1431–1440. doi: 10.1016/0028-3908(87)90110-9. [DOI] [PubMed] [Google Scholar]
- Cookson MR, Bandmann O. Parkinson's disease: insights from pathways. Hum Mol Genet. 2010;19:R21–R27. doi: 10.1093/hmg/ddq167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR, van der Brug M. Cell systems and the toxic mechanism(s) of alpha-synuclein. Exp Neurol. 2008;209:5–11. doi: 10.1016/j.expneurol.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa G, Abin-Carriquiry JA, Dajas F. Nicotine prevents striatal dopamine loss produced by 6-hydroxydopamine lesion in the substantia nigra. Brain Res. 2001a;888:336–342. doi: 10.1016/s0006-8993(00)03087-0. [DOI] [PubMed] [Google Scholar]
- Costa S, Iravani MM, Pearce RK, Jenner P. Glial cell line-derived neurotrophic factor concentration dependently improves disability and motor activity in MPTP-treated common marmosets. Eur J Pharmacol. 2001b;412:45–50. doi: 10.1016/s0014-2999(00)00933-x. [DOI] [PubMed] [Google Scholar]
- Costello S, Cockburn M, Bronstein J, Zhang X, Ritz B. Parkinson's disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am J Epidemiol. 2009;169:919–926. doi: 10.1093/aje/kwp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulom H, Birman S. Chronic exposure to rotenone models sporadic Parkinson's disease in Drosophila melanogaster. J Neurosci. 2004;24:10993–10998. doi: 10.1523/JNEUROSCI.2993-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossman AR. Primate models of dyskinesia: the experimental approach to the study of basal ganglia-related involuntary movement disorders. Neuroscience. 1987;21:1–40. doi: 10.1016/0306-4522(87)90322-8. [DOI] [PubMed] [Google Scholar]
- Crossman AR. Neural mechanisms in disorders of movement. Comp Biochem Physiol A Comp Physiol. 1989;93:141–149. doi: 10.1016/0300-9629(89)90201-6. [DOI] [PubMed] [Google Scholar]
- Crossman AR. A hypothesis on the pathophysiological mechanisms that underlie levodopa- or dopamine agonist-induced dyskinesia in Parkinson's disease: implications for future strategies in treatment. Mov Disord. 1990;5:100–108. doi: 10.1002/mds.870050203. [DOI] [PubMed] [Google Scholar]
- Crossman AR. Functional anatomy of movement disorders. J Anat. 2000;196:519–525. doi: 10.1046/j.1469-7580.2000.19640519.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtius HC, Wolfensberger M, Steinmann B, Redweik U, Siegfried J. Mass fragmentography of dopamine and 6-hydroxydopamine. Application to the determination of dopamine in human brain biopsies from the caudate nucleus. J Chromatogr. 1974;99:529–540. doi: 10.1016/s0021-9673(00)90882-3. [DOI] [PubMed] [Google Scholar]
- Davis GC, Williams AC, Markey SP, Ebert MH, Caine ED, Reichert CM, et al. Chronic Parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res. 1979;1:249–254. doi: 10.1016/0165-1781(79)90006-4. [DOI] [PubMed] [Google Scholar]
- Dawson L, Chadha A, Megalou M, Duty S. The group II metabotropic glutamate receptor agonist, DCG-IV, alleviates akinesia following intranigral or intraventricular administration in the reserpine-treated rat. Br J Pharmacol. 2000;129:541–546. doi: 10.1038/sj.bjp.0703105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson's disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decamp E, Schneider JS. Interaction between nicotinic and dopaminergic therapies on cognition in a chronic Parkinson model. Brain Res. 2009;1262:109–114. doi: 10.1016/j.brainres.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decamp E, Wade T, Schneider JS. Differential regulation of striatal dopamine D(1) and D(2) receptors in acute and chronic parkinsonian monkeys. Brain Res. 1999;847:134–138. doi: 10.1016/s0006-8993(99)02015-6. [DOI] [PubMed] [Google Scholar]
- Degkwitz R, Frowein R, Kulenkampff C, Mohs U. On the effects of L-dopa in man and their modification by reserpine, chlorpromazine, iproniazid and vitamin B6. Klin Wochenschr. 1960;38:120–123. doi: 10.1007/BF02189076. [DOI] [PubMed] [Google Scholar]
- Deshaies P, Bedard P, Falardeau P, Di Paolo T. Behavioral and biochemical evidence of apomorphine-induced supersensitivity of the striatal dopamine receptors. Neuropharmacology. 1984;23:1219–1222. doi: 10.1016/0028-3908(84)90243-0. [DOI] [PubMed] [Google Scholar]
- Drolet RE, Cannon JR, Montero L, Greenamyre JT. Chronic rotenone exposure reproduces Parkinson's disease gastrointestinal neuropathology. Neurobiol Dis. 2009;36:96–102. doi: 10.1016/j.nbd.2009.06.017. [DOI] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Chen Q, Gur TL, Hurtig H Stern MB, et al. Widespread nitration of pathological inclusions in neurodegenerative synucleinopathies. Am J Pathol. 2000;157:1439–1445. doi: 10.1016/S0002-9440(10)64781-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durif F, Vidailhet M, Bonnet AM, Blin J, Agid Y. Levodopa-induced dyskinesias are improved by fluoxetine. Neurology. 1995;45:1855–1858. doi: 10.1212/wnl.45.10.1855. [DOI] [PubMed] [Google Scholar]
- Duty S, Brotchie JM. Enhancement of the behavioral response to apomorphine administration following repeated treatment in the 6-hydroxydopamine-lesioned rat is temporally correlated with a rise in striatal preproenkephalin-B, but not preproenkephalin-A, gene expression. Exp Neurol. 1997;144:423–432. doi: 10.1006/exnr.1997.6431. [DOI] [PubMed] [Google Scholar]
- Eden RJ, Costall B, Domeney AM, Gerrard PA, Harvey CA, Kelly ME, et al. Preclinical pharmacology of ropinirole (SK&F 101468-A) a novel dopamine D2 agonist. Pharmacol Biochem Behav. 1991;38:147–154. doi: 10.1016/0091-3057(91)90603-y. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Galter D. The MitoPark Mouse – an animal model of Parkinson's disease with impaired respiratory chain function in dopamine neurons. Parkinsonism Relat Disord. 2009;15(Suppl 3):S185–S188. doi: 10.1016/S1353-8020(09)70811-9. [DOI] [PubMed] [Google Scholar]
- Elangbam CS. Drug-induced valvulopathy: an update. Toxicol Pathol. 2010;38:837–848. doi: 10.1177/0192623310378027. [DOI] [PubMed] [Google Scholar]
- Elliott PJ, Close SP, Walsh DM, Hayes AG, Marriott AS. Neuroleptic-induced catalepsy as a model of Parkinson's disease. I. Effect of dopaminergic agents. J Neural Transm Park Dis Dement Sect. 1990;2:79–89. doi: 10.1007/BF02260896. [DOI] [PubMed] [Google Scholar]
- Elsworth JD, Taylor JR, Sladek JR, Jr, Collier TJ, Redmond DE, Jr, Roth RH. Striatal dopaminergic correlates of stable Parkinsonism and degree of recovery in old-world primates one year after MPTP treatment. Neuroscience. 2000;95:399–408. doi: 10.1016/s0306-4522(99)00437-6. [DOI] [PubMed] [Google Scholar]
- Erzin-Waters C, Muller P, Seeman P. Catalepsy induced by morphine or haloperidol: effects of apomorphine and anticholinergic drugs. Can J Physiol Pharmacol. 1976;54:516–519. doi: 10.1139/y76-071. [DOI] [PubMed] [Google Scholar]
- Fahn S, The Parkingson Study Group Does levodopa slow or hasten the rate of progression of Parkinson's disease? J Neurol. 2005;252(Suppl 4):IV37–IV42. doi: 10.1007/s00415-005-4008-5. [DOI] [PubMed] [Google Scholar]
- Falardeau P, Bedard PJ, Di Paolo T. Relation between brain dopamine loss and D2 dopamine receptor density in MPTP monkeys. Neurosci Lett. 1988;86:225–229. doi: 10.1016/0304-3940(88)90575-7. [DOI] [PubMed] [Google Scholar]
- Feany MB, Bender WW. A Drosophila model of Parkinson's disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- Ferger B, Teismann P, Earl CD, Kuschunsky K, Oertel WH. The protective effects of PBN against MPTP toxicity are independent of hydroxyl radical trapping. Pharmacol Biochem Behav. 2000;65:425–431. doi: 10.1016/s0091-3057(99)00229-4. [DOI] [PubMed] [Google Scholar]
- Fernandez A, de Ceballos ML, Jenner P, Marsden CD. Striatal neuropeptide levels in Parkinson's disease patients. Neurosci Lett. 1992;145:171–174. doi: 10.1016/0304-3940(92)90014-x. [DOI] [PubMed] [Google Scholar]
- Fernandez HH, Chen JJ. Monoamine oxidase-B inhibition in the treatment of Parkinson's disease. Pharmacotherapy. 2007;27:174S–185S. doi: 10.1592/phco.27.12part2.174S. [DOI] [PubMed] [Google Scholar]
- Ferraro TN, Golden GT, DeMattei M, Hare TA, Fariello RG. Effect of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) on levels of glutathione in the extrapyramidal system of the mouse. Neuropharmacology. 1986;25:1071–1074. doi: 10.1016/0028-3908(86)90205-4. [DOI] [PubMed] [Google Scholar]
- Finberg JP, Youdim MB. Pharmacological properties of the anti-Parkinson drug rasagiline; modification of endogenous brain amines, reserpine reversal, serotonergic and dopaminergic behaviours. Neuropharmacology. 2002;43:1110–1118. doi: 10.1016/s0028-3908(02)00216-2. [DOI] [PubMed] [Google Scholar]
- Fine A, Reynolds GP, Nakajima N, Jenner P, Marsden CD. Acute administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine affects the adrenal glands as well as the brain in the marmoset. Neurosci Lett. 1985;58:123–126. doi: 10.1016/0304-3940(85)90340-4. [DOI] [PubMed] [Google Scholar]
- Flinn L, Mortiboys H, Volkmann K, Koster RW, Ingham PW, Bandmann O. Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio) Brain. 2009;132:1613–1623. doi: 10.1093/brain/awp108. [DOI] [PubMed] [Google Scholar]
- Fornai F, Schluter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, et al. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and α-synuclein. Proc Natl Acad Sci USA. 2005;102:3413–3418. doi: 10.1073/pnas.0409713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forno LS, Langston JW, DeLanney LE, Irwin I, Ricaurte GA. Locus ceruleus lesions and eosinophilic inclusions in MPTP-treated monkeys. Ann Neurol. 1986;20:449–455. doi: 10.1002/ana.410200403. [DOI] [PubMed] [Google Scholar]
- Fox SH, Lang AE, Brotchie JM. Translation of nondopaminergic treatments for levodopa-induced dyskinesia from MPTP-lesioned nonhuman primates to phase IIa clinical studies: keys to success and roads to failure. Mov Disord. 2006;21:1578–1594. doi: 10.1002/mds.20936. [DOI] [PubMed] [Google Scholar]
- Fox SH, Brotchie JM, Lang AE. Non-dopaminergic treatments in development for Parkinson's disease. Lancet Neurol. 2008;7:927–938. doi: 10.1016/S1474-4422(08)70214-X. [DOI] [PubMed] [Google Scholar]
- Fox SH, Visanji NP, Reyes G, Huot P, Gomez-Ramirez J, Johnston T, et al. Neuropsychiatric behaviors in the MPTP marmoset model of Parkinson's disease. Can J Neurol Sci. 2010;31:86–95. doi: 10.1017/s0317167100009707. [DOI] [PubMed] [Google Scholar]
- Fredriksson A, Archer T. Synergistic interactions between COMT-/MAO-inhibitors and L-Dopa in MPTP-treated mice. J Neural Transm Gen Sect. 1995;102:19–34. doi: 10.1007/BF01276562. [DOI] [PubMed] [Google Scholar]
- Fredriksson A, Plaznik A, Sundstrom E, Jonsson G, Archer T. MPTP-induced hypoactivity in mice: reversal by L-dopa. Pharmacol Toxicol. 1990;67:295–301. doi: 10.1111/j.1600-0773.1990.tb00833.x. [DOI] [PubMed] [Google Scholar]
- Fredriksson A, Danysz W, Quack G, Archer T. Co-administration of memantine and amantadine with sub/suprathreshold doses of L-Dopa restores motor behaviour of MPTP-treated mice. J Neural Transm. 2001;108:167–187. doi: 10.1007/s007020170086. [DOI] [PubMed] [Google Scholar]
- Fuchs J, Mueller JC, Lichtner P, Schulte C, Munz M, Berg D, et al. The transcription factor PITX3 is associated with sporadic Parkinson's disease. Neurobiol Aging. 2009;30:731–738. doi: 10.1016/j.neurobiolaging.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Fukuzaki K, Kamenosono T, Nagata R. Effects of ropinirole on various Parkinsonian models in mice, rats, and cynomolgus monkeys. Pharmacol Biochem Behav. 2000a;65:503–508. doi: 10.1016/s0091-3057(99)00240-3. [DOI] [PubMed] [Google Scholar]
- Fukuzaki K, Kamenosono T, Kitazumi K, Nagata R. Effects of ropinirole on motor behavior in MPTP-treated common marmosets. Pharmacol Biochem Behav. 2000b;67:121–129. doi: 10.1016/s0091-3057(00)00305-1. [DOI] [PubMed] [Google Scholar]
- Fuller RW, Hahn RA, Snoddy HD, Wikel JH. Depletion of cardiac norepinephrine in rats and mice by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Biochem Pharmacol. 1984;33:2957–2960. doi: 10.1016/0006-2952(84)90593-8. [DOI] [PubMed] [Google Scholar]
