Free-Energy Simulations Reveal that Both Hydrophobic and Polar Interactions Are Important for Influenza Hemagglutinin Antibody Binding (original) (raw)
Abstract
Antibodies binding to conserved epitopes can provide a broad range of neutralization to existing influenza subtypes and may also prevent the propagation of potential pandemic viruses by fighting against emerging strands. Here we propose a computational framework to study structural binding patterns and detailed molecular mechanisms of viral surface glycoprotein hemagglutinin (HA) binding with a broad spectrum of neutralizing monoclonal antibody fragments (Fab). We used rigorous free-energy perturbation (FEP) methods to calculate the antigen-antibody binding affinities, with an aggregate underlying molecular-dynamics simulation time of several microseconds (∼2 _μ_s) using all-atom, explicit-solvent models. We achieved a high accuracy in the validation of our FEP protocol against a series of known binding affinities for this complex system, with <0.5 kcal/mol errors on average. We then introduced what to our knowledge are novel mutations into the interfacial region to further study the binding mechanism. We found that the stacking interaction between Trp-21 in HA2 and Phe-55 in the CDR-H2 of Fab is crucial to the antibody-antigen association. A single mutation of either W21A or F55A can cause a binding affinity decrease of ΔΔG > 4.0 kcal/mol (equivalent to an ∼1000-fold increase in the dissociation constant Kd). Moreover, for group 1 HA subtypes (which include both the H1N1 swine flu and the H5N1 bird flu), the relative binding affinities change only slightly (< ±1 kcal/mol) when nonpolar residues at the _α_A helix of HA mutate to conservative amino acids of similar size, which explains the broad neutralization capability of antibodies such as F10 and CR6261. Finally, we found that the hydrogen-bonding network between His-38 (in HA1) and Ser-30/Gln-64 (in Fab) is important for preserving the strong binding of Fab against group 1 HAs, whereas the lack of such hydrogen bonds with Asn-38 in most group 2 HAs may be responsible for the escape of antibody neutralization. These large-scale simulations may provide new insight into the antigen-antibody binding mechanism at the atomic level, which could be essential for designing more-effective vaccines for influenza.
Introduction
Influenza A virus is one of the most fatal infectious diseases in humans and poultry (1,2). Each year, this virus causes hundreds of thousands of deaths in humans and tens of millions of deaths in birds worldwide (3–9). The recent H1N1 swine flu pandemic and the spread of highly pathogenic H5N1 avian flu caused a great public-health concern (10–18). Current vaccines usually respond to a limited number of strains and often fail to neutralize emerging new strains because of rapid genetic evolution (19–24). Although some neuraminidase (NA) inhibitors, such as oseltamivir (Tamiflu) and zanamivir (Relenza), have shown effective suppression against influenza viruses, their efficacy is often challenged by increasing drug resistance (25–32).
Therefore, broad and potent cross-protective host immunity is in great demand for influenza prevention and treatment. High-affinity neutralizing antibodies (nAbs), such as F10 and CR6261, have been selected by phage display on recombinant H5 HA (13–15). Both F10 and CR6261 have shown broad neutralization to all group 1 subtypes, including the H1N1 Spanish flu and the H5N1 bird flu. In general, 16 hemagglutinin (HA) subtypes of influenza A viruses are categorized into two major phylogenetic groups: group 1 (H1, H2, H5, H6, H8, H9, H11, H12, H13, and H16) and group 2 (H3, H4, H7, H10, H14, and H15). The cocrystal structures of H5 HA with antibodies reveal that both antibodies can block the viral infection by inserting their heavy chain into a conserved helical stem region in HA1 and HA2, thereby possibly preventing membrane fusion (13–15). We believe that the similar neutralization effect of these nAbs could result from some important common features that are critical for developing new broad-spectrum vaccines. However, several underlying key mechanisms remain unclear, and it remains to be determined at which molecular level antibodies have a broad range of neutralization, and why most group 2 influenza viruses (e.g., H3 and H7) cannot be neutralized by F10 or CR6261. To address these issues, we performed large-scale, free-energy perturbation (FEP) simulations to characterize key residues and their mutation effects on HA-Fab binding at the atomic level. The FEP method has been widely used to calculate binding affinities for a variety of chemical and biological systems, such as solvation free-energy calculation, ligand-receptor binding, protein–protein interaction, and protein-DNA (RNA) binding (17,18,33–44), and is often regarded as the most rigorous and reliable method for free-energy calculations. Many previous FEP calculations achieved high accuracy for various protein-protein and protein-ligand binding affinities when compared with experiments (37,45–47). Our previous work on H3N2 HA-antibody and H5N1 HA-receptor binding systems also showed excellent agreement between the calculated binding free energies and experimental values (17,18). Among several available computational methods developed in past years, the FEP method based on the all-atom explicit-solvent model is probably the most accurate approach for estimating the relative antigen-antibody binding affinity (45,46). In this study, we first validated our FEP protocol by comparing the simulated binding affinity changes with available experimental data. We then extended our FEP calculation to novel (to our knowledge) mutations at the interfacial region on either group 1 HA or monoclonal antibody fragment (Fab). We found that the stacking interaction is critical for HA-antibody recognition, and the nonspecific hydrophobic interaction is responsible for the broad neutralization of antibody. To understand why F10-like antibodies successfully neutralize most group 1 influenza subtypes but fail in group 2 subtypes, we further investigated the role of a highly conserved His residue observed in almost all important group 1 HA subtypes (e.g., H1 and H5) by mutating it to Asn in four of the six group 2 HA subtypes (i.e., H3, H7, H10, and H15). Our results show that such a computational approach can serve as a complementary tool for interpreting and predicting critical mutations for HA-antibody binding.
Method and System
Molecular systems
The H5 HA (Viet04/H5) with Fab F10 complex (PDB entry 3FKU) was used for antigen-antibody binding, where the HA (HA1 and HA2) monomer was bound to one Fab with both heavy and light chains (13). The HA-Fab complex was solvated in a 71.5 Å × 81.5 Å × 160.0 Å water box, and then seven sodium ions were added to neutralize the system, with a total number of ∼88,000 atoms. The solvated system was first energy-minimized by 20,000 steps, followed by a 500,000-step equilibration. The snapshot during the equilibration was randomly picked as the starting point for the FEP calculations. The unbound (free) state was modeled with the HA or F10 only, solvated in water, and equilibrated with a similar process. The particle-mesh Ewald method was used for the long-range electrostatic interactions with a cutoff distance of 12 Å (48). All underlying molecular-dynamics simulations, which are widely used to simulate biological systems (49–58), were performed using the NAMD2 (59) molecular modeling package with a 1.5 fs time step in the NPT ensemble at 1 atm and 300 K. The CHARMM22 force field (60) and TIP3P water model (61) were used.
