Molecular-level engineering of protein physical hydrogels for predictive sol-gel phase behavior (original) (raw)
. Author manuscript; available in PMC: 2012 Oct 10.
Published in final edited form as: Biomacromolecules. 2011 Sep 2;12(10):3406–3411. doi: 10.1021/bm200959e
Abstract
Predictable tuning of bulk mechanics from the molecular level remains elusive in many physical hydrogel systems due to the reliance on non-specific and non-stoichiometric chain interactions for network formation. We describe a Mixing-Induced Two-Component Hydrogel (MITCH) system, in which network assembly is driven by specific and stoichiometric peptide-peptide binding interactions. By integrating protein science methodologies with simple polymer physics model, we manipulate the polypeptide binding interactions and demonstrate the direct ability to predict the resulting effects on network crosslinking density, sol-gel phase behavior, and gel mechanics.
Keywords: physical hydrogels, protein engineering, biomaterials
Introduction
Recent innovations in polymer science have enabled the design of novel physical hydrogels for a range of biomedical applications including tissue engineering scaffolds and injectable cell- and drug-delivery vehicles. In these systems, the polymeric network is crosslinked by transient, non-covalent associations such as hydrophobic interactions, chain entanglements, hydrogen bonds, ionic bonds, or microcrystal formation.1 Bulk mechanical properties are governed at the molecular level by the strength and number of these transient interactions. While several strategies have been reported to exert control over these two parameters, few materials have been developed to allow the direct translation of molecular-level design parameters to macroscopic hydrogel viscoelasticity and sol-gel phase transitions. This is because most physical hydrogels utilize assembly interactions that are either non-specific (e.g. physical entanglement between poly(vinyl pyrrolidone) and chitosan derivatives2) or non-stoichiometric (e.g. host-guest complexation between ß-cyclodextrin and hydrophobic moieties,3,4 stereocomplexation of polylactic acid enantiomers5,6 and the aggregation of coiled-coil domains7).
We recently reported a protein-engineered physical hydrogel system termed MITCH (Mixing-Induced Two-Component Hydrogel) that assembles through specific and stoichiometric chain interactions.8 The hydrogels are composed of two recombinant protein polymers, each containing multiple repeats of the CC43 WW domain or its proline-rich PPxY ligand, which interact with a 1:1 stoichiometric ratio (Fig. 1a).9 Spontaneous network assembly is driven by specific recognition between these peptides, thus allowing sol-gel transition to occur by simple mixing. These materials were previously demonstrated to enable cell encapsulation at constant physiological conditions – a significant advantage over commonly used biomaterials (e.g. collagen and Matrigel™) that involve cell exposure to pH and temperature shifts for gel formation. Here, we combine protein science techniques and polymer physics theory to show that the bulk viscoelastic properties and sol-gel phase transitions of MITCH can be predicted by studying and controlling polypeptide binding interactions at the molecular level.
Figure 1.

(a) Schematic of MITCH. Protein polymers C7 and P9 contain repeats of the CC43 WW domain and proline-rich peptide. Mixing C7 and P9 results in hydrogel formation. (b) Microrheology of C7, P9, and C7:P9 (10.0% w/v). (c) Specific viscosity versus concentration of C7 and P9, showing scaling properties in the dilute, semi-dilute unentangled, and semi-dilute entangled regimes.
While a suite of protein science methodologies is routinely available for molecular biology research, its capability has not been deeply exploited in the field of polymer technology. Here we control chain-chain interactions through variations in temperature, stoichiometric ratio, and polymer concentration, and use protein science tools to interrogate the resulting polypeptide chain behavior. We further demonstrate how the coupling of these molecular-level measurements with polymer physics theory allows the predictable translation of these dynamic chain interactions into macroscopic viscoelastic properties.
