Optogenetic activation of LiGluR-expressing astrocytes evokes anion channel-mediated glutamate release (original) (raw)
Abstract
Non-technical summary
Ca2+ increases in astrocytes have been suggested to trigger the release of neuroactive compounds called gliotransmitters. To study the mechanism of gliotransmitter release, it is desirable to have a method able to selectively and reproducibly evoke Ca2+ increases in astrocytes. Here, we show that the optogenetic tool the light-gated glutamate receptor 6 (LiGluR) reproducibly evokes Ca2+ rises in cultured astrocytes. Combining LiGluR photoactivation and evanescent-field Ca2+ imaging, we explored the cellular mechanism of gliotransmitter release from astrocytes in an all-optical manner. The results suggest that high Ca2+ in astrocytes triggers the release of glutamate via anion channels rather than vesicular exocytosis.
Abstract
Increases in astrocyte Ca2+ have been suggested to evoke gliotransmitter release, however, the mechanism of release, the identity of such transmitter(s), and even whether and when such release occurs, are controversial, largely due to the lack of a method for selective and reproducible stimulation of electrically silent astrocytes. Here we show that photoactivation of the light-gated Ca2+-permeable ionotropic GluR6 glutamate receptor (LiGluR), and to a lesser extent the new Ca2+-translocating channelrhodopsin CatCh, evokes more reliable Ca2+ elevation than the mutant channelrhodopsin 2, ChR2(H134R) in cultured cortical astrocytes. We used evanescent-field excitation for near-membrane Ca2+ imaging, and epifluorescence to activate and inactivate LiGluR. By alternating activation and inactivation light pulses, the LiGluR-evoked Ca2+ rises could be graded in amplitude and duration. The optical stimulation of LiGluR-expressing astrocytes evoked probabilistic glutamate-mediated signalling to adjacent LiGluR-non-expressing astrocytes. This astrocyte-to-astrocyte signalling was insensitive to the inactivation of vesicular release, hemichannels and glutamate-transporters, and sensitive to anion channel blockers. Our results show that LiGluR is a powerful tool to selectively and reproducibly activate astrocytes.
Introduction
Electrically non-excitable astrocytes respond to neurotransmitters with Ca2+ elevations (Agulhon et al. 2008) that have been suggested to trigger the release of neuroactive gliotransmitters like glutamate and ATP, and to mediate communication between glia and between glia and neurons (Perea & Araque, 2007; Agulhon et al. 2008; Fiacco et al. 2009). Gliotransmission has emerged in recent years as an additional layer of cellular communication that could contribute to information processing by the brain (Halassa & Haydon, 2010). However, the Ca2+ signals required for gliotransmitter release, the release mechanism, and indeed the very existence of such release are highly controversial (Fiacco et al. 2009; Agulhon et al. 2010; Hamilton & Attwell, 2010).
One obstacle to a resolution of these questions is that astrocytes share many ligand-gated receptors with neurons (Fiacco et al. 2009). To activate astrocytes specifically, a transgenic mouse has been generated which selectively expresses a foreign G protein coupled receptor (GPCR) in astrocytes. Strikingly, in these mice, triggering specifically Ca2+ elevations in astrocytes with a ligand had an effect neither on synaptic transmission and plasticity nor on neuronal excitability (Fiacco et al. 2007; Agulhon et al. 2010). This contrasts with other results showing Ca2+-dependent gliotransmitter release (Perea & Araque, 2007; Andersson & Hanse, 2010; Gomez-Gonzalo et al. 2010; Halassa & Haydon, 2010). The reasons for these contradictory results are unclear (Agulhon et al. 2010; Halassa & Haydon, 2010).
The coupling between Ca2+ signals and gliotransmitter release is still debated. Astrocytic Ca2+ signals rely on plasma membrane receptors and channels, and on intracellular stores (Parpura et al. 2011) and near-membrane spontaneous and ATP-evoked Ca2+ signals depend on different Ca2+ sources (Shigetomi et al. 2010). This diversity of Ca2+ signals could explain why different Gq GPCRs are not equally competent to triggering glutamate release (Shigetomi et al. 2008). A better understanding of Ca2+-dependant gliotransmitter release will require the development of new specific tools for a reliable time-locked control of Ca2+ signals, which need to be associated with imaging techniques for the simultaneous monitoring of Ca2+ signals in electrically silent astrocytes. In an attempt to develop such methods, we set out to control astrocytic Ca2+ rises using the light-gated Ca2+-permeable ionotropic glutamate receptor 6 (LiGluR), the monovalent cationic channel channelrhodopsin 2 (ChR2), and the new Ca2+-translocating channelrhodopsin (CatCh). These channels have been used for a remote non-invasive activation of neurons with high spatio-temporal resolution (Szobota et al. 2007; Gradinaru et al. 2009; Wyart et al. 2009), and ChR2 has been used to activate astrocyte-dependent neuronal responses in vivo (Gradinaru et al. 2009; Gourine et al. 2010).
Using cultured cortical astrocytes, we first show that LiGluR and, to a lesser extent, CatCh, provide a more reliable means for triggering reproducible intracellular Ca2+ elevations than the enhanced ChR2 mutant (ChR2(H134R)). We then combined LiGluR activation with evanescent-field excitation Ca2+ imaging to study the responses of LiGluR non-expressing (LiGluR(−)) astrocytes to the photoactivation of their LiGluR-expressing (LiGluR(+)) neighbours. We find that light-evoked Ca2+ elevation in LiGluR(+) astrocytes triggers delayed short-lasting small-amplitude Ca2+ transients in adjacent LiGluR(−) astrocytes, and that this LiGluR-evoked astrocyte-to-astrocyte communication involves a glutamate-permeable anion channel. Our results demonstrate the utility of LiGluR-based optogenetic approaches for studying communication between electrically silent cells.