FEP protocol
When an antigenic variation occurs, the change in the HA binding affinity to a neutralizing antibody can be calculated by the FEP method (17,18,33–35,37–40). The Helmholtz free energy of a system can be expressed as
| G=−kTlnZ=kTln{∬dpdqexp[−βH(p,q)]}, | (1) |
|---|
where Z is the partition function; H (p,q) is the Hamiltonian of the system; p and q represent the momentum and the coordinate, respectively; k is the Boltzmann constant; T is the temperature; and β is equal to 1_/kT_. The binding free-energy change Δ_G_ due to a mutation in HA or antibody can then be calculated as
| ΔGλ=−kTln〈exp(−β[V(λ+Δλ)−V(λ)])〉λ | (2) |
|---|
where V(λ) = (1 − λ) _V_1 _+ λ V_2, with _V_1 representing the potential energy of the wild-type (WT) and _V_2 representing the potential energy of the mutant. The FEP parameter λ changes from 0 (_V_1) to 1 (_V_2) when the system changes from the WT to the mutant, and <…>λ represents the ensemble average at potential V(λ). In typical FEP calculations, to ensure a smooth transition from state A to state B, many perturbation windows have to be used. To avoid singularity at small interaction distances, when λ approaches 0 or 1 (a situation often referred to as endpoint-catastrophe), we used soft-core potentials for the Lennard-Jones interactions, with the 12-6 LJ function modified as follows (62,63):
| VvdW=εij[(Rij2rij2+δ(1−λ))6−(Rij2rij2+δ(1−λ))3], | (4) |
|---|
where εij is the depth of the potential well, Rij is the radius, rij is the distance between a pair of atoms, and δ is the shift parameter that allows a smooth transition from the original Lennard-Jones potential to zero or vice versa. The electrostatic interactions are handled with the normal Coulomb law but are switched on for the appearing atoms only after λ > 0.1, thus allowing the soft-core Lennard-Jones potentials to repel the possible overlapping before introducing the electrostatic interactions. Similarly, for the disappearing atoms, the electrostatic interactions are switched off after λ > (1 − 0.1) = 0.9.
In general, it is difficult to directly calculate the absolute binding affinity change Δ_GA_ for the binding process between a viral surface protein and an antibody due to the long timescale and complicated binding process. However, we can avoid this problem by designing a thermodynamic cycle to calculate the relative binding free-energy change, i.e., ΔΔ_GAB_. Instead of calculating the difficult direct binding energies Δ_GA_ and Δ_GB_, we calculate the free-energy changes for the same mutation in both the bound state (HA bound to antibody, Δ_G_1) and the free state (HA or antibody not bounded, Δ_G_2). Within a complete thermodynamic cycle, the total free-energy change should be zero, which gives the relative binding affinity due to the mutation from A → B as:
| ΔΔGbind=ΔGB−ΔGA=ΔG1−ΔG2. | (5) |
|---|
In the current setup, we adopted a 20-window scheme with a soft-core potential (λ = 0.00001, 0.0001, 0.001, 0.01, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 0.95, 0.99, 0.999, 0.9999, 0.99999, 1). For each mutation, we performed at least five independent runs starting from different initial configurations (taken from the equilibration) for better convergence. The simulation time for each window was 0.3 ns. In each FEP _λ_-window, the first 4000 steps were for the further equilibration. Larger window sizes and longer simulation durations were also tested in our previous studies, and we found that the current setup gives us a reasonable convergence in the final binding affinities. Therefore, at least 60 ns (20 windows × 0.3 ns × 5 runs × 2 states) of simulation time were generated for each mutation, and the total aggregate simulation time for this study was ∼2 _μ_s, which is much longer than most FEP calculations currently reported in the literature.
Despite the controversy in the literature about the meaningfulness of breaking down the total free energy into components, and the ambiguity associated with a path-dependent decomposition (64–67), we believe a breakup of the total binding free energy into its van der Waals (vdW) and electrostatic components can provide useful information about the underlying physical interactions involved in HA-Fab binding. In this study, we collected the contributions of free-energy change by vdW and electrostatic interactions separately. Because of the nonlinearity of the FEP formulation, there may be a small coupling term in this approach.
Results and Discussion
Validation of the FEP protocol with known experimental data
Mutagenesis studies of the H5-F10 complex have shown the importance of the binding region between the _α_A helix of H5 and Fab, which was proposed to prevent the conformational change of _α_A helix and the subsequent membrane fusion (13–15) (see Fig. 1). We noticed that some experimental data are available for mutagenesis studies in the _α_A helix region. Therefore, we first used these experimental data to validate our FEP protocol by calculating the relative binding free energies for mutations N502A, V522A, V522L, and V522E in the _α_A helix (subscripts 1 and 2 refer to HA1 and HA2, respectively, and the residue numbering system follows that of H3 (PDB entry: 2HMG)) (13).
Figure 1.
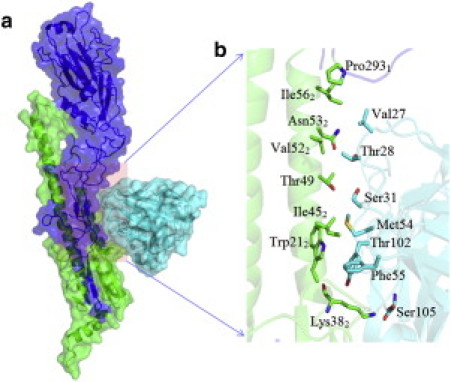
Molecular modeling system for HA protein binding with the antibody F10. (a) Overview of the HA-antibody complex structure. The HA and nAb are represented by surface and cartoon; HA1 and HA2 are colored blue and green, respectively, and both the heavy chain and light chain of the antibody are colored cyan. (b) Detailed view of the antigen-antibody binding interface, with the contact residues rendered with sticks. The total size of simulation system is 88,345 atoms for WT.