Materials and Methods
Synthesis and Purification of Engineered Proteins
The engineered proteins were cloned, synthesized, and purified as previously reported.8 Briefly, DNA sequences encoding the C7 and P9 block copolymers were cloned into the pET-15b vector (Novagen) and transformed into the BL21(DE3) Escherichia coli host strain (Novagen). Recombinant proteins were expressed following isopropyl β-D-1-thiogalactopyranoside (IPTG) induction, purified via specific binding of N-terminal polyhistidine tags to Ni-nitrilotriacetate resin (Qiagen), buffer exchanged, and concentrated in phosphate-buffered saline (PBS) by centrifugation across 10 kDa MWCO Amicon Ultracel-10K filter units (Millipore). Protein identity and purity were confirmed by gel electrophoresis, MALDI-TOF mass spectrometry, and amino acid compositional analysis.
Particle Tracking Microrheology
Monodisperse fluorescent polystyrene tracer microspheres (d = 0.2 μm, Molecular Probes) in buffer TN (100 mM TrisHCl and 100 mM NaCl, pH 8.0) were mixed with sample protein solutions to a final particle volume fraction of 1.8 × 10−3. The absence of tracer aggregation was confirmed through dynamic light scattering (SI Fig. S3). The mixtures (10 μL) were pippetted immediately between a microscope slide and a coverslip separated by a 120 μm-thick SecureSeal Imaging Spacer (Grace Bio Labs). Silicon sealing grease (Corning, Co.) was used to seal the coverslip edges to minimize sample dehydration. Samples were equilibrated for at least two hours prior to particle tracking. An optical microscope (40× oil-immersion objective, Carl Zeiss, Inc.) with a high-speed CCD camera (frame rate = 30 Hz, iXON DV897; Andor Technology) was used to record the Brownian dynamics of tracer particles for 16.6 s, at the resolution of 0.25 μm/pixel. Approximately 100 particles were seen in each image. Particle positions in each frame were determined to sub-pixel accuracy by computing the brightness-weighted centroid within a circular mask of 13 pixels. Static errors in centroiding were minimized by spreading the tracer image over a sufficient number of pixels to reasonably represent the particle's brightness distribution.10 This was achieved by keeping illumination levels near the maximum allowable by detector saturation. Dynamic errors arising from the underestimation of tracer motion were minimized by choosing an exposure time (0.00002 s) that was infinitesimal relative to the time interval between successive movie frames.11
Stacks of images were analyzed using IDL software (version 7.0) to determine the 2D particle trajectories, ensemble-averaged mean-squared displacement (MSD), and viscoelastic parameters following the algorithms of Crocker and Grier.12 Multiple measurements were obtained from independent samples to ensure data reproducibility.
Bulk Rheology
Dynamic oscillatory rheology experiments were performed on an Anton Paar Physica MCR 301 rheometer with a parallel-plate geometry (d = 8 mm, gap = 0.15 mm). Samples were prepared by mixing equal volumes of 10.0% w/v P9 and 10.0% w/v C7 solutions, and loaded immediately onto the instrument. A humidity chamber was placed around the samples to prevent dehydration, and the temperature was maintained at 25 ± 0.02 °C by a Peltier temperature control unit. Gel formation was monitored by dynamic time sweep experiments (frequency = 0.5 Hz, strain = 5 %). Once the storage (G') and loss (G”) moduli had reached plateau values, strain sweeps (5 – 15 %) were performed at constant angular frequency (0.5 Hz), which was verified to be in the linear viscoelastic regime. The frequency dependence of G' and G”(ω) in the range of 30 – 0.05 Hz was subsequently measured under a constant 5% strain.
Dynamic Light Scattering
The hydrodynamic dimensions of C7 and P9 in PBS were determined at 25 °C by dynamic light scattering using the Nano-Series Zeta Sizer (Nano-ZS ZEN3600, Malvern Instruments, Worcestershire, United Kingdom), equipped with a standard 633 nm laser. Polymer diffusion coefficient was calculated from auto-correlated light intensity data and converted to hydrodynamic radius (RH) using the ZetaSizer software according to the Stoke-Einstein equation. Subsequently, RH was used to compute the overlap concentration (c*) assuming close-packed non-interacting hard spheres.
