The Deubiquitinase Activity of the Salmonella Pathogenicity Island 2 Effector, SseL, Prevents Accumulation of Cellular Lipid Droplets (original) (raw)
Abstract
To cause disease, Salmonella enterica serovar Typhimurium requires two type III secretion systems that are encoded by Salmonella pathogenicity islands 1 and 2 (SPI-1 and -2). These secretion systems serve to deliver specialized proteins (effectors) into the host cell cytosol. While the importance of these effectors to promote colonization and replication within the host has been established, the specific roles of individual secreted effectors in the disease process are not well understood. In this study, we used an in vivo gallbladder epithelial cell infection model to study the function of the SPI-2-encoded type III effector, SseL. The deletion of the sseL gene resulted in bacterial filamentation and elongation and the unusual localization of Salmonella within infected epithelial cells. Infection with the Δ_sseL_ strain also caused dramatic changes in host cell lipid metabolism and led to the massive accumulation of lipid droplets in infected cells. This phenotype was directly attributable to the deubiquitinase activity of SseL, as a Salmonella strain carrying a single point mutation in the catalytic cysteine also resulted in extensive lipid droplet accumulation. The excessive buildup of lipids due to the absence of a functional sseL gene also was observed in murine livers during S. Typhimurium infection. These results suggest that SseL alters host lipid metabolism in infected epithelial cells by modifying the ubiquitination patterns of cellular targets.
INTRODUCTION
Salmonella enterica serovar Typhimurium has two type III secretion systems, encoded by the Salmonella pathogenicity islands SPI-1 and SPI-2, that are utilized to modify host cell function through the direct translocation of specialized bacterial proteins, termed effectors, into the host cell. Many genes associated with these secretion systems, encoding both structural and effector proteins, have been identified and linked directly to the virulence of S. Typhimurium (26). Genes within SPI-1 and SPI-2 were first identified by the characterization of mutant strains deficient in host cell invasion or highly attenuated in their ability to cause salmonellosis in mice (17, 24). The roles of SPI-1- and SPI-2-encoded proteins are crucial for Salmonella's ability to cause disease within its host, allowing for the invasion of and survival within host cells, respectively (16, 25).
Once intracellular, Salmonella remains within a vacuolar compartment that acquires lysosomal membrane glycoproteins and acidifies (18, 40). This compartment is termed the _Salmonella-_containing vacuole (SCV), a unique environment in which Salmonella organisms actively replicate. SPI-2 is induced within the acidified SCV, which results in the secretion of effector proteins into the host cytosol that promote intracellular replication through the alteration of host defense mechanisms, cellular trafficking events, and the integrity of the cytoskeleton (30). The proper maintenance and localization of this compartment are controlled in part by single SPI-2-secreted effectors, including SifA, SopD2, SseF, SseG, and SseJ (8, 14, 28, 29, 42, 46). The majority of work that has been carried out on the cell biology of individual effectors has been done in vitro using tissue culture infection models, as an in vivo epithelial cell infection model that allows us to study the molecular mechanisms of their action in the context of the infected host has been lacking.
A new in vivo epithelial cell infection model for Salmonella has been established recently within the gallbladder, enabling studies into novel aspects of Salmonella biology and pathogenesis (35). Briefly, wild-type Salmonella strains were found almost exclusively within the single epithelial cell layer of the gallbladder of orally infected mice, rarely translocating to the underlying lamina propria. This infection model recapitulates hallmarks of epithelial cell infection in tissue culture, such as the dependence of cell invasion upon a functional SPI-1 secretion system, the replication of Salmonella within a vacuole, and the colocalization of intracellular bacteria with lysosome-associated membrane glycoprotein 1 (LAMP-1). This model also revealed aspects of epithelial cell infection that could not otherwise be observed in vitro. For example, intracellular bacteria localized subnuclearly, displacing the nucleus to the apex of the cells while remaining contained within SCVs, where they actively replicated. This epithelial model can be exploited to examine previously inaccessible functional aspects of virulence factors of S. Typhimurium as they subvert host pathways to ensure the pathogen's survival.
In this study, we used the in vivo gallbladder epithelial cell infection model to identify novel features of the Salmonella effector, SseL. SseL is an SPI-2 effector that is encoded outside the SPI-2 pathogenicity island but is translocated into host cells via the SPI-2 secretion system (13). SseL has sequence similarities to cysteine proteases and possesses deubiquitinase activity (32, 43). Upon the infection of gallbladder epithelial cells in vivo, a Δ_sseL_ strain of Salmonella did not localize basolaterally, unlike wild-type Salmonella, and it also appeared filamentous and elongated in shape. Further investigation revealed that infection with Δ_sseL Salmonella_ greatly affected host cell lipid metabolism and that Δ_sseL-_infected host cells accumulated large amounts of lipids in the form of lipid droplets. This phenotype was restored upon complementation and was directly attributed to the deubiquitinase activity of SseL. This study identifies a new phenotype for the type III secreted effector, SseL, and suggests that SseL deubiquitinates target proteins to affect host cell lipid metabolism.
MATERIALS AND METHODS
Bacterial strains.
Salmonella enterica serovar Typhimurium strains SL1344 (Smr) and an unmarked, in-frame deletion mutant of sseL (13) were used in this study. The complementation of sseL was achieved by site-specific chromosomal insertion with Tn_7_ into S. Typhimurium SL1344 Δ_sseL_. A chloramphenicol-marked Tn_7_ delivery vector was created by subcloning a SacI frt-cat-frt, Klenow end-filled fragment from pFCM1 into the EcoRV site of pUC18R6KT-mini-Tn7T (11), yielding pMAC5, and then it was transformed into DH5αλpir. The orientation of the cat cassette was confirmed with EcoRI/NcoI restriction digests. The sseL gene was amplified from pWSK129-sseL_-2HA (13) and inserted into a cloning vector derived from pBC with a p15a origin of replication (pMAC2) using BamHI and XhoI restriction sites. The C-to-A mutation was introduced with the QuikChange site-directed mutagenesis kit (Stratagene) using primers described in reference 43. The genes encoding SseL-HA and SseL C/A-HA were subcloned from these vectors into the BamHI and XhoI sites of pMAC5. The Tn_7 vectors containing the sseL_-complementing constructs (in SM10λpir) were conjugated into S. Typhimurium SL1344 Δ_sseL in a 1:1:1 mating, along with SM10λpir pTNS2, for 6 h on LB agar. These matings were disrupted by vortexing, and dilutions were plated on LB agar containing chloramphenicol and streptomycin. Proper Tn_7_ transposition into the chromosome of SL1344 was checked as previously described (11). For a list of strains and plasmids used, see Table S3 in the supplemental material.