The first target site, Asn-502, is exposed to the solvent in the α_A helix, and hence is not in direct contact with Fab. Presumably, the mutation N502A will not affect the binding affinity significantly. Our simulations showed that the N502A mutation enhanced the binding affinity slightly, with ΔΔ_G = −0.28 ± 0.28 kcal/mol (equivalent to an ∼1.6-fold enhancement (decrease) in binding dissociation constant Kd; see Table 1), which agreed well with an experimental result of an ∼2-fold enhancement in Kd for the same mutation (13). For the second target site, Val-522, which is conserved in group 1 HA as directly interacting with the complementarity-determining region H1 (CDR-H1) of F10, we examined three different mutations: V522A, V522L, and V522E. An experimental site-directed mutagenesis study of V522A revealed an ∼10- to 20-fold increase in Kd (13) (equivalent to a 1.3∼1.6 kcal/mol decrease in binding free energy), which was comparable to our FEP result of ΔΔ_G_ = 0.91 ± 0.29 kcal/mol (see Table 1). However, a conservative mutation to Leu of similar size as Val-522 had little influence on the binding affinity. We observed only a slight reduction in the binding affinity (ΔΔ_G_ = 0.57 ± 0.60 kcal/mol), which was also consistent with experiments in which the concentration of the bound state was decreased by <10% (13). As one would expect for the V522E mutation, the inclusion of charged amino acid in the hydrophobic core at the binding interface severely interfered with the antibody association with the α_A helix of HA, resulting in a significant decrease in binding affinity, ΔΔ_G = 9.62 ± 0.93 kcal/mol. The V522E mutation on Fab F10 consistently failed to neutralize the HA in experiment (Table 1) (13).
Table 1.
Comparison of FEP simulation results with experimental data for the HA-nAb binding free-energy change due to the mutation in HA
| Mutation | Calculated _ΔΔ_G (kcal/mol) | Reverse _ΔΔ_G (kcal/mol) | Experimental ΔΔ_G_ (kcal/mol) |
|---|---|---|---|
| N502A | −0.28 ± 0.28 | 0.11 ± 0.48 (A502N) | −0.4 |
| V522L | 0.57 ± 0.60 | −0.20 ± 1.15 (L522V) | 0∼0.5 |
| V522A | 0.91 ± 0.29 | −1.08 ± 0.59 (A522V) | 1.3∼1.6 |
| V522E | 9.62 ± 0.93 | −8.22 ± 1.61 (E522V) | no bound states observed |
| I562A | 2.42 ± 0.45 | −1.81 ± 0.46 (A562I) | N/A |
We further validated our FEP approach by performing extra reverse FEP calculations in which five mutants (Ala-502, Leu-522, Ala-522, Glu-522, and Ala-562) were mutated back to their WTs (Asn-502, Val-522, Val-522, Val-522, and Ile-562), respectively (Table 1). We found that all five backward FEP simulations (mutants to WT) and forward FEP calculations (WT to mutants) showed similar binding affinities but with an opposite sign. The results of both methods are in good agreement with experimental results. Considering the large size of the HA-Fab system, our FEP calculation provided a relatively small deviation of <0.5 kcal/mol on average compared with available experimental data. Therefore, the FEP simulation protocol may be employed as a reliable tool for antigen-antibody complex binding affinity studies.
The stacking interaction and hydrophobic environment are crucial for HA binding to CDR-H2
Inspired by encouraging results from the above validation with known mutations, we extended our FEP method to novel (to our knowledge) mutations found in either HA or the Fab side at the binding interface. The cocrystal structures of both H5-F10 and H5-CR6261 complexes suggest that the interactions between the _α_A helix of HA and CDRs of the Fab may be important for neutralization (13,14). As a key interaction site, we first focused on Trp-212 and Ile-562, two conserved residues that are observed in all subtypes of HAs, which make hydrophobic interactions with Met-54 and Phe-55 in the CDR-H2 of Fab F10 (equivalent to Ile-53 and Phe-54, respectively, in antibody CR6261).
In addition to the general hydrophobic interaction, a stacking interaction between the indole ring of Trp-212 and phenyl group of Phe-55 (or Phe-54) of Fab F10 (or CR6261) is believed to play a critical role in binding recognition between the α_A helix of HA and the CDR-H2 of Fab F10 (i.e., by providing the binding specificity). Here, we performed computational mutagenesis for these sites to evaluate how interactions associated with the aromatic side chains (π_−_π stacking) aid in the recognition of their binding partners. We started by mutating Trp-212 to Ala using our FEP simulation. Knocking out Trp-212 with a smaller amino-acid Ala resulted in a binding affinity decrease of ΔΔ_G = 4.02 ± 0.34 kcal/mol, which corresponds to an ∼1000-fold increase in the dissociation constant Kd. Similarly, a counter mutation on the antibody's Phe-55 with Ala reduced the binding affinity by ΔΔ_G_ = 4.24 ± 0.76 kcal/mol, implying that the stacking interaction between two aromatic rings is indeed important, contributing ∼4 kcal/mol to the binding affinity. The mutation effect on the binding site structure is clearly depicted in Fig. 2 for the WT and W212A at the end of simulation. Due to the lack of an aromatic ring and a shorter side chain in the mutant Ala-212, the intermolecular hydrophobic cluster made of Trp-212, Phe-55, and Met-54 is largely disrupted. On the other hand, we noticed enhancement of an intramolecular interaction between Met-54 and Phe-55 in CDR-H2 of Fab. A free-energy decomposition confirmed the role of vdW interactions (see Method and System for details). Almost all (98%) of the total contribution to the binding affinity originated from the vdW interactions (4.11 kcal/mol), whereas the electrostatic interactions played only a minor role (−0.05 kcal/mol). The coupling term was 0.04 kcal/mol. Note that our estimated stacking interaction (∼4 kcal/mol) and the vdW component of the total free energy (4.11 kcal/mol) are very similar to the previously reported value of stacking interactions between two idealized benzene rings (4.08 kcal/mol) (68). Overall, our energetic analysis confirmed that the large free-energy loss in the W212A mutation is attributed to the destruction of stacking interactions between two aromatic residues from both HA and antibody. We further evaluated the effect of hydrophobic interactions with two nonconservative mutations: F55E (mutating to an acidic residue) and F55K (mutating to a basic residue). Much larger binding affinity decreases of 13.58 ± 0.55 kcal/mol and 7.66 ± 0.97 kcal/mol were found for F55E and F55K, respectively (Table 2), which supports the notion that the recognition of HA by nAb F10 is highly dependent on the hydrophobic interactions at the binding interface.