Circular Dichroism (CD)
Far-UV CD experiments were conducted on a model 202-01 circular dichroism spectrometer equipped with a Peltier temperature-controlled cell holder (Aviv Biomedical) to probe the WW domain and proline-rich peptide secondary structures. Temperature scans (4 °C – 95 °C) were performed on 0.125 mg/ml of CC43 WW domain (denoted as C1) and 0.125 mg/ml of proline-rich peptide (denoted as P1) in 50 mM phosphate buffer (pH 7.5) at 230 nm and 200 nm, respectively.
Isothermal Titration Calorimetry (ITC)
ITC experiments were carried out on an ITC200 isothermal titration calorimeter (Microcal, GE Healthcare). Calorimeters were electrically calibrated according to the manufacturer's instructions. In one set of experiments, 10 μM of C7 and 500 μM of P1 were loaded into the calorimeter cell and titration syringe, respectively. In a separate set of experiments, 50 μM of C7 and 500 μM of P9 were loaded to the cell and syringe, respectively. Titrations were carried out using 15–20 injections at 3–4 minute intervals, at a constant temperature of 25 °C. All proteins solutions were prepared using the same phosphate buffered saline (PBS) solution. Raw ITC thermographs were analyzed using the Origin software (version 7.0), assuming independent and identical binding site model.
Results and Discussion
Harnessing recombinant DNA technology, the C7 and P9 modular constructs (SI Fig. S1a) were expressed with high fidelity in Escherichiacoli, producing polymers with exact and monodisperse molecular weights (MW), as verified through gel electrophoresis and dynamic light scattering (DLS) (SI Figs. S1 and S2). For physical hydrogels, the scaling properties of the polymer chains in a particular solvent can dictate sol-gel phase transitions. Owing to the high occurrence of acidic amino acids in the hydrophilic spacers, C7 and P9 are both polyelectrolytes, with isoelectric points of 4.46 and 3.60, respectively. In pure water, polyelectrolyte chains assume an extended configuration because of intramolecular electrostatic repulsion. In physiological solvents of intermediate salt concentration such as phosphate-buffered saline (PBS), charges are screened by counterions, causing the polyelectrolyte chains to coil into an ideal random-walk configuration. The scaling properties of these screened polyelectrolytes are analogous to those of self-avoiding neutral polymers in a good solvent.13 In a dilute solution, individual chains behave as non-interacting spheres with a hydrodynamic volume of VH = (4/3)π_RH3_, where RH is the hydrodynamic radius. As measured by DLS, C7 and P9 have RH of 6.2 ± 0.7 nm and 5.3 ± 0.4 nm, respectively.
RH values enable estimation of the critical overlap concentration (c*) at which chains begin to touch, marking the transition from the dilute to the semi-dilute regime. Using the relation c* = MW/NAVH (where NA is Avogadro's number), C7 and P9 chains were predicted to start overlapping at 10.1 ± 3.5% w/v and 10.0 ± 2.3% w/v, respectively. Beyond the overlapping point, the chains will begin to entangle as the concentration exceeds ce, the critical concentration that separates the semi-dilute unentangled and semi-dilute entangled regimes. Within the observed experimental time scale, non-specific chain entanglements will alter network viscoelasticity, thus confounding the tuning of material mechanics by specific domain association. To prevent these non-specific interactions, C7 and P9 solutions were mixed at or below their respective c* values in all hydrogel preparations.