Methods
Cell preparation, solutions and recording conditions
All experiments followed the European Union and institutional guidelines for the care and use of laboratory animals (Council directive 86/609EEC). Cortical astrocytes were prepared from P0–1 (P0 being the day of birth) NMRI mice as previously described (Li et al. 2009). The neocortex was dissected and mechanically dissociated. Cells were plated and maintained on poly-ornithine-coated coverslips (no. 1, BK-7, 25 mm diameter, Menzel-Gläser GmbH) for 1 week to reach confluence. Then, 0.15 mm dibutyryl cAMP (Sigma) was added to induce astrocyte differentiation into a more stellate morphology and form non-confluent islands of several astrocytes. All cultures were kept at 37°C in a humidified 5% CO2 atmosphere. Primary astrocyte cultures were maintained in supplemented Dulbecco's modified Eagle's medium (DMEM, Invitrogen) with 5% fetal bovine serum (FBS), penicillin (5 U ml−1) and streptomycin (5 μg ml−1). The recordings were made during the following week.
For neuronal culture, cortical neurons and astrocytes were isolated from embryonic mice (E16). Cells were seeded on poly-l-lysine-treated coverslips and maintained in a medium containing minimum essential medium (MEM) with 5% FBS, 0.3% high glucose MEM, serum extender (1/1000) and penicillin/streptomycin (1/500). Cells were then maintained in half of this serum and half of medium of primary astrocytes containing growth factors for neurons. Cell media and supplements were from Invitrogen (Cergy Pontoise, France). Recordings were made at room temperature (RT) in solution containing (in mm): 140 NaCl, 5.5 KCl, 1.8 CaCl2, 1 MgCl2, 20 glucose, 10 Hepes (pH 7.3, adjusted with NaOH). Ca2+-free extracellular solutions contained nominally zero Ca2+ and 5 mm EGTA.
Fluorescent probes, reagents and plasmids
Cell media, supplements, Ca2+ indicators (Fluo-4 AM, Xrhod-1 AM), and dyes (FM4–64, calcein AM, acridine orange, the mitochondrial marker pyridinium, 4-(2-(4-(dimethylamino)phenyl)ethenyl)-1-methyl iodide (DASPMI)) were purchased from Invitrogen (Cergy Pontoise, France), P2 receptor antagonists (pyridoxal-phosphate-6-azophenyl-2′,4′-disulfonate (PPADS), suramin, 2′-deoxy-_N_6-methyladenosine 3′,5′-bisphosphate (MRS2179)), mGluR receptor antagonists (α-methyl-4-carboxyphenylglycine (MCPG), 2-methyl-6-(phenylethynyl)pyridine (MPEP)), glutamate transporter inhibitor (dl-threo-β-benzyloxyaspartic acid (TBOA)) and thapsigargin were from Tocris (Bristol, UK), bafilomycin A1 from Calbiochem (Merck, Lyon, France) and all other compounds from Sigma-Aldrich. For fluorescence immunostaining experiments, we used the following antibodies: rabbit polyclonal glutamate antibody (1:200, ab9440, Abcam Inc., Cambridge, MA, USA) conjugated to the secondary antibody Alexa 594 goat anti-rabbit IgG (1:500, Invitrogen), mouse glial fibrillary acid protein (GFAP) monoclonal antibody (1:400, MAB360, Chemicon, Temecula, CA, USA) conjugated to the secondary antibody Alexa 488 goat anti mouse IgG (1:500, Invitrogen). Astrocytes were fixed with 1% paraformaldehyde and 0.1% glutaraldehyde (10 min, RT). After permeabilization and blockage of unspecific sites with phosphate buffered saline (PBS) 1×, 0.3% Triton X-100 and 2% bovine serum albumin (BSA) (1 h, RT), astrocytes were probed with respective primary antibody in the same solution overnight at 4°C. After being washed with PBS three times at RT, cells were incubated with secondary antibodies (2 h, RT). After three final washes (PBS, 10 min, RT), cells were mounted with Vectashield onto microscope slides. Immunostained cells were imaged with a Zeiss Axiovert LSM 510 confocal microscope using a ×63, NA 1.4 oil objective. Transfection was performed using lipofectamine 2000 (Invitrogen). Astrocytes were labelled with FM4–64 by incubating cells in dye-containing (6.7 μm) solution for 15 min and rinsing the cells for 30 min prior to imaging. Ca2+ dyes were bulk-loaded into astrocytes as AM-esters in static dye-containing solutions (2 μm, 30 min for Fluo-4; 200 nm, 15 min for Xrhod-1). Another 30 min under continuous perfusion of dye-free solution allowed for the wash-off of extracellular dye and the complete de-esterification of the intracellular dye. During recording, cells were perfused at 0.5 ml min−1 with standard extracellular solution. The glutamate-evoked Ca2+ transients were induced by a brief (1 s) application of glutamate (100 μm) through a dual-channel local perfusion system, one continuously perfusing the control buffer and the other transiently delivering the glutamate-containing solution while the control channel was stopped. The solutions were delivered through plastic tubing (0.8 mm inner diameter, Tygon, Charny, France) to a multi-channel holder (AutoMate Scientific, Berkeley, CA, USA) connected to a small (250 μm ID) silica pipette (WPI, Saratosa, FL, USA).
