Graph accordance of next-generation sequence assemblies (original) (raw)
Abstract
Motivation: No individual assembly algorithm addresses all the known limitations of assembling short-length sequences. Overall reduced sequence contig length is the major problem that challenges the usage of these assemblies. We describe an algorithm to take advantages of different assembly algorithms or sequencing platforms to improve the quality of next-generation sequence (NGS) assemblies.
Results: The algorithm is implemented as a graph accordance assembly (GAA) program. The algorithm constructs an accordance graph to capture the mapping information between the target and query assemblies. Based on the accordance graph, the contigs or scaffolds of the target assembly can be extended, merged or bridged together. Extra constraints, including gap sizes, mate pairs, scaffold order and orientation, are explored to enforce those accordance operations in the correct context. We applied GAA to various chicken NGS assemblies and the results demonstrate improved contiguity statistics and higher genome and gene coverage.
Availability: GAA is implemented in OO perl and is available here: http://sourceforge.net/projects/gaa-wugi/.
Contact: lye@genome.wustl.edu
1 INTRODUCTION
The need for cost efficient whole genome assemblies has driven the adoption of many assembly programs exclusive to short sequence lengths, such as CABOG (Miller et al., 2008), Newbler (Roche 454 Life Sciences), ABYSS (Simpson et al., 2009), ALLPATHs-LG (Gnerre et al., 2011) and SOAPdenovo (Li et al., 2010). Compared to Sanger read-based assemblies, NGS assemblies are less representative of genome content, especially in regions with repetitive structure, for example segmental duplications (Alkan et al., 2010). Furthermore, fragmentation of assemblies can limit further annotation processes, such as gene prediction in silico.
Several factors present challenges for NGS assembly. However, the predominant barriers to sequence assembly are repeats and polymorphism in genomes. Typically, one assembly algorithm can outperform others in assembling certain genomic regions. Even the same assembler performs differently over varying parameter settings such as different k-mer sizes. A longer k-mer is better for resolving high coverage repetitive regions, whereas a shorter k-mer is better for finding paths in low-coverage polymorphic regions. The latter is analogous to what is a known hurdle for transcriptome assemblies, which is significantly unbalanced representation of each transcript due to expression level variation in different tissue types. Moreover, sequencing platforms have various biases (Harismendy et al., 2009), thus assemblies generated from different platforms can complement each other (DiGuistini et al., 2009; Nagarajan et al., 2010). Therefore, a combination of assemblies from different sequencing platforms or algorithms (or parameter settings for the same algorithm) is very promising to improve assembly qualities. Existing hybrid assembly and merging methods are mostly developed for small genomes, such as MIRA (http://sourceforge.net/apps/mediawiki/mira-assembler/index.php?title=Main_Page), Minimus2 (http://sourceforge.net/apps/mediawiki/amos/index.php?title=Minimus2) and MAIA (Nijkamp et al., 2010). Reconciliator (Zimin et al., 2008) and GAM (Casagrande et al., 2009) can only merge assemblies generated from the same dataset.
We have developed an efficient algorithm to generate an accordance assembly from two or more large genome assemblies. It uses the BLAT aligner (Kent, 2002) to align a query assembly to a target assembly. Based on the alignment, we construct an accordance graph, which is searched for maximal sub-paths. Contigs along each sub-path are candidates to be merged together. Besides weighing the edges in the graph using alignment scores, contig lengths and overhang sizes, scaffold information including order, orientation and gap sizes, are fully explored to validate each merging event. The query and target assemblies go through a misassembly detection stage, where potential breakpoints were evaluated by the ‘CE statistic’ (Zimin et al., 2008).
This algorithm is implemented as a program named GAA. It takes as inputs two draft assemblies, one as the target and the other as the query. The query is utilized to improve the target assembly.
2 METHODS
A novel data structure called accordance graph is proposed to capture the alignments between a target and a query assembly. In the accordance graph (Fig. 1), each node represents a contig in the target assembly, while each edge represents a link between two contigs in the target assembly. Links occur when one query contig maps to multiple target contigs. The length of an edge is the gap size between the two target contigs represented by the two nodes incident to the edge.
Fig. 1.

Graph accordance of assemblies. (A) Alignments between the target and query assemblies. (B) An accordance graph capturing the alignments with four maximal sub-paths included in dashed circles. T and Q represent contigs in the target and query assemblies, respectively. For example, T1.2 is the second contig of the first scaffold in the target assembly, and Q1.2 is the second contig of the first scaffold in the query assembly. M represents contigs in the merged assembly. Green edges represent consistent links, and red edges represent inconsistent links. (C) Target contigs are merged or bridged along each maximal sub-path with gap filling using query bases, and the merged contigs are renamed accordingly.
