Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment (original) (raw)
Abstract
A fat-enriched diet modifies intestinal microbiota and initiates a low-grade inflammation, insulin resistance and type-2 diabetes. Here, we demonstrate that before the onset of diabetes, after only one week of a high-fat diet (HFD), live commensal intestinal bacteria are present in large numbers in the adipose tissue and the blood where they can induce inflammation. This translocation is prevented in mice lacking the microbial pattern recognition receptors Nod1 or CD14, but overtly increased in Myd88 knockout and ob/ob mouse. This ‘metabolic bacteremia’ is characterized by an increased co-localization with dendritic cells from the intestinal lamina propria and by an augmented intestinal mucosal adherence of non-pathogenic Escherichia coli. The bacterial translocation process from intestine towards tissue can be reversed by six weeks of treatment with the probiotic strain Bifidobacterium animalis subsp. lactis 420, which improves the animals' overall inflammatory and metabolic status. Altogether, these data demonstrate that the early onset of HFD-induced hyperglycemia is characterized by an increased bacterial translocation from intestine towards tissues, fuelling a continuous metabolic bacteremia, which could represent new therapeutic targets.
Keywords: Bifidobacterium lactis 420, diabetes, inflammation, obesity, pathogen-associated molecular pattern receptors
INTRODUCTION
Over the last decades, the incidence of diabetes and obesity has risen dramatically and this unabated increase can be justly described as a world-wide epidemic. A dramatic change in dietary habits, characterized by an increased intake of lipids, has been suggested to be mainly responsible for the dramatic rise in metabolic diseases through mechanisms, which induce low grade inflammation (Hotamisligil, 2006; Shi et al, 2006; Shoelson et al, 2006). TNF-α continuously released by the adipose tissue during obesity has been shown to activate protein kinase C (PKC) and to increase the phosphorylation of insulin receptor substrates on serine residues such as Ser-307, leading to the inactivation of this insulin signalling molecule and hence to insulin resistance (Tanti et al, 2004). However, the origin of the antigens responsible for the inflammatory process has not been described. Recently, the causal role of the intestinal microbiota in weight gain has been demonstrated in experiments in which germ-free mice colonized with the intestinal microbiota from genetically obese ob/ob mice gained more weight than their counterparts colonized with microbiota from lean animals. Hence, obesity could be transferred by the intestinal microbiota (Backhed et al, 2004, 2005; Ley et al, 2005, 2006; Turnbaugh et al, 2006, 2007). Altogether, these data demonstrated that the intestinal microbiota and the interactions between the host and the microbiota are involved in the control of energy metabolism. A key question is to understand the mechanism through which the intestinal microflora could specifically affect tissue inflammation. An important observation in this respect is that intestinal phagocytes such as dendritic cells and macrophages capture bacterial intestinal antigens and transfer them into lysosomes for degradation (Sansonetti & Di Santo, 2007), thereby providing a direct cellular link between the intestinal microbiota and the host, named bacterial translocation. This process requires bacterial antigen receptors such as toll-like receptors (TLR) (Keita et al, 2008; Neal et al, 2006) and nod-like receptors (NLR) (Kufer et al, 2008), through which viable and dead bacteria and their components initiate the activation of innate immune cells. Furthermore, cells from the innate immune system infiltrate the adipose depots during metabolic diseases (Weisberg et al, 2003, 2006). Therefore, we suggest that during high-fat diet-induced diabetes, commensal intestinal bacteria translocate in a pathological manner from the intestine towards the tissues where they trigger a local inflammation.
The present data show for the first time that during the early onset of high-fat diet-induced diabetes, bacteria from the intestine are actively translocated into the mesenteric adipose tissue (MAT) and the blood. This translocation initiates a ‘low grade bacteremia’ which depends on CD14, Nod1, but not Nod2. It is further regulated by leptin, which identifies a new function for this hormone. This metabolic bacteremia was reversed by a treatment using the probiotic strain Bifidobacterium animalis subsp. lactis 420, which reduced the mucosal adherence and bacterial translocation of gram-negative bacteria from the Enterobacteriaceae group. Consequently, adipose tissue inflammation and several features characteristic of diabetes were normalized. Therefore, the control of intestinal bacterial translocation and mucosal dysbiosis could be considered as a new therapeutic strategy for the control of high-fat diet-induced diabetes and metabolic syndrome.
RESULTS
High-fat feeding increases the translocation of live gram-negative bacteria through intestinal mucosa to blood and mesenteric adipose tissue
To determine which tissues are targeted by the bacterial translocation, we first quantified the bacterial 16S rRNA DNA concentration in different tissues. The data show that bacterial DNA is detectable in the blood of normal chow fed mice (Fig 1A and D, Supporting information Fig S1A and C). However, there was roughly 1000 and 2000–10,000 times less bacterial DNA in MAT and blood, respectively, when compared to cæcal content (Supporting information Fig S1A). The overall microbial population was also evaluated by denaturing gradient gel electrophoresis (DGGE) analysis, and the number of bands showed that microbiota is less diverse in tissues than in the gut (Supporting information Fig S1B and C).
Figure 1. High-fat diet increases the concentration of bacterial DNA in blood, mesenteric adipose tissue (MAT), and corresponding lymph nodes (MLN) before the onset of diabetes. The pathogen recognition receptors Nod1 and CD14, but not Nod2, control high-fat diet-induced bacterial DNA accumulation in tissues. The regulatory role of leptin.
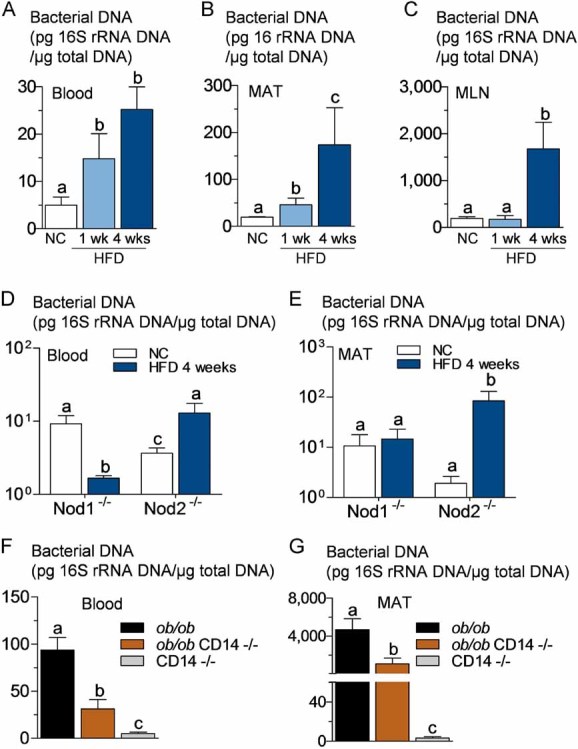
16S rRNA DNA concentration (pg/µg total DNA; qPCR):
- A,B,C. From total bacteria in blood (A), MAT (B) and MLN (C) of normal chow (NC)-fed, prediabetic (HFD 1 week), and diabetic mice (HFD 4 weeks) (mean ± SEM; n = 8).
- D,E. From total bacteria in blood (D), and MAT (E) of Nod1-deficient mice (Nod1−/−), and Nod2-deficient mice (Nod2−/−), after 4 weeks of a normal chow (NC) or a high-fat diet (HFD 4 weeks) (mean ± SEM; n = 11).
- F,G. From total bacteria in blood (F), and MAT (G), of ob/ob, ob/_ob_xCD14−/−, and CD14−/− mice (all NC-fed) (mean ± SEM; n = 8).
Data with identical superscript letters (A–G) do not differ from each other with p > 0.05.
To follow the bacterial translocation from intestine, a possible mechanism for the existence of bacterial DNA of intestinal origin found in tissues, we generated a fluorescently labelled, ampicillin-resistant Escherichia coli (GFP-E. coli) from a mouse E. coli. After only 1 week of a high-fat diet (HFD), GFP-E. coli was gavaged to mice. As a result, the colony forming units (cfu) equivalent of fluorescent E. coli (as quantified by RT-qPCR) was increased in blood, 2 h after gavage, when compared with normal chow (NC)-fed mice (Supporting information Fig S2C). This was similar to what was observed for wild-type E. coli DNA quantified by specific E. coli 16S qPCR already present in blood (Supporting information Fig S2B), total gram-negative bacteria DNA (Supporting information Fig S2A), and for total bacteria (Fig 1A).
After 4 weeks of HFD, when the diabetic state is established (Cani et al, 2008, 2009), the amounts of total bacterial DNA (Fig 1A), gram-negative bacterial DNA (Supporting information Fig S2A), and E. coli DNA (Supporting information Fig S2B) were still increasing in blood. We also quantified total bacterial DNA in MAT and in the corresponding mesenteric lymph nodes (MLN), since innate immune cells, which were suspected to have phagocytosed translocated intestinal bacteria, could accumulate in this tissue. After 1 week of HFD, the amount of bacterial DNA was increased in MAT but not in MLN (Fig 1B and C), and a similar observation was made in regard to the cfu quantification from ampicillin-resistant E. coli in these tissues 2 h after gavage (Supporting information Fig S2D). Once the diabetic state was established (4 weeks HFD), the amount of bacterial DNA was increased in both MAT and MLN (Fig 1B and C).