Mouse infections and sample collection.
Bacteria were grown overnight with shaking at 37°C in 10 ml LB broth supplemented with 100 μg/ml streptomycin. For inoculum preparation, 1 ml of culture was centrifuged at 7,000 × g for 5 min, and the pellet was resuspended in 1 ml of sterile 100 mM HEPES-0.9% NaCl, pH 8.0. For oral infections, washed bacteria were taken to a final volume of 10 ml with 100 mM HEPES-0.9% NaC, pH 8.0. Animals were housed at the UBC Animal Unit (University of British Columbia, Vancouver, Canada) and were provided with food and water ad libitum. Infections were performed in a biohazard facility in accordance with UBC animal protocols. Groups of 8-week-old female C57BL/6 mice (The Jackson Laboratory) were infected by oral gavage with 100-μl doses of 3 × 107 to 8 × 107 bacteria. Groups of five mice were sacrificed at 5 days postinfection, and tissue samples were taken for the quantification of bacteria. The gallbladders were collected and bile drained for bacterial enumeration. Tissue samples were placed in 1 ml sterile phosphate-buffered saline (PBS), weighed, and homogenized using a Mixer Mill MM 301 (Retsch GmbH, Germany) at a vibration frequency of 30/s for 10 min. The homogenates were placed on ice, and dilutions were plated on LB supplemented with streptomycin (100 μg/ml).
Metabolite extraction.
To extract metabolites from three groups of four pooled gallbladders each, including uninfected, wild-type infected, and Δ_sseL_ infected, 2 ml of chloroform-methanol (2:1) was added to each pool; the tissues then were homogenized using a tungsten bead and a Mixer Mill MM 301 (Retsch, Haan, Germany). The samples then were transferred to ice, and 1 ml of cold H2O was added. The samples were centrifuged at 3,250 × g for 10 min at 4°C. The upper phase was discarded, and another 500 μl of cold H2O was added. The samples were centrifuged again at 3,250 × g for 10 min at 4°C. The upper phase was discarded, and the lower phase was transferred to a new glass tube and dried under N2 gas. All extracts were kept at −80°C until further use.
FTICR MS.
Mass spectrometry (MS) analysis was performed essentially as previously described (4). Dried extracts were dissolved in 100 μl of 50% acetonitrile (in water) per 10 mg of the starting materials, vortexed, and cleared by centrifugation. All samples then were diluted 1:5 with 50% acetonitrile that contained either 0.2% formic acid (for positive-ion mode) or 0.5% ammonium hydroxide (for negative-ion mode) and a predefined amount of haloperidol, reserpine, and electrospray (ES) tuning mix solution as standard compounds for internal mass calibration. The resultant solutions subsequently were infused into an Apex-Qe 12-Tesla hybrid quadrupole Fourier transform ion cyclotron resonance MS (FTICR MS) (Bruker Daltonics, Billerica, MA) using a syringe pump (KD Scientific, Holliston, MA) at a flow rate of 2.5 μl/min. The FTICR MS system was equipped with an Apollo II electrospray ionization (ESI) source, a quadrupole mass filter, and a hexapole collision cell at the front end of the ICR cell. Full-scan mass spectra were recorded within m/z 150 to 1,000 with broadband detection and a transient size of 1,024 kilobytes per s. Other typical MS parameters included capillary electrospray voltage of 3,750 V, spray shield voltage of 3,450 V, source ion accumulation time of 0.1 s, and collision cell ion accumulation time of 0.2 s. Each sample was infused and detected in positive and negative-ion modes, and each mass spectrum was acquired from an accumulation of 200 scans with duplicate acquisitions.
Metabolomic data processing.
Mass spectra were batch processed using a custom VBA script (21) within the instrument vendor's data processing software package, DataAnalysis. First, all of the raw mass spectra acquired in positive and negative-ion modes were internally calibrated with the reference masses of the spiked standard compounds and a ubiquitous known contaminant, N-butylbenzensulfonamide. Monoisotopic peaks corresponding to the isotopic distribution patterns then were automatically determined, and those with signal-to-noise ratios above 3 were picked. The m/z values subsequently were converted to neutral masses by subtracting 1.007276 for positive-ion mode or adding 1.007276 for negative-ion mode. The resulting mass lists from the individual mass spectra within each paired sample group were further processed with another custom software program developed with LabVIEW (National Instruments, Austin, TX). The first step of this program is to remove the adduct ions, e.g., (M+Na)+ and/or (M+K)+ in positive-ion mode and (M+Cl)− in negative-ion mode, from the mass lists based on the expected mass differences for these ions within 2 ppm. The peak intensity of each unique monoisotopic neutral mass then was normalized to the intraspectrum total ion intensity. Masses observed in at least 50% of the spectra from each paired sample group were aligned into unique masses (metabolite features) from the masses that matched within 2 ppm. Finally, a two-dimensional data matrix (neutral mass versus peak intensity) was generated for each group and saved in a tab-delimited text format. To detect metabolite differences between the different sample groups, the masses were filtered in each list for metabolites that were present in one set of samples but not the other. Additionally, the intensities were averaged for each unique mass in each set of biological replicates, and we calculated the ratios between the sample groups. To assign possible metabolite identities to the masses present in only one set or showing at least 2-fold changes, the monoisotopic neutral masses of interest were queried against MassTrix (http://masstrix.org) software designed to incorporate mass queries into metabolic pathways (47). Masses were searched against the Mus musculus database within an error of 3 ppm.