Figure 2.
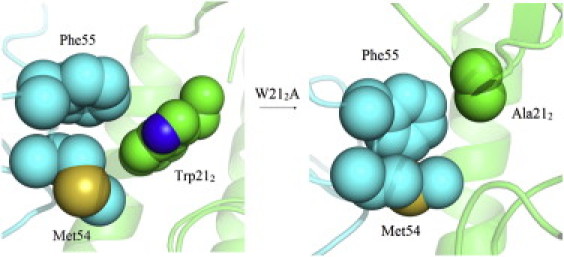
Structural comparison of F10 nAb bound to H5 HA around CDR-H2 due to the W212A mutation (HA, green; nAb, cyan). The overall complex is represented as a cartoon and the residues at interaction face are rendered with spheres. The interactions of Trp-212 (HA) with Met-54/Phe-55 (nAbs) are largely diminished by W212A substitution.
Table 2.
FEP simulation results for the HA-nAb binding free-energy change due to the mutation in HA2/CDR-H2
| Mutation | Calculated ΔΔ_G_ (kcal/mol) |
|---|---|
| W212A | 4.02 ± 0.34 |
| M54A | 2.13 ± 0.36 |
| M54L | 1.10 ± 0.31 |
| M54E | 10.02 ± 1.11 |
| M54K | 9.29 ± 1.20 |
| F55A | 4.24 ± 0.76 |
| F55E | 13.58 ± 0.55 |
| F55K | 7.66 ± 0.97 |
Given the crucial role of the hydrophobic interactions between these two aromatic residues (HA2's Trp-21 and F10's Phe-55), we further investigated the nearby nonpolar residue Met-54 in F10 (Ile-53 in CR6261), which displays strong contacts with Trp-212 and Ile-452 in HA2, forming a hydrophobic cluster. As expected, the nonconservative M54E or M54K mutations broke the hydrophobic core and caused ∼10 kcal/mol free-energy decreases in our FEP simulations (Table 2). However, substituting Met with Ile, as found in Fab CR6261, or with Ala, was shown to destabilize the protein association by ΔΔ_G_ = 1.10 ± 0.31 kcal/mol and 2.13 ± 0.36 kcal/mol, respectively (Table 2). Note that the free-energy decreases in conservative mutations of M54I or M54A are smaller than those of aromatic residue deletions in W212A or F55A. This indicates that the stacking interaction between aromatic residues is more important for the Fab F10 neutralization, even though a hydrophobic environment around these key aromatic residues is still required.
Overall, our FEP simulations revealed in detail the determining factors in the interaction between CDR-H2 of Fab and HA. The stacking interactions between the aromatic rings from both HA and Fab were essential for the antigen-antibody binding. Preserving a hydrophobic environment around these key aromatic residues was found to be important as well, i.e., the existence of nonpolar residues, such as Met-54 in F10 and Ile-53 in CR6261, also helped.
Nonspecific hydrophobic interactions are responsible for the broad antibody neutralization
In addition to the hydrophobic interactions between HA and CDR-H2, another hydrophobic core is formed in the α_A helix and the CDR-H1 (Fig. 3), where Pro-2931, Val-522, and Ile-562 in HA are in contact with Val-27 and the methyl group of Thr-28 in F10. We first investigated the role of Val-522 using two types of FEP mutations: one with other nonpolar residues to keep the hydrophobic environment intact, and one with charged ones to break the hydrophobic core. The FEP calculation results are listed in Table 3. In general, for mutations to similarly sized hydrophobic residues, minor binding free-energy changes could be expected, but for mutations to charged residues, significant decreases in binding affinity or even disassociations of the HA-Fabs complex could be expected. Indeed, our simulations showed that the polarity of the substituted residue at site 52 of HA2 is critical for preserving the WT binding affinity. Of more interest, the conservative hydrophobic mutations displayed more or less similar binding affinities, with an ∼±1 kcal/mol variation (Table 3), indicating that the exact hydrophobic residue type is less critical in this binding environment. The mutations to charged amino acids, on the other hand, clearly broke up the antigen-antibody association with a high free-energy penalty of 7.28∼15.47 kcal/mol (see Table 3). It should be noted that V522I substitution could have even stronger binding with Fab than the WT, where the binding affinity enhanced by ΔΔ_G = −1.08 ± 0.59 kcal/mol, indicating that Fab is even more effective for HAs that possess an I522 residue. It is interesting to observe that Ile, Leu, and Val are the most common residue types at site 52 of HA2 for all of the subtypes of group 1 HA: Ile and Leu are two dominant amino acids in subtype H13/H16 and H12, respectively, whereas Val is the majority amino acid in other subtypes of group 1 HAs (Table 4). Thus, the strong binding affinity and the limited sequence diversity (within aliphatic amino acids Ile, Leu, or Val) at site 52 explains why antibody F10 could have broad neutralization toward various HAs.
Figure 3.
Structural comparison of F10 nAb bound to H5 HA around CDR-H2 due to I562A and I562K mutations (HA, green; nAb, cyan). The overall complex is represented as cartoon and the residues at interaction face are rendered with spheres. The hydrophobic core (shown in spheres) is broken by the I562A mutation but is preserved in the I562K mutation.
Table 3.
FEP simulation results for the HA-nAb binding free-energy change due to the mutation in HA/CDR-H1
| Mutation | Calculated ΔΔ_G_ (kcal/mol) |
|---|---|
| H381N | 1.32 ± 0.78 |
| V522A | 0.91 ± 0.29 |
| V522I | −1.08 ± 0.59 |
| V522L | 0.57 ± 0.60 |
| V522F | 1.44 ± 0.87 |
| V522E | 10.43 ± 0.83 |
| V522D | 15.47 ± 1.09 |
| V522R | 13.45 ± 0.98 |
| V522K | 7.28 ± 1.51 |
| I562A | 2.42 ± 0.45 |
| I562V | −0.48 ± 0.46 |
| I562L | 0.84 ± 0.50 |
| I562E | 2.24 ± 1.59 |
| I562K | −0.38 ± 0.65 |
Table 4.
