Amygdala-enriched genes identified by microarray technology are restricted to specific amygdaloid subnuclei (original) (raw)
Abstract
Microarray technology represents a potentially powerful method for identifying cell type- and regionally restricted genes expressed in the brain. Here we have combined a microarray analysis of differential gene expression among five selected brain regions, including the amygdala, cerebellum, hippocampus, olfactory bulb, and periaqueductal gray, with_in situ_ hybridization. On average, 0.3% of the 34,000 genes interrogated were highly enriched in each of the five regions, relative to the others. In situ hybridization performed on a subset of amygdala-enriched genes confirmed in most cases the overall region-specificity predicted by the microarray data and identified additional sites of brain expression not examined on the microarrays. Strikingly, the majority of these genes exhibited boundaries of expression within the amygdala corresponding to cytoarchitectonically defined subnuclei. These results define a unique set of molecular markers for amygdaloid subnuclei and provide tools to genetically dissect their functional roles in different emotional behaviors.
Keywords: brain, cerebellum, hippocampus, olfactory bulb, gene chip
The mammalian brain is subdivided into cytoarchitectonically and physiologically distinct regions. Functional magnetic resonance imaging (fMRI) and lesioning studies have suggested that this anatomical parcellation reflects a modular functional organization (1). A major goal of modern neurobiology is to elucidate the functional roles of such brain modules, and of the neuronal subtypes that comprise them, in mediating specific behaviors. An important first step in applying the tools of molecular biology to this goal is to identify molecular markers for these subregions. Subtractive hybridization experiments have suggested that such brain subregion-restricted genes do exist (2) but have not been widely applied, perhaps because of their technical difficulty.
Microarray technology represents a potentially powerful approach to identifying genes specifically expressed in different cell or tissue types (3, 4). The application of microarray technology to the brain, however, poses problems of interpretation not encountered in more homogeneous cell populations, because of its complex anatomical organization and extreme cellular heterogeneity. This anatomical complexity necessitates that microarray analysis be integrated with systematic in situ hybridization studies to resolve the cellular distribution of identified transcripts.
Here we report the application of such an integrated analysis to the identification of genes expressed in the amygdala, a brain region implicated in emotional behaviors (5, 6). In situ hybridization has revealed that the majority of genes identified as amygdala-specific on microarrays exhibit intra-amygdaloid expression boundaries corresponding to cytoarchitectonically defined subnuclei. These results support the idea that brain subdivisions detectable by classical neuroanatomical methods reflect underlying differences in gene expression and demonstrate that systematic identification of molecular markers for such subregions is a feasible near-term goal.
Materials and Methods
Probe Preparation.
Five brain regions were chosen for analysis from 3-week-old male CD-1 mice: amygdala, cerebellum, hippocampus, olfactory bulb, and periaqueductal gray (PAG). For isolation of the amygdala and PAG, 34 mice were used. Thick sections (500–600 μm) were sliced with a vibratome, and the structures were dissected from these sections under a dissecting scope, following delineations from the mouse brain atlas (7). Dissected areas span approximately from −1.06 to −2.18 mm and from −2.92 to −4.24 mm with respect to bregma, for amygdala and PAG, respectively. Hippocampi, olfactory bulbs, and cerebella were dissected in their entirety from 17 brains. At least 5 μg of poly(A)+ RNA was purified from each brain region and converted to ≈20 μg of biotinylated cRNA hybridization probe, according to the Affymetrix manual.
Affymetrix Microarray Technology.
Oligonucleotide microarrays (ref. 8; also known as GeneChips) comprising 34,325 murine genes and expressed sequence tags (ESTs) were purchased from Affymetrix (1 set = Mu11kA, Mu11kB, Mu19kA, Mu19kB, and Mu19kC chips). Each gene or EST is represented on the GeneChips by ≈20 independent (nonoverlapping) “probe” sequences, each 25 nt in length. Each probe is located above a control probe containing a single-base mismatch. A score termed the “average difference” (Δ̄) is assigned to each gene, calculated as the average signal from the 20 perfect-match probes minus the average signal from the 20 corresponding mismatch probes. Note that such average difference values can therefore be >0 or <0.
Hybridization and scanning of GeneChips were carried out at a Howard Hughes Medical Institute facility at Stanford University (Stanford, CA). Because the purpose of the microarray analysis was to identify candidate genes for in situ hybridization analysis, rather than to provide accurate measurements of individual transcript abundance, a single set of microarrays (see above) was hybridized with each probe. However, independent studies have reported considerable reproducibility in replicate measurements using these arrays (9).