- Galter D, Pernold K, Yoshitake T, Lindqvist E, Hoffer B, Kehr J, et al. MitoPark mice mirror the slow progression of key symptoms and L-DOPA response in Parkinson's disease. Genes Brain Behav. 2010;9:173–181. doi: 10.1111/j.1601-183X.2009.00542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvey J, Petersen M, Waters CM, Rose SP, Hunt S, Briggs R, et al. Administration of MPTP to the common marmoset does not alter cortical cholinergic function. Mov Disord. 1986;1:129–134. doi: 10.1002/mds.870010207. [DOI] [PubMed] [Google Scholar]
- Gasmi M, Herzog CD, Brandon EP, Cunningham JJ, Ramirez GA, Ketchum ET, et al. Striatal delivery of neurturin by CERE-120, an AAV2 vector for the treatment of dopaminergic neuron degeneration in Parkinson's disease. Mol Ther. 2007;15:62–68. doi: 10.1038/sj.mt.6300010. [DOI] [PubMed] [Google Scholar]
- Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiol Dis. 2006;21:404–412. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- German DC, Dubach M, Askari S, Speciale SG, Bowden DM. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonian syndrome in Macaca fascicularis: which midbrain dopaminergic neurons are lost? Neuroscience. 1988;24:161–174. doi: 10.1016/0306-4522(88)90320-x. [DOI] [PubMed] [Google Scholar]
- German DC, Manaye K, Smith WK, Woodward DJ, Saper CB. Midbrain dopaminergic cell loss in Parkinson's disease: computer visualization. Ann Neurol. 1989;26:507–514. doi: 10.1002/ana.410260403. [DOI] [PubMed] [Google Scholar]
- German DC, Manaye KF, Sonsalla PK, Brooks BA. Midbrain dopaminergic cell loss in Parkinson's disease and MPTP-induced Parkinsonism: sparing of calbindin-D28k-containing cells. Ann N Y Acad Sci. 1992;648:42–62. doi: 10.1111/j.1749-6632.1992.tb24523.x. [DOI] [PubMed] [Google Scholar]
- Ghosh B, Antonio T, Reith ME, Dutta AK. Discovery of 4-(4-(2-((5-Hydroxy-1,2,3,4-tetrahydro-naphthalene-2-yl)(propyl)amino)ethyl) piperazin-1-yl)quinolin-8-ol and its analogues as highly potent dopamine D2/D3 agonists and as iron chelator: in vivo activity indicates potential application in symptomatic and neuroprotective therapy for Parkinson's disease. J Med Chem. 2010;53:2114–2125. doi: 10.1021/jm901618d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb WR, Lees AJ. Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra, and differential cell death in Parkinson's disease. J Neurol Neurosurg Psychiatry. 1991;54:388–396. doi: 10.1136/jnnp.54.5.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb WR, Lees AJ, Jenner P, Marsden CD. The dopamine neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) produces histological lesions in the hypothalamus of the common marmoset. Neurosci Lett. 1986;65:79–83. doi: 10.1016/0304-3940(86)90123-0. [DOI] [PubMed] [Google Scholar]
- Gibb WR, Terruli M, Lees AJ, Jenner P, Marsden CD. The evolution and distribution of morphological changes in the nervous system of the common marmoset following the acute administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Mov Disord. 1989;4:53–74. doi: 10.1002/mds.870040109. [DOI] [PubMed] [Google Scholar]
- Gibrat C, Saint-Pierre M, Bousquet M, Levesque D, Rouillard C, Cicchetti F. Differences between subacute and chronic MPTP mice models: investigation of dopaminergic neuronal degeneration and α-synuclein inclusions. J Neurochem. 2009;109:1469–1482. doi: 10.1111/j.1471-4159.2009.06072.x. [DOI] [PubMed] [Google Scholar]
- Glinka Y, Gassen M, Youdim MB. Mechanism of 6-hydroxydopamine neurotoxicity. J Neural Transm Suppl. 1997;50:55–66. doi: 10.1007/978-3-7091-6842-4_7. [DOI] [PubMed] [Google Scholar]
- Gnanalingham KK, Smith LA, Hunter AJ, Jenner P, Marsden CD. Alterations in striatal and extrastriatal D-1 and D-2 dopamine receptors in the MPTP-treated common marmoset: an autoradiographic study. Synapse. 1993;14:184–194. doi: 10.1002/syn.890140212. [DOI] [PubMed] [Google Scholar]
- Gnanalingham KK, Erol DD, Hunter AJ, Smith LA, Jenner P, Marsden CD. Differential anti-parkinsonian effects of benzazepine D1 dopamine agonists with varying efficacies in the MPTP-treated common marmoset. Psychopharmacology (Berl) 1995a;117:275–286. doi: 10.1007/BF02246102. [DOI] [PubMed] [Google Scholar]
- Gnanalingham KK, Hunter AJ, Jenner P, Marsden CD. The differential behavioural effects of benzazepine D1 dopamine agonists with varying efficacies, co-administered with quinpirole in primate and rodent models of Parkinson's disease. Psychopharmacology (Berl) 1995b;117:287–297. doi: 10.1007/BF02246103. [DOI] [PubMed] [Google Scholar]
- Goetz CG, Damier P, Hicking C, Laska E, Müller T, Olanow CW, et al. Sarizotan as a treatment for dyskinesias in Parkinson's disease: a double-blind placebo-controlled trial. Mov Disord. 2007;22:179–186. doi: 10.1002/mds.21226. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, et al. Nigrositratal dopaminergic deficits and hypokinesia causeb by inactivation of the familial Parkinsonism-linked gene, DJ-1. Neuron. 2005;45:489–496. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- Goldstein JM, Barnett A, Malick JB. The evaluation of anti-parkinson drugs on reserpine-induced rigidity in rats. Eur J Pharmacol. 1975;33:183–188. doi: 10.1016/0014-2999(75)90154-5. [DOI] [PubMed] [Google Scholar]
- Goto S, Hirano A, Matsumoto S. Met-enkephalin immunoreactivity in the basal ganglia in Parkinson's disease and striatonigral degeneration. Neurology. 1990;40:1051–1056. doi: 10.1212/wnl.40.7.1051. [DOI] [PubMed] [Google Scholar]
- Grealish S, Xie L, Kelly M, Dowd E. Unilateral axonal or terminal injection of 6-hydroxydopamine causes rapid-onset nigrostriatal degeneration and contralateral motor impairments in the rat. Brain Res Bull. 2008;77:312–319. doi: 10.1016/j.brainresbull.2008.08.018. [DOI] [PubMed] [Google Scholar]
- Greco B, Lopez S, van der Putten H, Flor PJ, Amalric M. Metabotropic glutamate 7 receptor subtype modulates motor symptoms in rodent models of Parkinson's disease. J Pharmacol Exp Ther. 2010;332:1064–1071. doi: 10.1124/jpet.109.162115. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Cannon JR, Drolet R, Mastroberardino PG. Lessons from the rotenone model of Parkinson's disease. Trends Pharmacol Sci. 2010;31:141–142. doi: 10.1016/j.tips.2009.12.006. author reply 142–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoire L, Samadi P, Graham J, Bedard PJ, Bartoszyk GD, Di Paolo T. Low doses of sarizotan reduce dyskinesias and maintain antiparkinsonian efficacy of L-Dopa in parkinsonian monkeys. Parkinsonism Relat Disord. 2009;15:445–452. doi: 10.1016/j.parkreldis.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Gudehithlu KP, Duchemin AM, Tejwani GA, Neff NH, Hadjiconstantinou M. Preproenkephalin mRNA and methionine-enkephalin increase in mouse striatum after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine treatment. J Neurochem. 1991;56:1043–1048. doi: 10.1111/j.1471-4159.1991.tb02027.x. [DOI] [PubMed] [Google Scholar]
- Gupta SP, Patel S, Yadav S, Singh AK, Singh S, Singh MP. Involvement of nitric oxide in maneb- and paraquat-induced Parkinson's disease phenotype in mouse: is there any link with lipid peroxidation? Neurochem Res. 2010;35:1206–1213. doi: 10.1007/s11064-010-0176-5. [DOI] [PubMed] [Google Scholar]
- Hadjiconstantinou M, Cavalla D, Anthoupoulou E, Laird HE, 2nd, Neff NH. N-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine increases acetylcholine and decreases dopamine in mouse striatum: both responses are blocked by anticholinergic drugs. J Neurochem. 1985;45:1957–1959. doi: 10.1111/j.1471-4159.1985.tb10558.x. [DOI] [PubMed] [Google Scholar]
- Halliday G, Herrero MT, Murphy K, McCann H, Ros-Bernal F, Barcia C, et al. No Lewy pathology in monkeys with over 10 years of severe MPTP Parkinsonism. Mov Disord. 2009;24:1519–1523. doi: 10.1002/mds.22481. [DOI] [PubMed] [Google Scholar]
- Hallman H, Lange J, Olson L, Stromberg I, Jonsson G. Neurochemical and histochemical characterization of neurotoxic effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine on brain catecholamine neurons in the mouse. J Neurochem. 1985;44:117–127. doi: 10.1111/j.1471-4159.1985.tb07120.x. [DOI] [PubMed] [Google Scholar]
- Hansard MJ, Smith LA, Jackson MJ, Cheetham SC, Jenner P. Dopamine reuptake inhibition and failure to evoke dyskinesia in MPTP-treated primates. Eur J Pharmacol. 2002a;451:157–160. doi: 10.1016/s0014-2999(02)02268-9. [DOI] [PubMed] [Google Scholar]
- Hansard MJ, Smith LA, Jackson MJ, Cheetham SC, Jenner P. Dopamine, but not norepinephrine or serotonin, reuptake inhibition reverses motor deficits in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated primates. J Pharmacol Exp Ther. 2002b;303:952–958. doi: 10.1124/jpet.102.039743. [DOI] [PubMed] [Google Scholar]
- Hansard MJ, Smith LA, Jackson MJ, Cheetham SC, Jenner P. The monoamine reuptake inhibitor BTS 74 398 fails to evoke established dyskinesia but does not synergise with levodopa in MPTP-treated primates. Mov Disord. 2004;19:15–21. doi: 10.1002/mds.10596. [DOI] [PubMed] [Google Scholar]
- Hardy J. Genetic analysis of pathways to Parkinson disease. Neuron. 2010;68:201–206. doi: 10.1016/j.neuron.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkavyi A, Whitton PS. Glucagon-like peptide receptor 1 stimulation as a means of neuroprotection. Br J Pharmacol. 2010;159:495–501. doi: 10.1111/j.1476-5381.2009.00486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington AJ, Hamamichi S, Caldwell GA, Caldwell KA. C. elegans as a model organism to investigate molecular pathways involved with Parkinson's disease. Dev Dyn. 2010;239:1282–1295. doi: 10.1002/dvdy.22231. [DOI] [PubMed] [Google Scholar]
- Hassani OK, Mouroux M, Feger J. Increased subthalamic neuronal activity after nigral dopaminergic lesion independent of disinhibition via the globus pallidus. Neuroscience. 1996;72:105–115. doi: 10.1016/0306-4522(95)00535-8. [DOI] [PubMed] [Google Scholar]
- Hassouna I, Wickert H, Zimmermann M, Gillardon F. Increase in bax expression in substantia nigra following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) treatment of mice. Neurosci Lett. 1996;204:85–88. doi: 10.1016/0304-3940(96)12323-5. [DOI] [PubMed] [Google Scholar]
- Hauser RA, Salin L, Juhel N, Konyago VL. Randomized trial of the triple monoamine reuptake inhibitor NS 2330 (tesofensine) in early Parkinson's disease. Mov Disord. 2007;22:359–365. doi: 10.1002/mds.21258. [DOI] [PubMed] [Google Scholar]
- Hauser RA, Shulman LM, Trugman JM, Roberts JW, Mori A, Ballerini R, et al. Study of istradefylline in patients with Parkinson's disease on levodopa with motor fluctuations. Mov Disord. 2008;23:2177–2185. doi: 10.1002/mds.22095. [DOI] [PubMed] [Google Scholar]
- Haywood AF, Staveley BE. Parkin counteracts symptoms in a Drosophila model of Parkinson's disease. BMC Neurosci. 2004;5:14. doi: 10.1186/1471-2202-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert G, Arsaut J, Dantzer R, Demotes-Mainard J. Time-course of the expression of inflammatory cytokines and matrix metalloproteinases in the striatum and mesencephalon of mice injected with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, a dopaminergic neurotoxin. Neurosci Lett. 2003;349:191–195. doi: 10.1016/s0304-3940(03)00832-2. [DOI] [PubMed] [Google Scholar]
- Heeringa MJ, Abercrombie ED. Biochemistry of somatodendritic dopamine release in substantia nigra: an in vivo comparison with striatal dopamine release. J Neurochem. 1995;65:192–200. doi: 10.1046/j.1471-4159.1995.65010192.x. [DOI] [PubMed] [Google Scholar]
- Hefti F, Melamed E, Sahakian BJ, Wurtman RJ. Circling behavior in rats with partial, unilateral nigro-striatal lesions: effect of amphetamine, apomorphine, and DOPA. Pharmacol Biochem Behav. 1980;12:185–188. doi: 10.1016/0091-3057(80)90353-6. [DOI] [PubMed] [Google Scholar]
- Heikkila RE, Cabbat FS, Manzino L, Duvoisin RC. Potentiation by deprenil of 1-dopa induced circling in nigral-lesioned rats. Pharmacol Biochem Behav. 1981;15:75–79. doi: 10.1016/0091-3057(81)90342-7. [DOI] [PubMed] [Google Scholar]
- Heinrich JN, Brennan J, Lai MH, Sullivan K, Hornby G, Popiolek M, et al. Aplindore (DAB-452), a high affinity selective dopamine D2 receptor partial agonist. Eur J Pharmacol. 2006;552:36–45. doi: 10.1016/j.ejphar.2006.08.063. [DOI] [PubMed] [Google Scholar]