Comparison of the sequence conservation among 16 HA subtypes
| Group | Cluster | Subtype | HA1 | HA2 | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 17 | 18 | 38 | 40 | 18 | 19 | 20 | 21 | 38 | 41 | 42 | 44 | 45 | 46 | 48 | 49 | 52 | 53 | 55 | 56 | 111 | |||
| Group 1 | H1a | H2 | Y | H | H | K | V | D | G | W | K | T | Q | A | I | D | I | T | V | N | V | I | H |
| H5 | Y | H | H | Q | V | D | G | W | Q | T | Q | A | I | D | V | T | V | N | I | V | H | ||
| H1 | Y | H | H | V | V | D | G | W | K | T | Q | A | I | D | I | T | V | N | V | I | H | ||
| H6 | Y | H | H | V | I | D | G | W | K | T | Q | A | I | D | I | T | V | N | I | I | H | ||
| H1b | H13 | Y | L | S | I | I | N | G | W | K | T | Q | A | I | D | I | T | I | N | I | I | H | |
| H16 | Y | L | S | V | I | N | G | W | K | T | Q | A | I | D | I | T | I | N | I | I | H | ||
| H11 | Y | L | S | V | I | N | G | W | K | T | Q | A | I | D | I | T | V | N | I | V | H | ||
| H9 | H8 | Y | Q | Q | M | I | D | G | W | Q | T | Q | A | I | D | I | T | V | N | I | I | H | |
| H12 | Y | Q | Q | E | V | A | G | W | R | T | Q | A | I | D | M | Q | L | N | V | I | H | ||
| H9 | Y | Q | H | K | V | A | G | W | R | T | Q | A | I | D | I | T | V | N | I | V | H | ||
| Group 2 | H3 | H4 | H | H | T | Q | I | D | G | W | L | T | Q | A | I | D | I | T | L | N | L | I | T |
| H14 | H | H | S | K | I | D | G | W | L | T | Q | A | I | D | I | N | L | N | L | I | T | ||
| H3 | H | H | N | T | V | D | G | W | L | T | Q | A | I | D | I | N | L | N | V | I | T | ||
| H15 | H15 | H | H | N | T | I | D | G | W | Y | T | Q | A | I | D | I | T | L | N | L | I | A | |
| H7 | H | H | N | T | I | D | G | W | Y | T | Q | A | I | D | I | T | L | N | L | I | A | ||
| H10 | H | H | N | T | V | D | G | W | Y | T | Q | A | I | D | I | T | L | N | L | I | A |
In contrast to the mutational effect at Val-522, which shows a clear binary dependence on residue polarity, the nearby hydrophobic residue Ile-562 displayed a more sophisticated pattern for the individual amino acid substitutions. For example, similarly sized I562V and I562L mutations showed similar binding activities as the WT, within ±1 kcal/mol, whereas mutation to a smaller but still hydrophobic Ala residue reduced the binding affinity by ∼2.42 ± 0.45 kcal/mol (Table 3). The free-energy decomposition analysis revealed that most of the free-energy loss (2.50 kcal/mol) was due to the weaker vdW interactions. This is clearly shown in Fig. 3, where the hydrophobic interaction in the WT Ile-562 with Pro-2931 in HA1 and Val-27 of Fab has been weakened by the short side chain of Ala, and the extra space is partially filled by two water molecules making contact with Ala.
A more dramatic difference was observed in the I562K mutation, where a nonconservative charged residue was introduced; however, the binding affinity was not affected or was slightly enhanced by ΔΔ_G_ = −0.38 ± 0.65 kcal/mol. Fig. 3 shows how the long side chain (also hydrophobic) and positively charged NH3+ group of Lys could be packed in the binding interface. The terminal amine group is pointing out from the hydrophobic binding interface and interacts with hydrophilic residues such as Asn-532 and Asn-602, whereas the long aliphatic chain of Lys still makes favorable contacts with hydrophobic residues Val-27, Val-522, and Pro-2931, thereby maintaining a strong interaction between the _α_A helix and CDR-H1.
Compared with the highly conservative aromatic residues in the binding region of HA and Fab-CDR-H2 discussed above, we found that the binding interface between HA and Fab-CDR-H1 exhibited relatively more variability in amino-acid selection in HA. Hydrophobic residues were usually required for site 522 to maintain the Fab binding. However, the binding interface near site 562 required a tightly packed hydrophobic environment. We found that the V522I or I562V mutation could actually increase the binding affinity by ∼1 kcal/mol and ∼0.5 kcal/mol, respectively. A strong but nonspecific binding between HA and antibody could be an important principle to consider in designing vaccines with a broad neutralization.
Asn-38 in group 2 HA1 may contribute to the antibody neutralization escape
The molecular mechanism that prevents most group 2 HAs (e.g., H3 and H7) from being neutralized by F10 or CR6261 is not fully understood. In previous studies, glycosylation at position 381 on group 2 HA was proposed as the main reason for neutralization escape (13,14). However, we found that two of the six subtypes in group 2 are not glycosylated at position 381, yet are able to escape the neutralization. That is, the glycosylation may not be the only mechanism to explain the immunity escape of group 2 subtypes. We investigated possible molecular mechanisms beyond the glycosylation by searching the sequence diversity around the HA-Fab binding interface. The sequences of all HA subtypes were collected from the National Center for Biotechnology Information Flu Database (Table 4). We hypothesized that a residue site in HA would be important for the antibody's escape from neutralization if the amino-acid types of that specific site were diversified over different groups but conserved within each group. That is, antibodies could neutralize HA subtypes in one group with a specific amino-acid type conserved at that site, whereas HA subtypes in the other group might escape from the same antibodies due to a different amino-acid type being conserved at the same site. For site 381, His is a well-conserved residue within group 1 (e.g., H1, H5, H2, H6, and H9), whereas Asn is a conserved residue within group 2 (in four out of six subtypes (H3, H7, H10, and H15)). Other sites, such as 401 and 382, are not fully conserved either across groups or within individual groups, indicating that they may play a less critical role in the neutralization (Table 4).