Translocation of intestinal bacteria, glucose metabolism and body weight are regulated by Nod1, leptin, Myd88 and CD14
We analysed first whether Nod1 and Nod2 were involved in the accumulation of tissue bacteria during HFD. When the Nod1−/− mice were fed with HFD for 1 month, fasting glycemia and glucose tolerance (Fig 2A and C), fat mass (Fig 2D) and body weight (Supporting information Fig S3A), fasted plasma insulin concentration (Fig 2E), and whole body insulin-sensitive glucose turnover rate (Fig 2F) were strictly similar between NC- and HFD-fed Nod1−/− mice as well as wild-type (WT) mice fed a normal chow, while WT mice fed a HFD were clearly sensitive to the dietary treatment for all parameters (Fig 2A, C, D, E and F, Supporting information Fig S3A). Conversely, Nod2−/− mice were still sensitive to HFD for all parameters studied (Fig 2B–F, Supporting information Fig S3A).
Figure 2. The pathogen recognition receptors Nod1 and CD14, but not Nod2, control high-fat diet-induced glucose intolerance, diabetes and fat mass gain. The regulatory role of leptin.
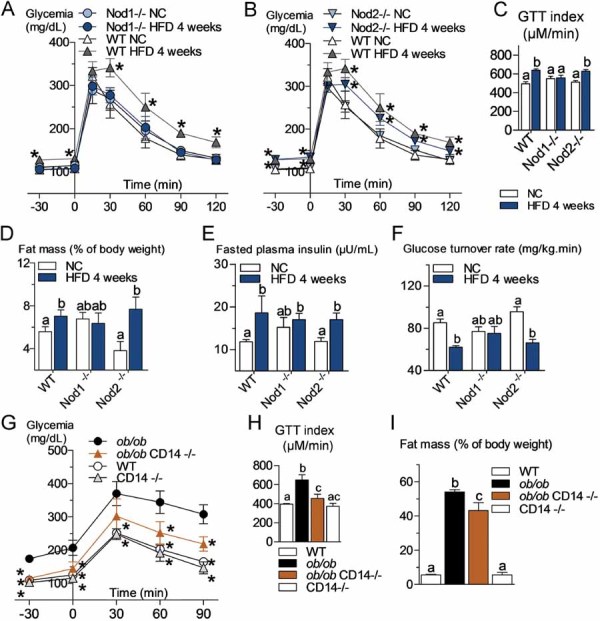
- A,B,C. Oral glucose tolerance test (OGTT) (1.5 g/kg) in Nod1-deficient mice (Nod1−/−) (A), Nod2-deficient mice (Nod2−/−) (B) and their corresponding wild-type controls (WT) after 4 weeks of a normal chow (NC) or a high-fat diet (HFD) and corresponding index (µM/min) from 15 to 120 min after glucose administration (C) (mean ± SEM; n = 11; *p < 0.05; data with identical superscript letters do not differ from each other with _p_ > 0.05).
- D,E,F. Fat mass (% of body weight; EchoMRI) (D), fasted plasma insulin (µU/ml) (E) and insulin sensitivity (glucose turnover rate in mg/kg min) (F), in Nod1−/− or Nod2−/− mice and their corresponding WT controls after 4 weeks of a NC or a HFD (mean ± SEM; n = 11 (D); 8 (E); or 5 (F); data with identical superscript letters do not differ from each other with p > 0.05).
- G. Intraperitoneal glucose tolerance test (IPGTT) (1 g/kg) in WT, ob/ob, ob/_ob_xCD14−/− and CD14−/− mice (all NC-fed) (mean ± SEM; n = 8; *p < 0.05 versus ob/ob).
- H. IPGTT corresponding index (µM/min) from 30 min before to 90 min after glucose administration (mean ± SEM; n = 8; data with identical superscript letters do not differ from each other with p > 0.05).
- I. Fat mass (% of body weight; EchoMRI) in WT, ob/ob, ob/_ob_xCD14−/− and CD14−/− mice (all NC-fed) (mean ± SEM; n = 8; data with identical superscript letters do not differ from each other with p > 0.05).
We also previously showed that CD14−/− mice resist the diabetic and obese phenotypes in response to HFD (Cani et al, 2008). We therefore mated leptin-deficient mice with CD14−/− mice, as described (Cani et al, 2008). In the double mutant mice, fasted glycemia and glucose tolerance (Fig 2G and H) as well as the fat mass percentage and whole body weight (Fig 2I, Supporting information Fig S3B) were improved when compared to the single mutant ob/ob mice.
Myd88 is one of the major signalling molecules involved in TLR activation (except activation of TLR3). Therefore, we compared the corresponding mutant mice with WT mice and showed that fasted glycemia and glucose intolerance (Fig 3A), body weight (Fig 3B), fat mass (Fig 3C), plasma insulin (Fig 3D) and insulin resistance (Fig 3E) were increased when compared to the corresponding WT mice.
Figure 3. Metabolic disturbances and increased bacterial translocation to mesenteric adipose tissue (MAT) in Myd88 knockout mice.
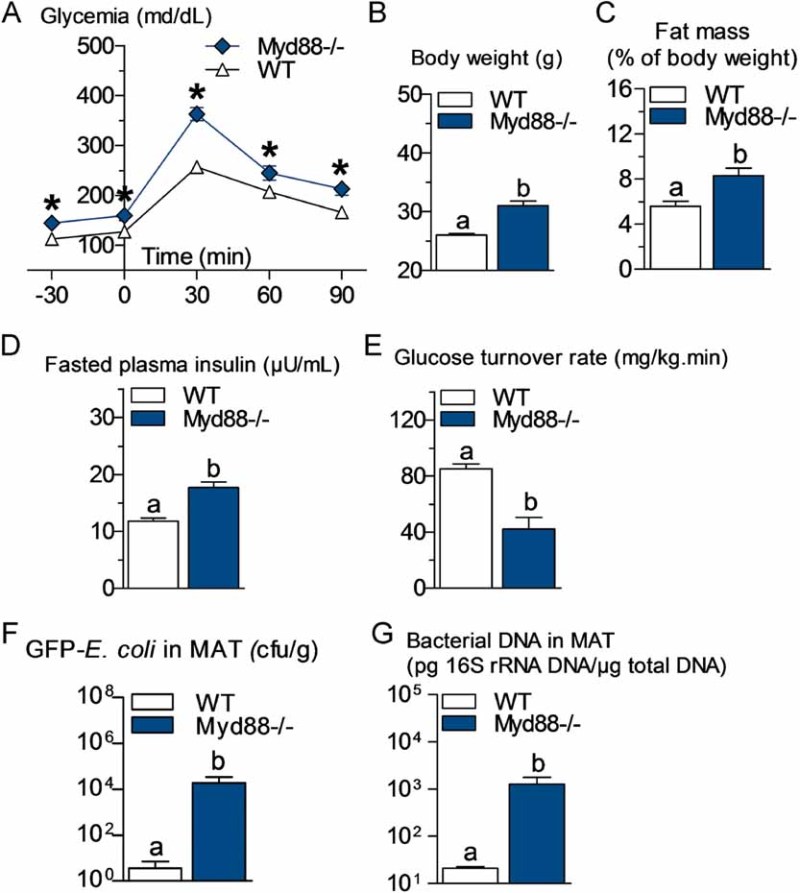
- A. Intraperitoneal glucose tolerance test (IPGTT) (1 g/kg) in Myd88-deficient mice (Myd88−/−) and their corresponding wild-type controls (WT) fed a normal chow (NC) diet (mean ± SEM; n = 7; *p < 0.05 versus controls).
- B,C,D,E. Body weight (B), fat mass (% of body weight; EchoMRI) (C), fasted plasma insulin (µU/ml) (D) and insulin sensitivity (glucose turnover rate in mg/kg min) (E), in Myd88−/− mice and their corresponding WT controls fed a NC diet (mean ± SEM; n = 5; data with identical superscript letters do not differ from each other with p > 0.05).
- F. Number of GFP-E. coli cfu per g of MAT in Myd88−/− mice and their corresponding WT controls fed a NC diet, 2 h post-gavage with 109 cfu of GFP-E. coli (mean ± SEM; n = 7; data with identical superscript letters do not differ from each other with p > 0.05).
- G. 16S rRNA DNA concentration (pg/µg total DNA; qPCR) from total bacteria in MAT of Myd88−/− mice and their corresponding WT controls fed a NC diet (mean ± SEM; n = 7; data with identical superscript letters do not differ from each other with p > 0.05).
Total bacterial DNA concentration, in blood and MAT, was unchanged or even reduced in Nod1−/− mice after HFD treatment, whereas it was conversely increased in Nod2−/− mice (Fig 1D and E, and compare with wild-type mice Fig 1A and B). The absence of leptin increased the total amount of bacterial DNA in blood (Fig 1F compared with Fig 1A) and MAT (Fig 1G compared with Fig 1B). A similar observation was made when GFP-E. coli was quantified in MAT 2 h following the gavage (Supporting information Fig S2E). In addition, the absence of CD14 in leptin-deficient mice decreased the bacterial DNA content in blood and MAT (Fig 1F and G), as well as the cfu count of GFP-E. coli in MAT 2 h after gavage (Supporting information Fig S2E) compared with ob/ob mice. In Myd88-deleted mice, the translocation of living E. coli (Fig 3F) and the total content of bacterial DNA (Fig 3G) were dramatically increased in MAT compared with WT mice.
