Hotspots organize clathrin-mediated endocytosis by efficient recruitment and retention of nucleating resources (original) (raw)
. Author manuscript; available in PMC: 2012 Dec 1.
Abstract
The formation of clathrin-coated pits (CCPs) at the plasma membrane has been reported to sometimes occur repeatedly at predefined sites. However, defining such CCP `hotspots' structurally and mechanistically has been difficult due to the dynamic and heterogeneous nature of CCPs. Here we explore the molecular requirements for hotspots using a global assay of CCP dynamics. Our data confirmed that a subset of CCPs is nucleated at spatially distinct sites. The degree of clustering of nucleation events at these sites is dependent on the integrity of cortical actin, and the availability of certain resources, including the adaptor protein AP-2 and the phospholipid PI(4,5)P2. We observe that modulation in the expression of FCHo1 and 2, which have been reported to initiate CCPs, affect only the number of nucleations. Modulation in the expression levels of other accessory proteins, such as SNX9, affects the spatial clustering of CCPs but not the number of nucleations. Based on these findings we distinguish two classes of accessory proteins in clathrin-mediated endocytosis (CME): nucleation factors and nucleation organizers. Finally, we observe that clustering of transferrin receptors spatially randomizes pit nucleation and thus reduces the role of hotspots. Based on these data, we propose that hotspots are specialized cortical actin patches that organize CCP nucleations from within the cell by more efficient recruitment and/or retention of resources required for CCP nucleation partially due to the action of nucleation organizers.
Keywords: clathrin, endocytosis, hotspot, SNX9, FCHo1, FCHo2
INTRODUCTION
Clathrin-mediated endocytosis (CME) constitutes the major route for selective cargo internalization in higher eukaryotes, and as such is also central to cellular homeostasis. In CME, clathrin-coated pits (CCPs) are nucleated at the plasma membrane by the recruitment of clathrin to adaptor proteins (e.g. AP-2), which bind to trans-membrane receptors and phosphoinositide-4,5-biphosphate (PIP2). CCP maturation then proceeds with progressive clathrin polymerization, CCP invagination, and, finally, membrane scission leading to internalization of clathrin-coated vesicles (CCVs) (1–3).
In neuronal cells, endocytosis is coordinated with synaptic vesicle exocytosis at specialized sites, termed active zones. Active zones are defined by a proteinaceous cytomatrix, whose assembly is mediated by multi-domain scaffolding proteins (4–6); endocytic nucleations are concentrated at actin-rich patches adjacent to these sites allowing the rapid recycling of synaptic vesicle components required for fast and repetitive neurotransmission (7).
In non-neuronal cells, less is known about the degree and significance of CCP organization at the membrane, and it is unclear which molecular factors might be implicated in spatially regulating CCP formation at specialized sites. Several studies have observed multiple sequential CCP nucleations at the same location, which were termed hotspots (8–11). One of these studies proposed that hotspots correspond to endocytic platforms at the plasma membrane, that are interconnected and positioned by the actin network, and that recruit and retain adaptor proteins and other coat components (8). Another study rejected the existence of preferred, structurally-defined nucleation sites, and instead proposed that the observed clustering of CCP nucleations was more consistent with `active' patches of random nucleation resulting from the compartmentalization of the plasma membrane (9).
While the spatial distribution of CCPs at the plasma membrane can be directly studied using fluorescently labeled coat components and live-cell imaging, the interpretation of the data has been complicated by the heterogeneity in dynamics and size of individual CCPs, as well as differences between cell types (9, 12, 13). Furthermore, interpreting the correlation of CCP nucleation sites is complicated by the fact that two CCPs could nucleate at the same site purely by coincidence. Thus, a systematic and quantitative investigation of CCP nucleation site clustering requires (a) a complete and global analysis of all nucleation events, and (b) use of robust statistical methods for spatial analysis combined with a large data sample. To accomplish this, we make use here of our automated detection and tracking assay (12, 14) allowing us to measure the dynamics and spatial distribution of all CCPs visible in total internal reflection fluorescence microscopy (TIR-FM), yielding a high-resolution analysis of the spatial and temporal distribution of CCP nucleation sites.
RESULTS AND DISCUSSION
Nucleation events are partially organized into hotspots
We imaged CCP dynamics using TIR-FM in live BSC1 cells expressing clathrin-light-chain-a conjugated to EGFP (EGFP-CLCa) (12). Figure 1A shows an example of a single frame from a representative time-lapse TIR-FM image sequence with all detected CCPs overlaid. BSC1 cells have been reported to be practically devoid of clathrin-coated plaques and non-terminal events (10). Therefore, all CCP nucleation events should have a single corresponding termination event. In order to correctly identify the sites and time points of CCP nucleation, accurate tracking of all CCPs is particularly important. The `breaking' of CCP trajectories into partial sub-trajectories, for example, would lead to erroneous multiplication of nucleation events at the same site, and this type of artifact could by itself generate apparent hotspots. We used the particle tracking approach introduced in (14), which distinguishes on a probabilistic basis between the short-term disappearance and reappearance of the same particle, and the bona fide formation of a new particle at the location of a distinct earlier particle. We extracted from TIR-FM sequences uninterrupted CCP trajectories, even for low contrast, i.e. small CCPs. We first analyzed trajectories from BSC1 cells expressing EGFP-CLCa (n = 113 cells; 2859±1410 CCP trajectories per cell). We excluded CCP trajectories with lifetimes under 8 s, since the very short-lived trajectories are more strongly affected by false-positive detection events and are also least likely to represent true maturing CCPs (12). This selection yielded an average of 1810±890 usable CCP tracks per cell.
FIGURE 1. CME is organized into hotspots.

(A) CCPs in BSC1 cells expressing EGFP-CLCa were visualized with TIR-FM and detected as in (12). Detection results are shown here for a single frame as red dots overlaid on the negative of the image. (B) All CCP nucleation events detected throughout the time lapse are projected onto the footprint of the cell. (C) Simulated point distribution with the same number of points as in B, but in a spatially random distribution. (D) Pair correlation function of all nucleation events averaged over 113 control movies (blue). The function shows a significant increase above the background of a random distribution (black), indicating spatial clustering of nucleation events over a length scale 0 – 300 nm. (E) The pair correlation function of nucleation events was calculated using different observation windows and the maxima of the pair correlation functions were plotted vs. the duration of the observation window. Scale bars: 2 μm.
To assay the spatial distribution of nucleation sites, we projected all nucleation sites onto the footprint of the cell (Fig. 1B). It should be noted that if we randomly re-arrange the same number of nucleation events on the same footprint, some points will appear clustered just by chance (see Fig. 1C). Thus, statistical methods are necessary to accurately distinguish between apparent clustering in a random distribution and significant `real' clustering of points. To accomplish this we used the pair correlation function (15, 16), which measures the expected density of neighboring points as a function of distance from a given point of the distribution. For a spatially random distribution of points, the pair correlation function is constant at all distances and equal to the average density of points. Therefore, we normalized the pair correlation function of projected nucleation sites by the average density. Accordingly, function values greater than 1 indicate the tendency of nucleation sites to cluster at a certain distance. The magnitude of the pair correlation function defines how many more CCPs nucleate at this distance than expected from a random distribution of the same density of CCPs. Conversely, values less than 1 indicate how many fewer CCPs nucleate at this distance, i.e. how strong the mutual exclusion of nucleation events would be.