To confirm our hypothesis from the above sequence analysis and to reveal the atomic detail of the binding specificity, we performed the FEP mutation for His-381 in our current group 1 H5N1 HA to Asn-38, mimicking the conservative amino acid in group 2 HA. The simulated FEP results showed that the binding affinity between HA and Fab decreased by 1.32 ± 0.78 kcal/mol, which is an ∼10-fold increase in the dissociation constant (Kd) value. When we compared the final structure with the WT, we found that the two native hydrogen bonds (nitrogen atoms at the side chain of His-381 to the hydroxyl group of Ser-30 or Gln-64 in Fab) were broken by the mutation (Fig. 4). The loss of side-chain hydrogen bonding was partially compensated by waters entered in the binding interface, but no direct contact was found between mutated Asn-381 and Fab. Therefore, the H381N mutation weakened the interaction between HA and Fab by breaking the hydrogen bonds between HA1 and the antibody. However, it should be noted that the binding affinity decrease of 1.32 ± 0.78 kcal/mol in the H381N mutation is not overwhelming, indicating that antibody neutralization is a complicated process, and that although this His-38 residue may be an important contributing factor, it is probably not the only factor (other factors, such as glycosylation, may contribute as well (13,14)). Indeed, several other group 1 subtypes that are bound/neutralized by F10, such as H8 and H11, do not have His at position 38, and instead have Gln or Ser, respectively (13). Nevertheless, our FEP calculations indicate that in addition to glycosylation, the existence of Asn at position 381 may also be an important contributing factor in neutralization escape in group 2 HA subtypes.
Figure 4.
Structural comparison of F10 nAb bound to H5 HA around CDR-H2 due to H381N mutations (HA, green; nAb, cyan). The overall complex is represented as cartoon and the residues at interaction face are rendered with sticks. The side-chain hydrogen bonds between His-381/Gln64 and His-381/Ser-30 (shown with yellow dashed line) are broken by H381N mutation.
Conclusion
In this work, we performed rigorous FEP calculations to estimate influenza antigen-antibody binding affinities (using H5 HA and F10 Fab as a template) and study the characteristics of antibodies with a broad neutralization capability (such as F10 and CR6261). The simulated binding affinities between HA and Fab were in excellent agreement with the currently available experimental data. We identified several key residues in the HA-Fab binding regions and further examined them by means of in silico mutagenesis studies to explore the molecular mechanism of HA-Fab binding.
We found that the stacking interaction of Trp-212 (HA) and Phe-55 (Fab) is critical for strong binding between the α_A helix and the CDR-H2 of antibody. Our FEP simulations suggest that either W212A in (HA side) or F55A (antibody side) will cause a significant binding affinity decrease of ΔΔ_G > 4.0 kcal/mol (equivalent to an ∼1000-fold increase in the binding dissociation constant Kd). In addition, neighboring hydrophobic residues were also required to preserve a stable hydrophobic network around the aromatic side chains. Furthermore, we observed more general hydrophobic interactions between HA and the CDR-H1 of Fab.
The HA residue sites 522 and 562 appeared to be more tolerable with various hydrophobic mutations with similar binding ability as the WT, which could explain the wide neutralization of Fabs among all group 1 subtypes. In addition, we found that the V522I and I562V substitutions could increase the binding affinity by ∼1 kcal/mol and ∼0.5 kcal/mol, respectively, which potentially could be used as a way to improve the efficiency of current antibodies.
In addition to the hydrophobic interactions, the hydrogen bonding between His-381 and Ser-30/Gln-64 were also found to be important for antibody neutralization. When His-381 was mutated to group 2-like Asn-381, two hydrogen bonds were lost, substituted by hydration around Asn-381 in between the HA and the Fabs, with a net decrease of ∼1.3 kcal/mol in binding affinity. This could be another important contributing factor for the neutralization escape in group 2 subtypes, in addition to the glycosylation.
Acknowledgments
We thank Bruce Berne, Payel Das, Ajay Royyuru, Pengyu Ren, Steven Turner, and David Topham for many useful discussions. We also thank Ian Wilson, Jim Paulson, and Peter Palese for helpful comments at the beginning of our influenza modeling effort.
References
- 1.Cox N.J., Subbarao K. Global epidemiology of influenza: past and present. Annu. Rev. Med. 2000;51:407–421. doi: 10.1146/annurev.med.51.1.407. [DOI] [PubMed] [Google Scholar]
- 2.Hilleman M.R. Realities and enigmas of human viral influenza: pathogenesis, epidemiology and control. Vaccine. 2002;20:3068–3087. doi: 10.1016/s0264-410x(02)00254-2. [DOI] [PubMed] [Google Scholar]
- 3.Stevens J., Blixt O., Wilson I.A. Glycan microarray analysis of the hemagglutinins from modern and pandemic influenza viruses reveals different receptor specificities. J. Mol. Biol. 2006;355:1143–1155. doi: 10.1016/j.jmb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Fleury D., Wharton S.A., Bizebard T. Antigen distortion allows influenza virus to escape neutralization. Nat. Struct. Biol. 1998;5:119–123. doi: 10.1038/nsb0298-119. [DOI] [PubMed] [Google Scholar]
- 5.Yang Z.Y., Wei C.J., Nabel G.J. Immunization by avian H5 influenza hemagglutinin mutants with altered receptor binding specificity. Science. 2007;317:825–828. doi: 10.1126/science.1135165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tumpey T.M., Maines T.R., García-Sastre A. A two-amino acid change in the hemagglutinin of the 1918 influenza virus abolishes transmission. Science. 2007;315:655–659. doi: 10.1126/science.1136212. [DOI] [PubMed] [Google Scholar]
- 7.Stevens J., Corper A.L., Wilson I.A. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science. 2004;303:1866–1870. doi: 10.1126/science.1093373. [DOI] [PubMed] [Google Scholar]
- 8.Stevens J., Blixt O., Wilson I.A. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science. 2006;312:404–410. doi: 10.1126/science.1124513. [DOI] [PubMed] [Google Scholar]