Data Analysis.
Before analysis, the data were normalized to correct for small differences in the amounts of each cRNA probe applied to the microarrays. Normalization factors were calculated (Affymetrixgenechips software) by comparing the mean fluorescent intensity of each array with respect to the corresponding amygdala array. On average, the mean Δ̄ value for each amygdala array was 1,160. Normalized average difference values were exported and analyzed with custom software (available athttp://www.its.caltech.edu/∼mariela/gene_screen.html; and see Appendixes A and B, which are published as supplemental data on the PNAS web site, www.pnas.org) written inmatlab (The MathWorks, Natick, MA). Two criteria were applied to identify genes enriched in each of the five brain regions: (i) the Δ̄ value for the gene in a given region; and (ii) the ratio (-fold difference) of its Δ̄ value in that region relative to each of the other four regions. For example, a given gene gi, with an average difference value in the amygdala of Δ̄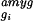 , was considered to be enriched relative to the other four regions if it satisfied the following constraints for these two criteria:
, was considered to be enriched relative to the other four regions if it satisfied the following constraints for these two criteria:
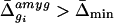 |
i |
|---|
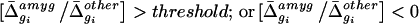 |
ii |
|---|
for all four other regions (cerebellum, hippocampus, olfactory bulb, and PAG).
In Situ Hybridization.
Male and female 3- to 4-week-old CD-1 mice were used. Clones were purchased from Research Genetics (Huntsville, AL) when available, or templates for probes were synthesized by PCR using specific primers and cDNA from mouse brain. For some genes, sense probes also were synthesized to control for nonspecific hybridization. Digoxigenin-labeled RNA probes were made and hybridization was performed essentially as described (10), with some modifications (see_Appendix A_). Adjacent sections were Nissl-stained for comparison. Images were collected with a Zeiss Axioskop or an Olympus IMT2 microscope attached to a charge-coupled device camera andneurolucida software (Microbrightfield, Colchester, VT), using 35 mm film or electronically acquired composite images, respectively.
Results
We used a custom algorithm to analyze the microarray data and iterated the analysis with in situ hybridization experiments to optimize search parameters (Fig. 1). Initial pairwise comparisons of the average difference values between each of the five brain regions, for all genes on the Mu11kA array, failed to identify obvious off-diagonal clusters indicative of differentially expressed genes (see Fig. 5, which is published as supplemental data on the PNAS web site). We therefore developed an algorithm to simultaneously compare relative gene expression levels among all five brain regions. We systematically varied different parameters (see Materials and Methods) in this algorithm to maximize the search for region-enriched genes. For example, we searched for genes whose average difference values were at least 3.5, 5, or 6 times higher in any given reference region as compared with the remaining four. Based on in situ hybridization experiments with genes identified in early iterations of this search, we concluded that a threshold ratio of 3.5 was optimal.
Figure 1.

Schematic diagram of the strategy used to identify region-enriched genes.
To filter out genes satisfying this ratio criterion, but whose absolute expression levels were likely to be too low to be detectable by our_in situ_ hybridization procedure, we empirically arrived at a minimum average difference value (Δ̄min). For genes enriched in the amygdala, for example, on the Mu11kA array Δ̄min was 110.4, corresponding to one-tenth of the mean Δ̄ value for all genes on this array and approximately 5-fold above the noise level of 22.5.
Analytical Characterization of Differentially Expressed Genes.
We found that only 455 of the 34,325 genes and ESTs analyzed (1.3%) fulfilled our selection criteria for enrichment in any one of the five brain regions, relative to the other four (Table1). Of these, 33 genes were enriched in the amygdala. On average, 0.3% of the sampled genes were highly enriched in any one of the five brain regions (Table 1). We also computed the number of genes that were “present” (i.e., had significant expression above background levels) in all five regions, as well as those that had no detectable expression. We found that 9,604 genes (28% of the genes on the array) were expressed in all areas examined, whereas 15,303 (45%) were present in none. Thus, of the 19,022 genes with detectable expression in one or more regions, half were present in all regions. Among the present genes, only 2.4% were differentially expressed in one region (455/19,022). A complete table with all 455 genes or ESTs and their corresponding Δ̄ values can be found in Table 3, which is published as supplemental data on the PNAS web site.
Table 1.