When we calculated the pair correlation function of projected nucleation events for control conditions we found values significantly greater than 1 for the distance range from 0 to 200 nm (Fig. 1D). This showed that a substantial number of nucleation events were succeeded within the 10-minute duration of our movies by at least one nucleation event located within a radius of 200 nm. Similar degrees of clustering were also observed in HeLa and NIH 3T3 cells (data not shown). Figure 1D displays the median of the individual pair correlation functions of 113 BSC1 cells. As expected, when we randomized the location of nucleation events within the footprint of each cell, the resulting median pair correlation function had a constant value of 1 (Fig. 1D). This confirmed that our data pool was large enough to identify the tendency of nucleation sites to tightly cluster.
To investigate whether this clustering depended on our choice of movie length, we measured the pair correlation for different observation periods. To do so, we re-analyzed the same data set by taking into account only CCP nucleations occurring within a given time frame (see Methods). We found that the maximum of the pair correlation function varied, but was significantly greater than 1, for all observation windows (Fig 1E). This suggests that CCP nucleations cluster over intervals as short as 30 s and over minutes.
Computer simulations confirm the existence of a CCP population nucleated at hotspots
To deduce the type of nucleation processes that might lead to the observed clustering of CCPs, we simulated the pair correlation functions of three nucleation configurations: 1) nucleation events that occur randomly at the membrane; 2) nucleation events that occur randomly within randomly dispersed circular active areas, i.e., CCPs would preferentially form in delimited membrane domains; and 3) nucleation events that occur within active areas, i.e. hotspots, where the probability of occurrence decays with distance from the center of the active area following a Gaussian bell curve. We obtained the best fit to the experimentally measured pair correlation by combining models 1) and 3) (Fig. 2A). In this configuration, the average number of nucleation repeats per hotspot and the ratio between nucleation events inside hotspots versus outside hotspots are strongly correlated, i.e., the simulation does not allow us to distinguish between the scenario where few hotspots induce nucleation repeats at a very high frequency or the scenario where more hotspots induce nucleation repeats at a more moderate frequency. It does, however, allow us to uniquely determine the radius of hotspots. Our analysis revealed that the locations of CCP nucleation events were distributed normally around hotspot locations with a full width at half maximum diameter of 268 nm. Interestingly, the best-fit diameter was reduced to 212 nm for data sampled at 400 ms intervals. We interpret this finding as an indication that CCP nucleation in hotspots is highly localized – below the 212 nm we can measure at fast frame-rates – and that the apparent positional jitter of nucleation events at slower sampling results from the diffusion of nascent CCPs over the time interval between formation and the acquisition of the subsequent image.
FIGURE 2. Models of point processes provide insight into the mechanism of hotspot regulation.

We simulated point processes and measured the pair correlation for these processes. Each simulation consisted of a total of 300 frames of 500 by 500 pixels. A total of 50 curves resulting from 50 iterations of the simulation were averaged to produce the curves displayed here. (A) Our experimental data is best fitted by a model in which 57% of the nucleations occur in invisible hotspots (`parent loci') randomly scattered throughout the frame. Spatially, nucleations were Gaussian-distributed around each parent locus with a full width at half maximum diameter of 268 nm. The remaining 43% points are randomly distributed. (B) Implementation of a spatial nucleation model (active random patches 200 nm in radius whose centers are separated by 400 nm) as described in Ehrlich et al. (9). We find that this model provides a less adequate fit, i.e. a higher sum of squares of the residuals, to the experimental data. (C, D, and E) We measured the pair correlation for different observation periods for a simulation as in (A). (C) Temporal dependencies between pits were modeled by introducing a Poisson-distributed probability for each parent locus to give rise to a nucleation with an expectancy value of 0.0022 nucleations/locus/frame. (D) Hotspots become incompetent for renewed nucleation for 16 s after a nucleation event. (E) Hotspots are forced to turn over after 160 s; to maintain a steady state density of hotspots the disappearance of a hotspot is accompanied by generation of a new hotspot in another location.
A previous study used a different model to fit their data, and proposed that the observed spatial distribution of nucleation sites is the result of random nucleation within endocytically active compartments of the plasma membrane that are 400 nm in diameter and whose centroids are separated by at least 400 nm (9). When we implemented this type of spatial model, we found that this diameter is too large to match our data (Fig. 2B), as evidenced by the fact that this model does not accurately reproduce the shape of the pair correlation at radii of about 200 nm. The difference in our results to this previous analysis likely originates in the faster sampling of our data, which leads to a more accurate localization of nucleation events.
Temporal characteristics of CCP nucleations at hotspots
The change in the peak of the pair correlation we observe with varying observation windows (Fig. 1E) contains information about the temporal relation between CCP nucleation events in any given hotspot. If nucleation events are temporally independent, the magnitude of the pair correlation function will be independent of the observation window, as shown in Fig. 2C. In contrast, our experimental data showed a strong increase in the magnitude of the pair correlation maximum as the duration of the observation window increased from 40 s to 160 s (Fig. 1E). This indicated that nucleations at hotspots are temporally coupled. We were able to simulate this rise in the magnitude by imposing an exclusion period of 160 s between subsequent nucleations at hotspots (Fig. 2D).
For observation windows longer than 160 s our data showed a slight decrease in the magnitude of the pair correlation for small radii (Fig. 1E). This behavior indicates a gradual loss of clustering, either because of a continuous turnover of hotspot sites, or because of cell movements. We favor the former interpretation because cell movement during our ten-minute acquisitions is minimal. Our model reproduced the observed decay in the pair correlation by enforcing turnover of hotspots after 160 s (Fig. 2E).
CCPs inside and outside hotspots have the same maturation kinetics
Hotspots were defined as groups of repeated nucleation events within a circular area of radius 164 nm as determined by the decay of the pair correlation function (Fig. 3A, see Methods). We found that 53% ±12% (n = 113 cells) of CCP nucleations in a cell occurred in hotspots, which is significantly higher (p < 0.001) than the fraction of coincidental nucleations within the same circular area for a spatially random population (41% ± 8%). To further validate the identification of hotspots we recalculated the pair correlation of all nucleation events relative to hotspots, rather than to other nucleation events, and found an eight-fold increase (Fig. 3B). This confirmed the high degree of clustering of CCP nucleation events around these hotspots.
FIGURE 3. Hotspots only affect the spatial organization of nucleation events.