Passive microrheology, a technique that measures material viscoelasticity by probing the thermal motion of embedded tracer particles,12,14,15 was used to experimentally observe the scaling properties of C7 and P9 polymers in different concentration regimes. The power-law dependence of particle mean-squared-displacement (MSD) with lag time (i.e. <Δ_r2_(τ)> ~ τα) can vary from α = 1 to 0, representing a continuum from purely diffusive (Newtonian) to purely elastic properties, respectively. Prior to mixing, individual C7 and P9 polymer solutions at 10.0% w/v exhibited sub-diffusive behaviors, having α values of 0.92 and 0.88, respectively (Fig. 1b). Deviations from the ideal Newtonian behavior (α = 1) were attributable to the storage of elastic energy as the polymers are disturbed from their equilibrium coiled configurations under shear.16 Conversely, tracer particles in the C7:P9 mixture exhibited restricted trajectories with nearly time-independent MSD (α = 0.01), demonstrating the transition into the elastic regime and the formation of a hydrogel. High throughput microrheology was used to probe the scaling behavior of specific viscosity (η_sp_ = η/η_o_ − 1, where η_o_ is the viscosity of pure solvent) across a range of C7 and P9 solution concentrations (Fig. 1c). In agreement with solution dynamics theory, C7 and P9 both appeared to traverse three regimes (dilute, semi-dilute unentangled, and semi-dilute entangled) as the solutions became more concentrated. In each regime, the experimentally-obtained scaling closely approximated the theoretical values for neutral polymers in a good solvent17 (η_sp_ ~ c1.0 for c < _c_*, η_sp_ ~ _c1.3_ for _c_* < _c_ < _ce_, and η_sp_ ~ _c3.9_ for _c_ > ce), confirming that the C7 and P9 polyelectrolytes behaved as neutral, self-avoiding random coils in physiological buffer. Below 10.0% w/v, C7 and P9 obeyed the scaling behavior of dilute polymers, in excellent agreement with the theoretical predictions of c* from DLS measurements of molecular radii.
The network structure of MITCH is held together by transient bonds (van der Waals interactions and hydrogen bonds) that are responsive to changes in temperature, pH, and ionic strength. In the clinical setting, variations in pH and ionic strength can be minimized by using buffered solutions. Temperature fluctuations, on the other hand, are more difficult to control and can trigger thermal denaturation of protein-based materials. We used circular dichroism (CD), a widely established method for identifying peptide secondary structures, to assess the operational temperature limits of MITCH. The far-UV CD spectrum of a WW domain exhibits a weak positive ellipiticity centered at 230 nm, characteristic of antiparallel ß-sheet secondary structures.18 Temperature scans of a single C domain at 230 nm demonstrated the maintenance of stable domain conformation up to 52 °C (Fig. 2a). The PPxY sequence in the P domain adopts a left-handed poly(_L_-proline) II helical structure, signified by a strong negative peak at 200 nm.19 CD analysis at 200 nm produced a linearly increasing thermal titration curve, reflecting a gradual transition towards an unordered structure. Based on these CD analyses, C7 and P9 solutions were predicted to remain active for hydrogel formation at temperatures below 52 °C.
Figure 2.
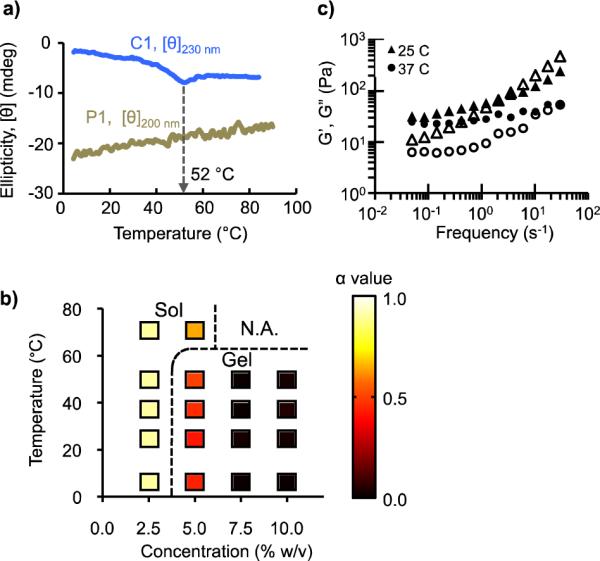
Hydrogel formation as a function of temperature. (a) Thermal stability of C and P domains monitored by circular dichroism (CD) at 230 nm and 200 nm, respectively. (b) Sol-gel phase diagram based on microrheology. The dotted lines were drawn as a visual guide to delineate sol formulations (α > 0.55) from gel formulations (α < 0.55). (c) Bulk oscillatory rheology of C7:P9 (10.0% w/v), showing storage (G', closed symbols) and loss (G”, open symbols) moduli.