Genes that are at least 3.5- or 5-fold enriched in each of the five areas
| Region | 3.5-fold relative to all four other areas | 5-fold relative to all four other areas | 5-fold relative to any three other areas |
|---|---|---|---|
| Amy | 33 (0.1) | 21 (0.1) | 65 (0.2) |
| Cb | 159 (0.5) | 86 (0.3) | 164 (0.5) |
| Hpc | 89 (0.3) | 57 (0.2) | 105 (0.3) |
| OB | 101 (0.3) | 68 (0.2) | 127 (0.4) |
| PAG | 73 (0.2) | 44 (0.1) | 95 (0.3) |
| Total | 455 (1.3) | 276 (0.8) | 556 (1.6) |
| Average | 91 (0.3) | 55 (0.16) | 111 (0.3) |
To investigate whether certain classes of genes were preferentially represented among these sequences, we classified all annotated differentially expressed genes based on their structure or function. Of the 455 sequences, 117 (26%) were annotated genes. In four cases, annotation was accomplished by using 5′ rapid amplification of cDNA ends (5′ RACE) to clone the coding region. The genes were classified among 21 different functional categories, following the Gene Ontology (GO) Consortium guidelines (11). The categories that were the most highly represented (contained >7% of the 117 genes) comprised signaling molecules (26%, n = 30), DNA binding molecules (17%, n = 20), enzymes (15%,n = 18), and structural proteins (9%,n = 10). Some examples of these are shown in Table2. (See Table 4, which is published as supplemental data on the PNAS web site, for a full list of the functional categories and the percentage of genes in each class for each of the five brain regions analyzed.)
Table 2.
Some examples of genes enriched at least 3.5-fold in each region
| Region | Functional category | ||||
|---|---|---|---|---|---|
| Signaling | DNA-binding | Structural | Enzyme- or ligand-binding | EST | |
| Amy | Vasopressin (M88354) | arp-1 (X76653) | Unconventional type myosin (TC37197)* | ND | TC35462 (activin receptor type II)* |
| Cb | Cerebellum P400 protein (X15373) | Neuro-D (U28068) | Pro-α-2 (I) collagen (Msa.2220.0) | Parvalbumin (X67141) | TC33451 |
| Hpc | ND | Friend of GATA-1 (FOG) (AF006492) | Dynactin (Msa.12975.0) | Neuropsin (D30785) | TC36417 |
| OB | B219/OB receptor (ET61693) | Dlx-1 (U51000) | Pro-collagen type V α-2 (Msa.544.0) | Tyrosine hydroxylase (M69200) | TC20543 |
| PAG | Angiotensinogen precursor (Msa.7127.0) | Gata-2 (AB000096) | ND | Angiotensin-converting enzyme (Msa.24687.0) | TC36249 |
Several of these genes had previously been reported to be enriched in their given areas. These included vasopressin (12, 13) and arp-1 (14) in the amygdala, P400 (15) and Neuro-D (16, 17) in the cerebellum, and tyrosine hydroxylase in the olfactory bulb (18). Of 13 genes identified as cerebellum-enriched in a recent GeneChip study (19), we independently identified 6. Of the remaining 7 genes, 2 were rejected by our algorithm because they had values <0, and 5 were rejected because they also were expressed at substantial levels in the olfactory bulb, a structure not analyzed in the earlier study. However, as that study included other brain regions such as the cortex, not examined in our experiments, the two data sets are complementary.
To compare the performance of our search algorithm to that of an independent method, we carried out a clustering analysis using thegenecluster program (20), which implements self-organizing maps (SOMs). All genes identified by our method also were included in SOM-derived clusters corresponding to single region-enriched genes. However, the total number of genes in each of these SOM clusters was about 6 to 10 times larger than the number of genes identified by using our algorithm, even with the use of stringent filters for SOMs. These additional genes were rejected by our algorithm because they either fell below Δ̄min, or because their -fold difference relative to the other four regions was too low. Nevertheless, several “best candidates” among these genes were selected for in situ hybridization analysis (see below).
Validation of GeneChip Results by in Situ Hybridization.
It was essential to validate the results of the microarray analysis by an independent method. We used in situ hybridization, rather than biochemical assays such as RNase protection, because the complex anatomical organization of the brain necessitates a method with high spatial resolution. Thirty-five genes were analyzed by in situ hybridization. Of these, approximately 60% were expressed in a manner consistent with the results of the microarray analysis, 20% did not show any signal, 13% hybridized everywhere, and 7% were inconsistent with the microarray results (i.e., hybridized more strongly to regions that were predicted to have lower abundance). Because it was impractical to optimize probe design and hybridization parameters for each gene, it is possible that the actual false negative and false positive rate is lower than we observed.