(A) Classification of CCP nucleation events into events occurring inside hotspots (red; hotspot areas indicated by circles) and outside hotspots (blue). (B) (Cross-) Pair correlation function of the different point populations identified in A. The pair correlation of nucleations relative to hotspot centroids (red), and the pair correlation of nucleations with respect to other nucleations (blue) are shown here. (C) Contributions and characteristic lifetime of early abortive, late abortive, and productive CCP populations nucleated inside hotspots and outside hotspots. Error bars indicate standard error of the mean. (D) Waiting times between subsequent nucleation events at each hotspot as a function of the lifetime of the preceding CCP. Statistical data in the text relies on n = 53610 nucleation events. For graphical purposes the data has been randomly subsampled reflecting n'= 1000 events. The black line represents the unity time. Data points below the line indicate nucleation events before termination of the preceding CCP. Data points above the line indicate nucleation events after termination of the preceding CCP. (E) Cumulative histogram of the time elapsed between two consecutive nucleation events in hotspots. Histograms are binned according to the lifetime of the first of the two CCPs.
In previous work, we decomposed the CCP population into early and late abortive, and productive CCPs based on lifetimes (12). We independently subjected the CCP subpopulations inside and outside hotspots to this decomposition analysis and found no significant difference in either their relative contributions or lifetimes (Fig. 3C). From this we conclude that nucleation within a hotspot does not enhance either the efficiency or the rate of CCP maturation.
The ability to identify hotspots allowed us to directly measure the temporal coordination 1) between subsequent nucleation events, and 2) relative to termination events, induced by either abortion or internalization. A graph of the lifetime of the preceding CCP in a hotspot versus the waiting time between its nucleation and the next nucleation event in the same hotspot (Fig. 3D) indicates that ~20% of CCP nucleations in hotspots occur before the termination of a preceding CCP (data points below the unity line). Thus, multiple CCPs can mature concurrently within a single hotspot, and nucleation of a CCP in a hotspot does not necessitate the termination of the preceding CCP as previously suggested (10, 17).
Given the broad heterogeneity in CCP lifetimes, we were interested in figuring out whether the lifetime of the preceding CCP influences the waiting time of the subsequent CCP. The graph in Fig. 3D displays an increasing fraction of data points below the unity line, suggesting that the longer the lifetime of a CCP in a hotspot the higher is the probability for a new nucleation in the same hotspot before its termination. To quantify this in more detail we binned CCPs into cohorts of different lifetime ranges and analyzed the cumulative histogram of the waiting times to the next nucleation (Fig. 3E). Indeed we find that while for a lifetime range of 20 – 40s only 9% of the subsequent nucleation events occur before termination. This fraction increases to 42% and 65% for lifetime ranges of 120 – 160s and 200s – 300s, respectively. However, the rightward shift in the cumulative histograms for bins containing longer-lived CCPs indicates that it takes longer for the hotspot to support nucleation of a new CCP after the production of a long-lived CCP. Longer-lived CCPs tend to be brighter, thus are larger and most likely bind more molecular resources (9, 12). Our previous simulations predicted that an exclusion period exists between consecutive nucleations at hotspots (Fig. 2D). Based on these data, we hypothesize that this exclusion period reflects a limited availability of resources for the initiation of CCPs, and that this exclusion period is prolonged after the nucleation of longer-lived CCPs, which exhaust more resources from the hotspot.
Nucleations at hotspots are regulated by actin and the availability of nucleation resources
The distribution of hotspots establishes a spatial memory for CME, which must somehow be imprinted onto the plasma membrane. The most likely candidate for this function is the actin cortex, which, according to the membrane matrix model, is the main structural network dividing the plasma membrane into microscopic parcels with potentially variable molecular properties (18). Earlier studies had indeed suggested a role for cortical actin in the maintenance and positioning of hotspots (8, 19). Based on these findings, we perturbed the structure of the actin cortex with the actin depolymerization agent Latrunculin A (LatA) (20). The drug was applied at a low concentration (50 nM) to avoid cell detachment from the substrate and/or uncontrolled side effects due to changes in cell morphology. Upon LatA treatment, we observed a decrease in the density of CCP nucleation events (Fig. 4A), as defined by the number of nucleation events observed in a given cell, divided by its footprint area. This is consistent with previous data suggesting a role for cortical actin in promoting CCP nucleation (19, 21). However, the clustering density, defined by the integration of the pair correlation function over the area of a hotspot (134 nm; see Methods), increased (Fig. 4B) in agreement with previous observations (19). We conclude that although perturbation of the actin cortex yields overall less CCPs, those remaining show an increased tendency to nucleate inside hotspots.
FIGURE 4. Clustering density of nucleation events depends on the actin cortex and nucleation resources.

(A) Normalized nucleation density (number of nucleation events divided by total area of the cell's footprint) and (B) normalized clustering density (integral of the pair correlation function over radius interval (0 – 134 nm) for control cells (n = 23; normalized to group's median), latrunculin treated cells (n = 5; normalized to median of 9 control wild-type cells) and jasplakinolide treated cells (n = 23; normalized to median of 18 control cells). Red bar, median; blue box, range between 25th and 75th percentiles; whiskers, range of data classified as inliers to the distribution. (C) Normalized nucleation density and (D) normalized clustering density for control cells (n = 23; normalized to group's median), cells treated with siRNA for PIP5Kα (n = 19; normalized to the median of 48 non-targeting siRNA treated cells), cells transfected with PIP5Kα tagged with mCherry (n = 23; normalized to the median of 29 cells transfected with mCherry alone), cells treated with PIP2 carried by neomycin (n = 10; normalized to median of 9 control neomycin treated cells), and μ2 (AP-2) siRNA treated cells (n = 14; normalized to median of 9 control wild-type cells). (E) Normalized nucleation density and (F) normalized clustering density for cells treated with non-targeting siRNA (n=8; normalized to group's median), with siRNA against FCHo1/2 (n = 8; normalized to the median of 8 non-targeting siRNA treated cells), and for cells transfected with tagRFP-T-FCHo2 (n = 6; normalized to the median of 10 wild-type cells). (G) Normalized nucleation density and (H) normalized clustering density for cells treated with non-targeting shRNA (n=6; normalized to group's median) and with shRNA against SNX9 (n =8; normalized to the median of 6 non-targeting shRNA treated cells). * is P < 0.05, ** is P < 0.01, *** is P < 0.001 according to paired t-test (see Methods). All conditions where compared to controls from the same user in order to account for user-to-user variability in acquisition conditions.
To differentiate between a requirement for a stable or a dynamic actin network in CCP nucleation and organization, we also treated cells with 125 nM Jasplakinolide, which prevents disassembly of actin filaments (22). Under this condition, cells displayed no significant change in either nucleation or clustering density (Fig. 4A and B), but did show changes in both CCP diffusion and lifetime (data not shown), which indicates that the dose of the treatment was sufficient to affect the function of a dynamic actin cytoskeleton in the later stages of CCP maturation. From this data we conclude that hotspots are defined by the presence, rather than the dynamic behavior, of a cortical actin network.