- Henry B, Crossman AR, Brotchie JM. Characterization of enhanced behavioral responses to L-DOPA following repeated administration in the 6-hydroxydopamine-lesioned rat model of Parkinson's disease. Exp Neurol. 1998;151:334–342. doi: 10.1006/exnr.1998.6819. [DOI] [PubMed] [Google Scholar]
- Henry B, Duty S, Fox SH, Crossman AR, Brotchie JM. Increased striatal pre-proenkephalin B expression is associated with dyskinesia in Parkinson's disease. Exp Neurol. 2003;183:458–468. doi: 10.1016/s0014-4886(03)00064-5. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Little MD, Bankiewicz K, Yang SC, Markey SP, Johannessen JN. Selective retention of MPP+ within the monoaminergic systems of the primate brain following MPTP administration: an in vivo autoradiographic study. Neuroscience. 1991;40:133–158. doi: 10.1016/0306-4522(91)90180-v. [DOI] [PubMed] [Google Scholar]
- Herrera AJ, Castano A, Venero JL, Cano J, Machado A. The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol Dis. 2000;7:429–447. doi: 10.1006/nbdi.2000.0289. [DOI] [PubMed] [Google Scholar]
- Herrero MT, Augood SJ, Hirsch EC, Javoy-Agid F, Luquin MR, Agid Y, et al. Effects of L-DOPA on preproenkephalin and preprotachykinin gene expression in the MPTP-treated monkey striatum. Neuroscience. 1995;68:1189–1198. doi: 10.1016/0306-4522(95)00120-8. [DOI] [PubMed] [Google Scholar]
- Herrero MT, Augood SJ, Asensi H, Hirsch EC, Agid Y, Obeso JA, et al. Effects of L-DOPA-therapy on dopamine D2 receptor mRNA expression in the striatum of MPTP-intoxicated parkinsonian monkeys. Brain Res Mol Brain Res. 1996a;42:149–155. doi: 10.1016/s0169-328x(96)00157-x. [DOI] [PubMed] [Google Scholar]
- Herrero MT, Levy R, Ruberg M, Javoy-Agid F, Luquin MR, Agid Y, et al. Glutamic acid decarboxylase mRNA expression in medial and lateral pallidal neurons in the MPTP-treated monkey and patients with Parkinson's disease. Adv Neurol. 1996b;69:209–216. [PubMed] [Google Scholar]
- Herrero MT, Levy R, Ruberg M, Luquin MR, Villares J, Guillen J, et al. Consequence of nigrostriatal denervation and L-dopa therapy on the expression of glutamic acid decarboxylase messenger RNA in the pallidum. Neurology. 1996c;47:219–224. doi: 10.1212/wnl.47.1.219. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S, Damier P, Brugg B, Faucheux BA, Michel PP, et al. Glial cell participation in the degeneration of dopaminergic neurons in Parkinson's disease. Adv Neurol. 1999;80:9–18. [PubMed] [Google Scholar]
- Hirsch EC, Breidert T, Rousselet E, Hunot S, Hartmann A, Michel PP. The role of glial reaction and inflammation in Parkinson's disease. Ann N Y Acad Sci. 2003;991:214–228. doi: 10.1111/j.1749-6632.2003.tb07478.x. [DOI] [PubMed] [Google Scholar]
- Hoglinger GU, Feger J, Prigent A, Michel PP, Parain K, Champy P, et al. Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. J Neurochem. 2003;84:491–502. doi: 10.1046/j.1471-4159.2003.01533.x. [DOI] [PubMed] [Google Scholar]
- Hubble JP. Long-term studies of dopamine agonists. Neurology. 2002;58:S42–S50. doi: 10.1212/wnl.58.suppl_1.s42. [DOI] [PubMed] [Google Scholar]
- Hudson JL, van Horne CG, Stromberg I, Brock S, Clayton J, Masserano J, et al. Correlation of apomorphine- and amphetamine-induced turning with nigrostriatal dopamine content in unilateral 6-hydroxydopamine lesioned rats. Brain Res. 1993;626:167–174. doi: 10.1016/0006-8993(93)90576-9. [DOI] [PubMed] [Google Scholar]
- Hung HC, Lee EH. The mesolimbic dopaminergic pathway is more resistant than the nigrostriatal dopaminergic pathway to MPTP and MPP+ toxicity: role of BDNF gene expression. Brain Res Mol Brain Res. 1996;41:14–26. doi: 10.1016/0169-328x(96)00062-9. [DOI] [PubMed] [Google Scholar]
- Hurley MJ, Jolkkonen J, Stubbs CM, Jenner P, Marsden CD. Dopamine D3 receptors in the basal ganglia of the common marmoset and following MPTP and L-DOPA treatment. Brain Res. 1996;709:259–264. doi: 10.1016/0006-8993(95)01309-1. [DOI] [PubMed] [Google Scholar]
- Hutchison WD, Lozano AM, Davis KD, Saint-Cyr JA, Lang AE, Dostrovsky JO. Differential neuronal activity in segments of globus pallidus in Parkinson's disease patients. Neuroreport. 1994;5:1533–1537. doi: 10.1097/00001756-199407000-00031. [DOI] [PubMed] [Google Scholar]
- Hutchison WD, Allan RJ, Opitz H, Levy R, Dostrovsky JO, Lang AE, et al. Neurophysiological identification of the subthalamic nucleus in surgery for Parkinson's disease. Ann Neurol. 1998;44:622–628. doi: 10.1002/ana.410440407. [DOI] [PubMed] [Google Scholar]
- Hwang D-Y, Ardayfio P, Kang UJ, Semina EV, Kim K-S. Selective loss of dopaminergic neurons in the substantia nigra of Pitx3-deficient aphakia mice. Mol Brain Res. 2003;114:123–131. doi: 10.1016/s0169-328x(03)00162-1. [DOI] [PubMed] [Google Scholar]
- Iczkiewicz J, Broom L, Cooper JD, Wong AM, Rose S, Jenner P. The RGD-containing peptide fragment of osteopontin protects tyrosine hydroxylase positive cells against toxic insult in primary ventral mesencephalic cultures and in the rat substantia nigra. J Neurochem. 2010;114:1792–1804. doi: 10.1111/j.1471-4159.2010.06896.x. [DOI] [PubMed] [Google Scholar]
- Inden M, Kitamura Y, Tamaki A, Yanagida T, Shibaike T, Yamamoto A, et al. Neuroprotective effect of the antiparkinsonian drug pramipexole against nigrostriatal dopaminergic degeneration in rotenone-treated mice. Neurochem Int. 2009;55:760–767. doi: 10.1016/j.neuint.2009.07.009. [DOI] [PubMed] [Google Scholar]
- Iravani MM, Costa S, Jackson MJ, Tel BC, Cannizzaro C, Pearce RK, et al. GDNF reverses priming for dyskinesia in MPTP-treated, L-DOPA-primed common marmosets. Eur J Neurosci. 2001;13:597–608. doi: 10.1046/j.1460-9568.2001.01408.x. [DOI] [PubMed] [Google Scholar]
- Iravani MM, Leung CC, Sadeghian M, Haddon CO, Rose S, Jenner P. The acute and the long-term effects of nigral lipopolysaccharide administration on dopaminergic dysfunction and glial cell activation. Eur J Neurosci. 2005;22:317–330. doi: 10.1111/j.1460-9568.2005.04220.x. [DOI] [PubMed] [Google Scholar]
- Jackson MJ, Smith LA, Al-Barghouthy G, Rose S, Jenner P. Decreased expression of l-dopa-induced dyskinesia by switching to ropinirole in MPTP-treated common marmosets. Exp Neurol. 2007;204:162–170. doi: 10.1016/j.expneurol.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Jackson MJ, Andree TH, Hansard M, Hoffman DC, Hurtt MR, Kehne JH, et al. The dopamine D(2) receptor partial agonist aplindore improves motor deficits in MPTP-treated common marmosets alone and combined with L-dopa. J Neural Transm. 2010;117:55–67. doi: 10.1007/s00702-009-0323-9. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Przedborski S. Protocol for the MPTP mouse model of Parkinson's disease. Nat Protoc. 2007;2:141–151. doi: 10.1038/nprot.2006.342. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Jakowec M, Burke RE, Przedborski S. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurodegeneration. 1995;4:257–269. doi: 10.1016/1055-8330(95)90015-2. [DOI] [PubMed] [Google Scholar]
- Jankovic J. Motor fluctuations and dyskinesias in Parkinson's disease: clinical manifestations. Mov Disord. 2005;20(Suppl 11):S11–S16. doi: 10.1002/mds.20458. [DOI] [PubMed] [Google Scholar]
- Jankovic J, Chen S, Le WD. The role of Nurr1 in the development of dopaminergic neurons and Parkinson's disease. Prog Neurobiol. 2005;77:128–138. doi: 10.1016/j.pneurobio.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Jarraya B, Boulet S, Ralph GS, Jan C, Bonvento G, Azzouz M, et al. Dopamine gene therapy for Parkinson's disease in a nonhuman primate without associated dyskinesia. Sci Transl Med. 2009;1:2ra4. doi: 10.1126/scitranslmed.3000130. [DOI] [PubMed] [Google Scholar]
- Javitch JA, D'Amato RJ, Strittmatter SM, Snyder SH. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc Natl Acad Sci USA. 1985;82:2173–2177. doi: 10.1073/pnas.82.7.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA. Pathology of Parkinson's disease. Changes other than the nigrostriatal pathway. Mol Chem Neuropathol. 1991;14:153–197. doi: 10.1007/BF03159935. [DOI] [PubMed] [Google Scholar]
- Jenner P. Clues to the mechanism underlying dopamine cell death in Parkinson's disease. J Neurol Neurosurg Psychiatry. 1989;52(Suppl):22–28. doi: 10.1136/jnnp.52.suppl.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P. The rationale for the use of dopamine agonists in Parkinson's disease. Neurology. 1995;45:S6–12. doi: 10.1212/wnl.45.3_suppl_3.s6. [DOI] [PubMed] [Google Scholar]
- Jenner P. Pharmacology of dopamine agonists in the treatment of Parkinson's disease. Neurology. 2002;58:S1–S8. doi: 10.1212/wnl.58.suppl_1.s1. [DOI] [PubMed] [Google Scholar]
- Jenner P. Dopamine agonists, receptor selectivity and dyskinesia induction in Parkinson's disease. Curr Opin Neurol. 2003a;16(Suppl 1):S3–S7. doi: 10.1097/00019052-200312001-00002. [DOI] [PubMed] [Google Scholar]
- Jenner P. The contribution of the MPTP-treated primate model to the development of new treatment strategies for Parkinson's disease. Parkinsonism Relat Disord. 2003b;9:131–137. doi: 10.1016/s1353-8020(02)00115-3. [DOI] [PubMed] [Google Scholar]
- Jenner P. Functional models of Parkinson's disease: a valuable tool in the development of novel therapies. Ann Neurol. 2008;64(Suppl 2):S16–S29. doi: 10.1002/ana.21489. [DOI] [PubMed] [Google Scholar]
- Jenner P. From the MPTP-treated primate to the treatment of motor complications in Parkinson's disease. Parkinsonism Relat Disord. 2009;15(Suppl 4):S18–S23. doi: 10.1016/S1353-8020(09)70829-6. [DOI] [PubMed] [Google Scholar]
- Jenner P, Rupniak NM, Rose S, Kelly E, Kilpatrick G, Lees A, et al. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in the common marmoset. Neurosci Lett. 1984;50:85–90. doi: 10.1016/0304-3940(84)90467-1. [DOI] [PubMed] [Google Scholar]
- Jeon BS, Jackson-Lewis V, Burke RE. 6-Hydroxydopamine lesion of the rat substantia nigra: time course and morphology of cell death. Neurodegeneration. 1995;4:131–137. doi: 10.1006/neur.1995.0016. [DOI] [PubMed] [Google Scholar]
- Jiang C, Wan X, He Y, Pan T, Jankovic J Le W. Age-dependent dopaminergic dysfunction in Nurr1 knockout mice. Exp Neurol. 2005;191:154–162. doi: 10.1016/j.expneurol.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Johannessen JN, Chiueh CC, Burns RS, Markey SP. Differences in the metabolism of MPTP in the rodent and primate parallel differences in sensitivity to its neurotoxic effects. Life Sci. 1985;36:219–224. doi: 10.1016/0024-3205(85)90062-1. [DOI] [PubMed] [Google Scholar]
- Johnels B. Locomotor hypokinesia in the reserpine-treated rat: drug effects from the corpus striatum and nucleus accumbens. Pharmacol Biochem Behav. 1982;17:283–289. doi: 10.1016/0091-3057(82)90082-x. [DOI] [PubMed] [Google Scholar]
- Johnson AM, Loew DM, Vigouret JM. Stimulant properties of bromocriptine on central dopamine receptors in comparison to apomorphine, (+)-amphetamine and L-DOPA. Br J Pharmacol. 1976;56:59–68. doi: 10.1111/j.1476-5381.1976.tb06959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LC, Jackson MJ, Rose S, McCreary AC, Jenner P. Pardoprunox reverses motor deficits but induces only mild dyskinesia in MPTP-treated common marmosets. Mov Disord. 2010;25:2059–2066. doi: 10.1002/mds.23249. [DOI] [PubMed] [Google Scholar]
- Johnston T, Duty S. GABA(B) receptor agonists reverse akinesia following intranigral or intracerebroventricular injection in the reserpine-treated rat. Br J Pharmacol. 2003;139:1480–1486. doi: 10.1038/sj.bjp.0705372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CA, Johnston LC, Jackson MJ, Smith LA, van Scharrenburg G, Rose S, et al. An in vivo pharmacological evaluation of pardoprunox (SLV308)–a novel combined dopamine D(2)/D(3) receptor partial agonist and 5-HT(1A) receptor agonist with efficacy in experimental models of Parkinson's disease. Eur Neuropsychopharmacol. 2010;20:582–593. doi: 10.1016/j.euroneuro.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Joyce JN, Marshall JF, Bankiewicz KS, Kopin IJ, Jacobowitz DM. Hemiparkinsonism in a monkey after unilateral internal carotid artery infusion of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is associated with regional ipsilateral changes in striatal dopamine D-2 receptor density. Brain Res. 1986;382:360–364. doi: 10.1016/0006-8993(86)91345-4. [DOI] [PubMed] [Google Scholar]