To determine the extent to which our algorithm conditions could be further relaxed, we performed in situ hybridization experiments for four best candidate genes identified bygenecluster that marginally failed to meet our selection criteria. Three of these did not show any signal, but one was indeed expressed in the amygdala (probe 41 in Fig.2C). However, this gene was identified by genecluster only with the use of a very lax filter that included many other genes that fell well below our selection criteria.
Figure 2.

In situ hybridization of amygdala-enriched genes. (A) Nissl staining of a coronal section (left side of the brain). To the right, a schematic representation of various amygdala subnuclei is shown. Cortical-like nuclei (lateral, basolateral, and cortical) are shown in blue. Striatal-like subdivisions (central and medial) are in yellow, and the basomedial region is in orange (BMP = basomedial, posterior; BMA = basomedial, anterior). (B–D) Low magnification pictures of the left hemibrain. Amygdala details are shown in the magnified area (boxes). To the right are computer-aided schematics of staining in the amygdaloid region. Note that the nuclear boundaries vary slightly depending on the axial level. Color boundaries of subnuclei follow the diagram from A. (B) Probe 29 (activin receptor type II, TIGR identifier TC35462). Intense labeling in the lateral, basomedial, and cortical amygdala is apparent (black arrows). Note that the medial nucleus is devoid of staining (white arrow). No signal was detected in the cerebellum or PAG. Very few cells were stained in the olfactory bulb (not shown). A sense probe (not shown) labeled the hippocampus and piriform cortex (arrowheads) in the same way as the antisense probe, so the signal in these regions may be mainly caused by nonspecific hybridization. (C) Probe 41 (laminin β3, GenBank accession no. U43298). Signal is visible in the medial amygdala (black arrow) and ventromedial hypothalamus (white arrow). No staining was detected in cerebellum, hippocampus, olfactory bulb, and PAG (not shown). (D) Probe 4 (arp-1, GenBank accession no. X76653). Strong signal is detected in the lateral and basolateral complexes (black arrow). Note also weaker signal in the medial amygdala (white arrow). The reticular thalamic nucleus also showed clear hybridization (arrowhead). No staining was detected in the other four regions examined on microarrays (not shown).
Strikingly, although our selection criteria required only a 3.5-fold difference in the level of expression in one region as compared with the others, in many cases this seemingly modest quantitative difference on the arrays translated into an apparent qualitative difference when examined by in situ hybridization. Thus, the expression of many amygdala-enriched genes simply was not detected by in situ hybridization in the other regions examined in the microarray analysis. This finding may reflect the fact that many of the genes had fairly low average difference values in the amygdala, so that a 3.5-fold lower level of expression in one of the other regions might be below the detection limit of the nonisotopic in situ method. As might be expected, most of the amygdala-enriched genes proved to be expressed in at least one other brain area not tested in the microarray experiment, such as the cortex (Fig.3C).
Figure 3.

Expression of amygdala-enriched genes in different amygdaloid subnuclei. (A) Probe 75 (unconventional type myosin, TIGR identifier TC37197). Note the sharp discontinuity in expression levels between the lateral (arrow), and basolateral (arrowhead) nuclei. Staining was also observed in cortical layers 2/3 (white arrow). No staining was detected in the cerebellum, olfactory bulb, or PAG (not shown). (B) Probe 45–6 (Lhx6, GenBank accession no.AB031040). Lhx 6 hybridized to many scattered cells in the forebrain and was particularly concentrated in the dorsal aspect of the medial amygdala (arrow); the cerebellum was unlabeled (not shown). Lhx 6 was not represented on the microarray, but was analyzed because of its coexpression with Lhx7 (28, 29), which also was enriched in the amygdala (not shown). (C) Probe 50 (neuronal pentraxin receptor, TIGR identifier TC18750). The expression domain matches the boundaries of the lateral and basolateral amygdala (arrow). Staining also was observed throughout cortex (arrowhead) and in hippocampus (not shown). No signal was detected in the cerebellum or PAG. The olfactory bulb had weak staining (not shown). (D) Probe 28 (plasma glutathione peroxidase, TIGR identifier TC31122). Intense labeling is apparent in the medial amygdala (arrow), hypothalamus, and PAG (not shown). Note also signal in a contiguous subregion of the basomedial amygdala (dotted line). Two other genes also showed expression in this same region (not shown). (E) Probe 68 [cerebrospinal fluid (CSF)-induced cysteine protease, TIGR identifier TC30215]. Hybridization in the basomedial amygdala (arrow) was detectable. Staining also was observed in the hippocampus but was absent in the remaining regions of study (not shown). (F) Probe 20 (Ccte chaperonin ɛ subunit, TIGR identifier TC30886). Signal was detected in the medial amygdala (arrow) and in the lateral, basolateral, and basomedial complexes (not shown). No staining was detected in the other four regions of study (not shown). Probe numbers are in parentheses.