The involvement of the actin cytoskeleton in defining hotspots further supports the notion that the observed decrease in the maximum of the pair correlation with longer observation windows (Fig 1F) is related to hotspot turnover. Indeed our simulations suggest that a hotspot turns over after approximately 160 s, which is similar to the 80–120 s it takes the actin network to transition locally from assembly to disassembly to reassembly (23).
It has been suggested that actin may play an indirect role in organizing nucleation events by corralling phosphatidylinositol-4,5-biphosphate (PIP2) (19), which is required to recruit adaptor proteins to the plasma membrane. To test whether PIP2 availability affects the organization of CCP nucleations we reduced the expression levels of type I phosphatidylinositol-4-phosphate 5-kinase α (PIP5K1-α), the major PI5-kinase isoform responsible for PIP2 production in BSC1 cells (24). Cells depleted of PIP5K1-α displayed a decrease in nucleation density ((24) and Fig. 4C), and a variable but significant increase in the clustering density (Fig. 4D). We suspect that the variability reflects cell-to-cell differences in the degree of PIP5K1-α knock-down. Correspondingly, overexpression of PIP5K1-α resulted in more nucleation events (Fig. 4C), consistent with a prior report of increased AP-2 binding to membranes under these conditions (25), and a reduced clustering density (Fig. 4D). To confirm that the effects of altering PIP5K1-α levels on CME were mediated through a change in PIP2 levels, we delivered exogenous PIP2 directly to cells via neomycin carriers. Cells treated in this manner exhibited the same increase in nucleations and decrease in clustering density seen in cells with increased expression levels of PIP5K1-α. These results are consistent with previous observations in that increased PIP2 levels lead to an increase in nucleations (19), and suggest that nucleation at hotspots is less sensitive to bulk PIP2 levels than random nucleation events.
The adaptor protein AP-2 plays a critical role in initiating CCP assembly (26). As previously reported (12), siRNA-mediated knock-down of the μ2 subunit of AP-2 by 50% resulted in a corresponding decrease in the number of CCPs. We show that this treatment also resulted in a 50% decrease in the number of nucleation events (Fig. 4C); interestingly, the clustering density increased (Fig. 4D). Thus CCP nucleation at hotspots is also less sensitive to bulk AP-2 levels than randomly nucleated events.
Taken together, these data are consistent with a model in which actin mesh elements define local chemical microenvironments with varying nucleation competencies based on the amount of nucleation resources each holds. When the global concentration of a given resource is reduced, a relative increase in nucleations is observed in the mesh elements that retain sufficient resources to support nucleation.
FCHo1/2 are not involved in the organization of nucleation sites
The BAR (Bin/Amphiphysin/RVS) domain-containing proteins FCHo1 and 2 have been recently implicated in CCP initiation (17). We therefore tested whether these proteins play a role in the organization of nucleation sites. To do this we knocked-down FCHo1 and 2 simultaneously (knock-down efficiencies of 20% and 55% respectively; Fig. 4E and Supplementary Fig. 1) because knock-down of only one isoform leads to a compensatory overexpression of the other (data not shown). Consistent with previous data (17), we observed a significant decrease in the number of nucleation events upon knock-down of FCHo1 and 2. However, this treatment did not affect CCP clustering. Because the observed knock-down efficiencies were relatively low (in agreement with previous reports (17)), we confirmed this result by overexpressing FCHo2. Correspondingly, we observed an increase in the number of nucleations, and no change in CCP clustering (Fig. 4E and F). Thus in contrast to modulations in the levels of other nucleation resources (e.g. AP-2 and PIP2), modulations in the levels of FCHo1 and 2 affect nucleations inside and outside hotspots to an equal extent.
SNX9 regulates the organization of CCPs and is recruited early to hotspots
In an effort to identify additional molecular components that might play a role in determining the differential nucleation inside and outside hotspots, we re-examined data from a study in which we reduced the expression levels of several endocytic accessory proteins by siRNA-mediated knock-down (27). Of these, only reduction in the expression levels of a subset of curvature generating and/or sensing proteins, including epsin1, SNX9 and endophilin, caused a weak but significant decrease in the degree of clustering of CCP initiation events (Supplementary Fig. 1D). Neither SNX9 nor endophilin affected CCP nucleation density (Supplementary Fig. 1C), suggesting that these proteins might play a more specific role in defining endocytic hotspots. Although the effect of knock-down of SNX9 and endophilin on CCP clustering was significant, at the levels of knock-down achieved in these experiments (35% and 50%, respectively (27)) it was not dramatic. This may reflect a functional redundancy between these two structurally related proteins and perhaps other BAR domain-containing accessory factors. Nonetheless, we focused on SNX9 because its depletion caused the strongest decrease in clustering, even though its knock-down was the least efficient (27).
To confirm that knock-down of SNX9 consistently leads to a reduction in the clustering of nucleation sites, we used an inducible shRNA construct to increase SNX9 knock-down efficiency. shRNA-mediated knock-down of SNX9 by 50% on average (knock-down efficiency varied between 40% and 80%; see Supplementary Fig. 1B for a representative blot) confirmed our previous observation, i.e. reduced spatial clustering of CCP nucleation sites without affecting the nucleation density (Fig 4G and H). While the magnitude of the effect did not change, the variability of the response was greatly reduced, reflecting our capability of selecting cells for filming that are expressing the shRNA via an mCherry reporter expressed in-line with the shRNA.
SNX9 contains an N-terminal Src homology 3 (SH3) domain, a phospholipid-binding PX domain, and a C-terminal, curvature generating/sensing BAR domain (28, 29). SNX9 can bind to clathrin, AP-2 and dynamin (30–32), and stimulates dynamin's GTPase and self-assembly activities (30, 32). SNX9 interacts with negatively charged phospholipids on membranes, in particular PIP2 and PIP3 (33, 34). It binds to and activates PIP5K (33), and also interacts with N-WASP or WASP, and Arp2/3, linking it to the actin cytoskeleton (34–37). SNX9, therefore, has many of the properties that might be expected of a protein involved in organizing the nucleation of CCPs at the interface between the actin cortex and the plasma membrane.
SNX9 has previously been shown to be recruited to productive CCPs during the late stages of their maturation and just prior to (30), or slightly after (38) their internalization. This kinetic behavior would be inconsistent with a role for SNX9 in organizing hotspots. Therefore, we reexamined the dynamics of mCherry-SNX9 relative to EGFP-CLCa-labeled CCPs using dual color TIR-FM. Neither nucleation density nor degree of clustering was altered in cells expressing mCherry-SNX9 (data not shown); thus SNX9 appears not to be a limiting component for either activity. To capture potentially different SNX9 recruitment profiles, we applied a previously described method (39) to determine SNX9-mCherry intensity traces at CCPs nucleated inside or outside hotspots (red and blue lines in Fig 5A respectively). Averaged intensity traces were calculated for a cohort of CCPs with lifetimes of 61–80 s (Fig. 5A; see Methods), which in large majority correspond to productive events. In agreement with previously observed behaviors (30), SNX9 reached its highest level of recruitment during the late stages of productive CCP maturation and declined concurrently with the loss of clathrin from the TIRF field (Fig. 5A), independently of whether CCPs nucleated inside or outside hotspots.