To test these predictions, we used microrheology to construct a sol-gel phase diagram mapping the effects of polymer concentration and temperature on material viscoelasticity (Fig. 2b). The sol-gel phase boundary can be defined using the criteria that at the critical gelation point, the MSD exhibits an invariant power law scaling over all lag times (i.e. <Δ_r2_(τ)> ~ τ_a_, where a is the critical relaxation exponent of the gel network) (refs 20,21). Precise determination of the critical exponent a requires the measurement of MSD over a much wider range of time scales than experimentally feasible. However, assuming that C7 and P9 behave as self-avoiding, dilute polymers with hydrodynamic interactions, the underlying MITCH dynamics can be approximated to obey Zimm-like behavior in a good solvent, for which sol-gel transitions occur at a = 0.55 (ref 21,22). Using this critical gelation criteria, samples whose MSD plots lie above and below <Δ_r2_(τ)> ~ τ_0.55_ were classified as sol and gel, respectively (SI Fig. S4a). As expected, the probability of gel formation correlated positively with concentration. At 2.5% w/v, C7:P9 mixtures behaved as free-flowing solutions (α ≈ 1). As concentration was increased, the α value quickly diminished, approaching elastic values (α ≈ 0) at 10.0% w/v. Weak gels formed at 5.0% w/v transitioned into the sol phase as the temperature was raised above 50 °C, in agreement with the molecular-level CD prediction. At this elevated temperature, the C domain begins its thermal unfolding and is no longer an active binding partner for gel formation. Domain unfolding exposes internal hydrophobic amino acid residues, which can act as nucleation sites for non-specific aggregation at high concentrations. Consistent with this, white precipitates were observed in the 7.5% and 10.0% w/v mixtures at 70 °C.
While microrheology is useful for rapid characterization of viscoelastic properties, the impracticality of tracking particles over infinite time intervals may introduce significant errors in the computation of G' and _G_”, especially near the frequency extremes.23 Bulk oscillatory rheology was thus performed in parallel to accurately compare the frequency dependence of G' and _G_” values at 25 °C and 37 °C (Fig. 2c). In the low frequency range, G' was greater than _G_” at both temperatures, reflecting a gel state. In the high frequency range, _G_” was observed to exceed G', highlighting the predominance of viscous dissipation by the solvent molecules over elastic storage by the network, as commonly observed for weak physical hydrogel systems such as MITCH.22,24 The low-frequency plateau storage modulus of 10.0% w/v C7:P9 gels at 25 °C was G' = 32 Pa, comparable to that of Matrigel, a cell transplantation vehicle widely-used in animal studies. As comparisons, other self-assembling peptide-based networks such as Puramatrix, leucine zipper hydrogels, and β-hairpin MAX1 hydrogels have storage modulus values of 5 Pa, 200 Pa, and 2500 Pa, respectively.7,25,26 Raising the temperature to 37 °C resulted in lower storage modulus across the measured frequency range, suggesting the weakening of gel network with temperature. Gel weakening was also observed through microrheology for 5.0% w/v mixtures across a temperature range of 4 – 50 °C (SI Fig. S4b). At higher temperatures, thermal excitation causes C:P interactions to be less stable and more transient, leading to a reduction in the number of crosslinks and consequently a decrease in storage modulus.