- Kanda T, Jackson MJ, Smith LA, Pearce RK, Nakamura J, Kase H, et al. Combined use of the adenosine A(2A) antagonist KW-6002 with L-DOPA or with selective D1 or D2 dopamine agonists increases antiparkinsonian activity but not dyskinesia in MPTP-treated monkeys. Exp Neurol. 2000;162:321–327. doi: 10.1006/exnr.2000.7350. [DOI] [PubMed] [Google Scholar]
- Karunakaran S, Saeed U, Mishra M, Valli RK, Joshi SD, Meka DP, et al. Selective activation of p38 mitogen-activated protein kinase in dopaminergic neurons of substantia nigra leads to nuclear translocation of p53 in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice. J Neurosci. 2008;28:12500–12509. doi: 10.1523/JNEUROSCI.4511-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenschlager R, Head J, Schrag A, Ben-Shlomo Y, Evans A, Lees AJ. Fourteen-year final report of the randomized PDRG-UK trial comparing three initial treatments in PD. Neurology. 2008;71:474–480. doi: 10.1212/01.wnl.0000310812.43352.66. [DOI] [PubMed] [Google Scholar]
- Kebabian JW, Britton DR, DeNinno MP, Perner R, Smith L, Jenner P, et al. A-77636: a potent and selective dopamine D1 receptor agonist with antiparkinsonian activity in marmosets. Eur J Pharmacol. 1992;229:203–209. doi: 10.1016/0014-2999(92)90556-j. [DOI] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Bjorklund A. Characterization of behavioral and neurodegenerative changes following partial lesions of the nigrostriatal dopamine system induced by intrastriatal 6-hydroxydopamine in the rat. Exp Neurol. 1998;152:259–277. doi: 10.1006/exnr.1998.6848. [DOI] [PubMed] [Google Scholar]
- Kirik D, Georgievska B, Rosenblad C, Bjorklund A. Delayed infusion of GDNF promotes recovery of motor function in the partial lesion model of Parkinson's disease. Eur J Neurosci. 2001;13:1589–1599. doi: 10.1046/j.0953-816x.2001.01534.x. [DOI] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE Muzycka N, et al. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci. 2002;22:2780–2791. doi: 10.1523/JNEUROSCI.22-07-02780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Annett LE, Burger C, Muyczka N, Mandel RJ, Bjorklund A. Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: a new primate model of Parkinson's disease. Proc Natl Acad Sci USA. 2003;100:2884–2889. doi: 10.1073/pnas.0536383100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile Parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, et al. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci USA. 2007;104:11441–11446. doi: 10.1073/pnas.0702717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Tong Y, Gautier CA, Shen J. Absence of nigral degeneration in aged parkin/DJ-1/PINK1 triple knockout mice. J Neurochem. 2009;111:696–702. doi: 10.1111/j.1471-4159.2009.06350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Araki T, Itoyama Y, Takeshita M, Ohta T, Oshima Y. Effects of L-dopa and bromocriptine on haloperidol-induced motor deficits in mice. Life Sci. 1997;61:2529–2538. doi: 10.1016/s0024-3205(97)01007-2. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Herzog CD, Dass B, Bakay RA, Stansell J, III, Gasmi M, et al. Delivery of neurturin by AAV2 (CERE-120)-mediated gene transfer provides structural and functional neuroprotection and neurorestoration in MPTP-treated monkeys. Ann Neurol. 2006a;60:706–715. doi: 10.1002/ana.21032. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Kanaan NM, Chu Y, Suresh BR, Stansell J, III, Terpstra BT, et al. Failure of proteasome inhibitor administration to provide a model of Parkinson's disease in rats and monkeys. Ann Neurol. 2006b;60:264–268. doi: 10.1002/ana.20935. [DOI] [PubMed] [Google Scholar]
- Kowall NW, Hantraye P, Brouillet E, Beal MF, McKee AC, Ferrante RJ. MPTP induces alpha-synuclein aggregation in the substantia nigra of baboons. Neuroreport. 2000;11:211–213. doi: 10.1097/00001756-200001170-00041. [DOI] [PubMed] [Google Scholar]
- Kulkarni SK, Bishnoi M, Chopra K. In vivo microdialysis studies of striatal level of neurotransmitters after haloperidol and chlorpromazine administration. Indian J Exp Biol. 2009;47:91–97. [PubMed] [Google Scholar]
- Kunikowska G, Jenner P. 6-Hydroxydopamine-lesioning of the nigrostriatal pathway in rats alters basal ganglia mRNA for copper, zinc- and manganese-superoxide dismutase, but not glutathione peroxidase. Brain Res. 2001;922:51–64. doi: 10.1016/s0006-8993(01)03149-3. [DOI] [PubMed] [Google Scholar]
- Kuoppamaki M, Al-Barghouthy G, Jackson M, Smith L, Zeng BY, Quinn N, et al. Beginning-of-dose and rebound worsening in MPTP-treated common marmosets treated with levodopa. Mov Disord. 2002;17:1312–1317. doi: 10.1002/mds.10263. [DOI] [PubMed] [Google Scholar]
- Kuoppamaki M, Al-Barghouthy G, Jackson MJ, Smith LA, Quinn N, Jenner P. L-dopa dose and the duration and severity of dyskinesia in primed MPTP-treated primates. J Neural Transm. 2007;114:1147–1153. doi: 10.1007/s00702-007-0727-3. [DOI] [PubMed] [Google Scholar]
- Kurkowska-Jastrzebska I, Wronska A, Kohutnicka M, Czlonkowski A, Czlonkowska A. The inflammatory reaction following 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine intoxication in mouse. Exp Neurol. 1999;156:50–61. doi: 10.1006/exnr.1998.6993. [DOI] [PubMed] [Google Scholar]
- Kuwahara T, Koyama A, Gengyo-Ando K, Masuda M, Kowa H, Tsunoda M, et al. Familial Parkinson mutant α-synuclein causes dopamine neuron dysfunction in transgenic Caenorhabditis elegans. J Biol Chem. 2006;281:334–340. doi: 10.1074/jbc.M504860200. [DOI] [PubMed] [Google Scholar]
- Lane EL, Cheetham SC, Jenner P. Repeated administration of the monoamine reuptake inhibitor BTS 74 398 induces ipsilateral circling in the 6-hydroxydopamine lesioned rat without sensitizing motor behaviours. Eur J Neurosci. 2005;21:179–186. doi: 10.1111/j.1460-9568.2004.03834.x. [DOI] [PubMed] [Google Scholar]
- Langston JW. MPTP: insights into the etiology of Parkinson's disease. Eur Neurol. 1987;26(Suppl 1):2–10. doi: 10.1159/000116349. [DOI] [PubMed] [Google Scholar]
- Langston JW, Ballard P. Parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): implications for treatment and the pathogenesis of Parkinson's disease. Can J Neurol Sci. 1984;11:160–165. doi: 10.1017/s0317167100046333. [DOI] [PubMed] [Google Scholar]
- Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- Langston JW, Forno LS, Rebert CS, Irwin I. Selective nigral toxicity after systemic administration of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyrine (MPTP) in the squirrel monkey. Brain Res. 1984;292:390–394. doi: 10.1016/0006-8993(84)90777-7. [DOI] [PubMed] [Google Scholar]
- Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Langston JW, Quik M, Petzinger G, Jakowec M, Di Monte DA. Investigating levodopa-induced dyskinesias in the parkinsonian primate. Ann Neurol. 2000;47:S79–S89. [PubMed] [Google Scholar]
- Lapointe N, St-Hilaire M, Martinoli MG, Blanchet J, Gould P, Rouillard C, et al. Rotenone induces non-specific central nervous system and systemic toxicity. FASEB J. 2004;18:717–719. doi: 10.1096/fj.03-0677fje. [DOI] [PubMed] [Google Scholar]
- Lau YS, Meredith GE. From drugs of abuse to Parkinsonism. The MPTP mouse model of Parkinson's disease. Methods Mol Med. 2003;79:103–116. doi: 10.1385/1-59259-358-5:103. [DOI] [PubMed] [Google Scholar]
- Lavoie B, Parent A, Bedard PJ. Effects of dopamine denervation on striatal peptide expression in parkinsonian monkeys. Can J Neurol Sci. 1991;18:373–375. doi: 10.1017/s0317167100032467. [DOI] [PubMed] [Google Scholar]
- Le W, Conneely OM, Zou L, He Y, Saucedo-Cardenas O, Jankovic J, et al. Selective agenesis of mesencephalic dopaminergic neurons in Nurr1-deficient mice. Exp Neurol. 1999a;159:451–458. doi: 10.1006/exnr.1999.7191. [DOI] [PubMed] [Google Scholar]
- Le W, Conneely OM, He Y, Jankovic J, Appel SH. Reduced Nurr1 expression increases the vulnerability of mesencephalic dopamine neurons to MPTP-induced injury. J Neurochem. 1999b;73:2218–2221. [PubMed] [Google Scholar]
- Le W, Pan T, Huang M, Xu P, Xie W, Zhu W, et al. Decreased NURR1 gene expression in patients with Parkinson's disease. J Neurol Sci. 2008;273:29–33. doi: 10.1016/j.jns.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, Sauer H, Bjorklund A. Dopaminergic neuronal degeneration and motor impairments following axon terminal lesion by instrastriatal 6-hydroxydopamine in the rat. Neuroscience. 1996;72:641–653. doi: 10.1016/0306-4522(95)00571-4. [DOI] [PubMed] [Google Scholar]
- Lee CS, Cenci MA, Schulzer M, Bjorklund A. Embryonic ventral mesencephalic grafts improve levodopa-induced dyskinesia in a rat model of Parkinson's disease. Brain. 2000;123(Pt 7):1365–1379. doi: 10.1093/brain/123.7.1365. [DOI] [PubMed] [Google Scholar]
- Lee SB, Kim W, Lee S, Chung J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem Biophys Res Commun. 2007;358:534–539. doi: 10.1016/j.bbrc.2007.04.156. [DOI] [PubMed] [Google Scholar]
- Lees AJ. Dopamine agonists in Parkinson's disease: a look at apomorphine. Fundam Clin Pharmacol. 1993;7:121–128. doi: 10.1111/j.1472-8206.1993.tb00226.x. [DOI] [PubMed] [Google Scholar]
- Lees AJ. Evidence-based efficacy comparison of tolcapone and entacapone as adjunctive therapy in Parkinson's disease. CNS Neurosci Ther. 2008;14:83–93. doi: 10.1111/j.1527-3458.2007.00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, et al. The ubiquitin pathway in Parkinson's disease. Nature. 1998;395:451–452. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- Li SJ, Jiang HK, Stachowiak MS, Hudson PM, Owyang V, Nanry K, et al. Influence of nigrostriatal dopaminergic tone on the biosynthesis of dynorphin and enkephalin in rat striatum. Brain Res Mol Brain Res. 1990;8:219–225. doi: 10.1016/0169-328x(90)90020-e. [DOI] [PubMed] [Google Scholar]
- Li X, Matsumoto K, Murakami Y, Tezuka Y, Wu Y, Kadota S. Neuroprotective effects of Polygonum multiflorum on nigrostriatal dopaminergic degeneration induced by paraquat and maneb in mice. Pharmacol Biochem Behav. 2005;82:345–352. doi: 10.1016/j.pbb.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Li Y, Perry T, Kindy MS, Harvey BK, Tweedie D, Holloway HW, et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and parkinsonism. Proc Natl Acad Sci USA. 2009;106:1285–1290. doi: 10.1073/pnas.0806720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Parasiadou L, Gu XL, Wang L, Shim H, Sun L, et al. Leucine-rich repeat kinase-2 regulates progression of neuropathology induced by Parkinson's disease-related mutant alpha-synuclein. Neuron. 2009;64:807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou HH, Chen RC, Chen TH, Tsai YF, Tsai MC. Attenuation of paraquat-induced dopaminergic toxicity on the substantia nigra by (-)-deprenyl in vivo. Toxicol Appl Pharmacol. 2001;172:37–43. doi: 10.1006/taap.2001.9130. [DOI] [PubMed] [Google Scholar]
- Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, et al. A Drosophila model for LRRK2-linked Parkinsonism. Proc Natl Acad Sci USA. 2008;105:2693–2698. doi: 10.1073/pnas.0708452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bianco C, Deglon N, Pralong W, Aebischer P. Lentiviral nigral delivery of GDNF does not prevent neurodegenration in a genetic rat model of Parkinson's disease. Neurobiol Dis. 2004;17:283–289. doi: 10.1016/j.nbd.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Lorenc-Koci E, Wolfarth S. Efficacy of pramipexole, a new dopamine receptor agonist, to relieve the parkinsonian-like muscle rigidity in rats. Eur J Pharmacol. 1999;385:39–46. doi: 10.1016/s0014-2999(99)00704-9. [DOI] [PubMed] [Google Scholar]
- Loschmann PA, Chong PN, Nomoto M, Tepper PG, Horn AS, Jenner P, et al. Stereoselective reversal of MPTP-induced Parkinsonism in the marmoset after dermal application of N-0437. Eur J Pharmacol. 1989;166:373–380. doi: 10.1016/0014-2999(89)90348-8. [DOI] [PubMed] [Google Scholar]