FIGURE 5. SNX9 is enriched in hotspots prior to CCP nucleation.

(A) Average SNX9 intensity trace for CCPs found to nucleate in hotspots (red) is compared to the average SNX9 intensity trace of CCPs found to nucleate outside hotspots (blue), and to the average clathrin intensity trace of all CCPs (black). 678 CCPs (430 hotspot CCPs, 345 outside nucleated CCPs) with a lifetime 60 – 80 s from 7 cells were used. SNX9 intensities were normalized to the maximal average SNX9 intensity of CCPs in hotspots; clathrin intensities were normalized to the maximal average clathrin intensity of the combined population of CCPs inside and outside hotspots. (B) Mean intensity (normalized by same factor as SNX9 traces in A) from 30 s to 6 s before CCP nucleation, and from 6 s to 30 s after CCP internalization for all CCPs (8 – 100 s in lifetime) found to nucleate in hotspots (hotspots), for CCPs that nucleate and remain in hotspots (remain in hotspots), for CCPs that nucleate in and then diffuse away from a hotspot (leave hotspots), and for CCPs that nucleate outside hotspots (random); n = 3797, 2003, 1794, and 3146 CCPs respectively. Error bars represent the standard error of the mean. (C) Average FCHo2 and clathrin intensities for CCPs with a lifetime 60 – 80 s (2606 hotspot CCPs, 1682 outside nucleated CCPs from 6 cells were used). (D) Average AP-2 (σ2-EGFP) and clathrin intensities for CCPs with a lifetime 60 – 80 s (1038 hotspot CCPs, 483 outside nucleated CCPs from 8 cells were used). AP-2 and FCHo2 intensity traces were normalized in the same manner as SNX9. SNX9, AP-2, and FCHo2 intensity traces in the background were obtained by sampling random partner intensity traces for each CCP at a distance of 134 nm.
EGFP-CLCa intensity profiles were identical in CCPs nucleated inside or outside hotspots (data not shown). In contrast, the intensity profiles for SNX9 differed for CCPs nucleated within or outside a hotspot. For all lifetime cohorts (Fig. 5A and data not shown) we could detect a substantial SNX9 signal at the nucleation site before the onset of clathrin assembly within hotspots, but not at random nucleation sites where SNX9 began to accumulate only after the onset of clathrin assembly. In addition, SNX9 was retained at higher levels after CCP internalization within hotspots than outside hotspots. Only a fraction of CCPs (47% ± 12%) that nucleate inside hotspots also terminate within the hotspot radius, others diffuse away before termination. When we compare the residual intensity of SNX9 at CCPs that terminate within and outside of hotspots we find that traces of the former return to an intensity comparable to the high pre-nucleation level (Fig. 5B), whereas traces of CCPs terminating outside hotspots tend to drop to the post-termination level of CCPs nucleated outside hotspots. This suggests that a portion of SNX9 is associated with the hotspot and does not turn over with the assembly and internalization of a CCP. Together, these observations are consistent with a role for SNX9 both in early events required for organizing repeated nucleation of CCPs within hotspots and late in CCP maturation.
To examine how the recruitment of SNX9 compares with the recruitment of nucleation resources that are not responsible for the spatial organization of hotspots, we also measured the intensity traces of FCHo2 and AP-2 at CCPs inside and outside hotspots. To measure FCHo2 levels, EGFP-CLCa expressing cells were transfected with tagRFP-T-FCHo2. To measure AP-2 levels, we transfected dTomato-CLCa into cells stably expressing the σ2 subunit of AP-2 tagged with EGFP. We found that the pre-nucleation and post-internalization intensities of FCHo2 were also higher at hotspot vs. non-hotspot CCPs (Fig. 5C and D), whereas AP-2 intensity profiles were the same for both populations. However, in contrast to SNX9, there was no difference in the shape of FCHo2 or AP-2 intensity profiles (Fig. 5C and D respectively) for CCPs nucleated inside or outside hotspots. Thus, of the proteins we examined SNX9 is unique in its differential behavior (i.e. early recruitment to and retention at CCPs) within hotspots.
Clustered transferrin receptors nucleate CCPs outside hotspots
Our data suggests that hotspots are organized by the actin cortex and associated nucleation factors, thus they represent a mechanism by which CME and its transmembrane cargo could be spatially organized in an inside-out manner. A contrasting mechanism would be one in which specific transmembrane receptor signals trigger the formation of endocytic structures in an outside-in manner. We recently showed that clustering of biotinylated transferrin receptor at the cell surface via fluorescent conjugates of multivalent streptavidin molecules triggers de novo CCP nucleation in an outside-in fashion (40). To determine whether these nucleation events occur within hotspots or are randomly distributed we reanalyzed the data published in (40) with respect to the spatial organization of CCP nucleation sites. When we compared the clustering of CCPs in cells treated with tetravalent cargo against cells treated with either monovalent cargo or cargo that cannot bind transferrin receptor, we found that CCP clustering is significantly reduced upon cross-linking of receptors (Fig. 6). This result suggests that clustering of non-signaling receptors by multivalent cargo can nucleate CCPs at randomized locations on the cell surface, i.e. outside hotspots.
FIGURE 6. Clustering cargo reduces CCP clustering.
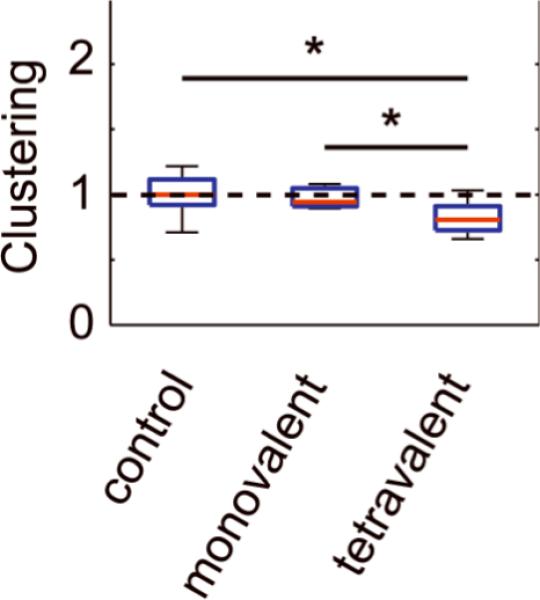
Clustering was measured for cells expressing biotinylated transferrin receptor and treated with a tetrameric streptavidin cargo in which each monomer contains a mutation and cannot bind biotin (control, n = 14), in which only one monomer can bind biotin (monovalent, n = 9), and in which all four monomers can bind biotin (tetravalent, n = 12). All conditions were normalized to the median of the cargo the control. * is P < 0.05 according to paired t-test (see Methods). Red bar, median; blue box, range between 25th and 75th percentiles; whiskers, range of data classified as inliers to the distribution.