High-fat feeding increases the adherence of a gram-negative bacteria species to intestinal mucosa and its co-localization with dendritic cells in lamina propria and mesenteric lymph nodes
Two hours after gavage with GFP-E. coli, adherence of the bacteria to mucosal surface of duodenum, jejunum, ileum and cæcum was increased in mice fed with a HFD for only 1 week, when compared with normal chow-fed mice (Fig 4A), while after 5 h the difference was more evident in cæcum (Supporting information Fig S4A). Importantly, 1 week of HFD is not sufficient to induce diabetes showing that bacterial translocation precedes the occurrence of diabetes. This increased adherence of GFP-E. coli to mucosa persisted significantly in ileum and cæcum until the 4th week of HFD (Fig 4A), by which time HFD-fed mice have developed diabetes. In addition, examination by fluorescence microscopy of ileal mucosa obtained by scraping 2 h after the oral gavage, confirmed the presence of fluorescent E. coli mainly in ileal mucosa of HFD-fed mice (Fig 4B and C). This was similar, although to a lower extent, after 5 h (Supporting information Fig S4B and C). Fluorescent bacteria could also be detected in sections of ileum, inside lamina propria, 2 h post-gavage (Fig 4D) and inside submucosa 5 h post-gavage (Supporting information Fig S4D). The number of transmucosal GFP-E. coli was estimated to be 5–10 times higher, 2 and 5 h post-gavage in HFD-fed mice compared to normal chow-fed mice (Fig 4E, Supporting information Fig S4E).
Figure 4. Intestinal mucosal adherence and transmucosal passage of bacteria are increased during high-fat diet (HFD) treatment before the onset of diabetes, and intestinal bacteria co-localize with dendritic cells of lamina propria and mesenteric lymph nodes (MLN).
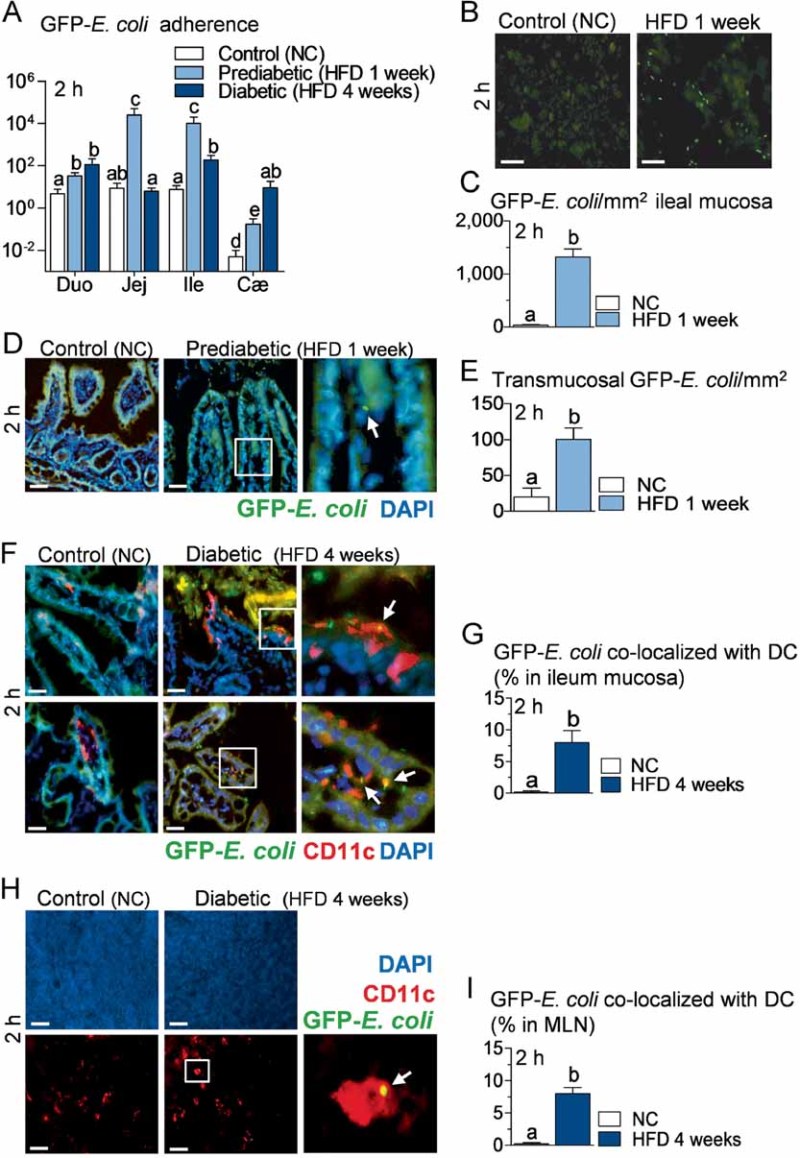
- A. Bacterial adherence (GFP-E. coli cfu per cm of mucosa/GFP-E. coli cfu per cm of lumen) in normal chow (NC)-fed (Control NC), prediabetic (HFD 1 week), and diabetic mice (HFD 4 weeks), 2 h after gavage with 109 cfu of GFP-E. coli (Duo, duodenum; Jej, jejunum; Ile, ileum; Cæ, cæcum) (mean ± SEM; n = 10–12; data with identical superscript letters do not differ from each other with p > 0.05).
- B. Fluorescence microscopy of scrapped ileal mucosa from NC-fed (Control NC) and prediabetic mice (HFD 1 week), 2 h after gavage with 109 cfu of GFP_-E. coli_. Bars = 20 µm.
- C. Corresponding number of GFP-E. coli/mm2 of scrapped ileal mucosa (fluorescence microscopy) (mean ± SEM; n = 6; data with identical superscript letters do not differ from each other with p > 0.05).
- D. Fluorescence microscopy of DAPI-counterstained cryosections of ileum from NC-fed (Control NC) and prediabetic mice (HFD 1 week), 2 h after gavage with 109 cfu of GFP_-E. coli_. Right panel corresponds to magnification of surrounded region. Bars = 20 µm.
- E. Corresponding number of transmucosal GFP-E. coli/mm2 of ileum section (fluorescence microscopy) (mean ± SEM; n = 6; data with identical superscript letters do not differ from each other with p > 0.05).
- F,H. Immunofluorescent labelling of CD11c-positive cells [dendritic cells (DC) in red] and co-localization with fluorescent GFP-E. coli (green) on DAPI-counterstained (nuclei in blue) cryosections of ileum (F), and MLN (H), from NC-fed (Control NC), and diabetic mice (HFD 4 weeks), 2 h after gavage with 109 cfu of GFP_-E. coli_. Arrows point co-localizations (yellow). Right panels correspond to magnification of surrounded regions. Bars = 20 µm.
- G,I. Corresponding quantification of CD11c-positive cells co-localized with GFP-E. coli (% of total DC) in ileum (G) and MLN (I) (mean ± SEM; n = 6; data with identical superscript letters do not differ from each other with p > 0.05).
After 4 weeks of HFD, when the diabetic state is established (Burcelin et al, 2002; Cook et al, 2004), we were able to detect co-localization of fluorescent bacteria with dendritic cells (DC) in the lamina propria. In normal chow-fed mice, DC were restricted inside the lamina propria, whereas after 4 weeks of HFD, DC were also seen between enterocytes, extending large dendrites towards the intestinal lumen. Importantly, fluorescent E. coli could be co-localized with CD11c-positive cells, i.e. DC, on the luminal side of villi and in lamina propria. These co-localizations were detected in HFD-fed mice only (Fig 4F). The percentage of DC that co-localized with fluorescent E. coli was estimated to be between 5 and 10% of all DC in lamina propria 2 h post-gavage in HFD-fed mice, whereas no co-localization was detectable in normal chow-fed controls (Fig 4G).
Interestingly, fluorescent E. coli also co-localized with DC in MLN, although to a lower extent than what was observed in the lamina propria (Fig 4H). The percentage of DC co-localizing with fluorescent E. coli in MLN was also estimated to be between 5 and 10% 2 h post-gavage in HFD-fed mice, while they were undetectable in normal chow-fed animals (Fig 4I).
To analyse whether there was a differential distribution of intestinal microbiota in the intestine, we quantified the mucosal and luminal bacterial DNA content by qPCR and showed that 1 week of HFD increased the microbial population in the lumen and the mucosa (Fig 5A). This was further reinforced in the mucosa after 4 weeks of HFD. The expression of markers of inflammation was quantified in MAT and showed a moderate, but significant increase after 4 weeks of HFD for TNF-α and IFN-γ (Fig 5B). Moreover, MAT TNF-α mRNA concentration was the only marker, which positively correlated with the luminal and mucosal bacterial content (Fig 5C and D).
Figure 5. TNF-α expression in mesenteric adipose tissue (MAT) correlates positively with bacterial DNA concentration in ileum before the onset of high-fat diet-induced diabetes.