- Loschmann PA, Smith LA, Lange KW, Jahnig P, Jenner P, Marsden CD. Motor activity following the administration of selective D-1 and D-2 dopaminergic drugs to MPTP-treated common marmosets. Psychopharmacology (Berl) 1992;109:49–56. doi: 10.1007/BF02245479. [DOI] [PubMed] [Google Scholar]
- Luchtman DW, Shao D, Song C. Behavior, neurotransmitters and inflammation in three regimens of the MPTP mouse model of Parkinson's disease. Physiol Behav. 2009;98:130–138. doi: 10.1016/j.physbeh.2009.04.021. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, Cenci MA. Pharmacological validation of behavioural measures of akinesia and dyskinesia in a rat model of Parkinson's disease. Eur J Neurosci. 2002;15:120–132. doi: 10.1046/j.0953-816x.2001.01843.x. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Vaudano E, Cenci MA. Cellular and behavioural effects of the adenosine A2a receptor antagonist KW-6002 in a rat model of l-DOPA-induced dyskinesia. J Neurochem. 2003;84:1398–1410. doi: 10.1046/j.1471-4159.2003.01632.x. [DOI] [PubMed] [Google Scholar]
- Luthman J, Fredriksson A, Lewander T, Jonsson G, Archer T. Effects of d-amphetamine and methylphenidate on hyperactivity produced by neonatal 6-hydroxydopamine treatment. Psychopharmacology (Berl) 1989;99:550–557. doi: 10.1007/BF00589907. [DOI] [PubMed] [Google Scholar]
- MacInnes N, Messenger MJ, Duty S. Activation of group III metabotropic glutamate receptors in selected regions of the basal ganglia alleviates akinesia in the reserpine-treated rat. Br J Pharmacol. 2004;141:15–22. doi: 10.1038/sj.bjp.0705566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maj J, Rogoz Z, Skuza G, Sowinska H, Superata J. Behavioural and neurochemical effects of Ro 40-7592, a new COMT inhibitor with a potential therapeutic activity in Parkinson's disease. J Neural Transm Park Dis Dement Sect. 1990;2:101–112. doi: 10.1007/BF02260898. [DOI] [PubMed] [Google Scholar]
- Maj J, Rogoz Z, Skuza G, Kolodziejczyk K. The behavioural effects of pramipexole, a novel dopamine receptor agonist. Eur J Pharmacol. 1997;324:31–37. doi: 10.1016/s0014-2999(97)00066-6. [DOI] [PubMed] [Google Scholar]
- Maneuf YP, Duty S, Hille CJ, Crossman AR, Brotchie JM. Modulation of GABA transmission by diazoxide and cromakalim in the globus pallidus: implications for the treatment of Parkinson's disease. Exp Neurol. 1996;139:12–16. doi: 10.1006/exnr.1996.0075. [DOI] [PubMed] [Google Scholar]
- Manning-Bog AB, Reaney SH, Chou VP, Johnston LC, McCormack AL, Johnston J, et al. Lack of nigrostriatal pathology in a rat model of proteasome inhibition. Ann Neurol. 2006;60:256–260. doi: 10.1002/ana.20938. [DOI] [PubMed] [Google Scholar]
- Manson AJ, Turner K, Lees AJ. Apomorphine monotherapy in the treatment of refractory motor complications of Parkinson's disease: long-term follow-up study of 64 patients. Mov Disord. 2002;17:1235–1241. doi: 10.1002/mds.10281. [DOI] [PubMed] [Google Scholar]
- Marin C, Aguilar E, Mengod G, Cortes R, Obeso JA. Concomitant short- and long-duration response to levodopa in the 6-OHDA-lesioned rat: a behavioural and molecular study. Eur J Neurosci. 2007;25:259–269. doi: 10.1111/j.1460-9568.2006.05265.x. [DOI] [PubMed] [Google Scholar]
- Marks WJ, Jr, Bartus RT, Siffert J, Davis CS, Lozano A, Boulis N, et al. Gene delivery of AAV2-neurturin for Parkinson's disease: a double-blind, randomised, controlled trial. Lancet Neurol. 2010;9:1164–1172. doi: 10.1016/S1474-4422(10)70254-4. [DOI] [PubMed] [Google Scholar]
- Mazzio EA, Reams RR, Soliman KF. The role of oxidative stress, impaired glycolysis and mitochondrial respiratory redox failure in the cytotoxic effects of 6-hydroxydopamine in vitro. Brain Res. 2004;1004:29–44. doi: 10.1016/j.brainres.2003.12.034. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Glial reactions in Parkinson's disease. Mov Disord. 2008;23:474–483. doi: 10.1002/mds.21751. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Schwab C, Parent A, Doudet D. Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Ann Neurol. 2003;54:599–604. doi: 10.1002/ana.10728. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Jenner P. Altered glial function causes neuronal death and increases neuronal susceptibility to 1-methyl-4-phenylpyridinium- and 6-hydroxydopamine-induced toxicity in astrocytic/ventral mesencephalic co-cultures. J Neurochem. 1999;73:2469–2476. doi: 10.1046/j.1471-4159.1999.0732469.x. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Jenner P. Dysfunction of rat forebrain astrocytes in culture alters cytokine and neurotrophic factor release. Neurosci Lett. 2000a;285:61–65. doi: 10.1016/s0304-3940(00)00982-4. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Jenner P. Extracellular accumulation of nitric oxide, hydrogen peroxide, and glutamate in astrocytic cultures following glutathione depletion, complex I inhibition, and/or lipopolysaccharide-induced activation. Biochem Pharmacol. 2000b;60:979–988. doi: 10.1016/s0006-2952(00)00415-9. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Jenner P. Proteasomal function is impaired in substantia nigra in Parkinson's disease. Neurosci Lett. 2001;297:191–194. doi: 10.1016/s0304-3940(00)01701-8. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Belizaire R, Jenner P, Olanow CW, Isacson O. Selective loss of 20S proteasome alpha-subunits in the substantia nigra pars compacta in Parkinson's disease. Neurosci Lett. 2002a;326:155–158. doi: 10.1016/s0304-3940(02)00296-3. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Bjorklund LM, Belizaire R, Isacson O, Jenner P, Olanow CW. Proteasome inhibition causes nigral degeneration with inclusion bodies in rats. Neuroreport. 2002b;13:1437–1441. doi: 10.1097/00001756-200208070-00018. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Mytilineou C, Jnobaptiste R, Yabut J, Shashidharan P, Jenner P, et al. Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. J Neurochem. 2002c;81:301–306. doi: 10.1046/j.1471-4159.2002.00821.x. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Perl DP, Brownell AL, Olanow CW. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson's disease. Ann Neurol. 2004;56:149–162. doi: 10.1002/ana.20186. [DOI] [PubMed] [Google Scholar]
- Meissner W, Prunier C, Guilloteau D, Chalon S, Gross CE, Bezard E. Time-course of nigrostriatal degeneration in a progressive MPTP-lesioned macaque model of Parkinson's disease. Mol Neurobiol. 2003;28:209–218. doi: 10.1385/MN:28:3:209. [DOI] [PubMed] [Google Scholar]
- Mela F, Marti M, Dekundy A, Danysz W, Morari M, Cenci NA. Antagonism of metabotropic glutamate receptor 5 attenuates L-DOPA induced dyskinesia and its molecular and neurochemical correlates in a rat model of Parkinson's disease. J Neurochem. 2007;101:483–497. doi: 10.1111/j.1471-4159.2007.04456.x. [DOI] [PubMed] [Google Scholar]
- Meredith GE, Totterdell S, Petroske E, Santa Cruz K, Callison RC, Jr, Lau YS. Lysosomal malfunction accompanies α-synuclein aggregation in a progressive mouse model of Parkinson's disease. Brain Res. 2002;956:156–165. doi: 10.1016/s0006-8993(02)03514-x. [DOI] [PubMed] [Google Scholar]
- Meredith GE, Totterdell S, Potashkin JA, Surmeier DJ. Modelling PD pathogenesis in mice: advantages of a chronic MPTP protocol. Parkinsonism Relat Disord. 2008;14(Suppl 2):112–115. doi: 10.1016/j.parkreldis.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith GE, Totterdell S, Beales M, Meshul CK. Impaired glutamate homeostasis and programmed cell death in a chronic MPTP mouse model of Parkinson's disease. Exp Neurol. 2009;219:334–340. doi: 10.1016/j.expneurol.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierau J, Schingnitz G. Biochemical and pharmacological studies on pramipexole, a potent and selective dopamine D2 receptor agonist. Eur J Pharmacol. 1992;215:161–170. doi: 10.1016/0014-2999(92)90024-x. [DOI] [PubMed] [Google Scholar]
- Millan MJ, Di Cara B, Hill M, Jackson M, Joyce JN, Brotchie J, et al. S32504, a novel naphtoxazine agonist at dopamine D3/D2 receptors: II. Actions in rodent, primate, and cellular models of antiparkinsonian activity in comparison to ropinirole. J Pharmacol Exp Ther. 2004;309:921–935. doi: 10.1124/jpet.103.062414. [DOI] [PubMed] [Google Scholar]
- Miller DB, Ali SF, O'Callaghan JP, Laws SC. The impact of gender and estrogen on striatal dopaminergic neurotoxicity. Ann N Y Acad Sci. 1998;844:153–165. [PubMed] [Google Scholar]
- Miller GW. Paraquat: the red herring of Parkinson's disease research. Toxicol Sci. 2007;100:1–2. doi: 10.1093/toxsci/kfm223. [DOI] [PubMed] [Google Scholar]
- Mitchell IJ, Cross AJ, Sambrook MA, Crossman AR. Sites of the neurotoxic action of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in the macaque monkey include the ventral tegmental area and the locus coeruleus. Neurosci Lett. 1985;61:195–200. doi: 10.1016/0304-3940(85)90424-0. [DOI] [PubMed] [Google Scholar]
- Miyagi M, Arai N, Taya F, Itoh F, Komatsu Y, Kojima M, et al. Effect of cabergoline, a long-acting dopamine D2 agonist, on reserpine-treated rodents. Biol Pharm Bull. 1996;19:1499–1502. doi: 10.1248/bpb.19.1499. [DOI] [PubMed] [Google Scholar]
- Miyoshi R, Kito S, Ishida H, Katayama S. Alterations of the central noradrenergic system in MPTP-induced monkey Parkinsonism. Res Commun Chem Pathol Pharmacol. 1988;62:93–102. [PubMed] [Google Scholar]
- Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett. 1994;165:208–210. doi: 10.1016/0304-3940(94)90746-3. [DOI] [PubMed] [Google Scholar]
- Mogi M, Togari A, Tanaka K, Ogawa N, Ichinose H, Nagatsu T. Increase in level of tumor necrosis factor-α in 6-hydroxydopamine-lesioned striatum in rats is suppressed by immunosuppressant FK506. Neurosci Lett. 2000;289:165–168. doi: 10.1016/s0304-3940(00)01275-1. [DOI] [PubMed] [Google Scholar]
- Montastruc JL, Rascol O, Senard JM. Current status of dopamine agonists in Parkinson's disease management. Drugs. 1993;46:384–393. doi: 10.2165/00003495-199346030-00005. [DOI] [PubMed] [Google Scholar]
- Monville C, Torres EM, Dunnett SB. Validation of the l-dopa-induced dyskinesia in the 6-OHDA model and evaluation of the effects of selective dopamine receptor agonists and antagonists. Brain Res Bull. 2005;68:16–23. doi: 10.1016/j.brainresbull.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Morissette M, Goulet M, Calon F, Falardeau P, Blanchet PJ, Bedard PJ, et al. Changes of D1 and D2 dopamine receptor mRNA in the brains of monkeys lesioned with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: correction with chronic administration of L-3,4-dihydroxyphenylalanine. Mol Pharmacol. 1996;50:1073–1079. [PubMed] [Google Scholar]
- Morissette M, Grondin R, Goulet M, Bedard PJ, Di Paolo T. Differential regulation of striatal preproenkephalin and preprotachykinin mRNA levels in MPTP-lesioned monkeys chronically treated with dopamine D1 or D2 receptor agonists. J Neurochem. 1999;72:682–692. doi: 10.1046/j.1471-4159.1999.0720682.x. [DOI] [PubMed] [Google Scholar]
- Moses D, Gross A, Finberg JP. Rasagiline enhances L-DOPA-induced contralateral turning in the unilateral 6-hydroxydopamine-lesioned guinea-pig. Neuropharmacology. 2004;47:72–80. doi: 10.1016/j.neuropharm.2004.02.016. [DOI] [PubMed] [Google Scholar]
- Müller T, Hefter H, Hueber R, Jost WH, Leenders KL, Odin P, et al. Is levodopa toxic? J Neurol. 2004;251(Suppl 6):VI/44–VI/46. doi: 10.1007/s00415-004-1610-x. [DOI] [PubMed] [Google Scholar]
- van den Munckhof P, Gilbert F, Chamberland M, Levesque D, Drouin J. Striatal neuroadaptation and rescue of locomotor defici by L-dopa in aphakia kice, a model of Parkinson's disease. J Neurochem. 2006;96:160–170. doi: 10.1111/j.1471-4159.2005.03522.x. [DOI] [PubMed] [Google Scholar]
- Nash JE, Hill MP, Brotchie JM. Antiparkinsonian actions of blockade of NR2B-containing NMDA receptors in the reserpine-treated rat. Exp Neurol. 1999;155:42–48. doi: 10.1006/exnr.1998.6963. [DOI] [PubMed] [Google Scholar]
- Nass R, Miller DM, Blakely RD. C. elegans: a novel pharmacogenetic model to study Parkinson's disease. Parkinsonism Relat Disord. 2001;7:185–191. doi: 10.1016/s1353-8020(00)00056-0. [DOI] [PubMed] [Google Scholar]