Conclusions
Using sensitive particle tracking methods and rigorous quantitative analysis we have established the existence of hotspots that organize clathrin-mediated endocytic events. Our data further suggest that accessory proteins implicated in the earliest stages of CCP formation can be functionally grouped into nucleation factors and nucleation organizers. Reduced expression of nucleation factors decreases the number of CCP initiation events, while increasing the clustering of remaining CCPs. From this we conclude that the recruitment efficiency of some of these nucleation factors, i.e. AP-2 and PIP2 is enhanced at hotspots, allowing for a nucleation advantage when these resources are scarce. Knockdown of FCHo1/2, which is also required for nucleation, does not affect CCP clustering, suggesting that its role is restricted to that of a nucleation factor and that it is not preferentially recruited to hotspots. In contrast, SNX9 appears to function as a nucleation organizer in that CCP clustering is decreased when its expression levels are reduced, whereas nucleation density is unaffected. Consistent with this role, SNX9 is selectively retained at hotspots after CCP internalization and selectively detected at hotspots before subsequent CCP assembly. SNX9 may function as a nucleation organizer by efficiently recruiting and/or retaining factors necessary to support multiple CCP nucleations at a specific site. The small, but significant effects of SNX9 knock-down on CCP clustering suggests that other factors, possibly redundant with SNX9, might also fulfill the function of a nucleation organizer. Our analysis of the siRNA-mediated knock-downs of various endocytic accessory proteins (see Suppl. Fig. 2) reveals that reduced expression of the BAR-domain containing protein endophilin also reduces CCP clustering without affecting nucleation. Further studies are required to identify other nucleation organizers.
Whether a CCP is nucleated inside or outside a hotspot has no effect on the efficiency or speed of CCP maturation. Instead, hotspots serve to organize CME spatially. Much like the organization of CME in neuronal active zones, the spatial regulation of CME in non-neuronal cells may play a crucial role in polarization of and/or functional organization of the cell surface (41). In particular, CME may be implicated in determining the spatial distribution of receptors at the plasma membrane to establish signaling gradients required for directed cell migration and morphogenesis. Few studies have attempted to link the polarization of signal transduction in these processes to a polarization in endocytosis (41), and even fewer have examined the mechanisms by which cells might spatially organize CME. Hotspots define a mechanism for the “inside-out” regulated nucleation of CCPs and their transmembrane cargo, which may be responsible for the spatial polarization of CME in response to intracellular cues. Alternatively, CME may be polarized by an “outside-in” mechanism where gradients in the clustering, concentration, and/or activation of receptors in response to extracellular signals establish spatial cues in CCP nucleation. We have presented evidence that both mechanisms can spatially regulate CCP initiation. It would therefore be of interest to scrutinize the importance of polarized CME and the role of hotspots in these cellular functions.
MATERIALS AND METHODS
Cell culture and life cell imaging via TIR-FM
BSC1 monkey kidney epithelial cells stably expressing rat brain clathrin-light-chain-a conjugated to EGFP or rat brain σ2-adaptin fused to EGFP (kindly provided by Dr. T. Kirchhausen, Harvard Medical School) were grown under 5% CO2 at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 20 mM HEPES, 10 μg/ml streptomycin, 66 μg/ml penicillin, 10% (vol/vol) fetal calf serum (FCS, Hyclone, Logan, UT) and 0.5 mg/ml geneticin (Invitrogen, Carlsbad, CA). Cells were imaged as previously described (12, 27).
Reagents and cell treatments
Latrunculin A and Blasticidine were bought from Sigma. Jasplakinolide was kindly provided by Dr. V. Fowler (The Scripps Research Institute). Doxycycline was bought from Clontech (Mountain View, CA). A primary antibody against FCHo2 was a gift from Dr. H. McMahon (Medical Research Council Laboratory of Molecular Biology); FCHo1 (ab84740) and α-tubulin (ab11304) were bought from Abcam; SNX9 (sc-166863) was bought from Santa Cruz. siRNA duplexes (Dharmacon) were the following: μ2: AAG UGG AUG CCU UUC GGG UCA (Motley et al., 2003); PIP5K1α: AAC TGC AGA GCT TCA AGA TAA; FCHO1: SMARTpool L-014114-02; FCHo2: SMARTpool L-024508-02; non-targeting D-001810-01; the rest of the sequences and siRNA-mediated protein depletion protocols were described previously (27). Addition of PIP2, using a shuttle PIP kit (Echelon Biosciences), was achieved by incubating cells with 20 μM PIP2/neomycin complex according to manufacturer instructions. For mCherry-SNX9 (35), PIP5K1-α-mCherry (from the mouse phosphatidylinositol-4-phosphate-5-kinase 1 (PIP5K) beta, which is the homolog of the human alpha isoform), FCHo1/2-tagRFP-T (a kind gift from Dr. H. McMahon), and dtTomato-CLa (cDNA for dsRed-CLCa obtained from Wolf Almers (Vollum Institute, Oregon Health and Science University) and subcloned in-frame with dtTomato) over-expression, cells were transfected with 6 μg DNA and 12 μl lipofectamine2000 transfection reagent (Invitrogen) in a total volume of 2 ml Opti-MEM, according to manufacturer's recommendations. After a 5-h incubation, cells were washed with culture medium and split if necessary. Cells were imaged by TIR-FM 24 hours after transfection.
shRNA against SNX9 (ctaagcactttgactggttata) or non-targeting (atctcgcttgggcgagagtaag) were cloned into Expression Arrest TRIPZ lentiviral shRNAmir (Open Biosystems), which was further modified to replace the puromycin resistance to blasticidine resistance by seamless cloning (42). Lentivirus was harvested from HEK293T cells cotransfected with the modified TRIPZ plasmid, psPAX2, and pMD2.G plasmids from addgene. BSC1 cells stably expressing EGFP-CLCa were infected with virus for 3 days, and then transferred into medium containing 2 mg/ml blasticidine for selection.
Image analysis
Custom-written software for single-particle detection and tracking was used to compute the trajectories of all CCPs visible in TIR-FM (14). The tracking algorithm incorporates a gap-closing scheme, which links partial trajectories resulting from the unstable EGFP-CLCa signal.
CCP lifetimes were decomposed into early and late abortive, and productive populations as previously described (12).
Pair Correlation
The quantitative analysis of spatial clustering of CCPs is based on Ripley's K-function (16), from which the pair correlation function is derived. The pair correlation function c(r) is the average normalized probability of finding a neighboring point at distance r. For a spatially random particle distribution c(r) = 1. Values c(r) < 1 indicate how much scarcer the point distribution is at this distance relative to a random distribution. Values c(_r_) > 1 indicate how much denser neighboring points are packed at this distance relative to a random distribution.