- 9.Gamblin S.J., Haire L.F., Skehel J.J. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science. 2004;303:1838–1842. doi: 10.1126/science.1093155. [DOI] [PubMed] [Google Scholar]
- 10.Lin T., Wang G., Yuen K.Y. The hemagglutinin structure of an avian H1N1 influenza A virus. Virology. 2009;392:73–81. doi: 10.1016/j.virol.2009.06.028. [DOI] [PubMed] [Google Scholar]
- 11.Igarashi M., Ito K., Takada A. Predicting the antigenic structure of the pandemic (H1N1) 2009 influenza virus hemagglutinin. PLoS ONE. 2010;5:e8553. doi: 10.1371/journal.pone.0008553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu R., Ekiert D.C., Wilson I.A. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science. 2010;328:357–360. doi: 10.1126/science.1186430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sui J., Hwang W.C., Marasco W.A. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 2009;16:265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ekiert D.C., Bhabha G., Wilson I.A. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Throsby M., van den Brink E., Goudsmit J. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS ONE. 2008;3:e3942. doi: 10.1371/journal.pone.0003942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russell R.J., Kerry P.S., Skehel J.J. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. USA. 2008;105:17736–17741. doi: 10.1073/pnas.0807142105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Das P., Li J., Zhou R. Free-energy simulations reveal a double mutant avian H5N1 virus hemagglutinin with altered receptor binding specificity. J. Comput. Chem. 2009;30:1654–1663. doi: 10.1002/jcc.21274. [DOI] [PubMed] [Google Scholar]
- 18.Zhou R., Das P., Royyuru A.K. Single mutation induced H3N2 hemagglutinin antibody neutralization: a free-energy perturbation study. J. Phys. Chem. B. 2008;112:15813–15820. doi: 10.1021/jp805529z. [DOI] [PubMed] [Google Scholar]
- 19.Chen R., Holmes E.C. Avian influenza virus exhibits rapid evolutionary dynamics. Mol. Biol. Evol. 2006;23:2336–2341. doi: 10.1093/molbev/msl102. [DOI] [PubMed] [Google Scholar]
- 20.Xia Z., Jin G., Zhou R. Using a mutual information-based site transition network to map the genetic evolution of influenza A/H3N2 virus. Bioinformatics. 2009;25:2309–2317. doi: 10.1093/bioinformatics/btp423. [DOI] [PubMed] [Google Scholar]
- 21.Smith D.J., Lapedes A.S., Fouchier R.A. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305:371–376. doi: 10.1126/science.1097211. [DOI] [PubMed] [Google Scholar]
- 22.Du X., Wang Z., Jiang T. Networks of genomic co-occurrence capture characteristics of human influenza A (H3N2) evolution. Genome Res. 2008;18:178–187. doi: 10.1101/gr.6969007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamada S., Suzuki Y., Kawaoka Y. Haemagglutinin mutations responsible for the binding of H5N1 influenza A viruses to human-type receptors. Nature. 2006;444:378–382. doi: 10.1038/nature05264. [DOI] [PubMed] [Google Scholar]
- 24.Apisarnthanarak A., Erb S., Mundy L.M. Seroprevalence of anti-H5 antibody among Thai health care workers after exposure to avian influenza (H5N1) in a tertiary care center. Clin. Infect. Dis. 2005;40:e16–e18. doi: 10.1086/427034. [DOI] [PubMed] [Google Scholar]
- 25.Mehta T., McGrath E., Alangaden G.J. Detection of oseltamivir resistance during treatment of 2009 H1N1 influenza virus infection in immunocompromised patients: utility of cycle threshold values of qualitative real-time reverse transcriptase PCR. J. Clin. Microbiol. 2010;48:4326–4328. doi: 10.1128/JCM.01190-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stephenson I., Democratis J., Zambon M. Neuraminidase inhibitor resistance after oseltamivir treatment of acute influenza A and B in children. Clin. Infect. Dis. 2009;48:389–396. doi: 10.1086/596311. [DOI] [PubMed] [Google Scholar]
- 27.Beigel J., Bray M. Current and future antiviral therapy of severe seasonal and avian influenza. Antiviral Res. 2008;78:91–102. doi: 10.1016/j.antiviral.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowen A.C., Palese P. Influenza virus transmission: basic science and implications for the use of antiviral drugs during a pandemic. Infect. Disord. Drug Targets. 2007;7:318–328. doi: 10.2174/187152607783018736. [DOI] [PubMed] [Google Scholar]
- 29.Fick J., Lindberg R.H., Olsen B. Antiviral oseltamivir is not removed or degraded in normal sewage water treatment: implications for development of resistance by influenza A virus. PLoS ONE. 2007;2:e986. doi: 10.1371/journal.pone.0000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bright R.A., Shay D.K., Klimov A.I. Adamantane resistance among influenza A viruses isolated early during the 2005-2006 influenza season in the United States. JAMA. 2006;295:891–894. doi: 10.1001/jama.295.8.joc60020. [DOI] [PubMed] [Google Scholar]
- 31.de Jong M.D., Tran T.T., Farrar J. Oseltamivir resistance during treatment of influenza A (H5N1) infection. N. Engl. J. Med. 2005;353:2667–2672. doi: 10.1056/NEJMoa054512. [DOI] [PubMed] [Google Scholar]
- 32.Kiso M., Mitamura K., Kawaoka Y. Resistant influenza A viruses in children treated with oseltamivir: descriptive study. Lancet. 2004;364:759–765. doi: 10.1016/S0140-6736(04)16934-1. [DOI] [PubMed] [Google Scholar]
- 33.Jorgensen W.L. Free-energy calculations—a breakthrough for modeling organic-chemistry in solution. Acc. Chem. Res. 1989;22:184–189. [Google Scholar]
- 34.Deng Y., Roux B. Calculation of standard binding free energies: aromatic molecules in the T4 lysozyme L99A mutant. J. Chem. Theory Comput. 2006;2:1255–1273. doi: 10.1021/ct060037v. [DOI] [PubMed] [Google Scholar]
- 35.Kollman P. Free-energy calculations—applications to chemical and biochemical phenomena. Chem. Rev. 1993;93:2395–2417. [Google Scholar]
- 36.Pathiaseril A., Woods R.J. Relative energies of binding for antibody-carbohydrate-antigen complexes computed from free-energy simulations. J. Am. Chem. Soc. 2000;122:331–338. doi: 10.1021/ja9914994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simonson T., Archontis G., Karplus M. Free-energy simulations come of age: protein-ligand recognition. Acc. Chem. Res. 2002;35:430–437. doi: 10.1021/ar010030m. [DOI] [PubMed] [Google Scholar]