- Nass R, Hall DH, Miller DM, 3rd, Blakely RD. Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2002;99:3264–3269. doi: 10.1073/pnas.042497999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neustadt BR, Liu H, Hao J, Greenlee WJ, Stamford AW, Foster C, et al. Potent and selective adenosine A2A receptor antagonists: 1,2,4-Triazolo[1,5-c]pyrimidines. Bioorg Med Chem Lett. 2009;19:967–971. doi: 10.1016/j.bmcl.2008.11.075. [DOI] [PubMed] [Google Scholar]
- Ng CH, Mok SZ, Koh C, Ouyang X, Fivaz ML, Tan EK, et al. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J Neurosci. 2009;29:11257–11262. doi: 10.1523/JNEUROSCI.2375-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklas WJ, Youngster SK, Kindt MV, Heikkila RE. MPTP, MPP+ and mitochondrial function. Life Sci. 1987;40:721–729. doi: 10.1016/0024-3205(87)90299-2. [DOI] [PubMed] [Google Scholar]
- Nisbet AP, Foster OJ, Kingsbury A, Eve DJ, Daniel SE, Marsden CD, et al. Preproenkephalin and preprotachykinin messenger RNA expression in normal human basal ganglia and Parkinson's disease. Neuroscience. 1995;66:361–376. doi: 10.1016/0306-4522(94)00606-6. [DOI] [PubMed] [Google Scholar]
- Niswender CM, Johnson KA, Weaver CD, Jones CK, Xiang Z, Luo Q, et al. Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol Pharmacol. 2008;74:1345–1358. doi: 10.1124/mol.108.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomoto M, Jenner P, Marsden CD. The D1 agonist SKF 38393 inhibits the antiparkinsonian activity of the D2 agonist LY 171555 in the MPTP-treated marmoset. Neurosci Lett. 1988;93:275–280. doi: 10.1016/0304-3940(88)90095-x. [DOI] [PubMed] [Google Scholar]
- Novikova L, Garris BL, Garris DR, Lau YS. Early signs of neuronal apoptosis in the substantia nigra pars compacta of the progressive neurodegenerative mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine/probenecid model of Parkinson's disease. Neuroscience. 2006;140:67–76. doi: 10.1016/j.neuroscience.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Nunes I, Tovmasian LT, Silva RM, Burke RE, Goff SP. Pitx3 is required for development of substantia nigra dopaminergic neurons. Proc Natl Acad Sci USA. 2003;100:4245–4250. doi: 10.1073/pnas.0230529100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuutila J, Kaakkola S, Mannisto PT. Potentiation of central effects of L-dopa by an inhibitor of catechol-O-methyltransferase. J Neural Transm. 1987;70:233–240. doi: 10.1007/BF01253600. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Olanow CW, Nutt JG. Levodopa motor complications in Parkinson's disease. Trends Neurosci. 2000a;23:S2–S7. doi: 10.1016/s1471-1931(00)00031-8. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Oroz MC, Chana P, Lera G, Rodriguez M, Olanow CW. The evolution and origin of motor complications in Parkinson's disease. Neurology. 2000b;55:S13–S20. [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Oroz MC, Rodriguez M, Lanciego JL, Artieda J, Gonzalo N, et al. Pathophysiology of the basal ganglia in Parkinson's disease. Trends Neurosci. 2000c;23:S8–19. doi: 10.1016/s1471-1931(00)00028-8. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Oroz MC, Rodriguez M, Arbizu J, Gimenez-Amaya JM. The basal ganglia and disorders of movement: pathophysiological mechanisms. News Physiol Sci. 2002;17:51–55. doi: 10.1152/nips.01363.2001. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Oroz MC, Itez-Temino B, Blesa FJ, Guridi J, Marin C, et al. Functional organization of the basal ganglia: therapeutic implications for Parkinson's disease. Mov Disord. 2008;23(Suppl 3):S548–S559. doi: 10.1002/mds.22062. [DOI] [PubMed] [Google Scholar]
- Oertel WH, Wolters E, Sampaio C, Gimenez-Roldan S, Bergamasco B, Dujardin M, et al. Pergolide versus levodopa monotherapy in early Parkinson's disease patients: the PELMOPET study. Mov Disord. 2006;21:343–353. doi: 10.1002/mds.20724. [DOI] [PubMed] [Google Scholar]
- Oestreicher E, Sengstock GJ, Riederer P, Olanow CW, Dunn AJ, Arendash GW. Degeneration of nigrostriatal dopaminergic neurons increases iron within the substantia nigra: a histochemical and neurochemical study. Brain Res. 1994;660:8–18. doi: 10.1016/0006-8993(94)90833-8. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Obeso JA. Preventing levodopa-induced dyskinesias. Ann Neurol. 2000;47:S167–S176. [PubMed] [Google Scholar]
- Olanow CW, Good PF, Shinotoh H, Hewitt KA, Vingerhoets F, Snow BJ, et al. Manganese intoxication in the rhesus monkey: a clinical, imaging, pathologic, and biochemical study. Neurology. 1996;46:492–498. doi: 10.1212/wnl.46.2.492. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Damier P, Goetz CG, Mueller T, Nutt J, Rascol O, et al. Multicenter, open-label, trial of sarizotan in Parkinson disease patients with levodopa-induced dyskinesias (the SPLENDID Study) Clin Neuropharmacol. 2004;27:58–62. doi: 10.1097/00002826-200403000-00003. [DOI] [PubMed] [Google Scholar]
- Olsson M, Nikkhah G, Bentlage C, Bjorklund A. Forelimb akinesia in the rat Parkinson model: differential effects of dopamine agonists and nigral transplants as assessed by a new stepping test. J Neurosci. 1995;15(5 Pt 2):3863–3875. doi: 10.1523/JNEUROSCI.15-05-03863.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillé V, Henry V, Lescaudron L, Brachet P, Damier P. Rat model of Parkinson's disease with bilateral motor abnormalities, reversible with levodopa, and dyskinesias. Mov Disord. 2007;22:533–539. doi: 10.1002/mds.21308. [DOI] [PubMed] [Google Scholar]
- Pan T, Zhu W, Zhao H, Deng H, Xie W, Jankovic J, et al. Nurr1 deficiency predisposes to lactacystin-induced dopaminergic neuron injury in vitro and in vivo. Brain Res. 2008;1222:222–229. doi: 10.1016/j.brainres.2008.05.022. [DOI] [PubMed] [Google Scholar]
- Pan-Montojo F, Anichtchik O, Dening Y, Knels L, Pursche S, Jung R, et al. Progression of Parkinson's disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS ONE. 2010;5:e8762. doi: 10.1371/journal.pone.0008762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa SM, Engber TM, Kask AM, Chase TN. Motor fluctuations in levodopa treated parkinsonian rats: relation to lesion extent and treatment duration. Brain Res. 1994;662:69–74. doi: 10.1016/0006-8993(94)90796-x. [DOI] [PubMed] [Google Scholar]
- Park J, Kim SY, Cha GH, Lee SB, Kim S, Chung J. Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene. 2005;361:133–139. doi: 10.1016/j.gene.2005.06.040. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA. 2000;284:1931–1938. doi: 10.1001/jama.284.15.1931. [DOI] [PubMed] [Google Scholar]
- Pearce RK, Banerji T, Jenner P, Marsden CD. De novo administration of ropinirole and bromocriptine induces less dyskinesia than L-dopa in the MPTP-treated marmoset. Mov Disord. 1998;13:234–241. doi: 10.1002/mds.870130207. [DOI] [PubMed] [Google Scholar]
- Pearce RK, Smith LA, Jackson MJ, Banerji T, Scheel-Kruger J, Jenner P. The monoamine reuptake blocker brasofensine reverses akinesia without dyskinesia in MPTP-treated and levodopa-primed common marmosets. Mov Disord. 2002;17:877–886. doi: 10.1002/mds.10238. [DOI] [PubMed] [Google Scholar]
- Pendleton RG, Parvez F, Sayed M, Hillman R. Effects of pharmacological agents upon a transgenic model of Parkinson's disease in Drosophila melanogaster. J Pharmacol Exp Ther. 2002;300:91–96. doi: 10.1124/jpet.300.1.91. [DOI] [PubMed] [Google Scholar]
- Perez FA, Palmiter RD. Parkin-deficinet mice are not a robust model of Parkinson's disease. Proc Natl Acad Sci USA. 2005;102:2174–2179. doi: 10.1073/pnas.0409598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Otano I, Oset C, Luquin MR, Herrero MT, Obeso JA, Del RJ. MPTP-induced Parkinsonism in primates: pattern of striatal dopamine loss following acute and chronic administration. Neurosci Lett. 1994;175:121–125. doi: 10.1016/0304-3940(94)91094-4. [DOI] [PubMed] [Google Scholar]
- Perumal AS, Gopal VB, Tordzro WK, Cooper TB, Cadet JL. Vitamin E attenuates the toxic effects of 6-hydroxydopamine on free radical scavenging systems in rat brain. Brain Res Bull. 1992;29:699–701. doi: 10.1016/0361-9230(92)90142-k. [DOI] [PubMed] [Google Scholar]
- Pesah Y, Burgess H, Middlebrooks B, Ronningen K, Prosser J, Tirunagaru V, et al. Whole-mount analysis reveals normal numbers of dopaminergic neurons following misexpression of alpha-synuclein in Drosophila. Genesis. 2005;41:154–159. doi: 10.1002/gene.20106. [DOI] [PubMed] [Google Scholar]
- Petroske E, Meredith GE, Callen S, Totterdell S, Lau YS. Mouse model of Parkinsonism: a comparison between subacute MPTP and chronic MPTP/probenecid treatment. Neuroscience. 2001;106:589–601. doi: 10.1016/s0306-4522(01)00295-0. [DOI] [PubMed] [Google Scholar]
- Pifl C, Schingnitz G, Hornykiewicz O. The neurotoxin MPTP does not reproduce in the rhesus monkey the interregional pattern of striatal dopamine loss typical of human idiopathic Parkinson's disease. Neurosci Lett. 1988;92:228–233. doi: 10.1016/0304-3940(88)90066-3. [DOI] [PubMed] [Google Scholar]
- Pinder RM, Brogden RN, Sawyer PR, Speight TM, Avery GS. Levodopa and decarboxylase inhibitors: a review of their clinical pharmacology and use in the treatment of Parkinsonism. Drugs. 1976;11:329–377. doi: 10.2165/00003495-197611050-00001. [DOI] [PubMed] [Google Scholar]
- Poirier LJ, Filion M, Larochelle L, Pechadre JC. Physiopathology of experimental Parkinsonism in the monkey. Can J Neurol Sci. 1975;2:255–263. doi: 10.1017/s0317167100020357. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Pothakos K, Kurz MJ, Lau YS. Restorative effect of endurance exercise on behavioral deficits in the chronic mouse model of Parkinson's disease with severe neurodegeneration. BMC Neurosci. 2009;10:6. doi: 10.1186/1471-2202-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prikhojan A, Brannan T, Yahr MD. Comparative effects of repeated administration of dopamine agonists on circling behavior in rats. J Neural Transm. 2000;107:1159–1164. doi: 10.1007/s007020070029. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Popilskis S, Kostic V, Levivier M, Fahn S, et al. Unilateral MPTP-induced Parkinsonism in monkeys. A quantitative autoradiographic study of dopamine D1 and D2 receptors and re-uptake sites. Neurochirurgie. 1991;37:377–382. [PubMed] [Google Scholar]
- Przedborski S, Levivier M, Jiang H, Ferreira M, Jackson-Lewis V, Donaldson D, et al. Dose-dependent lesions of the dopaminergic nigrostriatal pathway induced by intrastriatal injection of 6-hydroxydopamine. Neuroscience. 1995;67:631–647. doi: 10.1016/0306-4522(95)00066-r. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Naini AB, Jakowec M, Petzinger G, Miller R, et al. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a technical review of its utility and safety. J Neurochem. 2001;76:1265–1274. doi: 10.1046/j.1471-4159.2001.00183.x. [DOI] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, Police S, He L, Di Monte DA, Langston JW. Expression of D(3) receptor messenger RNA and binding sites in monkey striatum and substantia nigra after nigrostriatal degeneration: effect of levodopa treatment. Neuroscience. 2000;98:263–273. doi: 10.1016/s0306-4522(00)00130-5. [DOI] [PubMed] [Google Scholar]
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson's disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med. 2000;342:1484–1491. doi: 10.1056/NEJM200005183422004. [DOI] [PubMed] [Google Scholar]
- Rascol O, Arnulf I, Peyro-Saint Paul H, Brefel-Courbon C, Vidailhet M, Thalamas C, et al. Idazoxan, an α-2 antagonist, and L-DOPA-induced dyskinesias in patients with Parkinson's disease. Mov Disord. 2001;16:708–713. doi: 10.1002/mds.1143. [DOI] [PubMed] [Google Scholar]
- Rascol O, Poewe W, Lees A, Aristin M, Salin L, Juhel N, et al. Tesofensine (NS 2330), a monoamine reuptake inhibitor, in patients with advanced Parkinson disease and motor fluctuations: the ADVANS Study. Arch Neurol. 2008;65:577–583. doi: 10.1001/archneur.65.5.577. [DOI] [PubMed] [Google Scholar]
- Reavill C, Jenner P, Marsden CD. Differentiation of dopamine agonists using drug-induced rotation in rats with unilateral or bilateral 6-hydroxydopamine destruction of ascending dopamine pathways. Biochem Pharmacol. 1983;32:865–870. doi: 10.1016/0006-2952(83)90589-0. [DOI] [PubMed] [Google Scholar]