Immunohistochemistry and Oil-Red-O staining.
Tissue sections used for microscopy were prepared by Wax-it Histology Services Inc., Vancouver, Canada. Gallbladders from infected and uninfected control mice were removed and drained of bile. For immunofluorescent antibody localization, tissue samples were fixed in 3% paraformaldehyde (in PBS) for 3 h at room temperature (RT) and then washed three times with PBS. Five-μm-thick tissue sections were immediately prepared from frozen samples and mounted on glass slides. The slides were treated for 5 min in 0.2% Triton X-100 in PBS; this was followed by three 10-min washes with PBS. The tissues were blocked for 20 min with 5% normal goat serum (NGS)-0.5% bovine serum albumin (BSA) in PBS (GB-PBS) and then incubated with the primary antibodies overnight at 4°C in GB-PBS containing 0.1% Tween 20. The next day, the slides were washed three times for 10 min in 0.5% BSA-0.1% Tween 20 in PBS (TB-PBS) and incubated with fluorescent secondary antibodies for 2 h at RT and with BODIPY for 5 min at RT. The samples were washed as described above and mounted with ProLongGold hard-set mounting medium containing 4′,6′-diamidino-2-phenylindole (DAPI) (P36931; Invitrogen). The antibodies used in this study were anti-Salmonella rabbit polyclonal (1:1,000 in GB-PBS; 240984; BD Biosciences) and goat-anti-rabbit conjugated to Alexa 568 (1:1,000 in GB-PBS; Invitrogen). BODIPY 493/503 (D3922; Invitrogen) was diluted 1:1,000 in PBS from a saturated stock prepared in ethanol. Actin was visualized by staining with phalloidin conjugated to Alexa 488 as previously described (35). Fluorescence was visualized using an Olympus IX81 microscope. Tissues for Oil-Red-O staining were processed and stained by Wax-It Histology Services Inc. and analyzed with a Zeiss Axioskop 2 microscope.
Electron microscopy and toluidine blue staining.
Electron microscopy and toluidine blue staining were performed as previously described (19). Briefly, gallbladders were removed, the bile drained, and the organs placed in a 1.5% paraformaldehyde-1.5% glutaraldehyde fixative solution in 0.1 M cacodylate buffer (pH 7.3) for 3 h. Samples were washed three times with buffer and then postfixed with 1% OsO4 in 0.1% cacodylate for 1 h on ice. Samples were washed three times in double-distilled water (ddH2O), stained en bloc with 1% aqueous uranyl acetate for 1 h, and then washed again three times in ddH2O. The tissues were dehydrated through an ascending concentration series of ethanol solutions, infiltrated through propylene oxide-EPON 812 resin mixtures into 100% EPON 812, and then embedded in EPON 812. Sections were cut at 1-μm thickness, mounted on glass slides, stained with toluidine blue, and then photographed on a Zeiss Imager.A1 microscope. Thin sections were cut, mounted on grids, stained with lead citrate and uranyl acetate, and then visualized using a Phillips 300 electron microscope operated at 60 kV.
Chloroform-methanol extraction of liver samples.
Liver samples (∼500 mg) were placed in 1 ml sterile PBS, weighed, and homogenized using a Mixer Mill MM301 (Retsch GmbH, Germany) at a vibration frequency of 30/s for 10 min. Homogenates were extracted with a 3-fold volume excess of chloroform-methanol (2:1) by mixing in a Mixer Mill MM301 (for 10 min at a vibration frequency of 25/s; Retsch GmbH, Germany) and subsequent centrifugation at 10,000 × g for 5 min at room temperature. The organic phase was transferred to a new, weighed tube, and samples were dried overnight in a vacuum concentrator. Dried organic material was weighed using an analytical balance (Sartorius). Results were calculated as mg of dry chloroform-methanol-soluble material per mg of wet liver tissue.
FITCR MS data accession number.
The mass spectrometry data have been deposited in the GEO database (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE27604.
RESULTS
Lack of SseL results in elongated, filamentous intracellular Salmonella.
SseL is an SPI-2 type III effector that is translocated into host cells during intracellular infection; however, its exact role in the systemic phase of disease during murine infections has been contested (13, 32). To investigate the function of SseL in vivo, we performed side-by-side infections with wild-type Salmonella and a mutant strain lacking the SseL protein (Δ_sseL_). The Δ_sseL_ strain was able to reach and colonize systemic sites, including the liver, spleen, mesenteric lymph nodes (MLNs), and gallbladder, to wild-type levels after oral infection (Fig. 1A). These data suggest that SseL alone, along with many other individual SPI-2 effectors, is not essential for Salmonella colonization in vivo (10).
Fig. 1.

Δ_sseL_ mutant strain of Salmonella is able to colonize systemic sites to wild-type levels after oral infection and is characterized by an elongated, filamentous phenotype when intracellular. (A) Bacterial counts after oral infections are presented as CFU per mg of tissue. Error bars represent standard errors from the means; all mice were sacrificed on day 5 postinfection (n_=10 to 20). There are no statistical differences between wild-type and Δ_sseL strain counts in the liver, spleen, mesenteric lymph nodes (MLN), or bile. Bacterial counts of bile are used as an indication of gallbladder colonization levels. (B) Immunostaining of uninfected, wild-type-infected, and Δ_sseL_-infected gallbladder sections collected at day 5 postinfection. Bacteria are shown in red, actin in green, and cell nuclei in blue. Scale bars indicate 10 μm.