The main algorithm that we use to integrate two assemblies is as follows:
Aligning assemblies: the aligner module takes as input two assemblies. It uses BLAT (Kent, 2002) to align the query assembly to the target assembly. All unique high scoring matches are retained for the graph construction. For example, a score cutoff of 400 can be chosen to reduce sporadic matches.
Constructing the accordance graph: an accordance graph G is constructed to organize the relationship between target contigs and query contigs based on their alignments. In accordance graph G, each vertex v represents a target contig. For vertices _v_1 and _v_2, an edge e is added into graph G between _v_1 and _v_2 if there is a query contig mapped to both contigs represented by _v_1 and _v_2. The length of edge e, also called the observed gap size between _v_1 and _v_2, is the number of bases in the query contig between _v_1 and _v_2. We define the expected gap size as the gap size between _v_1 and _v_2 in the target assembly.
The construction of the accordance graph starts with sorting the target contigs within each query contig in the order of their start mapping positions in the query contig. In other words, the target contigs are anchored onto the query contigs. Then edges are drawn between two neighboring target contigs. Branches may occur due to non-proper mapping between target and query contigs, as shown in Figure 1A.
Finding maximal sub-paths: we define a path p in graph G as a maximal one if there are no more vertices can be appended to the path without introducing inconsistencies. The inconsistency can be due to non-proper matches, large difference between observed and expected gap sizes, or violation of original scaffold order or orientation. One alignment with long overhangs is a non-proper match, for example, the alignment between T1.3 and Q1.2 in Figure 1A. The length cutoff of an overhang is a user defined parameter with a default value of 90 bp. If a query contig can be used to close a gap in the target assembly, the observed gap size (length of edge e) should be close to the expect gap size estimated from mate pairs in the target assembly, otherwise, this gap cannot be closed due to the conflict. The order and orientation of contigs in the maximal sub-paths should be consistent with those in the original scaffolds. Thus, the contigs along a maximal sub-path are consistent and can be merged or bridged together based on the alignment of two assemblies. To find confident paths, edges are weighted based on the size of contigs, length of alignments and percentage of non-aligned overhangs. Preference of base and quality calling in the aligned regions can be given to the more accurate assembly.
A key feature of the graph model is that it can merge scaffolds if their contigs gear into each other in the alignment with a query contig. In other words, if the contigs of two scaffolds in the target assembly alternate along a maximal sub-path anchored by a query contig, they will be merged together. As a special case, scaffolds that contain only one contig can be dropped into the longer scaffolds completely.
Closing gaps: gaps are between contigs inside one scaffold. They can also occur inside contigs if there are deletion events in the alignments between target and query contigs. Gaps within contigs or scaffolds can be closed under correct contexts. For gaps inside contigs, we use the CE module to examine the gap regions in both target and query assemblies. Gaps in the target assembly can be closed by a corresponding query contig if the query contig has no CE statistics contradictions. For gaps inside a scaffold, we compare observed gap sizes with expected gap sizes provided by the target assembly. The merging operation is dropped if the two gap sizes diverge too much. One extra constraint in merging scaffolds is the consistency of neighboring contigs in both target and query scaffolds.
A CE module is implemented for detecting misassembled locations in query and target assemblies using mapped paired reads. In misassembled regions, the distribution of paired reads deviates from the entire genome. The module is an implementation of CE statistic as in the paper of Zimin et al. (2008). The statistic is defined as below
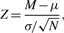
where M is the mean of the local insert sizes and is the library mean, N is the number of read pairs mapped to this location and is standard deviation of the library. It is based on the fact that the variance of the sum of independent random variables is the sum of their variances. When Z deviates from the library mean at a location, it is likely that the sample of inserts is compressed or expanded. These inserts variations are an indication of misassembly events, where a chunk of sequence might be deleted from or inserted into the contig at this location.
3 RESULTS
We applied GAA to the chicken genome (Gallus gallus) with an estimated genome size of 1.2 Gb. Two 454 de novo assemblies were generated with Newbler (Margulies et al., 2005) and CABOG (Miller et al., 2008) and one Illumina assembly was generated with SOAPdenovo (Li et al., 2010). The raw coverages of 454 data and Illumina data are 14- and 74-fold, respectively (Ye et al., 2011).