The absolute Δ̄ values obtained from the microarrays do not distinguish whether a given gene is expressed at high levels in a small subpopulation of cells or at lower levels in a larger population. Among the genes that we examined, one-fourth (25%) showed strong expression in relatively small, scattered cell populations, whereas the majority (75%) were expressed more broadly. Because the pieces of tissue we dissected for RNA isolation were relatively large and heterogeneous, it is likely that our analysis was biased against genes expressed at lower levels in small subpopulations of cells.
Amygdala-Enriched Genes Respect Subnuclear Boundaries.
The amygdala is a complex structure that can be anatomically subdivided into at least 13 distinct regions (21), such as the lateral, basolateral, medial, and central nuclei (Fig. 2A). This structural organization raises two questions: (i) Do the boundaries of amygdaloid nuclei reflect boundaries of gene expression domains?; and (ii) Do gene expression patterns reveal features of amygdaloid organization not visible by classical neuroanatomical techniques? To address these questions, we examined in detail the in situ hybridization pattern of 12 genes predicted by the microarray analysis to be enriched in the amygdala.
Surprisingly, the majority (75%) of these genes exhibited restricted, contiguous domains of expression, whose boundaries at least partly coincided with those of amygdaloid subnuclei (Fig.2A). (The remaining genes were expressed in scattered populations of cells.) Within this larger group of genes, approximately 50% completely respected nuclear boundaries (Figs. 2 B and C and 3 A, C, and_E_). The other half respected nuclear boundaries along part of their length, but in places extended beyond these boundaries into a well defined territory not coincident with any described amygdaloid subdivisions (e.g., Fig. 3D, dotted vs. dashed lines).
All of the amygdala-enriched genes we examined could be parsed into three groups, according to the distinct ontogenetic origins of the subnuclei in which they are expressed. One group of genes (five genes; 42%) were expressed in the lateral, basolateral, and cortical nuclei (Fig. 2A, blue), which are cortical-like structures embryologically derived from the pallium (22–24). Probes 29 (activin receptor type II; Fig. 2B) and 75 (unconventional type myosin; Fig. 3A) are characteristic of this group. The second group (five genes; 42%) was formed by genes expressed in the central and medial nuclei (Fig. 2A, yellow), which have subpallial (striatal or pallidal) origin. Probe 41 (laminin β3; Fig. 2C) is characteristic of this group. The third group (16%) consisted of two genes, the transcription factor arp-1 (Fig.2D) and Ccte chaperonin ɛ subunit (Fig. 3F), with widespread expression throughout the amygdala, including pallial and subpallial nuclei. Thus, the majority (84%) of the genes were expressed in either of two subsets of amygdaloid subnuclei related by a common developmental origin.
Genes in the first and second groups also shared some other features of their expression. For example, several of the genes in the first group (e.g., probes 75 and 50, Fig. 3 A and C) also were expressed to varying extents in the neocortex, consistent with the pallial origin of the amygdaloid regions in which this group is expressed. Conversely, a number of the genes in the second group (e.g., probe 28 and probe 41, Figs. 3D and 2_C_) also labeled the hypothalamus. Interestingly, all genes in the first group were expressed in contiguous cell populations. This observation may reflect the fact that the lateral and basolateral complexes are relatively homogeneous with respect to both cell type and neurotransmitter content (13, 25). By contrast, 80% of the genes in the second (striatal) group, such as the neuropeptide vasopressin, were expressed in scattered subpopulations of cells. This observation is consistent with the fact that amygdaloid neuropeptides are generally expressed in scattered cell populations (26), and also that the centromedial aspect of the amygdala is the most neuropeptide-rich region in the brain outside the hypothalamus (27). Other genes in this subgroup included the Lim homeodomain transcription factors Lhx-6 (Fig.3B) and -7. It is possible that these factors are involved in the regulation of amygdaloid neuropeptide gene expression.