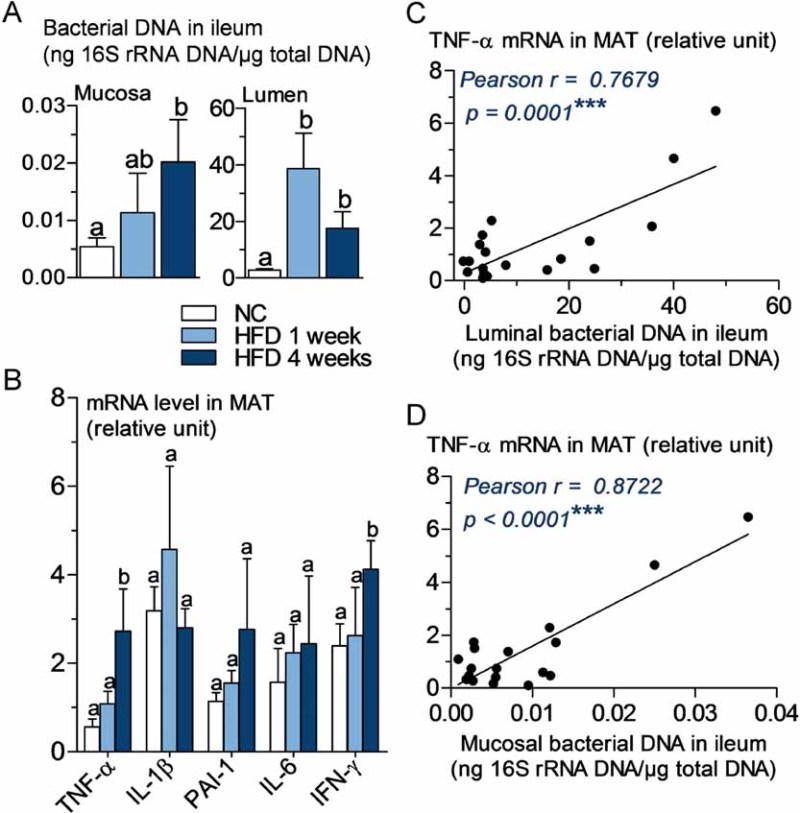
- A. 16S rRNA DNA concentration (ng/µg total DNA; qPCR) from total bacteria in mucosa and lumen of ileum, in normal chow (NC)-fed, prediabetic (HFD 1 week), and diabetic mice (HFD 4 weeks) (mean ± SEM; n = 6; data with identical superscript letters do not differ from each other with p >0.05).
- B. Concentration of mRNA coding for the inflammatory markers TNF-α, IL-1β, PAI-1, IL-6 and IFN-γ in MAT of normal chow (NC)-fed, prediabetic and diabetic mice (RT-qPCR) (mean ± SEM; n = 6; data with identical superscript letters do not differ with p > 0.05; expressions of the different markers were not compared between themselves).
- C,D. Correlations between TNF-α expression in MAT and bacterial concentration (ng 16S rRNA DNA/µg total DNA; qPCR), of ileal lumen (C), and ileal mucosa (D), in NC-fed, prediabetic (HFD 1 week), and diabetic mice (HFD 4 weeks).
Intestinal leptin delivery regulates intestinal bacterial adherence, translocation and metabolism
An increased bacterial translocation was characteristic for the ob/ob mice. To determine whether leptin regulates bacterial translocation, we administered a probiotic, which produces and releases leptin (Lactococcus lactis leptin) aiming to target intestinal metabolism. We treated HFD and ob/ob mice for 8 weeks. In the leptin-deficient mice, the circulating leptin concentration remained undetectable suggesting only a possible local effect of the leptin-producing probiotic. Body weight gain (Fig 6A) and fat mass gain (Fig 6B) were reduced over the time course of the treatment. Glucose intolerance (Fig 6C and D) was slightly reduced although fasted plasma insulin concentration (Fig 6E) remained mostly unchanged. This was associated with a dramatic reduction in the mucosal/lumen ratio of adherent GFP-E. coli in the ileum only, whereas no change in the cæcum were observed (Fig 6F). Bacterial translocation of live bacteria was reduced by the leptin treatment (Fig 6G) while total dead and live bacteria remained unaffected (Fig 6H). This was associated with a modest reduction of adipose tissue inflammation as shown by the reduction of the concentration of some cytokine mRNAs (Fig 6I). In a second set of experiments, ob/ob mice were treated similarly. Body weight gain (Fig 7A) and fat mass gain (Fig 7B), fasted glycemia (Fig 7C), glucose intolerance (Fig 7C and D), and fasted plasma insulin concentration (Fig 7E) were reduced by the leptin treatment. The mucosal bacterial adherence (Fig 7F), the content of live GFP-E. coli in MAT (Fig 7G), and bacterial mRNA (Fig 7I) were reduced by the leptin treatment, whereas the total bacterial content remained the same in MAT (Fig 7H). The mRNA concentration of some (PAI-1, IL-6), but not all (TNF-α, IL-1β, IFN-γ), inflammatory markers was reduced in the mesenteric adipose depot of mice treated with leptin (Fig 7J).
Figure 6. Probiotic-mediated intestinal leptin delivery reverses high-fat diet (HFD)-metabolic disturbances, bacterial adherence and translocation, and mesenteric adipose tissue (MAT) inflammation.
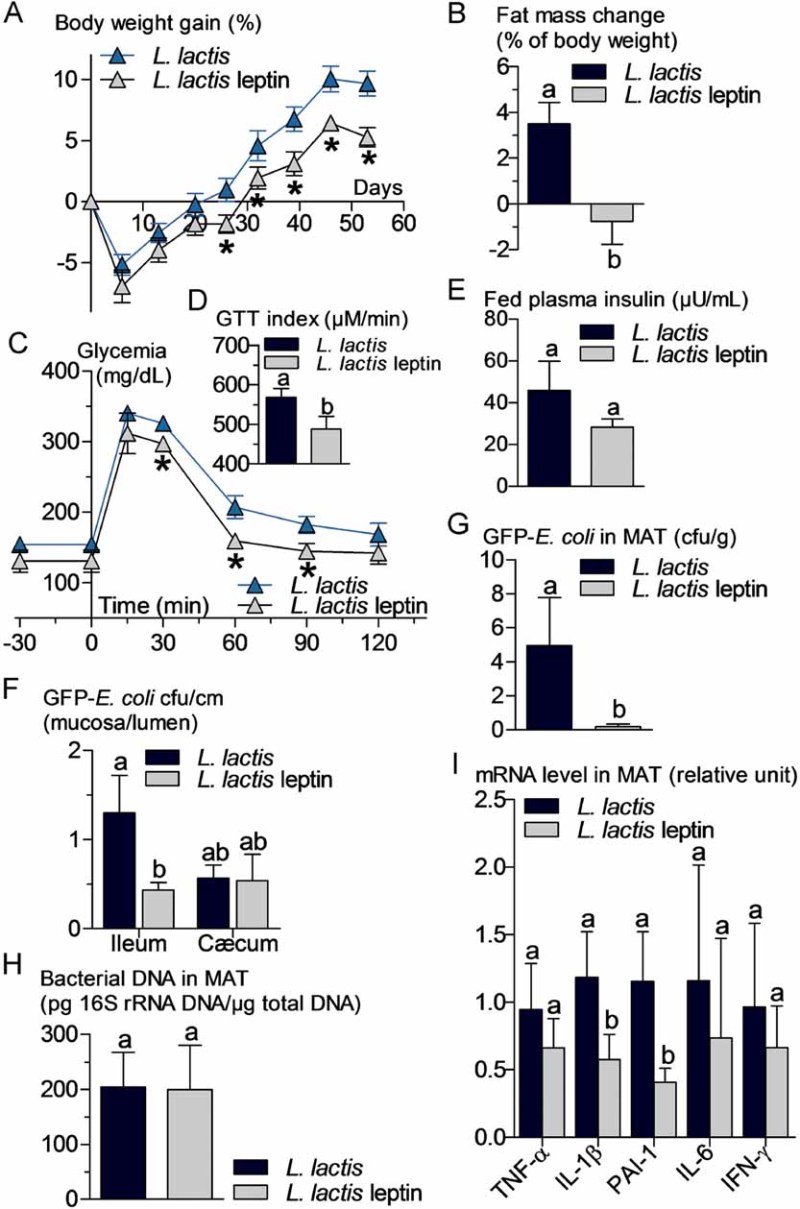
HFD-fed WT mice were orally treated with 109 cfu/day of leptin-producing Lactococcus lactis (L. lactis leptin) and their corresponding controls orally treated with 109 cfu/day of Lactococcus lactis (L. lactis).
- A,B. Body weight gain (% of initial weight) (A), fat mass change during treatment in % of body weight (EchoMRI) (B) (mean ± SEM; n = 6; *p < 0.05 _versus_ controls (A); data with identical superscript letters do not differ from each other with _p_ > 0.05 (B)).
- C,D. Oral glucose tolerance test (OGTT) (1.5 g/kg) (C), and corresponding index (µM/min) from 30 min before to 120 min after glucose administration (D) (mean ± SEM; n = 6; *p < 0.05 _versus_ controls (C); data with identical superscript letters do not differ from each other with _p_ > 0.05 (D)).
- E,F,G,H. Fed plasma insulin (µU/ml) (E), GFP-E. coli adherence (GFP-E. coli cfu per cm of mucosa/GFP-E. coli cfu per cm of lumen) in ileum and cæcum, 2 h post-gavage with 109 cfu of GFP-E. coli (F), number of GFP-E. coli cfu per g of MAT, 2 h post-gavage with 109 cfu of GFP-E. coli (G), and 16S rRNA DNA concentration (pg/µg total DNA; qPCR) from total bacteria in MAT (H) (mean ± SEM; n = 6; data with identical superscript letters do not differ from each other with p > 0.05).