A key advantage of MITCH is the ability to predict how variations in molecular composition affect macroscopic gel behavior. In the C7:P9 gel network, crosslinking density is synonymous to the concentration of P domains that are bound ([_P_]bound). Crosslinking density is also inversely related to the molecular weight between crosslinks (Mc), which in turn is inversely proportional to the storage modulus (G') of a hydrogel.17 Thus, the storage modulus (G') of the MITCH system is directly proportional to [_P_]bound, which by virtue of 1:1 binding stoichiometry is numerically identical to [_C_]bound.
Utilizing this direct connection between molecular-level binding and gel modulus, we predicted how variations in protein concentration and C:P domain stoichiometric ratio would affect gel viscoelasticity by estimating [_P_]bound for two limiting cases. We first considered the case of “independent domains”, with the assumption that domain concatemerization does not impose geometric restrictions. In this model, all C and P domains are freely accessible for binding and have an equilibrium association constant (Kb) of 5.88 ×105 M−1, identical to the monomeric domains.9 Treating the C and P domains as receptor and ligand, respectively, the non-cooperative equilibrium binding model (Eq. 1) (ref 27) was used to calculate [_P_]bound for mixtures with varying concentrations of C7 and P9 polymers and hence different relative proportions of independent C and P domains (denoted as C:P ratios).
Here, r is the fraction of bound ligand per total receptor, [_L_]free is the concentration of free ligand, and n is the number of binding sites on the receptor. For the independent domains case, r = [_P_]bound/[_C_]total, [_L_]free = [_P_]total − [_P_]bound, and n = 1. Using this model, the maximum extent of binding [_P_]bound, max was calculated to occur at a C:P ratio of 1.00 (Fig. 3a, solid black line), suggesting that if all domains were freely accessible, mixtures with equal numbers of C and P domains would yield the stiffest hydrogels. This prediction is intuitively consistent with the inherent 1:1 binding stoichiometry, as any deviation from this ratio would lead to excess binding domains that do not contribute to the network structure.
Figure 3.

Hydrogel formation as a function of C:P ratio. (a) Comparison of network structure (10.0% w/v). Left axis: predictions of crosslink density assuming independent domains (solid black line) or chains (dotted black line). Right axis: microrheology measurements (red squares). Numerical values give the maxima. (b) Sol gel phase diagram. The dotted lines were drawn as a visual guide to delineate sol formulations (α > 0.55) from gel formulations (α < 0.55).
In a real protein hydrogel, the assumption of “independent domains” may not be realized because receptor ligand interactions are influenced by spatial context. Concatemerization of C and P domains into C7 and P9 polymers may cause the effective C:P binding stoichiometry to depart from the ideal, freely diffusible scenario. Owing to constraints such as chain connectivity and spacer rigidity, the binding domains have limited degrees of kinetic freedom and may be sterically inaccessible for binding. Modeling C7 as a receptor with identical but independent binding sites, isothermal titration calorimetry (ITC) analysis with single P peptides (P1) revealed 7.46 apparent binding sites per C7 molecule (Table 1). Thus, all C domains in C7 were functional and available for binding to free ligand. However, when titration was performed against P9, only 2.32 effective binding sites in C7 were observed, supporting the hypothesized reduction of binding accessibility that would ensue from domain concatemerization. Taking into account the domain repeat frequency within each polymer, the apparent C:P binding stoichiometry in C7:P9 mixtures was calculated to be 0.34. That is, approximately one in three P domains in P9 is available for effective interaction with each of the C domains in C7.
Table 1.