Discussion
The modular functional organization of the mammalian brain is likely to reflect, at least in part, its anatomical parcellation into distinct substructures. We have used microarray analysis in conjunction with in situ hybridization to identify molecular markers of this anatomical regionalization. By using commercially available microarrays, we identified in each of five selected brain regions, on average, 91 genes that were highly enriched. This estimate is very close to that arrived at in a previous study employing subtractive hybridization (2), which estimated the number of transcripts highly enriched in the hypothalamus to be on the order of 100. Our figure constitutes 0.3% of the ≈34,000 genes interrogated, and 0.5% of all genes expressed in at least one of the five areas (91/19,022). Similar values were recently reported by Sandberg et al. (19), who analyzed the expression of about 13,000 genes and ESTs in a different subset of brain regions than we examined. These values may, however, be an underestimate because genes expressed at low levels in small subsets of cells may have been systematically excluded by both analyses.
Among the differentially expressed genes with known function, 67% fell into 4 of 21 functional categories, comprising signaling molecules, transcription factors, enzymes, or structural proteins. However, the majority (72%) of the differentially expressed genes were unannotated ESTs, making it difficult to draw firm conclusions about categorical representation. It is also likely that many other unknown region-specific genes exist, which were not interrogated by the Affymetrix GeneChips. Other microarray methods that do not rely on previous knowledge of sequences may prove useful in identifying these.
Analytic Considerations.
For simply identifying region-specific or highly enriched genes, our custom algorithm proved more efficient than SOMs cluster analysis (20). That is because our program permits the explicit specification of multiple criteria for “enriched” genes. In contrast,genecluster identifies collections of genes that share similar features. Therefore, no constraint about the ratio of expression in one brain region relative to all of the others can be independently set. However, SOM analysis is designed for gene-profiling studies, where the comparison of expression patterns among a large collection of samples is sought (20). This analysis is fundamentally different from positively selecting highly enriched genes that fulfill specific ratio criteria.
Validation of Microarray Data.
A recent study (19) also used Affymetrix GeneChips to characterize region-specific gene expression in the brain, but did not validate the microarray results by in situ hybridization. Our results suggest that in situ hybridization is essential to confirm GeneChip data. Of the 35 genes we tested, 80% yielded detectable_in situ_ hybridization signals. Of these, approximately 25% exhibited patterns apparently inconsistent with the microarray data. Thus, 60% of the 35 genes examined were validated by in situ hybridization. Of the 14 cases of inconsistency, most (65%) reflected probes that hybridized everywhere. These cases may simply represent suboptimal probe design rather than any inherent inaccuracy of the GeneChip method. The remaining cases, however, constituted probes that gave strong in situ signals in regions predicted to be weak or negative by the microarrays. It is possible that replicate microarray experiments with independently prepared samples and chips would have lowered the number of false positives. However, considering that at least 17 mice were used to prepare cRNA probes from each brain region, it is unlikely that the discrepancies we observed are attributable to inconsistent tissue dissection or to biological differences between the animals used to prepare microarray probes and those used for in situ hybridization.
Even for those genes whose in situ pattern was consistent with the predictions of the microarray data, in situ hybridization was also essential to identify sites of expression not included among the original five samples. This is important, as it is technically impossible to analyze every brain region or nucleus in a given microarray experiment. Our in situ analysis also revealed how extrapolating mRNA abundance levels based on Δ̄ values from the microarrays is not necessarily informative, because this value reflects both the abundance of a given transcript within expressing cells as well as the proportion of cells expressing the transcript in a given brain region. We have found examples of genes with low Δ̄ values that were expressed at very high levels in a few cells, and conversely, genes expressed broadly at more modest levels that yielded high Δ̄ values.
Toward a Molecular Anatomy of the Amygdala.
The amygdala, a brain region implicated in emotional learning (5, 6), lies at the interface between the cortex and subcortical structures such as the striatum and hypothalamus, and therefore is well positioned to integrate computational and neuromodulatory functions. Accordingly, the amygdala is structurally heterogeneous, consisting of over a dozen subnuclei (13, 21, 22). We made no special effort to microdissect such subnuclei in preparing the microarray hybridization probe; rather, a relatively crude dissection of the entire amygdala was used. Thus, it is striking that 75% of the amygdala-enriched genes that we examined by in situ hybridization (n = 12) exhibited expression boundaries at least partly coinciding with those of one or more subnuclei (Fig. 4v).A priori, this need not have been the case. At least four other kinds of expression patterns could have been obtained (Fig. 4): (i) uniform expression throughout the amygdala; (ii) contiguous subdomains bearing no relationship to classically defined subnuclei; (iii) scattered expression in cells contained within specific subnuclei; and (iv) scattered expression not respecting subnuclear boundaries. It is striking that no genes fell into either of the first two categories. These data suggest not only that the boundaries of amygdaloid subnuclei reflect gene expression boundaries, but moreover that the majority of amygdala-enriched genes may respect such boundaries. The genes we have identified should, therefore, provide useful markers for amygdaloid subnuclei, some of which can be difficult to visualize by Nissl staining on thin histological sections.