Fluorescence imaging
All combinations of excitation wavelengths and dichroic and emission filters used are listed in Supplemental Table 1. A custom-built inverted and upright microscope was used for bright-field, polychromatic epifluorescence imaging and through-the-objective (PlanApo TIRFM, ×60, 1.45 NA, Olympus, Hamburg, Germany) total internal reflection fluorescence microscopy (TIRFM). A Polychrome II (TILL Photonics, Gräfelfing, Germany) provided monochromatic (18 nm FWHM) epifluorescence (EPI) illumination. The 488 and 568 nm lines used for TIRFM were isolated from the beam of an Ar/Kr multi-line laser (CVI Melles Griot, Carlsbad, CA, USA) with an acousto-optical tuneable filter (AA.Opto, Saint Rémy de Chevreuse, France) and directed onto the glass–water interface at a super-critical angle. We estimated the effective penetration depth (1/e2-intensity decay) of the order of 200 nm (Nadrigny et al. 2007). Fluorescence images were further magnified (×2) and projected on an electron multiplying charge-coupled device (EMCCD, QuantEM 512, Princeton Instruments, Trenton, NJ, USA). All devices were controlled by Metamorph (Molecular Devices, Downingtown, PA, USA). The effective pixel size in the sample plane was 133 nm. Time-lapse image stacks were taken at 0.5 Hz with 50–300 ms exposure times unless otherwise indicated.
Non-ratiometric Ca2+ indicators (Fluo-4 and Xrhod-1) and TIRFM were used to monitor near-membrane Ca2+ increases throughout this study, unless otherwise indicated. TIRFM confines illumination to the near-membrane, providing the conditions for sensitive detection of local Ca2+ signals. Time-lapse fluorescence changes are plotted as Ca2+-dependent fluorescence measured in 2 μm × 2 μm (15 pixel × 15 pixel) regions of interest (ROIs) normalized to the pre-stimulation baseline (Δ_F_/F_0). The Ca2+ elevations in LiGluR(−) cells were searched for by scanning such ROIs across the LiGluR(−) cell. To avoid a possible interference with light-evoked Ca2+ rises in LiGluR(+) cells, Ca2+ transients in LiGluR(−) cells were searched for in regions more than 2 μm away from the visible interface. Signalling events were identified as Δ_F/_F_0 increases larger than three times the standard deviation (SD) of the baseline. Traces were corrected for photobleaching as follows: individual reference traces were separately obtained by imaging dye-loaded cells without photoactivation, from which the mean decay time constant was obtained from a mono-exponential fit and then applied to correct the Ca2+ traces in photostimulation experiments. Ca2+ signals in astrocytes co-labelled with LiGluR–green fluorescent protein (GFP) and FM4–64 were imaged with Xrhod-1 following the triple-colour detection scheme as described previously (Li et al. 2008). Fluo-4 AM was used for Ca2+ imaging of cells labelled with LiGluR–red fluorescent protein (RFP) and calcein. Ca2+ imaging was performed ∼5–10 min after pharmacological treatment of astrocytes unless otherwise stated. For the experiments done in zero Ca2+, cells were kept in Ca2+-free buffer for less than 5 min to avoid an interference with internal Ca2+ stores (Li et al. 2008). Finally, Ca2+ elevations were recorded only from cultured astrocytes showing no spontaneous Ca2+ elevations.
LiGluR, ChR2 and CatCh photoactivation
The engineering of LiGluR and synthesis of the tethered photoswitch maleimide-azobenzene-glutamate (MAG) have been previously described (Volgraf et al. 2006; Gorostiza et al. 2007; Numano et al. 2009). Cultured astrocytes were transfected with LiGluR-GFP, LiGluR-RFP, or cotransfected with LiGluR-RFP and AsRed under the control of the cytomegalovirus (CMV) promoter, using lipofectamine 2000 following the standard protocol. Cells were maintained in culture for 1–2 days after transfection to allow the proper targeting of LiGluR to the cell surface. Stock MAG solutions of 5–10 mm were prepared in anhydrous dimethyl sulfoxide (≥99.9%, Sigma) and stored at −20°C. To efficiently conjugate the photoswitch to LiGluR, MAG was pre-illuminated with 385 nm light (0.3 mW mm-2) for 1 min (Gorostiza et al. 2007) before dilution in standard imaging solution to a final concentration of 10 μm. Transfected cells were then incubated in MAG-containing solution for 30 min in the dark followed by another 30 min washing with control buffer. During incubation, 5 μm concanavalin A (Sigma) was added to the medium to block LiGluR desensitisation (Numano et al. 2009). To switch on LiGluR, a 385 nm LED source (Thorlabs, Maisons Lafitte, France) was focused through an air objective (×10, NA 0.25, Olympus) to excite the cells in a 2 mm wide field, and the light pulse duration was 50–200 ms, unless otherwise stated. Where indicated, 488 nm light from the monochromator was used to EPI-illuminate the target cell and switch off LiGluR (39.1 mW mm-2). Astrocytes and neurons were transfected with ChR2(H134R)-GFP using the same protocol as used for LiGluR, and examined 1–2 days after transfection. CatCh–yellow fluorescent protein (YFP) plasmid was provided by Dr Ernst Bamberg (MPI für Biophysik, Frankfurt, Germany). The red Ca2+ dye Xrhod-1 (200 nm, 15 min) was used for Ca2+ imaging of cells expressing ChR2 or CatCh. ChR2 and CatCh were photoactivated by 458 nm light (27.3 mW mm-2 and 15.1 mW mm-2, respectively) generated by the monochromator and delivered through the inverted epifluorescence pathway. For all light-gated channels, Ca2+ imaging was temporally ceased during photoactivation and deactivation.
Statistics
All data are expressed as mean ± standard deviation (SD), and Student's t test was used for testing the significance of P values. Non-normally distributed data were compared using their median ± absolute deviation and non-parametric tests (Kolmogorov–Smirnov, KS). All statistical operations used Matlab (The MathWorks, Natick, MA, USA). *P < 0.05, **P < 0.01, ***P < 0.001.