Differences between intracellular wild-type and Δ_sseL_ Salmonella strains were investigated in the gallbladder epithelium, a site of active Salmonella replication (35). Sections of wild-type- and Δ_sseL_-infected gallbladders were analyzed by immunofluorescent microscopy. We observed the abnormal localization of Δ_sseL Salmonella_ within infected cells (Fig. 1B). Whereas wild-type Salmonella organisms typically cluster and localize to the basolateral area of the cell, thereby displacing the nucleus toward the apex (35), in Δ_sseL_-infected cells the bacteria displayed a filamentous, elongated phenotype, and cell nuclei remained basolaterally located (Fig. 1B). The elongation and filamentation of bacterial cells is a classic indicator of bacterial stress (22). Although it does not affect the overall number of bacteria in vivo, this suggested that the lack of SseL prevents intracellular bacteria from creating optimal environmental conditions. For example, infection with Δ_sseL_ Salmonella could lead to the lack of a signaling molecule or nutrient important for bacterial cell division/survival, to an increased presence of reactive oxygen species, or simply to an unfavorable nutritional environment. We decided to follow up on the possibility that the observed phenotype was the result of modifications to the metabolic environment during colonization and further analyzed the metabolic composition of Δ_sseL_- and wild-type-infected tissues.
SseL directly affects lipid composition and lipid levels within infected tissues.
To examine potential differences in metabolite levels and compositions in uninfected, wild-type-, and Δ_sseL_-infected cells, we used direct-infusion Fourier transform ion cyclotron resonance mass spectrometry (FTICR MS) to establish a comprehensive analysis of metabolites within gallbladder samples. FTICR MS creates a metabolic footprint of infected host tissues through the isolation and identification of individual metabolites to directly compare biochemical changes occurring in response to infection (4). Uninfected, wild-type-, and Δ_sseL_-infected gallbladders were isolated, and chloroform-methanol extracts from these tissues were analyzed by FTICR MS. The average intensities of all identified metabolites were calculated, and values from these samples were compared. Metabolites that showed changes of 2-fold or greater between sample groups were combined with those that were observed in wild-type- or Δ_sseL_-infected mice, but not in uninfected mice, for further analyses. Compared to uninfected gallbladders, wild-type-infected gallbladders showed a >2-fold enrichment in 152 nonredundant metabolites, and Δ_sseL_-infected gallbladders were >2-fold enriched in 163 nonredundant metabolites (see Table S1 in the supplemental material). Furthermore, a combined total of 131 nonredundant metabolites were identified between the two infection conditions directly (see Table S2 in the supplemental material). Identified metabolites ranged in mass from 158 to 947 Da.
The presumptive identities of masses of interest were assigned using MassTrix (http://masstrix.org), which relies on data from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/) (47). The examination of the identities of individual metabolites revealed a striking effect of both wild-type and Δ_sseL_ infection on lipid metabolism (Fig. 2). In particular, there were notable increases in the abundance of phospholipids and fatty acids within infected tissues. The Δ_sseL_ strain caused greater accumulation of phospholipids, whereas the wild-type strain had a stronger effect on fatty acids (Fig. 2). This trend was confirmed in a direct comparison of metabolites enriched in infections by one of the strains versus the other (Fig. 3). These data show that Salmonella infection drastically affects the lipid composition of infected gallbladders, and that lipid metabolism is differentially affected in gallbladders infected with the Δ_sseL_ strain, indicating that SseL has a direct influence on gallbladder lipid metabolism.
Fig. 2.

Metabolites enriched in wild-type- or Δ_sseL_-infected gallbladders and uninfected gallbladders. Pie charts indicate the categories and total numbers of metabolites that are >2-fold enriched in wild-type-infected or Δ_sseL_-infected gallbladders after direct comparison to uninfected gallbladders. PC, phosphatidylcholine; PS, phosphatidylserine; PE, phosphatidylethanolamine.
Fig. 3.

Metabolites enriched in wild-type- or Δ_sseL_-infected gallbladders. Pie charts indicate the categories and total numbers of metabolites that are >2-fold enriched in wild-type-infected or Δ_sseL_-infected gallbladders after direct comparison to each other. “Others” includes metabolites that could not be annotated using the databases outlined in Materials and Methods. PC, phosphatidylcholine; PS, phosphatidylserine; PI, phosphatidylinositol; PE, phosphatidylethanolamine.
In the liver, lipid accumulation previously has been described as a characteristic of Salmonella infection (20). Indeed, upon the visual inspection of centrifuged liver homogenates from infected animals, we observed what appeared to be lipid layers in both wild-type- and Δ_sseL_-infected samples. Interestingly, these potential lipid layers were considerably larger in homogenates from Δ_sseL_-infected livers (Fig. 4A). To confirm this observation and to quantify this effect, we used a mixture of chloroform and methanol to extract lipids from liver homogenates. The extracted organic fraction then was dried and weighed. In these experiments, uninfected control tissue contained ∼0.02 mg dry chloroform-methanol-soluble material per mg of wet liver tissue. After infection with either wild-type or Δ_sseL Salmonella_, this value was increased to ∼0.039 and 0.07 mg/mg tissue, respectively (Fig. 4B). This suggests that SseL also affects lipid accumulation in infected livers. To further analyze the impact of SseL on host lipid biology, we pursued a microscopic examination of intracellular lipid distribution within infected gallbladder epithelial cells in vivo.
Fig. 4.