- I. Concentration of mRNA coding for the inflammatory markers TNF-α, IL-1β, PAI-1, IL-6 and IFN-γ in MAT (RT-qPCR) (mean ± SEM; n = 6; data with identical superscript letters do not differ with p > 0.05; expressions of the different markers were not compared between themselves).
Figure 7. Probiotic-mediated intestinal leptin delivery reverses metabolic disturbances, bacterial adherence and translocation, and mesenteric adipose tissue (MAT) inflammation in ob/ob mice.
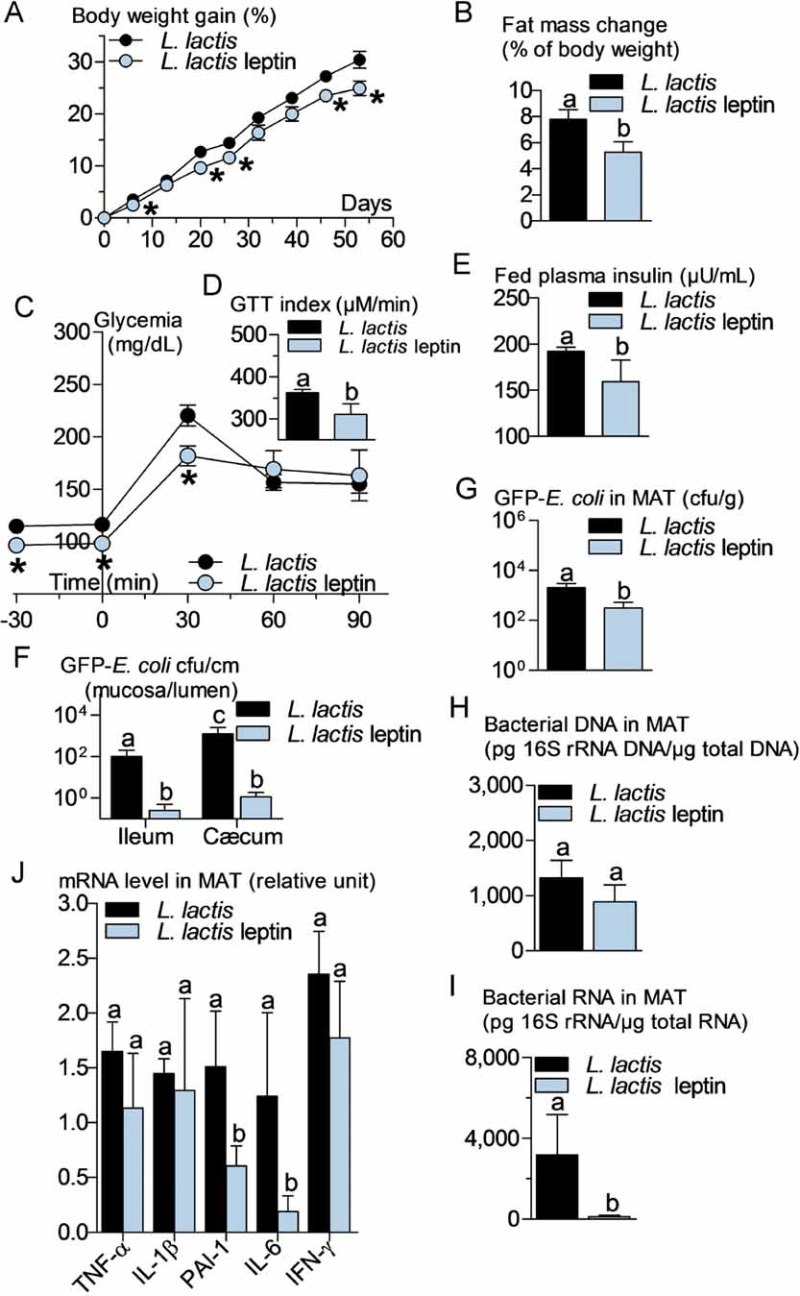
ob/ob mice were orally treated with 109 cfu/day of leptin-producing Lactococcus lactis (L. lactis leptin) and their corresponding controls orally treated with 109 cfu/day of Lactococcus lactis (L. lactis).
- A,B. Body weight gain (% of initial weight) (A), fat mass change during treatment time course (% of body weight; EchoMRI) (B) (mean ± SEM; n = 6; *p < 0.05 _versus_ controls (A); data with identical superscript letters do not differ from each other with _p_ > 0.05 (B)).
- C,D. Intraperitoneal glucose tolerance test (IPGTT) (1 g/kg) (C), and corresponding index (µM/min) from 30 min before to 90 min after glucose administration (D) (mean ± SEM; n = 6; *p < 0.05 _versus_ controls (C); data with identical superscript letters do not differ from each other with _p_ > 0.05 (D)).
- E,F,G,H,I. Fed plasma insulin (µU/ml) (E), GFP-E. coli adherence (GFP-E. coli cfu per cm of mucosa/GFP-E. coli cfu per cm of lumen) in ileum and cæcum, 2 h post-gavage with 109 cfu of GFP-E. coli (F), number of GFP-E. coli cfu per g of MAT, 2 h post-gavage with 109 cfu of GFP-E. coli (G), 16S rRNA DNA concentration (pg/µg total DNA; qPCR) from total bacteria in MAT (H), 16S rRNA concentration (pg/µg total RNA; RT-qPCR) from total bacteria in MAT (I) (mean ± SEM; n = 6; data with identical superscript letters do not differ from each other with p > 0.05).
- J. Concentration of mRNA coding for the inflammatory markers TNF-α, IL-1β, PAI-1, IL-6 and IFN-γ in MAT (RT-qPCR, 2−ΔΔCt) (mean ± SEM; n = 6; data with identical superscript letters do not differ with p > 0.05; expressions of the different markers were not compared between themselves).
A probiotic treatment reduces mucosal dysbiosis, bacterial translocation and improves glucose metabolism
To determine if a change in bacterial translocation could affect HFD-induced diabetes, we treated mice with the probiotic Bifidobacterium animalis subsp. lactis 420 (B420) (Supporting information Fig S5A), previously shown to bind to and exclude pathogenic bacteria from intestinal mucosa in vitro (Collado et al, 2007; Collado & Sanz, 2007). A 1-month daily treatment of HFD-induced diabetic mice with 109 cfu of B420 effectively reduced the number of GFP-E. coli in different segments of small intestine mucosa (Fig 8A and B). Concomitantly, the quantity of Bifidobacterium spp. was slightly, although not significantly, increased in ileal mucosa (Supporting information Fig S5B). We further analysed some of the major bacterial groups in MAT and showed that the number of Enterobacteriaceae, which was strongly increased in mice fed with a HFD when compared to normal chow-fed controls, was decreased by the probiotic treatment whereas the other groups were mostly unaffected (Fig 8C). Bifidobacterium spp. was not detected in MAT at any time point. In addition, the expression of the major pro-inflammatory cytokines TNF-α, IL-1β, PAI-1 and IL-6, was reduced in MAT, liver and muscle of treated HFD-fed mice when compared with untreated HFD-fed control mice (Fig 8D, Supporting information Fig S5C and D). The probiotic treatment also impacted on the overall metabolism since glucose intolerance was moderately blunted although fasting glycemia remained unaffected (Supporting information Fig S5E). In addition, insulin sensitivity and fasting hyperinsulinemia were completely normalized by the probiotic treatment (Fig 8E, Supporting information Fig S5F).
Figure 8. A probiotic treatment with Bifidobacterium lactis 420 reverses high-fat diet (HFD)-induced bacterial adherence and translocation, adipose tissue (MAT) inflammation and insulin resistance.
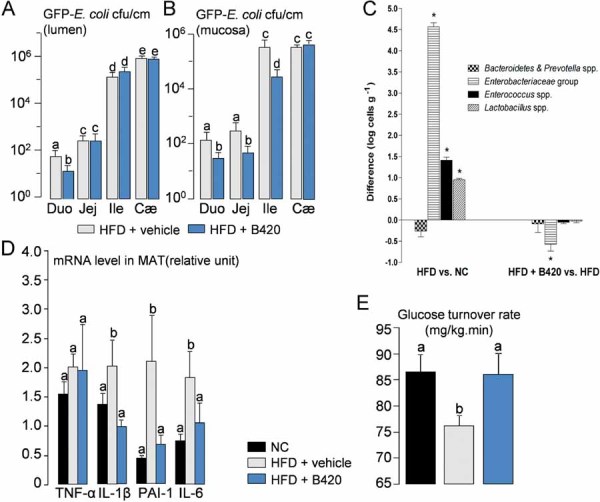
- A,B. Luminal (A) and mucosal (B) number of colony forming units (cfu) per cm of duodenum (Duo), jejunum (Jej), ileum (Ile), and cæcum (Cæ), 2 h following oral gavage with 109 cfu of GFP-E. coli.) (mean ± SEM; n = 8; data with identical superscript letters do not differ with p > 0.05).
- C. Difference in major groups of bacteria in MAT (qPCR), between HFD and NC groups (left panel), and HFD + B420 and HFD groups (right panel) (mean ± SEM; n = 8; *p < 0.05).
- D. Concentration of mRNA coding for the inflammatory markers TNF-α, IL-1β, PAI-1 and IL-6 in MAT (RT-qPCR) (mean ± SEM; n = 8–10; data with identical superscript letters do not differ with p > 0.05; expressions of the different markers were not compared between themselves).