Figure 4.

Possible gene expression patterns in the amygdala and the percentage of amygdala-enriched genes examined that exhibited such patterns. (i) Contiguous, panamygdaloid expression. (ii) Contiguous expression in subdomains whose boundaries bear no relationship to those of classically defined amygdaloid subnuclei. (iii) Expression in scattered cells contained within specific subnuclei. (iv) Expression in scattered cells not respecting subnuclear boundaries. (v) Contiguous expression in subdomains whose boundaries match, at least in part, those of amygdaloid subnuclei. The majority of genes examined (75%) exhibited pattern (v).
Our data also indicate, however, that not all gene expression boundaries correspond precisely to boundaries of amygdaloid subnuclei. For example, we observed three genes that had a similar, well defined expression domain that included the medial amygdala, but which extended into a limited subregion of the adjacent basomedial amygdala (Fig.3D, dotted line). Thus, gene expression domains do not simply validate classically defined anatomical units, but also may reveal organizational features not easily visualized by existing staining techniques.
At present, the rate-limiting step in the analysis of microarray data derived from the brain is its validation by in situ hybridization. When efficient, large-scale, high-throughput, automated_in situ_ hybridization procedures for adult brain sections become available, it should be possible to exploit microarray data to generate a “molecular brain atlas” in which each structure also is delineated by its molecular repertoire. The results presented here demonstrate that such a long-term goal is, in principle, feasible. The genes identified by such an exercise, moreover, are not simply markers, but also will provide tools to genetically dissect the roles of such brain substructures in specific behaviors.
Supplementary Material
Supplemental Data
Acknowledgments
We thank R. Mongeau for dissecting the PAG; A. Smith and J. Xiao (Stanford University/Howard Hughes Medical Institute) for performing microarray hybridizations; G. Meissner, C. Hsu, and S. Pintchovski for help with in situ hybridization; G. Miller for help with Fig. 2; and G. Mosconi for managerial assistance. We also acknowledge M. Zylka and B. Wold for helpful discussions. This work was supported by National Institute of Mental Health Grant MH62825, a gift from Merck Inc., and a grant from the Mettler Fund on Autism. D.J.A. is a Howard Hughes Medical Institute Investigator.
Abbreviations
Δ̄
average difference
PAG
periaqueductal gray
EST
expressed sequence tag
SOM
self-organizing map
Footnotes
†
To whom reprint requests should be addressed. E-mail: c/o mancusog@caltech.edu.
References
- 1.Kandel E, Schwartz J, Jessell T. Principles of Neural Science. New York: McGraw–Hill; 2000. pp. 1–5. [Google Scholar]
- 2.Gautvik K, deLecea L, Gautvik V, Danielson P, Tranque P, Dopazo A, Bloom F, Sutcliffe J. Proc Natl Acad Sci USA. 1996;93:8733–8738. doi: 10.1073/pnas.93.16.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown P, Botstein D. Nat Genet. 1999;21:33–37. doi: 10.1038/4462. [DOI] [PubMed] [Google Scholar]
- 4.Lipshutz R, Fodor S, Gingeras T, Lockhart D. Nat Genet. 1999;21:20–24. doi: 10.1038/4447. [DOI] [PubMed] [Google Scholar]
- 5.Davis M. Annu Rev Neurosci. 1992;15:353–375. doi: 10.1146/annurev.ne.15.030192.002033. [DOI] [PubMed] [Google Scholar]
- 6.LeDoux J E. Annu Rev Psychol. 1995;46:209–235. doi: 10.1146/annurev.ps.46.020195.001233. [DOI] [PubMed] [Google Scholar]
- 7.Franklin K, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. San Diego: Academic; 1997. [Google Scholar]