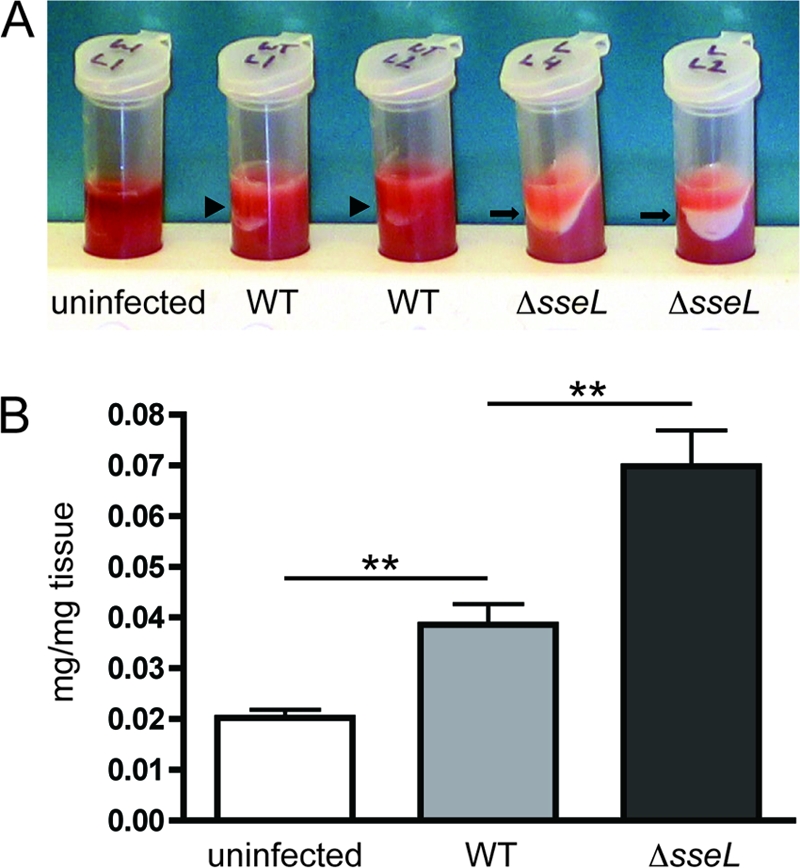
SseL prevents accumulation of lipids in the liver. (A) Photograph of centrifuged liver homogenates from uninfected, wild-type-infected, and Δ_sseL_-infected mice collected at day 5 postinfection. The lipid layer is indicated by arrowheads for the wild type and arrows for Δ_sseL_-infected tissues. (B) Weight of dry chloroform-methanol-soluble material (in mg) per weight of wet liver tissue (in mg) for uninfected, wild-type-infected, and Δ_sseL_-infected mice. Animals were infected orally, and livers were collected at day 5 postinfection; each group contained four mice. Error bars represent standard errors from the means. **, P < 0.01.
Lack of SseL causes accumulation of lipid droplets in infected epithelial cells in vivo.
Toluidine blue staining of infected gallbladder tissue allowed for the effective visualization of lipids within infected cells. In the case of wild-type Salmonella, tan-colored bodies indicative of lipids accumulated within infected cells but were not seen in an uninfected control (Fig. 5A and B). Further investigations revealed that upon infection with a Δ_sseL_ strain of Salmonella, cells accumulated massive amounts of lipids (Fig. 5C and D); in some cases, the entire cytoplasm was filled with lipid. This phenotype was apparent only within infected cells, further correlating it with the direct absence of the SseL effector protein and demonstrating that this phenomenon is cell endogenous and is not the result of a general host response to infection.
Fig. 5.

Absence of the secreted SseL effector protein results in massive accumulation of lipids within infected cells. Toluidine blue-stained gallbladder sections from an uninfected mouse (A), a wild-type-infected mouse at day 5 postinfection (B), and two representative Δ_sseL_ mutant strain-infected mice at day 5 postinfection (C and D). Arrows indicate lipids induced upon infection, and arrowheads indicate intracellular bacteria. The scale bar indicates 10 μm.
To confirm the identity of these lipid structures, gallbladder tissue sections were stained with Oil-Red-O, a lysochrome diazo dye used to stain neutral lipids, and the lipid droplet marker BODIPY, a fluorophore specific for cellular lipid droplets. Indeed, neutral lipid bodies of various sizes and shapes accumulated within Δ_sseL_-infected cells, as revealed by staining with both BODIPY and Oil-Red-O (Fig. 6B and C). The identity of these lipid bodies was confirmed as lipid droplet organelles by electron microscopy (Fig. 6A), as each lipid body was spherical in shape, uniformly stained gray, and not surrounded by a distinct intracellular membrane. These data show that infection with Salmonella leads to the formation of lipid droplets in infected gallbladder epithelial cells in vivo, and that this process is exacerbated in the absence of the SseL effector protein. This indicates that Salmonella organisms affect lipid droplet pathways, resulting in the redirection of lipids during colonization and/or replication, and that SseL influences these processes.
Fig. 6.

Lipids accumulating within Δ_sseL_-infected gallbladder epithelial cells are in the form of lipid droplets. (A) Electron micrographs of uninfected, wild-type-infected, and Δ_sseL_-infected gallbladder epithelial cells at day 5 postinfection. Arrows indicate lipid droplets; arrowheads indicate intracellular bacteria. Scale bars indicate 1 μm. (B) Immunostainings of gallbladder sections collected at day 5 postinfection. Bacteria are shown in red, BODIPY in green, and cell nuclei in blue; scale bars indicate 10 μm. (C) Oil-Red-O stainings of uninfected, wild-type-infected, and Δ_sseL_-infected gallbladder sections collected at day 5 postinfection. Lipid droplets stain red; scale bars indicate 50 μm.
The deubiquitinase activity of SseL inhibits lipid droplet accumulation in infected cells.
SseL has sequence similarities to cysteine proteases that have deubiquitinating activity and has been shown to specifically hydrolyze mono- and polyubiquitinated substrates in vitro, functioning as an active deubiquitinase within infected cells (32, 43). This deubiquitinase activity is dependent upon a catalytic cysteine, as the replacement of this cysteine residue with alanine abolishes the enzyme's ability to cleave ubiquitin (32, 43). To investigate the role of the deubiquitinase activity of SseL in the lipid droplet accumulation phenotype seen in infected gallbladder epithelial cells, a complemented strain of Δ_sseL_ was created that carries a chromosomal copy of sseL with a C-to-A point mutation under the control of its own promoter (referred to as Δ_sseL-sseL_ C/A). This point mutant strain was directly compared to a Δ_sseL_ strain equally complemented with a wild-type copy of the sseL gene (Δ_sseL-sseL_). Both strains were able to colonize susceptible mice to wild-type levels in an oral infection model and reached the gallbladder by day 5 postinfection (see Fig. S1 in the supplemental material).