3.1 Assembly contiguity
We generated the N50 statistics for all the de novo and merged assemblies (Table 1). The N50 statistics is defined as the largest length L, such that 50% of all nucleotides are contained in contigs/scaffolds of size at least L. Here, we calculated N50 statistics based on the size (1.1 Gb) of the _Gallus_gallus_-2.1 reference assembly (Consortium, 2004). GAA significantly improved assembly contiguities overall (Table 1). Comparing with the target 454/Newbler assembly, the merged 454/Newbler and 454/Cabog assembly (NC) has an N50 contig length 37% longer and scaffold length 27% longer, whereas, the merged 454/Newbler and Illumina/SOAP assembly (NS) has an N50 contig length about 3-fold longer and an N50 scaffold length about 2-fold longer. We also merged two 454 assemblies with Reconciliator as it can only merge assemblies generated from the same dataset. Reconciliator improved about 10% of N50 contig length.
Table 1.
Chicken assemblies
| Statistics | 454/Newbler | 454/Cabog | Illumina/SOAP | NC | NS | Reconciliator |
|---|---|---|---|---|---|---|
| Contig N50 (kb) | 13 | 11 | 10 | 18 | 38 | 15 |
| Scaffold N50 (kb) | 353 | 44 | 263 | 448 | 752 | 358 |
| Assembled size (Mb, > 500 bp) | 930 | 900 | 948 | 957 | 975 | 969 |
| Gene coverage (%) | 54.8 | 42.4 | 66.1 | 61.1 | 69.1 | 56.7 |
| BAC coverage (%) | 96.0 | 89.8 | 95.6 | 97.4 | 98.2 | 97.3 |
| Base error (‰) | 0.568 | 1.028 | 0.08 | 0.827 | 0.561 | 0.579 |
| Mis-assembly (%) | 3.90 | 4.30 | 4.54 | 4.30 | 4.40 | 4.12 |
| Supporting pairs (%) | 79.1 | 72.4 | 83.6 | 82.6 | 86.8 | 79.0 |
3.2 Gene representation
We used 17 934 unspliced G. gallus gene transcripts from Ensembl 59 to evaluate gene coverage of each assembly. We use BLAT (Kent, 2002) to align gene transcripts to each assembly. To evaluate the quality of the assemblies, we define a gene is covered if its best match can cover >90% of the gene with a percent identity over 95%. Comparing with the target 454/Newbler assembly, 6.3 and 14.3% more genes were found in merged NC and NS assemblies, respectively (Table 1), whereas, only 3.5% more genes were found in the merged assembly from Reconiliator.
We further examined the cases where some genes are not detected in original target assemblies but show up in the merged assemblies. Several explanations emerge: (i) a gene is completely missing from the target assembly and the query assembly brings the gene in after merging the contig containing this gene to a target contig (Fig. 2A), (ii) a gene spans two target contigs, which have a small overlap in the gene region but are not assembled together.
Fig. 2.

Gene recovery in merged assemblies. Blue represents target contigs, purple query contigs and red genes. ‘−’ represents the reverse complement of a contig.
A query contig overlaps with both contigs and merge them into one contig (Fig. 2B). (iii) A gene spans two contigs inside one scaffold, between which there is a gap in the target assembly. GAA filled this gap with query contig bases and the gene shows up in the merged assembly (Figure 2C).
3.3 Assembly validation
We evaluated merged assemblies using 193 finished BAC sequences, _Gallus-gallus_-2.1 reference assembly and 126 million short jump reads (~2 kb insert size). We estimated base accuracy, including substitution, insertion and deletion, by aligning finished BACs to each assembly. Finished BAC clones were aligned against each assembly using WU-BLASTN, then unique regions were refined using Cross-Match with default parameters. Both NC and NS have a higher coverage than original assemblies. As expected, higher error rates were observed for the assemblies involving 454 sequences (Table 1). Thus, it is necessary to correct the errors using Illumina sequences (Otto et al., 2010). Base error rates of both merged assemblies falls between the target and query assemblies.
To estimate misassembly rates, each assembly was broken into 1-kb chunks and aligned to the _Gallus-gallus_-2.1 reference assembly. Only those 1-kb chunks that can be uniquely placed on the reference are retained for the analysis. The chunks that are <1 kb were discarded. We define that an 1-kb chunk has misassembly events if the error rate is >1% (Gnerre et al., 2011). Both NC and NS have an error rate not higher than their corresponding query assemblies, but higher than their corresponding target assemblies. Reconciliator has a lower error rate, which is slightly higher than the target assembly.
We further examined long-range connectivity by mapping jump reads from a 2-kb insert library onto each assembly using the Burrows–Wheeler Alignment tool BWA (Li and Durbin, 2009). To assess the accuracy of an assembly, we calculated the percentage of supporting mate pairs that are within six standard deviation of the library insert size in the mapping results. The NC and NS assemblies have 3.5 and 7.7% more supporting pairs than the target assembly, respectively and 10.2 and 3.2% more than their corresponding query assemblies, respectively. The NC assembly has ~ 3.6% more supporting pairs than Reconciliator.