To calculate Ripley's K-function or the pair-correlation function, the numbers and distances of all neighboring points are determined for each point in the image. For points located at the edge of the image - which may have additional 'invisible' close neighbor points located outside of the image - a variety of edge correction methods can be used to account for the missing observations. We used Ripley's correction to adjust the number of visible neighbors in a circle of radius r centered on the point located at (x,y) by a correction factor derived from the fraction of the circular circumference that falls inside the field of view/region of interest. For a rectangular region of interest, this correction factor c(x,y,r) can be determined analytically (15), and is given by
c(de,r)={π+2sin−1(de∕r)2πwherer>de1elsewhere}
where de denotes Euclidian distance of a given point location from the cell edge. For a non-rectangular area of interest, such as a cell footprint, we can use the same correction factor by assuming that the edge of this region is straight where it intersects with the circle centered on a point at (x,y). Based on random distributions, we find that this approximation is accurate for distances up to several microns.
In order to measure the pair correlation for different observation windows, we calculated the pair correlation for CCP nucleations that occurred within a given time frame in a 10-minute movie. To illustrate, for an observation window of 100 s the pair correlation was calculated taking into account CCP nucleations observed within the first 100 s of a movie. This pair correlation was then averaged with another six pair correlations calculated from the nucleations occurring within 100 s time intervals shifted by 40 s each (i.e. nucleations occurring from t = 40s to t = 140s, t = 80 to t = 180s, etc.) to give the 100 s pair correlation for a given movie.
We derived the clustering density parameter from the integration of the pair correlation up to a radius of 134 nm. Taking such a small radius as the limit of integration ensures that clustering is measured well within the typical cluster size for all conditions.
Classification of CCP nucleation events into inside and outside hotspots
Hotspots were identified by a newly developed point clustering algorithm. For a given data set, the algorithm first determines a clustering diameter derived from the pair correlation. This diameter is calculated by measuring the distance for which the pair correlation acquires a value of one over the average value at large distances, which is defined as the average value of the pair correlation function between distances ranging from 10 pixels (0.67 μm) to 20 pixels (1.34 μm). Doubling this distance gives the clustering diameter. The algorithm then finds all the neighbors of each nucleation that can be fit within this diameter. Each set of neighbors defines a possible cluster. The largest possible cluster, in terms of number of neighbors, is chosen as a true cluster. The members of this cluster are removed from further consideration. The algorithm then iterates, finding neighbors and choosing clusters. The iteration stops when no neighbors within the clustering distance can be found for the remaining nucleations. The cluster with the smallest average nucleation-to-cluster-centroid distance is chosen if two clusters of the same size are found that have nucleations in common.
Intensity Profile Averaging
CCP intensity profiles were calculated as described in (39): For a given channel, a given CCP, and a given frame we average the pixel intensities within a radius of 134 nm (2 pixels) from the centroid of the CCP as detected in the EGFP-CLCa channel. We repeat this for each frame to form an intensity trace for a given CCP. For each trace, we subtract the average intensity trace of 8 neighboring background pixels. In order to average traces for CCPs of different lifetimes, we calculate a weighted average between the traces first aligned relative to the frame of CCP appearance and then aligned relative to the frame of CCP disappearance. The weighting function is set so that intensity values at early time points are dominated by the traces lined up to the first frame and intensity values in later time points are dominated by traces lined up to the final frame. This results in average intensity traces with the highest uncertainty in middle time points.
Supplementary Material
Supp Figure S1
ACKNOWLEDGEMENTS
This work was funded by a National Institutes of Health research grant R01 GM073165 to GD and SLS. MM was supported by a Postdoctoral Fellowship from the American Heart Association (Western Affiliates). The authors thank the Nikon Imaging Center at Harvard Medical School for help with light microscopy.
Abbreviations
CME
clathrin-mediated endocytosis
CCP
clathrin-coated pit
SNX9
sorting nexin 9
REFERENCES
- 1.Brett TJ, Traub LM. Molecular structures of coat and coat-associated proteins: function follows form. Curr Opin Cell Biol. 2006;18:395–406. doi: 10.1016/j.ceb.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 2.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 3.Kirchhausen T, Clathrin Annu Rev Biochem. 2000;69:699–727. doi: 10.1146/annurev.biochem.69.1.699. [DOI] [PubMed] [Google Scholar]
- 4.Schoch S, Gundelfinger ED. Molecular organization of the presynaptic active zone. Cell Tissue Res. 2006;326:379–391. doi: 10.1007/s00441-006-0244-y. [DOI] [PubMed] [Google Scholar]
- 5.Owald D, Sigrist SJ. Assembling the presynaptic active zone. Curr Opin Neurobiol. 2009;19:311–318. doi: 10.1016/j.conb.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 6.Bai J, Hu Z, Dittman JS, Pym EC, Kaplan JM. Endophilin functions as a membrane-bending molecule and is delivered to endocytic zones by exocytosis. Cell. 2010;143:430–441. doi: 10.1016/j.cell.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brodin L, Shupliakov O. Giant reticulospinal synapse in lamprey: molecular links between active and periactive zones. Cell Tissue Res. 2006;326:301–310. doi: 10.1007/s00441-006-0216-2. [DOI] [PubMed] [Google Scholar]
- 8.Gaidarov I, Santini F, Warren RA, Keen JH. Spatial control of coated-pit dynamics in living cells. Nat Cell Biol. 1999;1:1–7. doi: 10.1038/8971. [DOI] [PubMed] [Google Scholar]
- 9.Ehrlich M, Boll W, Van Oijen A, Hariharan R, Chandran K, Nibert ML, Kirchhausen T. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell. 2004;118:591–605. doi: 10.1016/j.cell.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 10.Saffarian S, Cocucci E, Kirchhausen T. Distinct dynamics of endocytic clathrin-coated pits and coated plaques. PLoS Biol. 2009;7:e1000191. doi: 10.1371/journal.pbio.1000191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keyel PA, Watkins SC, Traub LM. Endocytic adaptor molecules reveal an endosomal population of clathrin by total internal reflection fluorescence microscopy. J Biol Chem. 2004;279:13190–13204. doi: 10.1074/jbc.M312717200. [DOI] [PubMed] [Google Scholar]