Toluidine blue and BODIPY staining revealed lipid accumulation in gallbladders infected with the Δ_sseL-sseL_ strain to levels comparable to those seen during wild-type infections, confirming that the phenotype can be attributed directly to the presence of SseL (Fig. 7). In contrast, the C/A point mutant strain (Δ_sseL-sseL_ C/A) caused exaggerated lipid droplet accumulation, as was seen with the full-deletion Δ_sseL_ strain (Fig. 7). Again, lipid droplets only accumulated within infected cells. These data establish that lipid droplet accumulation is directly dependent on the deubiquitinase activity of SseL, suggesting that SseL targets ubiquitinated proteins to alter lipid metabolism during infection.
Fig. 7.

Deubiquitinase activity of SseL is essential to prevent accumulation of lipid droplets in infected cells. (A to C) Toluidine blue-stained sections of gallbladders from an uninfected mouse (A), a representative orally Δ_sseL_-_sseL_-infected mouse at day 5 postinfection (B), and a representative orally Δ_sseL_-sseL C/A-infected mouse at day 5 postinfection (C). Arrows indicate lipid droplets induced upon infection; the scale bar indicates 25 μm. (D) Immunostainings of the gallbladder epithelial cell layer. Tissues were collected at day 5 after oral infection. Bacteria are shown in red, and BODIPY is in green. Scale bars indicate 10 μm. L, lumen; LP, lamina propria.
DISCUSSION
More than a dozen SPI-2-encoded effectors have been identified in Salmonella; however, in many instances, the loss of a single effector has no apparent effect on the overall virulence of Salmonella in vivo (10). In this study, we show that a Salmonella strain lacking the SPI-2 effector, SseL, is able to colonize the host in a manner comparable to that of wild-type bacteria. However, at the cellular level, SseL deficiency results in an elongated, filamentous bacterial phenotype, accompanied by the accumulation of lipid droplets in infected gallbladder epithelial cells in vivo.
Lipid droplets are cellular organelles that can be found in nearly all eukaryotic organisms, including yeast, plants, and mammals (34). Recent studies have suggested that they are involved in various cellular functions, including cell signaling, membrane trafficking and biogenesis, the regulation of lipid metabolism, vesicular transport, and intracellular cholesterol dynamics (7, 27). Lipid droplets contain a hydrophobic core of neutral lipids, primarily triacylglycerols and sterol esters, and are surrounded by a monolayer of phospholipids with embedded proteins (38, 48). Proteomic studies have identified more than 100 lipid-droplet-associated proteins (6), which include Rab, SNARE, and motor and cytoskeletal proteins, supporting the active role of these organelles in various cellular functions (33).
Lipid droplets have previously been implicated in infectious disease. Several laboratories have shown that the hepatitis C core protein localizes to lipid droplets, and that this association is crucial for the proper assembly of viral particles (9, 36, 45). Similar results also have been reported for dengue virus (44). In addition, dengue virus has been shown to induce the autophagy-dependent breakdown of lipid droplets to raise cellular levels of ATP, a process necessary for efficient viral replication (23). It has furthermore been reported that some proteins secreted by the intracellular bacterium Chlamydia trachomatis localize to lipid droplets, and that lipid droplets are translocated into the lumen of the bacterial vacuole (inclusion) during infection, most likely as a means of bacterial nutrient acquisition (12, 31). For Salmonella, the SPI-2-secreted effector SseJ has been implicated in enhancing lipid droplet formation in tissue culture, a phenotype that has been attributed to this effector's cholesterol esterase activity (39). In this study, we showed that infection with wild-type Salmonella results in lipid droplet formation in vivo; however, this process is magnified in the absence of a functional SPI-2 effector, SseL. Interestingly, this effect is directly dependent on the deubiquitinase activity of SseL, suggesting that the deubiquitination of target proteins by SseL either prevents excessive cellular lipid droplet accumulation or serves to break down lipid droplets that are triggered as a general response to Salmonella infection.
We have recently shown that SseL interacts with human oxysterol-binding protein 1 (OSBP), a protein involved in various cellular processes, including cholesterol transport, signaling, lipid metabolism, and vesicular trafficking (41). Interestingly, it has been demonstrated that OSBP is also bound by the hepatitis C protein NS5A (1, 5). Thus, both Salmonella and hepatitis C virus (HCV) extensively modify lipid metabolism during the intracellular stages of infection and have dedicated proteins to the recognition of human OSBP. This suggests that OSBP-dependent processes are specifically targeted by these pathogens to generate a lipid environment favorable for pathogen survival inside the host. In the case of HCV, it could be shown that the host phosphorylation of OSBP negatively affects HCV replication, thus OSBP binding by NS5A could serve to reduce OSBP phosphorylation (2). Our present study demonstrates that deubiquitination plays a role in the alteration of lipid metabolism by Salmonella. However, we were unable to demonstrate the direct deubiquitination of OSBP by SseL (data not shown). It remains unclear whether the deubiquitination-mediated reduction of lipid droplets by SseL and its OSBP-binding capabilities represent independent functions or whether OSBP serves to guide SseL to its deubiquitination targets within the host cell.
The metabolomic analysis of infected gallbladders demonstrated a pronounced effect of Salmonella infection on host lipid metabolism. These data confirm a differential regulation of cellular lipid composition between wild-type- and _ΔsseL_-infected tissues. In line with the lipid droplet phenotype, we found a large number of phospholipid species enriched in _ΔsseL_-infected gallbladders (Fig. 2 and 3). Phospholipids are major components of the lipid droplet envelope, representing an important constituent of lipid droplets (37). Lipid droplets are known to protect cells from lipotoxic effects of unesterified lipids. In the presence of excess fatty acids and/or sterols, cells esterify and then sequester these potentially toxic lipids into lipid droplets (15). More fatty acids were detected during wild-type infection than from _ΔsseL_-infected gallbladders (Fig. 3). Further experiments showed that wild-type-infected gallbladder epithelial cells also contained significantly lower levels of intracellular lipid droplets than _ΔsseL_-infected cells. Therefore, wild-type Salmonella, through the action of SseL, may be diverting this pathway for a selective advantage. This could serve several purposes, including access by Salmonella to the host cell's lipid stores and/or the prevention of lipid droplet buildup within the cell, thereby preventing subsequent host inflammatory and/or stress responses.