Results
Photoactivation of LiGluR triggers astrocytic Ca2+ rises
LiGluR is a light-gated channel consisting of a synthetic cysteine-reactive photoisomerizable agonist MAG covalently attached to a cysteine-substituted ionotropic glutamate receptor GluR6 (Volgraf et al. 2006; Numano et al. 2009). To examine the ability of LiGluR to control astrocytic Ca2+ increase, mouse cortical astrocytes in culture were transfected with LiGluR-RFP to which MAG was covalently attached. Short pulses of 385 nm light (0.3 mW mm-2, 50 ms) evoked robust and reliable Ca2+ rises monitored with the green-fluorescent Ca2+ indicator Fluo-4 (Δ_F_/F_0= 29.9 ± 4.0%, n = 23 trials from 7 cells; Fig. 1_A_). Light-evoked Ca2+ responses were neither observed in the absence of LiGluR-RFP (−0.1 ± 1.5%, n = 5 cells), nor in cells expressing LiGluR without MAG (0.3 ± 2.0%, n = 5 cells), nor in cells lacking LiGluR and MAG (Δ_F/F_0= 0.1 ± 1.6%, n = 5 cells) (Supplemental Fig. 1_A). The LiGluR-evoked Ca2+ rises recorded in the thin processes of LiGluR(+) astrocytes were synchronized and their amplitude was maintained along the process as expected for a direct activation of LiGluR targeted to the process membrane (Fig. 1_B_). Finally, the amplitude of LiGluR-elicited Ca2+ signals correlated with the duration of illumination (Supplemental Fig. 1_B_).
Figure 1. LiGluR evokes precisely timed and shaped Ca2+ rises in astrocytes.
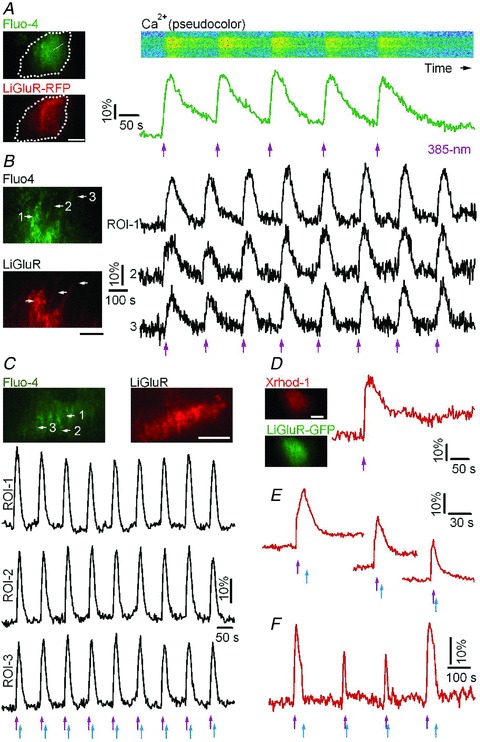
A, dual-colour TIRF image of a cultured cortical astrocyte transfected with LiGluR-RFP conjugated with the photoswitch MAG and loaded with the green-fluorescent Ca2+ indicator Fluo-4 (left). Dashed line shows the contour of the cell. The pseudocolour kymograph and green trace illustrate reproducible Ca2+ rises evoked by 385 nm light pulses (violet arrows, 0.3 mW mm-2, 50 ms). B, LiGluR photoactivation evoked repetitive and synchronized Ca2+ rises in astrocytic soma (ROI-1) and processes (ROI-2 and ROI-3). C, temporal shaping of astrocytic Ca2+ rises by switching LiGluR on and off with alternate 385 nm (violet arrows) and 488 nm (blue arrows, 39.1 mW mm-2, 200 ms) EPI light pulses. D–F, astrocytic Ca2+ elevations induced by LiGluR-GFP photoactivation and monitored with the red-fluorescent Ca2+ indicator Xrhod-1. The Ca2+ signals were shaped by switching off LiGluR with variable delay using 488 nm EPI light from the monochromator. Bars, 10 μm.
Besides being activated by UV light, LiGluR can be switched off with 488 nm blue light (Szobota et al. 2007). Because the spectral band for deactivating LiGluR overlaps with the 488 nm excitation wavelength of Fluo-4, epifluorescence Ca2+ imaging quickly terminates LiGluR-evoked responses (Supplemental Fig. 1_C_ and D). To minimize imaging-light induced LiGluR deactivation, and to shape the Ca2+ increases with a pulse of EPI-illumination deactivation, we confined Ca2+ imaging to the near-membrane cellular compartment, using total internal reflection fluorescence microscopy (TIRFM) (Supplemental Fig. 1_E_). In these conditions, alternating pulses of 385 nm and 488 nm light evoked reproducible Ca2+ transients (Fig. 1_C_). To minimize LiGluR deactivation during Ca2+ imaging, we also used a LiGluR-GFP construct with the red-fluorescent Ca2+ indicator Xrhod-1 AM. This combination allowed us to monitor LiGluR-evoked Ca2+ responses (Δ_F_/_F_0= 31.3 ± 6.0%, n = 12 trials from 5 cells), and to shape the Ca2+ rise by altering the interval between the activating and deactivating light pulses (Fig. 1_D_–F). Together, these results indicate that photoactivation of LiGluR provides a robust, graded and reliable control of Ca2+ signalling in astrocytes.
Opsin-derived proteins are less efficient than LiGluR in triggering astrocytic Ca2+ rises
Previous reports indicate that ChR2 can activate astrocytes (Gradinaru et al. 2009; Gourine et al. 2010); therefore we decided to compare the ability of ChR2 and LiGluR to elicit Ca2+ elevations in astrocytes.
We first confirmed our ability to activate ChR2-expressing neurons in culture by showing that the repetitive photoactivation of ChR2(H134R) (Nagel et al. 2005) with 458 nm light pulses (27.3 mW mm-2, 500 ms) evoked Ca2+ transients (Fig. 2_A_) in 9 of 14 cortical neurons expressing ChR2(H134R)-GFP. Light-evoked Ca2+ transients were absent in control neurons without ChR2(H134R)-GFP (Fig. 2_B_), and were inhibited by cobalt, a non-selective blocker of voltage-gated Ca2+ channels (VGCCs) (Fig. 2_C_), indicating that neuronal Ca2+ rise is mainly due to a secondary entry via VGCCs.