- 12.Loerke D, Mettlen M, Yarar D, Jaqaman K, Jaqaman H, Danuser G, Schmid SL. Cargo and dynamin regulate clathrin-coated pit maturation. PLoS Biol. 2009;7:e57. doi: 10.1371/journal.pbio.1000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merrifield CJ, Feldman ME, Wan L, Almers W. Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat Cell Biol. 2002;4:691–698. doi: 10.1038/ncb837. [DOI] [PubMed] [Google Scholar]
- 14.Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, Danuser G. Robust single-particle tracking in live-cell time-lapse sequences. Nat Methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haase P. Spatial pattern-analysis in ecology based on Ripley k-function - Introduction and methods of edge correction. Journal of Vegetation Science. 1995;6:575–582. [Google Scholar]
- 16.Ripley BD. 2nd-order analysis of stationary point processes. J Appl Probab. 1976;13:255–266. [Google Scholar]
- 17.Henne WM, Boucrot E, Meinecke M, Evergren E, Vallis Y, Mittal R, McMahon HT. FCHo proteins are nucleators of clathrin-mediated endocytosis. Science. 2010;328:1281–1284. doi: 10.1126/science.1188462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujiwara T, Ritchie K, Murakoshi H, Jacobson K, Kusumi A. Phospholipids undergo hop diffusion in compartmentalized cell membrane. J Cell Biol. 2002;157:1071–1081. doi: 10.1083/jcb.200202050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boucrot E, Saffarian S, Massol R, Kirchhausen T, Ehrlich M. Role of lipids and actin in the formation of clathrin-coated pits. Exp Cell Res. 2006;312:4036–4048. doi: 10.1016/j.yexcr.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coue M, Brenner SL, Spector I, Korn ED. Inhibition of actin polymerization by latrunculin A. FEBS Lett. 1987;213:316–318. doi: 10.1016/0014-5793(87)81513-2. [DOI] [PubMed] [Google Scholar]
- 21.Yarar D, Waterman-Storer CM, Schmid SL. A dynamic actin cytoskeleton functions at multiple stages of clathrin-mediated endocytosis. Mol Biol Cell. 2005;16:964–975. doi: 10.1091/mbc.E04-09-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bubb MR, Senderowicz AM, Sausville EA, Duncan KL, Korn ED. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J Biol Chem. 1994;269:14869–14871. [PubMed] [Google Scholar]
- 23.Ponti A, Matov A, Adams M, Gupton S, Waterman-Storer CM, Danuser G. Periodic patterns of actin turnover in lamellipodia and lamellae of migrating epithelial cells analyzed by quantitative Fluorescent Speckle Microscopy. Biophys J. 2005;89:3456–3469. doi: 10.1529/biophysj.104.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antonescu CN, Aguet F, Danuser G, Schmid SL. Phosphatidylinositol-(4,5)-bisphosphate regulates clathrin-coated pit initiation, stabilization and size. Mol. Biol. Cell. 2011;22:2588–2600. doi: 10.1091/mbc.E11-04-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Padron D, Wang YJ, Yamamoto M, Yin H, Roth MG. Phosphatidylinositol phosphate 5-kinase Ibeta recruits AP-2 to the plasma membrane and regulates rates of constitutive endocytosis. J Cell Biol. 2003;162:693–701. doi: 10.1083/jcb.200302051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kirchhausen T. Adaptors for clathrin-mediated traffic. Annu Rev Cell Dev Biol. 1999;15:705–732. doi: 10.1146/annurev.cellbio.15.1.705. [DOI] [PubMed] [Google Scholar]
- 27.Mettlen M, Stoeber M, Loerke D, Antonescu CN, Danuser G, Schmid SL. Endocytic accessory proteins are functionally distinguished by their differential effects on the maturation of clathrin-coated pits. Mol Biol Cell. 2009;20:3251–3260. doi: 10.1091/mbc.E09-03-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Worby CA, Dixon JE. Sorting out the cellular functions of sorting nexins. Nat Rev Mol Cell Biol. 2002;3:919–931. doi: 10.1038/nrm974. [DOI] [PubMed] [Google Scholar]
- 29.Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJ, Evans PR, McMahon HT. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- 30.Soulet F, Yarar D, Leonard M, Schmid SL. SNX9 regulates dynamin assembly and is required for efficient clathrin-mediated endocytosis. Mol Biol Cell. 2005;16:2058–2067. doi: 10.1091/mbc.E04-11-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lundmark R, Carlsson SR. The beta-appendages of the four adaptor-protein (AP) complexes: structure and binding properties, and identification of sorting nexin 9 as an accessory protein to AP-2. Biochem J. 2002;362:597–607. doi: 10.1042/0264-6021:3620597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lundmark R, Carlsson SR. Sorting nexin 9 participates in clathrin-mediated endocytosis through interactions with the core components. J Biol Chem. 2003;278:46772–46781. doi: 10.1074/jbc.M307334200. [DOI] [PubMed] [Google Scholar]
- 33.Shin N, Ahn N, Chang-Ileto B, Park J, Takei K, Ahn SG, Kim SA, Di Paolo G, Chang S. SNX9 regulates tubular invagination of the plasma membrane through interaction with actin cytoskeleton and dynamin 2. J Cell Sci. 2008;121:1252–1263. doi: 10.1242/jcs.016709. [DOI] [PubMed] [Google Scholar]
- 34.Yarar D, Surka MC, Leonard MC, Schmid SL. SNX9 activities are regulated by multiple phosphoinositides through both PX and BAR domains. Traffic. 2008;9:133–146. doi: 10.1111/j.1600-0854.2007.00675.x. [DOI] [PubMed] [Google Scholar]
- 35.Yarar D, Waterman-Storer CM, Schmid SL. SNX9 couples actin assembly to phosphoinositide signals and is required for membrane remodeling during endocytosis. Dev Cell. 2007;13:43–56. doi: 10.1016/j.devcel.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 36.Badour K, McGavin MK, Zhang J, Freeman S, Vieira C, Filipp D, Julius M, Mills GB, Siminovitch KA. Interaction of the Wiskott-Aldrich syndrome protein with sorting nexin 9 is required for CD28 endocytosis and cosignaling in T cells. Proc Natl Acad Sci U S A. 2007;104:1593–1598. doi: 10.1073/pnas.0610543104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shin N, Lee S, Ahn N, Kim SA, Ahn SG, YongPark Z, Chang S. Sorting nexin 9 interacts with dynamin 1 and N-WASP and coordinates synaptic vesicle endocytosis. J Biol Chem. 2007;282:28939–28950. doi: 10.1074/jbc.M700283200. [DOI] [PubMed] [Google Scholar]
- 38.Taylor MJ, Perrais D, Merrifield CJ. A high precision survey of the molecular dynamics of Mammalian clathrin-mediated endocytosis. PLoS biology. 2011;9:e1000604. doi: 10.1371/journal.pbio.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loerke D, Mettlen M, Schmid SL, Danuser G. Measuring the Hierarchy of Molecular Events During Clathrin-Mediated Endocytosis. Traffic. 2011;12:815–825. doi: 10.1111/j.1600-0854.2011.01197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu AP, Aguet F, Danuser G, Schmid SL. Local clustering of transferrin receptors promotes clathrin-coated pit initiation. J Cell Biol. 2010;191:1381–1393. doi: 10.1083/jcb.201008117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scita G, Di Fiore PP. The endocytic matrix. Nature. 2010;463:464–473. doi: 10.1038/nature08910. [DOI] [PubMed] [Google Scholar]
- 42.Lu Q. Seamless cloning and gene fusion. Trends Biotechnol. 2005;23:199–207. doi: 10.1016/j.tibtech.2005.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp Figure S1