- E. Insulin sensitivity (glucose turnover rate in mg/kg min). (mean ± SEM; n = 8–10; data with identical superscript letters do not differ with p > 0.05).
DISCUSSION
We have identified for the first time that adipose tissue and blood from diabetic mice fed with HFD contain live bacteria, which originate from intestine and are linked to low-grade inflammation. Nod1, CD14 and Myd88 are required for this process. Furthermore, leptin inhibits mucosal bacterial adherence and the translocation of live bacteria from the intestine towards tissues. At the early onset of diabetes, we further showed that intestinal bacteria from the lumen reach the mucosal layer where dendritic cells may phagocytose the bacteria, which then may migrate inside the body towards metabolically active tissues. Importantly, a probiotic treatment, which prevents bacterial adherence and translocation, protects also against HFD-induced inflammation, insulin resistance and diabetes.
It is now well accepted that HFD-induced metabolic diseases are associated with a low-grade inflammation occurring, among other tissues, in adipose depots. This inflammatory process is characterized by an increased production of cytokines and infiltration of macrophages (Weisberg et al, 2003, 2006). Furthermore, the genetic deletion of the tissue inhibitor of metalloproteinase 3 (Timp3), which is a natural inhibitor of tumour necrosis factor-alpha-converting enzyme (TACE) (Serino et al, 2007), has been recently shown to catalyse HFD-induced metabolic diseases (Federici et al, 2005; Menghini et al, 2009). Therefore, these data reinforce the role of inflammation as an early factor responsible for insulin resistance before the onset of obesity. The nature of antigens present in adipose depots and responsible for the inflammatory process is not fully understood. In light of the recently increasing amount of data showing the role of intestinal microflora in the control of metabolic diseases, we determined here the microbial content of the adipose depot. To this aim, we have isolated a commensal E. coli from mouse intestinal microbiota and labelled it with a fluorophore and an ampicillin reporter gene. As quickly as 2 h after gavage with fluorescent E. coli this commensal bacterium dramatically accumulated in mucosa of the different segments of intestine of HFD-fed mice only. This was also true for the overall bacterial content, suggesting that the intestinal mucosa from HFD-fed mice exhibits properties, which are different from the mucosa of normal chow-fed mice and which facilitate the bacterial adherence. Indeed, leptin resistance could lead to an impaired intestinal barrier function since the hormone modulates the expression of secreted and membrane-associated mucins in colonic epithelial cells by targeting PKC, phosphoinositide 3-kinase (PI3K), and mitogen-activated protein kinase (MAPK) pathways (El Homsi et al, 2007).
Although the mechanism of mucosal adherence remains to be elucidated, the accumulation of bacteria close to the mucosa could certainly facilitate their translocation through the epithelium. We analysed this last hypothesis and showed that HFD led to an increase in the E. coli co-localization with DC in the intestinal lamina propria. The bacteria, probably remaining inside DC, then rapidly disseminated into MAT and corresponding MLN where again the fluorescent E. coli co-localized with DC. The putative rate-limiting role of DC cannot be fully and causally explored in this model of bacterial translocation. However, their role in the control of glucose metabolism has been demonstrated elsewhere (Macia et al, 2006; Nguyen et al, 2007) by means of the secretion of CXCL14 required for the regulatory role of DC on lymphocytes (Hara & Nakayama, 2009) and for other inflammatory diseases such as inflammatory bowel diseases (Niess, 2008). Interestingly, our data show that the presence of various bacteria in MAT and the corresponding MLN represents a physiological mechanism that is exacerbated during HFD-induced metabolic diseases. It is noteworthy that HFD increases the accumulation of gram-negative bacteria that produce lipopolysaccharides (LPS), which are highly inflammatogenic molecules. Therefore, our data show that this metabolic translocation of gram-negative bacteria and LPS provides a rationale for HFD-induced inflammation and insulin resistance. We previously documented that in humans and mice, an increased proportion of fat in the diet moderately augmented, by two to three times, the plasma concentration of gram-negative bacteria cell wall components LPS (Amar et al, 2008; Cani et al, 2007a). Hence, we described this increase in plasma LPS as 'metabolic endotoxemia'. In addition, we showed that antibiotics and dietary fibres, most likely acting by changing the intestinal microbiota of HFD-fed mice, can reduce metabolic endotoxemia, inflammation, and improve the overall impaired metabolism (Cani et al, 2007b, 2008; Membrez et al, 2008).
The mechanisms through which bacterial factors could reach adipose tissue are unknown but there could be several hypotheses. Firstly, we initially suggested that a HFD is associated with an increased intestinal tight junction permeability (Cani et al, 2008, 2009) by which bacterial fragments could diffuse. Secondly, here we suggest that a translocation of intestinal bacteria through the intestinal epithelium could be exacerbated during the development of metabolic diseases. Similarly, commensal bacteria have been proposed to translocate into the host mammary glands via MLN and Peyer's patches in lactating mice (Hase et al, 2009; Perez et al, 2007; Sansonetti & Phalipon, 1999).
Intraluminal microbial detection requires the recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs) that are distributed on the cell surface and within the cytosol of innate immune cells. Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (Nod)-like receptor (NLRs) families function as extracellular and intracellular PRR, respectively, that trigger innate immune responses (Kufer & Sansonetti, 2007; Lundin et al, 2008). Therefore, molecular signalling during the bacterial translocation process could involve both the LPS receptors CD14/TLR4 and intracellular receptors for various other antigens such as Nod1 and Nod2 (Fritz et al, 2007; Kufer & Sansonetti, 2007). In this study, we showed that in absence of Nod1 but not Nod2, a HFD could no longer induce glucose intolerance and diabetes, suggesting that Nod1, which detects d-glutamyl-meso-diaminopimelic acid (meso-DAP)-containing peptidoglycan (PGN) found principally in gram-negative bacteria, was required for the control of metabolic diseases. The bacterial translocation process was reduced accordingly. Similar results were obtained in CD14 knockout mice. In agreement with other reports (Paulos et al, 2007), the internalization of gram-negative bacteria through intestinal epithelium in vivo and their transport towards MAT and MLN was significantly lower in mice with the LPS co-receptor CD14 deletion when compared with wild-type (WT) mice. Our data demonstrate that the recognition of bacterial fragments by specialized cells is mandatory for the induction of inflammation and the triggering of metabolic disease. Furthermore, the fact that both CD14 and Nod1 were necessary for the induction of diabetes in response to the fat-enriched diet suggests that a crosstalk between the two non-redundant pathways might be involved, which synergizes for a full inflammatory response to the translocated gram-negative bacteria. Myd88 is a good candidate; however, surprisingly the intolerance to glucose as well as the fasted glycemia were higher in the knock-out model than in wild-type mice. This phenotype was associated with an increased bacterial translocation. This last set of data suggests that a bacterial receptor linked to Myd88 signalling would be protecting from HFD-induced metabolic diseases. Our data suggest that conversely to Nod1, Nod2 could be associated with this protective function and hence the ablation of Myd88 would prevent the protecting role of Nod2.
Other antigen receptors such as the nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (Nlrp3) inflammasome, belonging to the NLR family, could also be involved in bacterial translocation. Nlrp3 inflammasome senses lipotoxicity-associated increases in intracellular ceramide to induce caspase-1 cleavage in macrophages and adipose tissue (Stienstra et al, 2010; Vandanmagsar et al, 2011). Therefore, complex lipids of bacterial origin could trigger inflammation through the binding of Nlrp3. Hence, our data sets the scene for the discovery of numerous other receptors involved in the triggering of inflammation in response to a fat-enriched diet.
Our data support a similar conclusion regarding body weight gain. This conclusion is further reinforced by the fact that obese/diabetic ob/ob mice are also characterized by an increased bacterial translocation as described here. Therefore, this mechanism does not seem to be restricted to HFD but more widely distributed in different animal models. Furthermore, deletion of CD14 within ob/ob mice reduced bacterial translocation and improved the metabolic and inflammatory phenotypes as previously described (Cani et al, 2008). These data also demonstrate a new role for leptin: the control of intestinal bacterial translocation. Importantly, leptin regulates the translocation of live bacteria into the tissues since the local intestinal leptin treatment did not reduce the total tissue content of whole bacteria. Our data also suggest that the role of leptin on intestinal bacterial adherence and translocation is directed to the intestine. The treatment using the leptin-producing probiotic did not modify the circulating leptin concentration, suggesting that the hormone had a role on the intestinal metabolism. However, we cannot rule out that this original role of leptin might be indirect through its effect on body weight or other of the many aspects regulated by leptin included that of the immune system.