Figure 2. Ca2+ signalling in astrocytes evoked by photoactivation of the mutant channelrhodopsin ChR2(H134R) and the Ca2+-permeable channelrhodopsin CatCh.
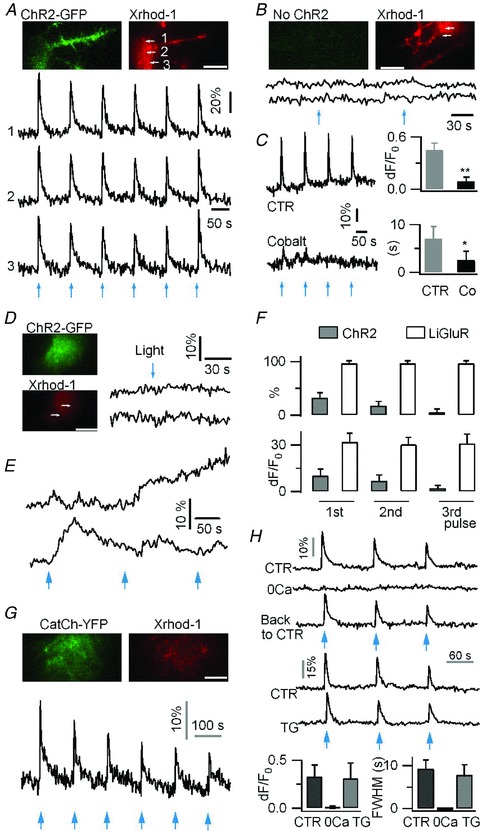
A, in a neuron expressing ChR2(H134R)-GFP and loaded with the red Ca2+ indicator Xrhod-1, 458 nm EPI pulses (27.3 mW mm−2, 500 ms) evoked reproducible Ca2+ rises (Δ_F_/F_0= 46.0 ± 13.2%, n = 9 cells). B, absence of light-evoked Ca2+ increases in neurons not expressing ChR2 (Δ_F/F_0= 1.3 ± 6%, n = 4 cells). C, neuronal ChR2-evoked Ca2+ rises were inhibited by the non-selective voltage-gated Ca2+ channel (VGCC) blocker, cobalt (Co, 1 mm, Δ_F/F_0= 9.2 ± 4.7%, FWHM = 2.5 ± 1.9 s; vs. control Δ_F/_F_0= 45.4 ± 8.3%, P < 0.01, FWHM = 7.0 ± 2.6 s, P < 0.05, n = 8 trials from 4 cells per condition). D, in astrocytes, short photoactivation (458 nm, 27.3 mW mm-2, 500 ms) of ChR2 did not evoke any near-membrane Ca2+ elevation. E, example Ca2+ responses evoked by longer light pulse (458 nm, 1 s) in ChR2-expressing astrocytes. F, percentage of astrocytes showing light-gated Ca2+ rises, and Ca2+ response amplitude in LiGluR-expressing (60/63 cells) and ChR2-expressing (19/60 cells) astrocytes. The comparison was made using three successive light pulses (385 nm, 100 ms for LiGluR; 458 nm, 1 s for ChR2) applied every 150 s. G, in astrocytes, the photoactivation (1 s, 458 nm) of CatCh evokes fast Ca2+ rises which fade upon repetitive photoactivation. H, CatCh-evoked Ca2+ elevations in astrocytes were absent in 0 mm Ca2+ extracellular solution (0 Ca, solution change was achieved in less than 30 s), and were unaffected by depleting the endoplasmic reticulum Ca2+ store by thapsigargin (TG, 2 μm, 20 min). Recordings were done on the same cells in different conditions. CatCh photoactivation, 2 s 458 nm (CTR, 15 trials, 5 cells; 0 Ca, 6 trials, 3 cells; TG, 18 trials, 6 cells). Bars, 10 μm.
We then expressed ChR2(H134R)-GFP in astrocytes. The average ChR2(H134R)-GFP fluorescence intensity was found to be comparable in astrocytes and neurons, suggesting that in our conditions, similar expression levels are achieved in both cell types (Supplemental Fig. 2_A_). The same light intensity used for neurons, however, evoked small Ca2+ changes in cultured cortical astrocytes expressing ChR2(H134R) (Δ_F_/F_0= 5.1 ± 4.2%, n = 5, Fig. 2_D_), which were not significantly different from the control responses of ChR2-non-expressing cells (Δ_F/F_0= 2.2 ± 3.0%, n = 5, P = 0.3). Longer (1 s) 458 nm light pulse occasionally evoked Ca2+ increases in a subset of astrocytes, but responses were variable in duration and amplitude, as imaged with TIRFM (Fig. 2_E_) and epifluorescence (Supplemental Fig. 2_B). In comparison with LiGluR, ChR2(H134R) was significantly less effective in driving astrocytic Ca2+ elevation, in terms of light intensity and duration needed, percentage of responsive cells, and amplitude of light-evoked Ca2+ responses (Fig. 2_F_). Our inability to trigger reliable Ca2+ rises in astrocyte following ChR2(H134R) photoactivation could be due to a malfunction of this channel when expressed in astrocytes. To investigate this possibility, we compared the performance of ChR2(H134R) in astrocytes and neurons. Since ChR2 is highly permeable to H+ (Nagel et al. 2003), the H+ influx induced by its photoactivation is expected to quench the fluorescence of ChR2(H134R)-GFP (Nadrigny et al. 2006). We observed that ChR2(H134R) photoactivation induced a similar degree of quenching of intracellular ChR2(H134R)-GFP fluorescence in astrocytes and neurons (Supplemental Fig. 2_C_). This result indicates that ChR2(H134R) is as well-expressed and functional in astrocytes as it is in neurons. Thus, the relative inefficacy of ChR2 to trigger Ca2+ rise in astrocytes might be due to its low Ca2+ permeability. We therefore tested a recently developed Ca2+-permeable channelrhodopsin mutant called CatCh (Kleinlogel et al. 2011). Ca2+ rises could be triggered in astrocytes by optical activation of CatCh, although this required ∼50 times more power and ∼10 times longer illumination (15.1 mW mm-2, 1 s) than with LiGluR and, unlike the reproducible LiGluR-evoked responses, the CatCh-evoked responses faded in amplitude with repeated stimulation (Fig. 2_G_). Our results indicate that Ca2+-permeable LiGluR and, to a lesser extent, CatCh are more efficient than ChR2 in controlling astrocytic Ca2+ elevation.