We have recently published a metabolomic analysis of the Salmonella infection of the intestinal tract and liver and have shown that Salmonella infection disrupts lipid metabolism in all tissues tested (3). Although the exact compounds affected vary depending on the infected tissue, it is clear that the modulation of host lipid metabolism is a common theme during infection with this pathogen. The data presented here shed light on one of the mechanisms involved in this phenomenon; further studies will likely reveal other strategies used by Salmonella to modulate host lipid metabolic pathways.
In summary, using an in vivo epithelial cell infection model, we identified a single _Salmonella_-secreted SPI-2 type III effector, SseL, as a major regulator of lipid metabolism. This is the first study revealing a role for SseL in these processes, reinforcing the advancements this model provides. We found that SseL prevents the accumulation of lipid droplets in infected cells, and that this phenotype depends on the deubiquitinase activity of this effector. This suggests a mechanism by which Salmonella can intercept host lipid homeostasis by the direct modification of cellular ubiquitination patterns. This work provides new perspectives on how Salmonella is able to subvert cellular functions.
Supplementary Material
Supplemental Material
ACKNOWLEDGMENTS
We thank Kristie Keeney for helpful suggestions, as well as members of the Finlay laboratory for valuable discussions. Special thanks go to Aruna Somasiri (Wax-It Histology Services Inc.) for the preparation of microscopic slides.
E.T.A. is supported by a University Graduate Fellowship from the Armauer Hansen Memorial Fellowship (UBC). S.D.A. is supported by a postdoctoral fellowship from the Human Frontier Science Program Organization. L.C.M.A. is supported by a postdoctoral fellowship from the Canadian Institutes of Health Research. M.A.C. is supported by a Canadian Association of Gastroenterology/Canadian Institutes of Health Research/Ferring Pharmaceuticals Fellowship. J.A.G. is a CIHR New Investigator. This work was supported by the Canadian Institutes of Health Research through operating grants and also the Genome Canada and Genome British Columbia through platform funding.
B.B.F. is an HHMI International Research Scholar and the University of British Columbia Peter Wall Distinguished Professor.
Footnotes
▿
Published ahead of print on 29 August 2011.
REFERENCES
- 1.Amako Y., Sarkeshik A., Hotta H., Yates J., III, Siddiqui A. 2009. Role of oxysterol binding protein in hepatitis C virus infection. J. Virol. 83:9237–9246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amako Y., Syed G. H., Siddiqui A. 2011. Protein kinase D negatively regulates hepatitis C virus secretion through phosphorylation of oxysterol-binding protein and ceramide transfer protein. J. Biol. Chem. 286:11265–11274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antunes L. C., et al. 2011. Metabolomics reveals phospholipids as important nutrient sources during Salmonella growth in bile in vitro and in vivo. J. Bacteriol. 193:4719–4725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antunes L. C., et al. 2011. Impact of salmonella infection on host hormone metabolism revealed by metabolomics. Infect. Immun. 79:1759–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Auweter S. D., et al. 2011. Quantitative mass spectrometry catalogues salmonella pathogenicity island-2 effectors and identifies their cognate host binding partners. J. Biol. Chem. 286:24023–24035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartz R., et al. 2007. Dynamic activity of lipid droplets: protein phosphorylation and GTP-mediated protein translocation. J. Proteome Res. 6:3256–3265 [DOI] [PubMed] [Google Scholar]
- 7.Beller M., Thiel K., Thul P. J., Jackle H. 2010. Lipid droplets: a dynamic organelle moves into focus. FEBS Lett. 584:2176–2182 [DOI] [PubMed] [Google Scholar]
- 8.Beuzón C. R., et al. 2000. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 19:3235–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boulant S., Targett-Adams P., McLauchlan J. 2007. Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J. Gen. Virol. 88:2204–2213 [DOI] [PubMed] [Google Scholar]
- 10.Buckner M. M. C., Croxen M. A., Arena E. T., Finlay B. B. 2011. A comprehensive study of the contribution of Salmonella enterica serovar Typhimurium SPI2 effectors to bacterial colonization, survival, and replication in typhoid fever, macrophage, and epithelial cell infection models. Virulence 2:208–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi K. H., et al. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2:443–448 [DOI] [PubMed] [Google Scholar]
- 12.Cocchiaro J. L., Kumar Y., Fischer E. R., Hackstadt T., Valdivia R. H. 2008. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc. Natl. Acad. Sci. U. S. A. 105:9379–9384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coombes B. K., et al. 2007. SseL is a salmonella-specific translocated effector integrated into the SsrB-controlled salmonella pathogenicity island 2 type III secretion system. Infect. Immun. 75:574–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deiwick J., et al. 2006. The translocated Salmonella effector proteins SseF and SseG interact and are required to establish an intracellular replication niche. Infect. Immun. 74:6965–6972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farese R. V., Jr., Walther T. C. 2009. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell 139:855–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galán J. E. 1996. Molecular genetic bases of Salmonella entry into host cells. Mol. Microbiol. 20:263–271 [DOI] [PubMed] [Google Scholar]