To further validate the role of bacterial translocation on the control of glucose metabolism, we took advantage of a treatment with the probiotic strain Bifidobacterium lactis 420, which has been shown to exclude other bacteria from the mucosa (Collado et al, 2007) and to improve the integrity of the epithelial cell layer (Putaala et al, 2008). Hence, we expected to reduce the bacterial translocation process by the probiotic treatment. A one-month treatment markedly reduced the gram-negative bacteria E. coli adherence to intestinal mucosa and the translocation of Enterobacteriaceae, the bacterial group to which E. coli belongs, in particular into MAT. Accordingly, inflammation, insulin sensitivity, and fasted hyperinsulinemia were almost completely normalized. This impacted the overall glycemic control since glucose intolerance was reduced, although moderately, in the probiotic-treated HFD-fed mice when compared with non-treated HFD-fed mice. Our data suggest that the reversal of diabetes by probiotic treatment required first a normalization of mucosal dysbiosis, which was followed by reduced bacterial translocation, tissue inflammation, insulin resistance, and secondarily glycemia. Our results further validate the conclusion that a reduction in bacterial translocation and mucosal dysbiosis helps to control the development of metabolic diseases and opens the way to a new probiotic strategy for the treatment of diabetes and obesity. However, we did not observe a dramatic increase in the concentration of Bifidobacterium spp. in the mucosa of HFD-fed treated mice when compared with non-treated mice. This was expected since we previously described that fat-diet strongly reduces the amount of the Bifidobacterium genus (Cani et al, 2008). Therefore, the mechanisms through which the probiotic regulates glucose metabolism do not seem to be related to an increased mucosal concentration of the probiotic and hence remain yet to be discovered. Recent data showed that some Bifidobacterium species can increase production of acetate and inhibit the translocation of the pathogenic E. coli O157:H7 Shiga toxin from the gut lumen to the blood (Fukuda et al, 2011). Our data here show that the bacterial translocation of commensal bacteria is indeed reduced by the probiotic treatment arguing for a similar mechanism, which has been described in other instances (Fukuda et al, 2011). We cannot rule out that an acute administration of probiotic could have a direct impact on inflammation, therefore a similar reasoning could be made with regards to the gavage of E. coli. However, we do not have any evidence for this assumption.
In conclusion, we have discovered that a fat-enriched diet induces a low-grade infection before the onset of diabetes. This infection targets the MAT through a mechanism described as bacterial translocation. This mechanism requires the recognition of PAMPs by CD14, signalling through Myd88 and is leptin-regulated. Therefore, our data demonstrate that the increased bacterial translocation of gram-negative bacteria into the adipose tissue is mostly responsible for the continuous fuelling of inflammatory antigens thus characterizing the origin of the low-grade inflammation during diabetes and obesity. This concept opens up avenues for the treatment of metabolic diseases using probiotic strategies.
MATERIALS AND METHODS
Animals and diets
C57bl6, ob/ob, CD14−/−, ob/obxCD14−/−, Myd88−/−, Nod1−/− or Nod2−/− mice were fed with either a normal chow diet (NC) or a HFD (Safe, Augy, France) and the diabetic phenotype studied. This diet was previously demonstrated to generate fasting hyperglycemia, glucose intolerance, and insulin resistance following one month of feeding (Burcelin et al, 2002; Riant et al, 2009). All of the animal experimental procedures were validated by the local ethical committee of the Rangueil Hospital.
Generation of ampicillin-resistant GFP-E. coli
An Escherichia coli was isolated from mouse cæcum (Inserm U1048) and transformed with the plasmid pZE1R-GFP (gift from J. Oberto, CNRS UMR8621, Orsay, France) to induce the expression of green fluorescent protein (GFP) and β-lactamase conferring resistance to 100 µg/ml of ampicillin.
The paper explained
PROBLEM
We are facing an epidemic of metabolic diseases, which the classical pharmacological strategies have failed to overcome. Hence, new paradigms are required. We have previously shown that bacterial fragments such as lipopolysaccharides (LPS) are present in increased amounts in the blood of diabetic mice, and induce adipose tissue inflammation, which is the first step leading to insulin resistance and adipose tissue expansion, resulting in obesity. Our previous data suggested that targeting mechanisms involved in the recognition of bacterial fragments could be an original therapeutic strategy to overcome the low-grade inflammation and the development of metabolic diseases. Here, we aimed at identifying such mechanisms.
RESULTS
We show that at the early onset of HFD-induced diabetes and obesity, the mucosal adherence of commensal bacteria is increased dramatically in the mouse intestine. This is accompanied by an augmentation of the translocation of these bacteria into the adipose tissue. These living bacteria then co-localized with dendritic cells of the intestinal lamina propria and the mesenteric lymph nodes when diabetes is established. Importantly, we demonstrate that this mechanism requires CD14 and Nod1, which are receptors that recognize different fragments of gram-negative bacteria. Furthermore, leptin reduces intestinal mucosal adherence and translocation of bacteria, since in the absence of the hormone, the mice are dramatically obese and inflamed. Finally, to overcome the bacterial translocation, we used a probiotic treatment and showed that glucose metabolism was indeed improved, demonstrating the importance of the control of intestinal adherence and translocation of commensal bacteria in the regulation of metabolic diseases.
IMPACT
We thus propose a new paradigm for the treatment of metabolic disease. We suggest that targeting intestinal bacterial adherence, bacterial translocation, or receptors of bacterial fragments would be an original and novel strategy to prevent or reverse the occurrence of metabolic diseases. Therefore, new probiotics, which aim to target this function, can be defined.
Generation and preparation of L. lactis leptin and L. lactis
The bacterium strain Lactococcus lactis leptin (L. lactis leptin), expressing and secreting active human leptin, was generated by Bermúdez-Humarán and colleagues (INRA, Jouy en Josas, France) as previously described (Bermudez-Humaran et al, 2007). Briefly, the food-grade lactic acid bacterium Lactococcus lactis strain NZ9000 was transformed with a plasmid allowing the expression of mature human leptin under the control of inducible promoter (P_nisA_). As a negative control, the same L. lactis strain was transformed with the same plasmid lacking only the leptin-coding sequence.
Lactococcus lactis and L. Lactis leptin were grown in M17 (Sigma-Aldrich, France) supplemented with 1% glucose at 30°C without shaking. Mice were orally treated with 109 cfu/day of L. Lactis leptin or L. Lactis resuspended in sterile PBS.
Quantification of GFP-E. coli translocation towards mesenteric adipose tissue and mesenteric lymph nodes and adherence to intestinal mucosa
Two or five hours after gavage with 109 GFP-E. coli, mice were sacrificed. Mesenteric adipose tissue (MAT) and corresponding lymph nodes (MLN) were harvested, luminal and mucosal contents of each intestinal segment were separated. Tissues were then homogenized in Luria Broth (LB), plated onto ampicillin-supplemented (100 µg/ml) LB agar, and yellow colonies were enumerated after overnight incubation at 37°C.
Quantification of bacterial DNA in intestine, blood and mesenteric adipose tissue by qPCR
Genomic DNA was isolated from blood, MAT, MLN or intestine (contents and mucosa). All bacterial DNA was quantified by quantitative real-time PCR targeting conserved regions of the 16S rRNA gene, with bacterial DNA as standard template for absolute quantification (for details see Supporting information).
Quantification of pro-inflammatory markers expression by RT-qPCR
Total RNAs from liver, muscle (vast lateralis) and MAT were prepared, reverse transcripted, and submitted to qPCR targeting TNF-α, IL-1β, PAI-1, IFN-γ and IL-6 genes, with RPL19 as housekeeping gene for relative quantification (for details see Supporting information).
Statistical analysis
Data were analysed by using Prism GraphPad version 5.01 (GraphPad Software Inc., California, USA). All data are expressed as mean ± standard error of the mean (SEM). For comparison of two groups, Student's _t_-test was used. For comparison of more than two groups, ANOVA was performed followed by posthoc with Bonferroni's multiple comparison test to determine significance between groups. For linear regression analysis, significance of the correlation was determined by Pearson's test. Statistical difference was considered significant when p < 0.05.
Acknowledgments
We would like to thank Jacques Oberto (Institut de Génétique et Microbiologie, Université Paris-Sud), Sandra Handgraaf and Jason Iacovoni for their useful contribution, Ivo G. Boneca (Institut Pasteur, France) for providing Nod1 and Nod2 knockout mice and John Woodley for editing the manuscript. We also thank Jaana Larsson-Leskelä and Sofia Forssten (Danisco, Kantvik, Finland) for skilful technical assistance. RB is the recipient of grants from the Agence Nationale de la Recherche (ANR Floradip, Transflora, & Vaiomer), and the coordinator of the seventh framework program F7P (Florinash). This work was supported in part by the European Commission's Seventh Framework programme under grant agreement N° 241913 (FLORINASH).
Supporting information is available at EMBO Molecular Medicine online.
Conflict of interest statement: Sampo Lahtinen, Arthur Ouwehand and Nina Rautonen are employees of Danisco and have a conflict of interest. Thierry Sulpice is an employee of Physiogenex and has a conflict of interest.