We next examined the cellular mechanisms of the Ca2+ increase evoked in astrocytes by the photoactivation of CatCh and LiGluR. CatCh is a light-sensitive Ca2+-permeable ChR2 (Kleinlogel et al. 2011), and CatCh-evoked Ca2+ transients were abolished in 0 mm Ca2+ (Fig. 2_H_). The endoplasmic reticulum (ER) Ca2+ stores contribute to Ca2+ signalling in astrocytes (Agulhon et al. 2008), but do not seem to be involved in CatCh-evoked Ca2+ rises, since the application of thapsigargin (TG, 1 μm), which induced long-lasting cytoplasmic Ca2+ rises reflecting the leak of Ca2+ from ER stores (Δ_F_/_F_0= 71.7 ± 10.4%, n = 3; Fig. 3_D_), had no effect on the subsequent CatCh-evoked Ca2+ rises (Fig. 2_H_). These results suggest that CatCh-evoked Ca2+ transients depend only on Ca2+ influx.
Figure 3. LiGluR-evoked near-membrane Ca2+ rises in astrocytes depend on Ca2+ influx and uptake into the endoplasmic reticulum.
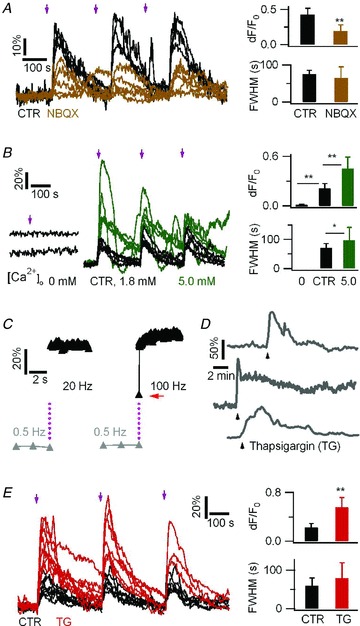
A, LiGluR-evoked Ca2+ responses in the absence (control, 12 trials from 4 cells) and in the presence (15 trials, 5 cells) of a competitive GluR6 antagonist NBQX (1 mm). LiGluR-RFP and the green Ca2+ indicator Fluo-4 were used. B, LiGluR-evoked Ca2+ responses in zero Ca2+ (5 trials, 5 cells, left), 1.8 mm Ca2+ (control, 15 trials from 5 cells, middle, black traces), and 5 mm Ca2+ (15 trials, 5 cells; green traces). C, LiGluR activation triggers a rapid near-membrane Ca2+ increase (grey triangles, 0.5 Hz imaging prior to photoactivation. Black triangles, 20 and 100 Hz imaging after 10 ms activation light pulse, purple dots). An intermediate rising step, indicated by a red arrow, was revealed at 100 Hz. Similar results were obtained for six astrocytes. D, the Ca2+-ATPase inhibitor thapsigargin (TG, 1 μm) led to Ca2+ elevation due to Ca2+ release from the internal store. Black arrowhead indicates the beginning of TG application, which was maintained throughout the recording. Each trace depicts the response of a single astrocyte. E, inhibition of Ca2+-ATPase by TG (1 μm, 15 min) enhanced LiGluR-evoked Ca2+ increases (red, 17 trials, 7 cells, vs. control, black, 19 trials, 8 cells. n.s. P > 0.16).
LiGluR is derived from the kainate-type glutamate receptor GluR6 (Volgraf et al. 2006), and in line with previous reports (Gorostiza et al. 2007; Numano et al. 2009), the competitive AMPA/kainate receptor antagonist NBQX attenuated the LiGluR-evoked Ca2+ increase (Δ_F_/F_0= 19.3 ± 9.7%, n = 12 trials from 4 cells, vs. control, 43.2 ± 8.5%, n = 15 trials from 5 cells, P < 0.01; Fig. 3_A_). As predicted by the high Ca2+ permeability of GluR6 (Egebjerg & Heinemann, 1993), the photoactivation of LiGluR in 0 mm Ca2+ failed to trigger detectable Ca2+ rises (Δ_F/F_0= 0.32 ± 1.4%, n = 5), while the Ca2+ rises evoked in 5 mm extracellular Ca2+ (Δ_F/F_0= 45.2 ± 14.3%, FWHM = 97.1 ± 43.8 s, n = 15 trials from 5 cells) were larger than the control response evoked in 1.8 mm Ca2+ (Δ_F/F_0= 21.6 ± 6.7%, P < 0.01; FWHM = 71.3 ± 14.7 s, P < 0.05; n = 15 trials from 5 cells; Fig. 3_B_). The Ca2+ influx through the ion channel should lead to a rapid rise of the near-membrane Ca2+ concentration detectable with TIRFM (Demuro & Parker, 2005). Sampling LiGluR-evoked Ca2+ rises at 100 Hz, we captured an intermediate Δ_F/F_0 step one frame after light activation (Fig. 3_C_, red arrow), suggesting that the Ca2+ rise occurs within 10 ms after LiGluR activation. We then tested a possible involvement of the Ca2+ stores in LiGluR-evoked Ca2+ signalling using thapsigargin (TG, 1 μm, 15 min) to block the ER Ca2+-ATPase and deplete the Ca2+ stores prior to LiGluR activation. Surprisingly, we found that LiGluR-evoked Ca2+ rises were enhanced in TG-treated cells (Δ_F/_F_0= 56.3 ± 16.8%, n = 17 trials from seven cells, vs. control, 23.4 ± 6.2%, n = 19 trials from 8 cells, P < 0.01; Fig. 3_E_). These results indicate that LiGluR-evoked Ca2+ elevations rely on Ca2+ influx, and that Ca2+ uptake into the ER via the Ca2+-ATPase limits the LiGluR-evoked cytosolic Ca2+ increase.