- 8.Lockhart D, Dong H, Byrne M, Follettie M, Gallo M, Chee M, Mittmann M, Wang C, Kobayashi M, Horton H, Brown E. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 9.Wodicka L, Dong H, Mittmann M, Ho M, Lockhart D. Nat Biotechnol. 1997;15:1359–1367. doi: 10.1038/nbt1297-1359. [DOI] [PubMed] [Google Scholar]
- 10.Henrique D, Adam J, Myat A, Chitnis A, Lewis J, Ish-Horowicz D. Nature (London) 1995;375:787–790. doi: 10.1038/375787a0. [DOI] [PubMed] [Google Scholar]
- 11.Ashbuner M, Ball C, Blake J, Botstein D, Butler H, Cherry J, Davis A, Dolinski K, Dwight S, Eppig J, et al. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caffe A, Vanleeuwen F. Cell Tissue Res. 1983;233:23–33. doi: 10.1007/BF00222229. [DOI] [PubMed] [Google Scholar]
- 13.Price J, Russchen F, Amaral D. In: Handbook of Chemical Neuroanatomy. Bjorklund A, Hokfelt T, Swanson L, editors. Vol. 5. Amsterdam: Elsevier; 1987. pp. 279–388. [Google Scholar]
- 14.Dasilva S, Cox J, Jonk L, Kruijer W, Burbach J. Mol Brain Res. 1995;30:131–136. doi: 10.1016/0169-328x(94)00289-q. [DOI] [PubMed] [Google Scholar]
- 15.Nakanishi S, Maeda N, Mikoshiba K. J Neurosci. 1991;11:2075–2086. doi: 10.1523/JNEUROSCI.11-07-02075.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee J E, Hollenberg S M, Snider L, Turner D L, Lipnick N, Weintraub H. Science. 1995;268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- 17.Schwab M, Druffel-Augustin S, Gass P, Jung M, Klugmann M, Bartholomae A, Rossner M, Nave K. J Neurosci. 1998;18:1408–1418. doi: 10.1523/JNEUROSCI.18-04-01408.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hokfelt T, Martensson R, Bjorklund A, Kleinau S, Goldstein M. In: Handbook of Chemical Neuroanatomy. Bjorklund A, Hokfelt T, editors. Vol. 2. Amsterdam: Elsevier; 1984. pp. 277–379. [Google Scholar]
- 19.Sandberg R, Yasuda R, Pankratz D, Carter T, Del Rio J, Wodicka L, Mayford M, Lockhart D, Barlow C. Proc Natl Acad Sci USA. 2000;97:11038–11043. doi: 10.1073/pnas.97.20.11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander E S, Golub T S. Proc Natl Acad Sci USA. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pitkänen A, Savander V, LeDoux J E. Trends Neurosci. 1997;20:517–523. doi: 10.1016/s0166-2236(97)01125-9. [DOI] [PubMed] [Google Scholar]
- 22.Puelles L, Kuwana E, Puelles E, Rubenstein J. Eur J Morphol. 1999;37:139–150. doi: 10.1076/ejom.37.2.139.4756. [DOI] [PubMed] [Google Scholar]
- 23.Puelles L, Kuwana E, Puelles E, Bulfone A, Shimamura K, Keleher J, Smiga S, Rubenstein J. J Comp Neurol. 2000;424:409–438. doi: 10.1002/1096-9861(20000828)424:3<409::aid-cne3>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 24.Swanson L, Petrovich G. Trends Neurosci. 1998;21:323–331. doi: 10.1016/s0166-2236(98)01265-x. [DOI] [PubMed] [Google Scholar]
- 25.McDonald A. In: The Amygdala: Neurobiological Aspects of Emotion, Memory and Mental Dysfunction. Aggleton J, editor. New York: Wiley–Liss; 1992. pp. 67–96. [Google Scholar]
- 26.Roberts G, Woodhams P, Polak J, Crow T. Neuroscience. 1982;7:99–131. doi: 10.1016/0306-4522(82)90156-7. [DOI] [PubMed] [Google Scholar]
- 27.Roberts G. In: The Amygdala: Neurobiological Aspects of Emotion, Memory and Mental Dysfunction. Aggleton J, editor. New York: Wiley–Liss; 1992. pp. 115–142. [Google Scholar]
- 28.Zhao Y, Guo Y, Tomac A, Taylor N, Grinberg A, Lee E, Huang S, Westphal H. Proc Natl Acad Sci USA. 1999;96:15002–15006. doi: 10.1073/pnas.96.26.15002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grigoriou M, Tucker A, Sharpe P, Pachnis V. Development (Cambridge, UK) 1998;125:2063–2074. doi: 10.1242/dev.125.11.2063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data