Author contribution
AW, PK, CV, NS and MB performed the experiments. LGBH, PL, TS, AO and NR contributed to discussion. PJS designed the experiments and analysed the data. RB, SL and JA designed the experiments, analysed the data, contributed to discussion and wrote the paper. CC designed and performed the experiments, analysed the data, contributed to discussion, wrote and edited the paper.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Amar J, Burcelin R, Ruidavets J, Cani P, Fauvel J, Alessi M, Chamontin B, Ferrieres J. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr. 2008;87:1219–1223. doi: 10.1093/ajcn/87.5.1219. [DOI] [PubMed] [Google Scholar]
- Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- Bermudez-Humaran LG, Nouaille S, Zilberfarb V, Corthier G, Gruss A, Langella P, Issad T. Effects of intranasal administration of a leptin-secreting Lactococcus lactis recombinant on food intake, body weight, and immune response of mice. Appl Environ Microbiol. 2007;73:5300–5307. doi: 10.1128/AEM.00295-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burcelin R, Crivelli V, Dacosta A, Roy-Tirelli A, Thorens B. Heterogeneous metabolic adaptation of C57BL/6J mice to high-fat diet. Am J Physiol Endocrinol Metab. 2002;282:E834–E842. doi: 10.1152/ajpendo.00332.2001. [DOI] [PubMed] [Google Scholar]
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007a;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, Gibson GR, Delzenne NM. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia. 2007b;50:2374–2383. doi: 10.1007/s00125-007-0791-0. [DOI] [PubMed] [Google Scholar]
- Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, Geurts L, Naslain D, Neyrinck AM, Lambert DM, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut. 2009;58:1091–1103. doi: 10.1136/gut.2008.165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado MC, Meriluoto J, Salminen S. Role of commercial probiotic strains against human pathogen adhesion to intestinal mucus. Lett Appl Microbiol. 2007;45:454–460. doi: 10.1111/j.1472-765X.2007.02212.x. [DOI] [PubMed] [Google Scholar]
- Collado MC, Sanz Y. Induction of acid resistance in Bifidobacterium: a mechanism for improving desirable traits of potentially probiotic strains. J Appl Microbiol. 2007;103:1147–1157. doi: 10.1111/j.1365-2672.2007.03342.x. [DOI] [PubMed] [Google Scholar]
- Cook S, Hugli O, Egli M, Menard B, Thalmann S, Sartori C, Perrin C, Nicod P, Thorens B, Vollenweider P, et al. Partial gene deletion of endothelial nitric oxide synthase predisposes to exaggerated high-fat diet-induced insulin resistance and arterial hypertension. Diabetes. 2004;53:2067–2072. doi: 10.2337/diabetes.53.8.2067. [DOI] [PubMed] [Google Scholar]
- El Homsi M, Ducroc R, Claustre J, Jourdan G, Gertler A, Estienne M, Bado A, Scoazec JY, Plaisancie P. Leptin modulates the expression of secreted and membrane-associated mucins in colonic epithelial cells by targeting PKC, PI3K, and MAPK pathways. Am J Physiol Gastrointest Liver Physiol. 2007;293:G365–G373. doi: 10.1152/ajpgi.00091.2007. [DOI] [PubMed] [Google Scholar]
- Federici M, Hribal ML, Menghini R, Kanno H, Marchetti V, Porzio O, Sunnarborg SW, Rizza S, Serino M, Cunsolo V, et al. Timp3 deficiency in insulin receptor-haploinsufficient mice promotes diabetes and vascular inflammation via increased TNF-alpha. J Clin Invest. 2005;115:3494–3505. doi: 10.1172/JCI26052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz JH, Le Bourhis L, Sellge G, Magalhaes JG, Fsihi H, Kufer TA, Collins C, Viala J, Ferrero RL, Girardin SE, et al. Nod1-mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity. 2007;26:445–459. doi: 10.1016/j.immuni.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T, et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. 2011;469:543–547. doi: 10.1038/nature09646. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakayama Y. CXCL14 and insulin action. Vitam Horm. 2009;80:107–123. doi: 10.1016/S0083-6729(08)00605-5. [DOI] [PubMed] [Google Scholar]
- Hase K, Kawano K, Nochi T, Pontes GS, Fukuda S, Ebisawa M, Kadokura K, Tobe T, Fujimura Y, Kawano S, et al. Uptake through glycoprotein 2 of FimH(+) bacteria by M cells initiates mucosal immune response. Nature. 2009;462:226–230. doi: 10.1038/nature08529. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Keita AV, Salim SY, Jiang T, Yang PC, Franzen L, Soderkvist P, Magnusson KE, Soderholm JD. Increased uptake of non-pathogenic E. coli via the follicle-associated epithelium in longstanding ileal Crohn's disease. J Pathol. 2008;215:135–144. doi: 10.1002/path.2337. [DOI] [PubMed] [Google Scholar]
- Kufer TA, Kremmer E, Adam AC, Philpott DJ, Sansonetti PJ. The pattern-recognition molecule Nod1 is localized at the plasma membrane at sites of bacterial interaction. Cell Microbiol. 2008;10:477–486. doi: 10.1111/j.1462-5822.2007.01062.x. [DOI] [PubMed] [Google Scholar]
- Kufer TA, Sansonetti PJ. Sensing of bacteria: NOD a lonely job. Curr Opin Microbiol. 2007;10:62–69. doi: 10.1016/j.mib.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Lundin A, Bok CM, Aronsson L, Bjorkholm B, Gustafsson JA, Pott S, Arulampalam V, Hibberd M, Rafter J, Pettersson S. Gut flora, Toll-like receptors and nuclear receptors: a tripartite communication that tunes innate immunity in large intestine. Cell Microbiol. 2008;10:1093–1103. doi: 10.1111/j.1462-5822.2007.01108.x. [DOI] [PubMed] [Google Scholar]
- Macia L, Delacre M, Abboud G, Ouk TS, Delanoye A, Verwaerde C, Saule P, Wolowczuk I. Impairment of dendritic cell functionality and steady-state number in obese mice. J Immunol. 2006;177:5997–6006. doi: 10.4049/jimmunol.177.9.5997. [DOI] [PubMed] [Google Scholar]
- Membrez M, Blancher F, Jaquet M, Bibiloni R, Cani PD, Burcelin RG, Corthesy I, Mace K, Chou CJ. Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. Faseb J. 2008;22:2416–2426. doi: 10.1096/fj.07-102723. [DOI] [PubMed] [Google Scholar]
- Menghini R, Menini S, Amoruso R, Fiorentino L, Casagrande V, Marzano V, Tornei F, Bertucci P, Iacobini C, Serino M, et al. Tissue inhibitor of metalloproteinase 3 deficiency causes hepatic steatosis and adipose tissue inflammation in mice. Gastroenterology. 2009;136:663–672. doi: 10.1053/j.gastro.2008.10.079. [DOI] [PubMed] [Google Scholar]
- Neal MD, Leaphart C, Levy R, Prince J, Billiar TR, Watkins S, Li J, Cetin S, Ford H, Schreiber A, et al. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J Immunol. 2006;176:3070–3079. doi: 10.4049/jimmunol.176.5.3070. [DOI] [PubMed] [Google Scholar]
- Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, Liu-Bryan R, Glass CK, Neels JG, Olefsky JM. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. 2007;282:35279–35292. doi: 10.1074/jbc.M706762200. [DOI] [PubMed] [Google Scholar]
- Niess JH. Role of mucosal dendritic cells in inflammatory bowel disease. World J Gastroenterol. 2008;14:5138–5148. doi: 10.3748/wjg.14.5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulos CM, Wrzesinski C, Kaiser A, Hinrichs CS, Chieppa M, Cassard L, Palmer DC, Boni A, Muranski P, Yu Z, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest. 2007;117:2197–2204. doi: 10.1172/JCI32205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez PF, Dore J, Leclerc M, Levenez F, Benyacoub J, Serrant P, Segura-Roggero I, Schiffrin EJ, Donnet-Hughes A. Bacterial imprinting of the neonatal immune system: lessons from maternal cells. Pediatrics. 2007;119:e724–e732. doi: 10.1542/peds.2006-1649. [DOI] [PubMed] [Google Scholar]
- Putaala H, Salusjarvi T, Nordstrom M, Saarinen M, Ouwehand AC, Bech Hansen E, Rautonen N. Effect of four probiotic strains and Escherichia coli O157:H7 on tight junction integrity and cyclo-oxygenase expression. Res Microbiol. 2008;159:692–698. doi: 10.1016/j.resmic.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Riant E, Waget A, Cogo H, Arnal JF, Burcelin R, Gourdy P. Estrogens protect against high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology. 2009;150:2109–2117. doi: 10.1210/en.2008-0971. [DOI] [PubMed] [Google Scholar]
- Sansonetti P, Di Santo J. Debugging how bacteria manipulate the immune response. Immunity. 2007;26:149–161. doi: 10.1016/j.immuni.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Sansonetti PJ, Phalipon A. M cells as ports of entry for enteroinvasive pathogens: mechanisms of interaction, consequences for the disease process. Semin Immunol. 1999;11:193–203. doi: 10.1006/smim.1999.0175. [DOI] [PubMed] [Google Scholar]
- Serino M, Menghini R, Fiorentino L, Amoruso R, Mauriello A, Lauro D, Sbraccia P, Hribal ML, Lauro R, Federici M. Mice heterozygous for tumor necrosis factor-alpha converting enzyme are protected from obesity-induced insulin resistance and diabetes. Diabetes. 2007;56:2541–2546. doi: 10.2337/db07-0360. [DOI] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson S, Lee J, Goldfine A. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stienstra R, Joosten LA, Koenen T, van Tits B, van Diepen JA, van den Berg SA, Rensen PC, Voshol PJ, Fantuzzi G, Hijmans A, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010;12:593–605. doi: 10.1016/j.cmet.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanti J, Gual P, Gremeaux T, Gonzalez T, Barres R, Le Marchand-Brustel Y. Alteration in insulin action: role of IRS-1 serine phsophorylation in the retroregulation of insulin signalling. Ann Endocrinol. 2004;65:43–48. doi: 10.1016/s0003-4266(04)95629-6. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Ferrante AW., Jr CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116:115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.