- Riachi NJ, LaManna JC, Harik SI. Entry of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine into the rat brain. J Pharmacol Exp Ther. 1989;249:744–748. [PubMed] [Google Scholar]
- Robledo P, Feger J. Acute monoaminergic depletion in the rat potentiates the excitatory effect of the subthalamic nucleus in the substantia nigra pars reticulata but not in the pallidal complex. J Neural Transm Gen Sect. 1991;86:115–126. doi: 10.1007/BF01250572. [DOI] [PubMed] [Google Scholar]
- Rondot P, Ziegler M. Activity and acceptability of piribedil in Parkinson's disease: a multicentre study. J Neurol. 1992;239(Suppl 1):S28–S34. doi: 10.1007/BF00819564. [DOI] [PubMed] [Google Scholar]
- Rose S, Nomoto M, Jenner P, Marsden CD. Transient depletion of nucleus accumbens dopamine content may contribute to initial akinesia induced by MPTP in common marmosets. Biochem Pharmacol. 1989a;38:3677–3681. doi: 10.1016/0006-2952(89)90572-8. [DOI] [PubMed] [Google Scholar]
- Rose S, Nomoto M, Kelly E, Kilpatrick G, Jenner P, Marsden CD. Increased caudate dopamine turnover may contribute to the recovery of motor function in marmosets treated with the dopaminergic neurotoxin MPTP. Neurosci Lett. 1989b;101:305–310. doi: 10.1016/0304-3940(89)90550-8. [DOI] [PubMed] [Google Scholar]
- Rozas G, Lopez-Martin E, Guerra MJ, Labandeira-Garcia JL. The overall rod performance test in the MPTP-treated-mouse model of Parkinsonism. J Neurosci Methods. 1998;83:165–175. doi: 10.1016/s0165-0270(98)00078-8. [DOI] [PubMed] [Google Scholar]
- Rylander D, Ideberg H, Li Q, Dekundy A, Zhang J, Li H, et al. A mGluR5 antagonist under clinical development improves L-DOPA-induced dyskinesia in parkinsonian rats and monkeys. Neurobiol Dis. 2010;39:352–361. doi: 10.1016/j.nbd.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Saha S, Guillily MD, Ferree A, Lanceta J, Chan D, Ghosh J, et al. LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J Neurosci. 2009;29:9210–9218. doi: 10.1523/JNEUROSCI.2281-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Pierre M, Tremblay ME, Sik A, Gross RE, Cicchetti F. Temporal effects of paraquat/maneb on microglial activation and dopamine neuronal loss in older rats. J Neurochem. 2006;98:760–772. doi: 10.1111/j.1471-4159.2006.03923.x. [DOI] [PubMed] [Google Scholar]
- Sakai K, Gash DM. Effect of bilateral 6-OHDA lesions of the substantia nigra on locomotor activity in the rat. Brain Res. 1994;633:144–150. doi: 10.1016/0006-8993(94)91533-4. [DOI] [PubMed] [Google Scholar]
- Sanberg PR. Haloperidol-induced catalepsy is mediated by postsynaptic dopamine receptors. Nature. 1980;284:472–473. doi: 10.1038/284472a0. [DOI] [PubMed] [Google Scholar]
- Sanberg PR, Bunsey MD, Giordano M, Norman AB. The catalepsy test: its ups and downs. Behav Neurosci. 1988;102:748–759. doi: 10.1037//0735-7044.102.5.748. [DOI] [PubMed] [Google Scholar]
- Saporito MS, Thomas BA, Scott RW. MPTP activates c-Jun NH(2)-terminal kinase (JNK) and its upstream regulatory kinase MKK4 in nigrostriatal neurons in vivo. J Neurochem. 2000;75:1200–1208. doi: 10.1046/j.1471-4159.2000.0751200.x. [DOI] [PubMed] [Google Scholar]
- Saravanan KS, Sindhu KM, Senthilkumar KS, Mohanakumar KP. L-deprenyl protects against rotenone-induced, oxidative stress-mediated dopaminergic neurodegeneration in rats. Neurochem Int. 2006;49:28–40. doi: 10.1016/j.neuint.2005.12.016. [DOI] [PubMed] [Google Scholar]
- Sauer H, Oertel WH. Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6-hydroxydopamine: a combined retrograde tracing and immunocytochemical study in the rat. Neuroscience. 1994;59:401–415. doi: 10.1016/0306-4522(94)90605-x. [DOI] [PubMed] [Google Scholar]
- Savola JM, Hill M, Engstrom M, Merivuori H, Wurster S, McGuire SG, et al. Fipamezole (JP-1730) is a potent alpha2 adrenergic receptor antagonist that reduces levodopa-induced dyskinesia in the MPTP-lesioned primate model of Parkinson's disease. Mov Disord. 2003;18:872–883. doi: 10.1002/mds.10464. [DOI] [PubMed] [Google Scholar]
- Sawin ER, Ranganathan R, Horvitz HR. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron. 2000;26:619–631. doi: 10.1016/s0896-6273(00)81199-x. [DOI] [PubMed] [Google Scholar]
- Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST. CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, Parkinsonism and spinal cord injury. Neuropharmacology. 2000;39:777–787. doi: 10.1016/s0028-3908(00)00005-8. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Cleeter MW, Muddle JR, Workman JM, Cooper JM, King RH. Proteasomal inhibition causes loss of nigral tyrosine hydroxylase neurons. Ann Neurol. 2006;60:253–255. doi: 10.1002/ana.20934. [DOI] [PubMed] [Google Scholar]
- Schmidt WJ, Lebsanft H, Heindl M, Gerlach M, Gruenblatt E, Riederer P, et al. Continuous versus pulsatile administration of rotigotine in 6-OHDA-lesioned rats: contralateral rotations and abnormal involuntary movements. J Neural Transm. 2008;115:1385–1392. doi: 10.1007/s00702-008-0102-z. [DOI] [PubMed] [Google Scholar]
- Schneider JS, Dacko S. Relative sparing of the dopaminergic innervation of the globus pallidus in monkeys made hemi-parkinsonian by intracarotid MPTP infusion. Brain Res. 1991;556:292–296. doi: 10.1016/0006-8993(91)90318-p. [DOI] [PubMed] [Google Scholar]
- Schneider JS, Kovelowski CJ., II Chronic exposure to low doses of MPTP I: Cognitive deficits in motor asymptomatic monkeys. Brain Res. 1990;519:122–128. doi: 10.1016/0006-8993(90)90069-n. [DOI] [PubMed] [Google Scholar]
- Schneider JS, Yuwiler A, Markham CH. Selective loss of subpopulations of ventral mesencephalic dopaminergic neurons in the monkey following exposure to MPTP. Brain Res. 1987;411:144–150. doi: 10.1016/0006-8993(87)90691-3. [DOI] [PubMed] [Google Scholar]
- Sedelis M, Schwarting RK, Huston JP. Behavioral phenotyping of the MPTP mouse model of Parkinson's disease. Behav Brain Res. 2001;125:109–125. doi: 10.1016/s0166-4328(01)00309-6. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, et al. Mechanism of toxicity in rotenone models of Parkinson's disease. J Neurosci. 2003a;23:10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Kim JH, Greenamyre JT. Selective microglial activation in the rat rotenone model of Parkinson's disease. Neurosci Lett. 2003b;341:87–90. doi: 10.1016/s0304-3940(03)00172-1. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Kim JH, Betarbet R, Greenamyre JT. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and α-synuclein aggregation. Exp Neurol. 2003c;179:9–16. doi: 10.1006/exnr.2002.8072. [DOI] [PubMed] [Google Scholar]
- Shimizu K, Ohtaki K, Matsubara K, Aoyama K, Uezono T, Saito O, et al. Carrier-mediated processes in blood – brain barrier penetration and neural uptake of paraquat. Brain Res. 2001;906:135–142. doi: 10.1016/s0006-8993(01)02577-x. [DOI] [PubMed] [Google Scholar]
- Shimoji M, Zhang L, Mandir AS, Dawson VL, Dawson TM. Absence of inclusion bosy formation in the MPTP mouse model of Parkinson's disease. Brain Res Mol Brain Res. 2005;134:103–108. doi: 10.1016/j.molbrainres.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Shiosaki K, Jenner P, Asin KE, Britton DR, Lin CW, Michaelides M, et al. ABT-431: the diacetyl prodrug of A-86929, a potent and selective dopamine D1 receptor agonist: in vitro characterization and effects in animal models of Parkinson's disease. J Pharmacol Exp Ther. 1996;276:150–160. [PubMed] [Google Scholar]
- Shiozaki S, Ichikawa S, Nakamura J, Kitamura S, Yamada K, Kuwana Y. Actions of adenosine A2A receptor antagonist KW-6002 on drug-induced catalepsy and hypokinesia caused by reserpine or MPTP. Psychopharmacology (Berl) 1999;147:90–95. doi: 10.1007/s002130051146. [DOI] [PubMed] [Google Scholar]
- Shook BC, Rassnick S, Osborne MC, Davis S, Westover L, Boulet J, et al. In vivo characterization of a dual adenosine A(2A)/A(1) receptor antagonist in animal models of Parkinson's disease. J Med Chem. 2010;53:8104–8115. doi: 10.1021/jm100971t. [DOI] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Haron-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverdale MA, Nicholson SL, Ravenscroft P, Crossman AR, Millan MJ, Brotchie JM. Selective blockade of D(3) dopamine receptors enhances the anti-parkinsonian properties of ropinirole and levodopa in the MPTP-lesioned primate. Exp Neurol. 2004;188:128–138. doi: 10.1016/j.expneurol.2004.03.022. [DOI] [PubMed] [Google Scholar]
- Skuza G, Rogoz Z, Quack G, Danysz W. Memantine, amantadine, and L-deprenyl potentiate the action of L-dopa in monoamine-depleted rats. J Neural Transm Gen Sect. 1994;98:57–67. doi: 10.1007/BF01277594. [DOI] [PubMed] [Google Scholar]
- Smith LA, Gordin A, Jenner P, Marsden CD. Entacapone enhances levodopa-induced reversal of motor disability in MPTP-treated common marmosets. Mov Disord. 1997;12:935–945. doi: 10.1002/mds.870120616. [DOI] [PubMed] [Google Scholar]
- Smith LA, Jackson MJ, Hansard MJ, Maratos E, Jenner P. Effect of pulsatile administration of levodopa on dyskinesia induction in drug-naive MPTP-treated common marmosets: effect of dose, frequency of administration, and brain exposure. Mov Disord. 2003;18:487–495. doi: 10.1002/mds.10394. [DOI] [PubMed] [Google Scholar]
- Smith LA, Jackson MJ, Johnston L, Kuoppamaki M, Rose S, Al-Barghouthy G, et al. Switching from levodopa to the long-acting dopamine D2/D3 agonist piribedil reduces the expression of dyskinesia while maintaining effective motor activity in MPTP-treated primates. Clin Neuropharmacol. 2006;29:112–125. doi: 10.1097/01.WNF.0000220818.71231.DF. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Heikkila RE. Neurotoxic effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and methamphetamine in several strains of mice. Prog Neuropsychopharmacol Biol Psychiatry. 1988;12:345–354. doi: 10.1016/0278-5846(88)90054-1. [DOI] [PubMed] [Google Scholar]
- Sourkes TL, Poirier LJ. Neurochemical bases of tremor and other disorders of movement. Can Med Assoc J. 1966;94:53–60. [PMC free article] [PubMed] [Google Scholar]
- Speiser Z, Levy R, Cohen S. Effects of N-propargyl-1-(R)aminoindan (rasagiline) in models of motor and cognition disorders. J Neural Transm Suppl. 1998;52:287–300. doi: 10.1007/978-3-7091-6499-0_29. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanic D, Finkelstein DI, Bourke DW, Drago J, Horne MK. Timecourse of striatal re-innervation following lesions of dopaminergic SNpc neurons of the rat. Eur J Neurosci. 2003;18:1175–1188. doi: 10.1046/j.1460-9568.2003.02800.x. [DOI] [PubMed] [Google Scholar]
- Stocchi F, Vacca L, Ruggieri S, Olanow CW. Intermittent vs continuous levodopa administration in patients with advanced Parkinson disease: a clinical and pharmacokinetic study. Arch Neurol. 2005;62:905–910. doi: 10.1001/archneur.62.6.905. [DOI] [PubMed] [Google Scholar]
- Stockwell KA, Virley DJ, Perren M, Iravani MM, Jackson MJ, Rose S, et al. Continuous delivery of ropinirole reverses motor deficits without dyskinesia induction in MPTP-treated common marmosets. Exp Neurol. 2008;211:172–179. doi: 10.1016/j.expneurol.2008.01.019. [DOI] [PubMed] [Google Scholar]
- Stockwell KA, Scheller D, Rose S, Jackson MJ, Tayarani-Binazir K, Iravani MM, et al. Continuous administration of rotigotine to MPTP-treated common marmosets enhances anti-parkinsonian activity and reduces dyskinesia induction. Exp Neurol. 2009;219:533–542. doi: 10.1016/j.expneurol.2009.07.011. [DOI] [PubMed] [Google Scholar]
- Sundstrom E, Fredriksson A, Archer T. Chronic neurochemical and behavioral changes in MPTP-lesioned C57BL/6 mice: a model for Parkinson's disease. Brain Res. 1990;528:181–188. doi: 10.1016/0006-8993(90)91656-2. [DOI] [PubMed] [Google Scholar]
- Tansey MG, Goldberg MS. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taquet H, Nomoto M, Rose S, Jenner P, Javoy-Agid F, Mauborgne A, et al. Levels of Met-enkephalin, Leu-enkephalin, substance P and cholecystokinin in the brain of the common marmoset following long term 1-methyl-4-phenyl-1,2,3,6,-tetrahydropyridine treatment. Neuropeptides. 1988;12:105–110. doi: 10.1016/0143-4179(88)90039-x. [DOI] [PubMed] [Google Scholar]