Quantification of C7 binding to P1 or P9.
| Ligand | Kb | ΔH | ΔS | Apparent binding sites | Apparent C:P binding stoichiometry |
|---|---|---|---|---|---|
| (M−1 × 103) | (cal/mol × 103) | (cal/mol/K) | (#/C7 polymer) | ||
| P1 | 8.52 ± 6.36 | −7.15 ± 0.51 | −6.0 | 7.46 ± 2.96 | 0.94 ± 0.62 |
| P9 | 814 ± 258 | −14.77 ± 0.50 | −22.5 | 2.32 ± 0.06 | 0.34 ± 0.01 |
With this insight, Eq. 1 was used to calculate [P]bound for the second limiting case of “independent chains”, which takes into account potential constraints due to domain connectivity. Here, C7 was treated as a receptor with n = 2.32 effective binding sites, each occupied by one P9 ligand (i.e. r = [P9]bound/[C7]total). With this binding model, [P]bound, max was predicted to occur at a C:P ratio of 0.34 for C7:P9 gels at 10.0% w/v (Fig. 3a, dotted black line). This ratio is numerically equal to the apparent C:P binding stoichiometry measured by ITC, reiterating the intuition that gels of highest storage modulus are formed when neither binding partner is in excess. Since the non cooperative equilibrium binding model (Eq. 1) always assumes that the ligand is monovalent, the limiting case of “independent chains” neglects the ability of P9 to bind more than one C7 receptor at a time. The disregard of P9 multivalency in this limiting case provides an underestimate of the effective C:P binding stoichiometry observed in actual C7:P9 networks.
From these theoretical analyses of molecular-level binding dynamics, we hypothesized that the stiffest hydrogels would form at a C:P ratio intermediate between the two limiting cases above. To verify, a sol-gel phase diagram mapping various C:P ratios and concentrations was constructed by microrheology (Fig. 3b). The observed phase boundary had an extremum at a C:P ratio of 0.50. Consistent with our hypothesis, this ratio was shifted downward from the ideal “independent domains” ratio of 1.00, but was higher than the 0.34 ratio predicted from the “independent chains” model. Across all concentrations, mixtures with a C:P ratio of 0.50 not only exhibited the highest degree of elasticity (i.e. having the lowest α values), but also formed the strongest network (i.e. having the lowest MSD values and the lowest creep compliance J(τ), SI Fig. S5). In an elastic material (α ≈ 0), MSD is inversely proportional to the storage modulus G'; thus, MSD−1 provides a measure of gel stiffness. The maximum gel stiffness for 10.0% w/v C7:P9 was found to occur at a C:P ratio of 0.50 (Fig. 3a, red squares), which is between the two ideal predictions. The agreement between these microrheological observations and the molecular binding dynamics predictions demonstrates MITCH as a platform that enables the direct translation of molecular-level interactions to macroscopic level sol-gel mechanics.
Conclusions
While many novel physical hydrogel crosslinking strategies have been developed, the interaction mechanisms typically lack molecular specificity. Consequently, the mechanical properties of the resulting hydrogels often rely on empirical tuning. Protein-engineered block copolymers are ideal model systems for tapping the synergy between protein science and polymer theory in the creation of hydrogels with molecular-level control of bulk mechanics. As an example, the MITCH system, which is designed to have specific and stoichiometric receptor-ligand interactions, enables direct bottom-up predictions of crosslinking density, sol-gel phase behavior, and gel mechanics. The MITCH materials are currently being explored for applications in tissue engineering and cell delivery, where viscoelasticity is a critical parameter that dictates cellular function.
Supplementary Material
1_si_001
ACKNOWLEDGMENT
W.M. is supported by a Stanford Graduate Fellowship. We acknowledge funding from NIH, grants 1DP2OD006477-01 and R01DK085720-01.