3.4 Computational resources
We run GAA on a computer having 2.8 GHz Dual-Core AMD Opteron Model 8220 processors and 256 GB of RAM. GAA finished NC and NS in 10 and 13 h, respectively, with <10 GB of RAM. Majority of the running time is spent on assembly alignment, and about 1/5 on graph construction and analysis. However, Reconciliator uses about 24 h and more than 100 GB of RAM to merge the 454/Newbler and 454/Cabog assemblies.
4 DISCUSSION
In the era of NGS assemblies with fragmented representation all methods need to be explored that will improve sequence contig and scaffold length. A post-assembly that will blend the strengths of different assembly algorithms or sequencing platforms is one option we have pursued here. GAA can significantly improve contiguity of NGS assemblies and resolve most assembly mistakes in the less stringent assembly. The proposed algorithm can also recover missing genes. Our previous results showed that both 454 and Illumina de novo assemblies can reveal some novel sequences that are missing in the Sanger reference assembly (Ye et al., 2011). The significant improvement in the merged NS assembly further shows the advantages of cross-platform merging.
The target assembly is the master and lays out the backbone of the final assembly. Then the query assembly is explored to piece together fragmented regions in the target assembly. Some errors from the query assembly could be introduced into the final assembly. If the query assembly is more accurate at the base level, we can update GAA in the future version to generate a more accurate merged assembly by swapping target assembly bases with query assembly bases. The underlying algorithm of GAA uses evidences from gap sizes, mate pairs to enforce the correct context and prevent query errors. There are more mate pairs that are satisfied in the merged assemblies, which shows the strength of GAA to generate better connectivity by merging assemblies in the context besides overlaps. Since the GAA algorithm can detect and avoid most misassembled regions due to the lower stringency of query assemblies, we suggest choosing the more stringent assembly as the target and using the relaxed assembly to improve it.
In summary, we have implemented a graph accordance assembly program, which can significantly improve the contiguity and gene coverage of NGS assemblies. It provides an efficient method for reducing biases of different sequencing platforms. GAA can also be easily adapted to combine transcriptome or metagenomic assemblies generated with different k-mer sizes as suggested by Surget-Groba and Montoya-Burgos (2010).
Funding: National Human Genome Research Institute (grant HG004968 to G.M.W.).
Conflict of Interest: none declared.
Footnotes
† The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
REFERENCES
- Alkan C., et al. Limitations of next-generation genome sequence assembly. Nat. Methods. 2010;8:61–65. doi: 10.1038/nmeth.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casagrande A., et al. IEEE International Conference on Bioinformatics and Biomedicine (BIBM). Washington, DC: 2009. GAM: genomics assemblies merger: a graph based method to integrate different assemblies; pp. 321–326. [Google Scholar]
- Consortium I.C.G.S. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- DiGuistini S., et al. De novo genome sequence assembly of a filamentous fungus using Sanger, 454 and Illumina sequence data. Genome Biol. 2009;10:R94. doi: 10.1186/gb-2009-10-9-r94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnerre S., et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl Acad. Sci. USA. 2011;108:1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harismendy O., et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009;10:R32. doi: 10.1186/gb-2009-10-3-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R., et al. The sequence and de novo assembly of the giant panda genome. Nature. 2010;463:311–317. doi: 10.1038/nature08696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M., et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.R., et al. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 2008;24:2818–2824. doi: 10.1093/bioinformatics/btn548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan H., et al. De Novo assembly of the complete genome of an enhanced electricity-producing variant of Geobacter sulfurreducens using only short reads. PLoS One. 2010;5:e10922. doi: 10.1371/journal.pone.0010922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijkamp J., et al. Integrating genome assemblies with MAIA. Bioinformatics. 2010;26:i433–i439. doi: 10.1093/bioinformatics/btq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T.D., et al. Iterative Correction of reference nucleotides (iCORN) using second generation sequencing Technology. Bioinformatics. 2010;26:1704–1707. doi: 10.1093/bioinformatics/btq269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson J.T., et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surget-Groba Y., Montoya-Burgos J.I. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010;20:1432–1440. doi: 10.1101/gr.103846.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L., et al. A vertebrate case study of the quality of assemblies derived from next-generation sequences. Genome Biol. 2011;12:R31. doi: 10.1186/gb-2011-12-3-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin A.V., et al. Assembly reconciliation. Bioinformatics. 2008;24:42–45. doi: 10.1093/bioinformatics/btm542. [DOI] [PubMed] [Google Scholar]