All optical probing of astrocyte-to-astrocyte communication with LiGluR
Having established LiGluR as a tool for controlling astrocytic Ca2+ in culture, we investigated if LiGluR-evoked Ca2+ increases can trigger gliotransmitter release. After transfection, only a subset of astrocytes express LiGluR, while the entire population of astrocytes can be loaded with acetoxymethyl ester (AM) derivative of Ca2+ indicators (Fig. 4_A_). This permitted us to explore astrocyte-to-astrocyte signalling in an all-optical manner, in which light was used to drive Ca2+ elevation in LiGluR expressing (LiGluR(+)) cells as well as to report Ca2+ signals in both the LiGluR(+) and adjacent LiGluR(−) astrocytes. To avoid interference with spontaneous Ca2+ oscillations, recordings were made from astrocytes without spontaneous activity.
Figure 4. All optical probing of astrocyte-to-astrocyte communication using LiGluR and TIRF.
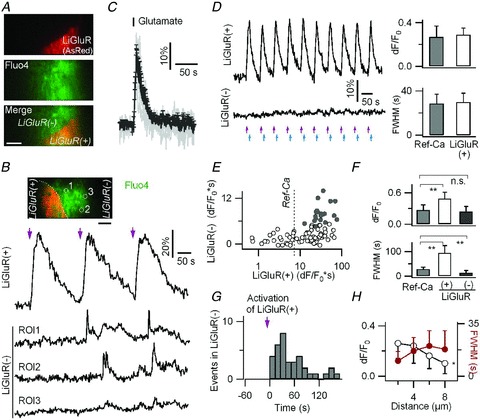
A, AsRed was co-transfected with LiGluR-RFP to identify the border between LiGluR-RFP/AsRed-expressing (LiGluR(+)) and LiGluR-RFP/AsRed-non-expressing (LiGluR(−)) astrocytes. Fluo-4 AM was used to monitor Ca2+ activity in LiGluR(+) and LiGluR(−) astrocytes. B, light-evoked near-membrane Ca2+ rises in a LiGluR(+) astrocyte induced asynchronous short-lasting small-amplitude Ca2+ rises in an adjacent LiGluR(−) astrocyte. Light pulses are indicated by violet arrows. C, astrocytic Ca2+ elevations (individual traces in grey; average ± SD in black) evoked by glutamate application (100 μm, 1 s, 8 cells). The average response was designated as the reference Ca2+ response (Ref-Ca). D, repetitive light activation tuned to evoke Ca2+ rises in a LiGluR(+) astrocyte with peak amplitude (Δ_F_/F_0) and half-duration (FWHM) matching the Ref-Ca (right, P > 0.7) did not trigger measurable near membrane Ca2+ responses in the neighbouring LiGluR(−) astrocyte. E, relative distribution of the integral over time (Δ_F/F*0s) of the light-evoked Ca2+ increases in LiGluR(+) astrocytes and LiGluR(−) astrocytes (85 cell pairs). Filled circles represent pairs in which Ca2+ transients were detected in LiGluR(−) cells (n = 20). The dotted line indicates the integral of the Ref-Ca signal. F, in comparison with the Ref-Ca signal, the LiGluR-evoked Ca2+ rises in LiGluR(+) astrocytes was larger and longer lasting, while the Ca2+ transients detected in LiGluR(−) astrocytes showed a similar amplitude but a shorter duration (19 events, 5 cells; n.s., P = 0.8). G, temporal distribution of the LiGluR-evoked Ca2+ transients detected in LiGluR(−) astrocytes relative to the initiation of photoactivated Ca2+ rises in LiGluR(+) astrocytes (32 events, 14 cells, bi_n_ = 8 s). H, peak amplitude (Δ_F_/_F_0) and half-duration (FWHM) of the LiGluR-evoked Ca2+ transients in LiGluR(−) astrocytes as a function of the distance from the visible interface between LiGluR(+) and LiGluR(−) astrocytes (n = 4–12 per condition). Bars, 5 μm.
Photoactivation of LiGluR(+) was obtained using a UV 385 nm light-emitting diode (LED) and a ×10, NA 0.25 objective. All LiGluR(+) and LiGluR(−) astrocytes within a region of ∼2 mm diameter were exposed to UV light. Simultaneously, Ca2+ signals in both LiGluR(+) cells and LiGluR(−) neighbouring cells were imaged using the ×60, NA 1.45 TIRF objective in a field of view of 50 × 50 μm, divided in two by a dual viewer. We found that the light-gated Ca2+ rises in LiGluR(+) astrocytes triggered asynchronous Ca2+ transients in adjacent LiGluR(−) astrocytes (Fig. 4_B_) in ∼35% of cell pairs (21/60). Tuning the amplitude and duration of the light-evoked Ca2+ increases in the LiGluR(+) astrocytes over a wide range, we found that the threshold Ca2+ integral in the LiGluR(+) astrocyte needed to trigger Ca2+ transients in LiGluR(−) neighbours was larger than a reference Ca2+ signal (Ref-Ca) evoked by a 1 s application of 100 μm glutamate (Fig. 4_C_ and E), a concentration that is in the physiological range of the glutamate encountered by astrocytes in situ (Dzubay & Jahr, 1999). A LiGluR-evoked Ca2+ signal mimicking Ref-Ca did not trigger statistically significant Ca2+ transients in adjacent LiGluR(−) cells (Δ_F_/_F_0= 3.8 ± 3.9%, n = 15, vs. control in non-transfected cell pairs, 0.9 ± 2.3%, n = 5, P = 0.15; Fig. 4_D_).