- Tatton NA, Kish SJ. In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice using terminal deoxynucleotidyl transferase labelling and acridine orange staining. Neuroscience. 1997;77:1037–1048. doi: 10.1016/s0306-4522(96)00545-3. [DOI] [PubMed] [Google Scholar]
- Tayarani-Binazir K, Jackson MJ, Rose S, McCreary AC, Jenner P. The partial dopamine agonist pardoprunox (SLV308) administered in combination with l-dopa improves efficacy and decreases dyskinesia in MPTP treated common marmosets. Exp Neurol. 2010;226:320–327. doi: 10.1016/j.expneurol.2010.09.007. [DOI] [PubMed] [Google Scholar]
- Taylor AE, Saint-Cyr JA, Lang AE. Frontal lobe dysfunction in Parkinson's disease: the cortical focus of neostriatal outflow. Brain. 1986;109:845–883. doi: 10.1093/brain/109.5.845. [DOI] [PubMed] [Google Scholar]
- Taylor MD, de Ceballos ML, Rose S, Chong PN, Jenner P, Marsden CD. Neuropeptide levels in the basal ganglia of aged common marmosets following prolonged treatment with MPTP. J Neural Transm Park Dis Dement Sect. 1991;3:99–108. doi: 10.1007/BF02260885. [DOI] [PubMed] [Google Scholar]
- Tel BC, Zeng BY, Cannizzaro C, Pearce RK, Rose S, Jenner P. Alterations in striatal neuropeptide mRNA produced by repeated administration of L-DOPA, ropinirole or bromocriptine correlate with dyskinesia induction in MPTP-treated common marmosets. Neuroscience. 2002;115:1047–1058. doi: 10.1016/s0306-4522(02)00535-3. [DOI] [PubMed] [Google Scholar]
- Temlett JA, Chong PN, Oertel WH, Jenner P, Marsden CD. The D-1 dopamine receptor partial agonist, CY 208-243, exhibits antiparkinsonian activity in the MPTP-treated marmoset. Eur J Pharmacol. 1988;156:197–206. doi: 10.1016/0014-2999(88)90322-6. [DOI] [PubMed] [Google Scholar]
- Temlett JA, Quinn NP, Jenner PG, Marsden CD, Pourcher E, Bonnet AM, et al. Antiparkinsonian activity of CY 208-243, a partial D-1 dopamine receptor agonist, in MPTP-treated marmosets and patients with Parkinson's disease. Mov Disord. 1989;4:261–265. doi: 10.1002/mds.870040307. [DOI] [PubMed] [Google Scholar]
- Thiruchelvam M, Brockel BJ, Richfield EK, Baggs RB, Cory-Slechta DA. Potentiated and preferential effects of combined paraquat and maneb on nigrostriatal dopamine systems: environmental risk factors for Parkinson's disease? Brain Res. 2000;873:225–234. doi: 10.1016/s0006-8993(00)02496-3. [DOI] [PubMed] [Google Scholar]
- Tornwall M, Mannisto PT. Effects of three types of catechol O-methylation inhibitors on L-3,4-dihydroxyphenylalanine-induced circling behaviour in rats. Eur J Pharmacol. 1993;250:77–84. doi: 10.1016/0014-2999(93)90623-p. [DOI] [PubMed] [Google Scholar]
- Um JW, Park HJ, Song J, Jeon I, Lee G, Lee PH, et al. Formation of parkin aggregates and enhanced PINK1 accumulation during the pathogenesis of Parkinson's disease. Biochem Biophys Res Commun. 2010;393:824–828. doi: 10.1016/j.bbrc.2010.02.090. [DOI] [PubMed] [Google Scholar]
- Ungerstedt U. 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur J Pharmacol. 1968;5:107–110. doi: 10.1016/0014-2999(68)90164-7. [DOI] [PubMed] [Google Scholar]
- Varastet M, Riche D, Maziere M, Hantraye P. Chronic MPTP treatment reproduces in baboons the differential vulnerability of mesencephalic dopaminergic neurons observed in Parkinson's disease. Neuroscience. 1994;63:47–56. doi: 10.1016/0306-4522(94)90006-x. [DOI] [PubMed] [Google Scholar]
- Ved R, Saha S, Westlund B, Perier C, Burnam L, Sluder A, et al. Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of α-synuclein, parkin, and DJ-1 in Caenorhabditis elegans. J Biol Chem. 2005;280:42655–42668. doi: 10.1074/jbc.M505910200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velayati A, Yu WH, Sidransky E. The role of glucocerebrosidase mutations in Parkinson disease and Lewy body disorders. Curr Neurol Neurosci Rep. 2010;10:190–198. doi: 10.1007/s11910-010-0102-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon AC, Croucher MJ, Dexter DT. Additive neuroprotection by metabotropic glutamate receptor subtype-selective ligands in a rat Parkinson's model. Neuroreport. 2008;19:475–478. doi: 10.1097/WNR.0b013e3282f602df. [DOI] [PubMed] [Google Scholar]
- Viaro R, Marti M, Morari M. Dual motor response to l-dopa and nociceptin/orphanin FQ receptor antagonists in 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP) treated mice: Paradoxical inhibition is relieved by D(2)/D(3) receptor blockade. Exp Neurol. 2010;223:473–484. doi: 10.1016/j.expneurol.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Vila M, Herrero MT, Levy R, Faucheux B, Ruberg M, Guillen J, et al. Consequences of nigrostriatal denervation on the gamma-aminobutyric acidic neurons of substantia nigra pars reticulata and superior colliculus in parkinsonian syndromes. Neurology. 1996a;46:802–809. doi: 10.1212/wnl.46.3.802. [DOI] [PubMed] [Google Scholar]
- Vila M, Levy R, Herrero MT, Faucheux B, Obeso JA, Agid Y, et al. Metabolic activity of the basal ganglia in parkinsonian syndromes in human and non-human primates: a cytochrome oxidase histochemistry study. Neuroscience. 1996b;71:903–912. doi: 10.1016/0306-4522(95)00549-8. [DOI] [PubMed] [Google Scholar]
- Vila M, Levy R, Herrero MT, Ruberg M, Faucheux B, Obeso JA, et al. Consequences of nigrostriatal denervation on the functioning of the basal ganglia in human and nonhuman primates: an in situ hybridization study of cytochrome oxidase subunit I mRNA. J Neurosci. 1997;17:765–773. doi: 10.1523/JNEUROSCI.17-02-00765.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila M, Jackson-Lewis V, Vukosavic S, Djaldetti R, Liberatore G, Offen D, et al. Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl- 4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Proc Natl Acad Sci USA. 2001;98:2837–2842. doi: 10.1073/pnas.051633998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visanji NP, Gomez-Ramirez J, Johnston TH, Pires D, Moon V, Brotchie JM, et al. Pharmacological characterisation of psychosis-like behavior in the MPTP-lesioned nonhuman primate model of Parkinson's disease. Mov Dis. 2006a;21:1879–1891. doi: 10.1002/mds.21073. [DOI] [PubMed] [Google Scholar]
- Visanji NP, O'Neill MJ, Duty S. Nicotine, but neither the α4beta2 ligand RJR2403 nor an α7 nAChR subtype selective agonist, protects against a partial 6-hydroxydopamine lesion of the rat median forebrain bundle. Neuropharmacology. 2006b;51:506–516. doi: 10.1016/j.neuropharm.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Visanji NP, Orsi A, Johnston TH, Howson PA, Dixon K, Callizot N, et al. PYM50028, a novel orally active, nonpeptide neurotrophic factor inducer, prevents and reverses neuronal damage induced by MPP+ in mesencephalic neurons and by MPTP in a mouse model of Parkinson's disease. FASEB J. 2008;22:2488–2497. doi: 10.1096/fj.07-095398. [DOI] [PubMed] [Google Scholar]
- Visanji NP, Fox SH, Johnston TH, Millan MJ, Brotchie JM. a-1 Adrenoceptors mediate dihydroxyphenylalanine-induced activity in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaques. J Pharmacol Exp Ther. 2009;328:276–283. doi: 10.1124/jpet.108.144097. [DOI] [PubMed] [Google Scholar]
- Wang D, Qian L, Xiong H, Liu J, Neckameyer WS, Oldham S, et al. Antioxidants protect PINK1-dependent dopaminergic neurons in Drosophila. Proc Natl Acad Sci USA. 2006a;103:13520–13525. doi: 10.1073/pnas.0604661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XF, Li S, Chou AP, Bronstein JM. Inhibitory effects of pesticides on proteasome activity: implication in Parkinson's disease. Neurobiol Dis. 2006b;23:198–205. doi: 10.1016/j.nbd.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Wang C, Lu R, Ouyang X, Ho MW, Chia W, Yu F, et al. Drosophila overexpressing parkin R275W mutant exhibits dopaminergic neuron degeneration and mitochondrial abnormalities. J Neurosci. 2007;27:8563–8570. doi: 10.1523/JNEUROSCI.0218-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L, Wei W, Gu W, Huang P, Ren X, Zhang Z, et al. Visualization of monoaminergic neurons and neurotoxicity of MPTP in live transgenic zebrafish. Dev Biol. 2008;314:84–92. doi: 10.1016/j.ydbio.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Whitton PS. Inflammation as a causative factor in the aetiology of Parkinson's disease. Br J Pharmacol. 2007;150:963–976. doi: 10.1038/sj.bjp.0707167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth AJ, Theodore DA, Greene JC, Benes H, Wes PD, Pallanck LJ. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. Proc Natl Acad Sci USA. 2005;102:8024–8029. doi: 10.1073/pnas.0501078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth AJ, Wes PD, Pallanck LJ. Drosophila models pioneer a new approach to drug discovery for Parkinson's disease. Drug Discov Today. 2006;11:119–126. doi: 10.1016/S1359-6446(05)03693-7. [DOI] [PubMed] [Google Scholar]
- Winkler C, Sauer H, Lee CS, Bjorklund A. Short-term GDNF treatment provides long-term rescue of lesioned nigral dopaminergic neurons in a rat model of Parkinson's disease. J Neurosci. 1996;16:7206–7215. doi: 10.1523/JNEUROSCI.16-22-07206.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y, Ryan J, Noble S, Yu M, Yilbas AE, Ekker M. Impaired dopaminergic neuron development and locomotor function in zebrafish with loss of pink1 function. Eur J Neurosci. 2010;31:623–633. doi: 10.1111/j.1460-9568.2010.07091.x. [DOI] [PubMed] [Google Scholar]
- Yang YX, Wood NW, Latchman DS. Molecular basis of Parkinson's disease. Neuroreport. 2009;20:150–156. doi: 10.1097/WNR.0b013e32831c50df. [DOI] [PubMed] [Google Scholar]
- Yoshimura N, Mizuta E, Kuno S, Sasa M, Yoshida O. The dopamine D1 receptor agonist SKF 38393 suppresses detrusor hyperreflexia in the monkey with Parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Neuropharmacology. 1993;32:315–321. doi: 10.1016/0028-3908(93)90151-r. [DOI] [PubMed] [Google Scholar]
- Yoshimura N, Mizuta E, Yoshida O, Kuno S. Therapeutic effects of dopamine D1/D2 receptor agonists on detrusor hyperreflexia in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned parkinsonian cynomolgus monkeys. J Pharmacol Exp Ther. 1998;286:228–233. [PubMed] [Google Scholar]
- You ZB, Herrera-Marschitz M, Pettersson E, Nylander I, Goiny M, Shou HZ, et al. Modulation of neurotransmitter release by cholecystokinin in the neostriatum and substantia nigra of the rat: regional and receptor specificity. Neuroscience. 1996;74:793–804. doi: 10.1016/0306-4522(96)00149-2. [DOI] [PubMed] [Google Scholar]
- Young WS, 3rd, Bonner TI, Brann MR. Mesencephalic dopamine neurons regulate the expression of neuropeptide mRNAs in the rat forebrain. Proc Natl Acad Sci USA. 1986;83:9827–9831. doi: 10.1073/pnas.83.24.9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebrowska-Lupina I, Porowska A, Pietrasiewicz T. Interaction of antidepressants with antiparkinsonian agents in rats. Pol J Pharmacol Pharm. 1985;37:865–874. [PubMed] [Google Scholar]
- Zeng BY, Medhurst AD, Jackson M, Rose S, Jenner P. Proteasomal activity in brain differs between species and brain regions and changes with age. Mech Ageing Dev. 2005;126:760–766. doi: 10.1016/j.mad.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Zetler G. Anticataleptic actions of amantadine hydrochloride. Naunyn Schmiedebergs Arch Pharmakol. 1970;266:276–278. doi: 10.1007/BF00997288. [DOI] [PubMed] [Google Scholar]
- Zetterstrom RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science. 1997;276:248–250. doi: 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
- Zhang J, Fitsanakis VA, Gu G, Jing D, Ao M, Amarnath V, et al. Manganese ethylene-bis-dithiocarbamate and selective dopaminergic neurodegeneration in rat: a link through mitochondrial dysfunction. J Neurochem. 2003;84:336–346. doi: 10.1046/j.1471-4159.2003.01525.x. [DOI] [PubMed] [Google Scholar]
- Zuch CL, Nordstroem VK, Briedrick LA, Hoernig GR, Granholm AC, Bickford PC. Time course of degenerative alterations in nigral dopaminergic neurons following a 6-hydroxydopamine lesion. J Comp Neurol. 2000;427:440–454. doi: 10.1002/1096-9861(20001120)427:3<440::aid-cne10>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]