- 17.Galán J. E., Curtiss III R. 1989. Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc. Natl. Acad. Sci. U. S. A. 86:6383–6387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia-del Portillo F., Finlay B. B. 1995. Targeting of Salmonella typhimurium to vesicles containing lysosomal membrane glycoproteins bypasses compartments with mannose 6-phosphate receptors. J. Cell Biol. 129:81–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guttman J. A., et al. 2006. Attaching and effacing pathogen-induced tight junction disruption in vivo. Cell Microbiol. 8:634–645 [DOI] [PubMed] [Google Scholar]
- 20.Hall G. A. 1975. A comparative study of liver changes produced by inoculating pregnant rats with Salmonella dublin or with its endotoxin. Br. J. Exp. Pathol. 56:216–222 [PMC free article] [PubMed] [Google Scholar]
- 21.Han J., et al. 2008. Towards high-throughput metabolomics using ultrahigh-field Fourier transform ion cyclotron resonance mass spectrometry. Metabolomics 4:128–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hazeleger W. C., Dalvoorde M., Beumer R. R. 2006. Fluorescence microscopy of NaCl-stressed, elongated Salmonella and Listeria cells reveals the presence of septa in filaments. Int. J. Food Microbiol. 112:288–290 [DOI] [PubMed] [Google Scholar]
- 23.Heaton N. S., Randall G. 2010. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 8:422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hensel M., et al. 1995. Simultaneous identification of bacterial virulence genes by negative selection. Science 269:400–403 [DOI] [PubMed] [Google Scholar]
- 25.Hensel M., et al. 1998. Genes encoding putative effector proteins of the type III secretion system of Salmonella pathogenicity island 2 are required for bacterial virulence and proliferation in macrophages. Mol. Microbiol. 30:163–174 [DOI] [PubMed] [Google Scholar]
- 26.Hueck C. J. 1998. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol. Mol. Biol. Rev. 62:379–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikonen E. 2008. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 9:125–138 [DOI] [PubMed] [Google Scholar]
- 28.Jiang X., et al. 2004. The related effector proteins SopD and SopD2 from Salmonella enterica serovar Typhimurium contribute to virulence during systemic infection of mice. Mol. Microbiol. 54:1186–1198 [DOI] [PubMed] [Google Scholar]
- 29.Kuhle V., Hensel M. 2002. SseF and SseG are translocated effectors of the type III secretion system of Salmonella pathogenicity island 2 that modulate aggregation of endosomal compartments. Cell Microbiol. 4:813–824 [DOI] [PubMed] [Google Scholar]
- 30.Kuhle V., Jackel D., Hensel M. 2004. Effector proteins encoded by Salmonella pathogenicity island 2 interfere with the microtubule cytoskeleton after translocation into host cells. Traffic 5:356–370 [DOI] [PubMed] [Google Scholar]
- 31.Kumar Y., Cocchiaro J., Valdivia R. H. 2006. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr. Biol. 16:1646–1651 [DOI] [PubMed] [Google Scholar]
- 32.Le Negrate G., et al. 2008. Salmonella secreted factor L deubiquitinase of Salmonella typhimurium inhibits NF-kappaB, suppresses IkappaBalpha ubiquitination and modulates innate immune responses. J. Immunol. 180:5045–5056 [DOI] [PubMed] [Google Scholar]
- 33.Liu P., et al. 2004. Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J. Biol. Chem. 279:3787–3792 [DOI] [PubMed] [Google Scholar]
- 34.Martin S., Parton R. G. 2006. Lipid droplets: a unified view of a dynamic organelle. Nat. Rev. Mol. Cell Biol. 7:373–378 [DOI] [PubMed] [Google Scholar]
- 35.Menendez A., et al. 2009. Salmonella infection of gallbladder epithelial cells drives local inflammation and injury in a model of acute typhoid fever. J. Infect. Dis. 200:1703–1713 [DOI] [PubMed] [Google Scholar]
- 36.Miyanari Y., et al. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9:1089–1097 [DOI] [PubMed] [Google Scholar]
- 37.Murphy D. J. 2001. The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog. Lipid Res. 40:325–438 [DOI] [PubMed] [Google Scholar]
- 38.Murphy D. J., Vance J. 1999. Mechanisms of lipid-body formation. Trends Biochem. Sci. 24:109–115 [DOI] [PubMed] [Google Scholar]
- 39.Nawabi P., Catron D. M., Haldar K. 2008. Esterification of cholesterol by a type III secretion effector during intracellular Salmonella infection. Mol. Microbiol. 68:173–185 [DOI] [PubMed] [Google Scholar]
- 40.Rathman M., Sjaastad M. D., Falkow S. 1996. Acidification of phagosomes containing Salmonella typhimurium in murine macrophages. Infect. Immun. 64:2765–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raychaudhuri S., Prinz W. A. 2010. The diverse functions of oxysterol-binding proteins. Annu. Rev. Cell Dev. Biol. 26:157–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruiz-Albert J., et al. 2002. Complementary activities of SseJ and SifA regulate dynamics of the Salmonella typhimurium vacuolar membrane. Mol. Microbiol. 44:645–661 [DOI] [PubMed] [Google Scholar]
- 43.Rytkönen A., et al. 2007. SseL, a Salmonella deubiquitinase required for macrophage killing and virulence. Proc. Natl. Acad. Sci. U. S. A. 104:3502–3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Samsa M. M., et al. 2009. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 5:e1000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shavinskaya A., Boulant S., Penin F., McLauchlan J., Bartenschlager R. 2007. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J. Biol. Chem. 282:37158–37169 [DOI] [PubMed] [Google Scholar]
- 46.Stein M. A., Leung K. Y., Zwick M., Garcia-del Portillo F., Finlay B. B. 1996. Identification of a Salmonella virulence gene required for formation of filamentous structures containing lysosomal membrane glycoproteins within epithelial cells. Mol. Microbiol. 20:151–164 [DOI] [PubMed] [Google Scholar]
- 47.Suhre K., Schmitt-Kopplin P. 2008. MassTRIX: mass translator into pathways. Nucleic Acids Res. 36:W481–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zweytick D., Athenstaedt K., Daum G. 2000. Intracellular lipid particles of eukaryotic cells. Biochim. Biophys. Acta 1469:101–120 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material