- 38.Tembe B.L., McCammon J.A. Ligand receptor interactions. Comput. Chem. 1984;8:281–283. [Google Scholar]
- 39.Warshel A. Simulating the energetics and dynamics of enzymatic reactions. Pontif. Acad. Sci. Scripta Varia. 1984;55:60–81. doi: 10.1073/pnas.81.2.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warshel A., Sharma P.K., Parson W.W. Modeling electrostatic effects in proteins. Biochim. Biophys. Acta. 2006;1764:1647–1676. doi: 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 41.Xia Z., Zhu Z., Zhou R. Recognition mechanism of siRNA by viral p19 suppressor of RNA silencing: a molecular dynamics study. Biophys. J. 2009;96:1761–1769. doi: 10.1016/j.bpj.2008.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng L., Chen M., Yang W. Random walk in orthogonal space to achieve efficient free-energy simulation of complex systems. Proc. Natl. Acad. Sci. USA. 2008;105:20227–20232. doi: 10.1073/pnas.0810631106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiao D., Golubkov P.A., Ren P. Calculation of protein-ligand binding free-energy by using a polarizable potential. Proc. Natl. Acad. Sci. USA. 2008;105:6290–6295. doi: 10.1073/pnas.0711686105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chodera J.D., Mobley D.L., Pande V.S. Alchemical free-energy methods for drug discovery: progress and challenges. Curr. Opin. Struct. Biol. 2011;21:150–160. doi: 10.1016/j.sbi.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Almlöf M., Aqvist J., Brandsdal B.O. Probing the effect of point mutations at protein-protein interfaces with free-energy calculations. Biophys. J. 2006;90:433–442. doi: 10.1529/biophysj.105.073239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brandsdal B.O., Smalås A.O. Evaluation of protein-protein association energies by free-energy perturbation calculations. Protein Eng. 2000;13:239–245. doi: 10.1093/protein/13.4.239. [DOI] [PubMed] [Google Scholar]
- 47.Wang J., Deng Y., Roux B. Absolute binding free-energy calculations using molecular dynamics simulations with restraining potentials. Biophys. J. 2006;91:2798–2814. doi: 10.1529/biophysj.106.084301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Darden T.A., York D.M., Pedersen L.G. Particle mesh Ewald: an NlogN method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 49.Eleftheriou M., Germain R.S., Zhou R. Thermal denaturing of mutant lysozyme with both the OPLSAA and the CHARMM force fields. J. Am. Chem. Soc. 2006;128:13388–13395. doi: 10.1021/ja060972s. [DOI] [PubMed] [Google Scholar]
- 50.Liu P., Huang X., Berne B.J. Observation of a dewetting transition in the collapse of the melittin tetramer. Nature. 2005;437:159–162. doi: 10.1038/nature03926. [DOI] [PubMed] [Google Scholar]
- 51.Zhou R., Eleftheriou M., Berne B.J. Destruction of long-range interactions by a single mutation in lysozyme. Proc. Natl. Acad. Sci. USA. 2007;104:5824–5829. doi: 10.1073/pnas.0701249104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou R., Huang X., Berne B.J. Hydrophobic collapse in multidomain protein folding. Science. 2004;305:1605–1609. doi: 10.1126/science.1101176. [DOI] [PubMed] [Google Scholar]
- 53.Gao Y.Q., Yang W., Karplus M. A structure-based model for the synthesis and hydrolysis of ATP by F1-ATPase. Cell. 2005;123:195–205. doi: 10.1016/j.cell.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 54.Hummer G., Rasaiah J.C., Noworyta J.P. Water conduction through the hydrophobic channel of a carbon nanotube. Nature. 2001;414:188–190. doi: 10.1038/35102535. [DOI] [PubMed] [Google Scholar]
- 55.Kamberaj H., van der Vaart A. An optimized replica exchange molecular dynamics method. J. Chem. Phys. 2009;130:074906. doi: 10.1063/1.3077857. [DOI] [PubMed] [Google Scholar]
- 56.Karplus M., Gao Y.Q., Yang W. Protein structural transitions and their functional role. Philos. Transact. A Math. Phys. Eng. Sci. 2005;363:331–355. doi: 10.1098/rsta.2004.1496. discussion 355–356. [DOI] [PubMed] [Google Scholar]
- 57.Hua L., Huang X., Berne B.J. Nanoscale dewetting transition in protein complex folding. J. Phys. Chem. B. 2007;111:9069–9077. doi: 10.1021/jp0704923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li X., Li J., Zhou R. Hydration and dewetting near fluorinated superhydrophobic plates. J. Am. Chem. Soc. 2006;128:12439–12447. doi: 10.1021/ja057944e. [DOI] [PubMed] [Google Scholar]
- 59.Kumar S., Huang C., Kale L.V. Scalable molecular dynamics with NAMD on Blue Gene/L. IBM J. Res. Dev. 2008;52:177–188. [Google Scholar]
- 60.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 61.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 62.Beutler T.C., Mark A.E., Gunsteren W.F.v. Avoiding singularities and numerical instabilities in free-energy calculations based on molecular simulations. Chem. Phys. Lett. 1994;222:529–539. [Google Scholar]
- 63.Zacharias M., Straatsma T.P., McCammon J.A. Separation-shifted scaling, a new scaling method for Lennard-Jones interactions in thermodynamic integration. J. Chem. Phys. 1994;100:9025–9031. [Google Scholar]
- 64.Boresch S., Karplus M. The meaning of component analysis: decomposition of the free-energy in terms of specific interactions. J. Mol. Biol. 1995;254:801–807. doi: 10.1006/jmbi.1995.0656. [DOI] [PubMed] [Google Scholar]
- 65.Brady G.P., Sharp K.A. Decomposition of interaction free energies in proteins and other complex systems. J. Mol. Biol. 1995;254:77–85. doi: 10.1006/jmbi.1995.0600. [DOI] [PubMed] [Google Scholar]
- 66.Bren M., Florian J., Bren U. Do all pieces make a whole? Thiele cumulants and the free-energy decomposition. Theor. Chem. Acc. 2007;117:535–540. [Google Scholar]
- 67.Mark A.E., van Gunsteren W.F. Decomposition of the free-energy of a system in terms of specific interactions. Implications for theoretical and experimental studies. J. Mol. Biol. 1994;240:167–176. doi: 10.1006/jmbi.1994.1430. [DOI] [PubMed] [Google Scholar]
- 68.Golubkov P.A., Ren P.Y. Generalized coarse-grained model based on point multipole and Gay-Berne potentials. J. Chem. Phys. 2006;125:64103. doi: 10.1063/1.2244553. [DOI] [PubMed] [Google Scholar]