Footnotes
Supporting Information Available. Contents: amino acid sequences, further polymer characterizations. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Tsitsilianis C. Soft Matter. 2010;6:2372. [Google Scholar]
- (2).Abashzadeh S, Hajimiri MH, Atyabi F, Amini M, Dinarvand R. Journal of Applied Polymer Science [Google Scholar]
- (3).van de Manakker F, van der Pot M, Vermonden T, van Nostrum CF, Hennink WE. Macromolecules. 2008;41:1766. [Google Scholar]
- (4).Koopmans C, Ritter H. Macromolecules. 2008;41:7418. [Google Scholar]
- (5).Lim DW, Park TG. Journal of Applied Polymer Science. 2000;75:1615. [Google Scholar]
- (6).de Jong SJ, van Eerdenbrugh B, van Nostrum CF, Kettenes-van den Bosch JJ, Hennink WE. J Control Release. 2001;71:261. doi: 10.1016/s0168-3659(01)00228-0. [DOI] [PubMed] [Google Scholar]
- (7).Petka WA, Harden JL, McGrath KP, Wirtz D, Tirrell DA. Science. 1998;281:389. doi: 10.1126/science.281.5375.389. [DOI] [PubMed] [Google Scholar]
- (8).Wong Po Foo CT, Lee JS, Mulyasasmita W, Parisi Amon A, Heilshorn SC. Proc Natl Acad Sci U S A. 2009;106:22067. doi: 10.1073/pnas.0904851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Russ WP, Lowery DM, Mishra P, Yaffe MB, Ranganathan R. Nature. 2005;437:579. doi: 10.1038/nature03990. [DOI] [PubMed] [Google Scholar]
- (10).Crocker JC, Hoffman BD. Methods in Cell Biology. Vol. 83. Elsevier; 2007. [DOI] [PubMed] [Google Scholar]
- (11).Savin T, Doyle PS. Biophys J. 2005;88:623. doi: 10.1529/biophysj.104.042457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Crocker JC, Grier DG. Journal of Colloid and Interface Science. 1996;179:298. [Google Scholar]
- (13).Dobrynin AV, Colby RH, Rubinstein M. Macromolecules. 1995;28:1859. [Google Scholar]
- (14).Mason TG, Ganesan K, vanZanten JH, Wirtz D, Kuo SC. Physical Review Letters. 1997;79:3282. [Google Scholar]
- (15).Schultz KM, Baldwin AD, Kiick KL, Furst EM. Soft Matter. 2009;5:740. doi: 10.1039/b818178k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Rouse PE. Journal of Chemical Physics. 1953;21:1272. [Google Scholar]
- (17).Rubinstein M, Colby RH. Polymer Physics. 1 ed. Oxford University Press; USA: 2003. [Google Scholar]
- (18).Koepf EK, Petrassi HM, Sudol M, Kelly JW. Protein Sci. 1999;8:841. doi: 10.1110/ps.8.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bochicchio B, Tamburro AM. Chirality. 2002;14:782. doi: 10.1002/chir.10153. [DOI] [PubMed] [Google Scholar]
- (20).Larsen TH, Furst EM. Phys Rev Lett. 2008;100:146001. doi: 10.1103/PhysRevLett.100.146001. [DOI] [PubMed] [Google Scholar]
- (21).Furst EM, Veerman C, Rajagopal K, Palla CS, Pochan DJ, Schneider JP. Macromolecules. 2006;39:6608. [Google Scholar]
- (22).Doi M, Edwards SF. The Theory of Polymer Dynamics. Clarendon; Oxford: 1989. [Google Scholar]
- (23).Mason TG. Rheologica Acta. 2000;39:371. [Google Scholar]
- (24).Wirtz D. Annu Rev Biophys. 2009;38:301. doi: 10.1146/annurev.biophys.050708.133724. [DOI] [PubMed] [Google Scholar]
- (25).Allen P, Melero-Martin J, Bischoff J. J Tissue Eng Regen Med. 2011;5:e74. doi: 10.1002/term.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Schneider JP, Ozbas B, Kretsinger J, Rajagopal K, Pochan DJ. Macromolecules. 2004;37:7331. [Google Scholar]
- (27).Klotz IM, Hunston DL. Biochemistry. 1971;10:3065. doi: 10.1021/bi00792a013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1_si_001