Unlike the direct LiGluR-evoked Ca2+ signals in the processes of LiGluR(+) astrocytes which were time-locked with the light pulses, the Ca2+ transients detected in adjacent LiGluR(−) astrocytes were local, asynchronous (Fig. 4_B_), smaller in peak amplitude, shorter lasting (Fig. 4_F_), and occurred after a variable delay (36.0 ± 26.2 s, median ± absolute deviation, n = 37 events in 15 cells; Fig. 4_G_). The amplitude and width of the Ca2+ responses in LiGluR(−) astrocytes also varied with the distance from the boundary between two cells (Fig. 4_H_). Such localized astrocytic Ca2+ signals resemble those seen in situ (Wang et al. 2006; Gordon et al. 2009; Agulhon et al. 2010), suggesting that LiGluR-evoked Ca2+ rises in cultured astrocytes mediate a chemical communication to adjacent LiGluR(−) astrocytes.
LiGluR-evoked astrocyte-to-astrocyte communication is due to glutamate release
Having observed what appears to be a LiGluR-evoked signalling between adjacent astrocytes, we sought to identify the cellular mechanism underlying this astrocyte-to-astrocyte communication.
Second messengers such as IP3 or Ca2+ are suggested to diffuse between astrocytes through gap junctions (Scemes & Giaume, 2006). The gap junction blocker carbonoxolone had no effect on the light-evoked Ca2+ rises in LiGluR(+) astrocytes (Supplemental Table 2), and did not interfere with the Ca2+ transients in adjacent LiGluR(−) cells (Fig. 5_A_), suggesting that the LiGluR-evoked astrocyte-to-astrocyte signal is not due to gap junction communication but to the release of a transmitter into the external medium.
Figure 5. LiGluR-evoked astrocyte-to-astrocyte communication is mediated by glutamate release.
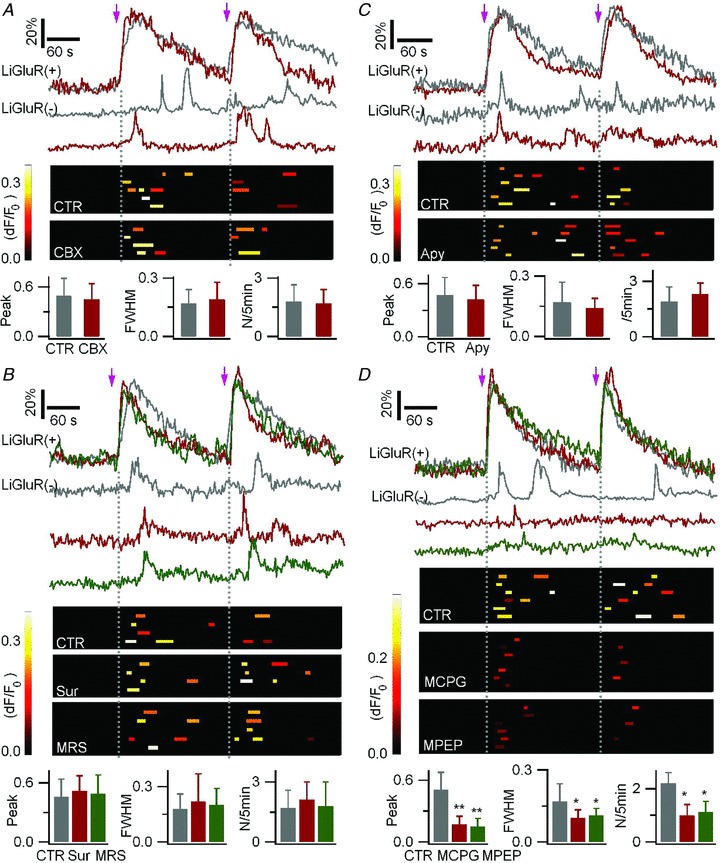
To monitor Ca2+ activity in LiGluR(+) and LiGluR(−) cells, astrocytes were co-transfected with LiGluR-RFP and AsRed, and loaded with Fluo-4 AM. A, the gap junction blocker carbenoxolone (CBX) had no effect on the LiGluR-evoked Ca2+ rises in LiGluR(−) astrocytes. Light pulses are indicated by arrows. Top, Δ_F_/_F_0 sample traces in a LiGluR(+) cells and an adjacent LiGluR(−) cells in control (CTR, grey traces) and in carbenoxolone (CBX, 100 μm, red traces). Middle, summary plots of the near-membrane Ca2+ rises detected in LiGluR(−) cells. Each row represents a single cell, each bar represents a single event (larger than 3SD of the background noise), its location on the x_-axis reflects its timing, bar length reflects its half-duration (FWHM), and its peak amplitude (Δ_F/_F_0) is encoded in pseudocolour. Bottom, summary data of the relative peak amplitude and FWHM, both normalized to the corresponding Ca2+ rises in LiGluR(+) cells, and the occurrence frequency (N/5 min) of the Ca2+ rises in LiGluR(−) astrocytes in CTR (12 events, 5 cells) and CBX (9 events, 4 cells; P > 0.6). B, top, sample traces in control (grey traces), in the presence of the P2 purinergic receptor antagonist suramin (Sur, 100 μm, red traces), and P2Y1 receptor antagonist MRS2179 (MRS, 25 μm, green traces). Middle, summary plots of the Ca2+ events in LiGluR(−) cells as defined in A. Bottom, summary data in control (n = 9 events, 4 cells), suramin (11 events, 4 cells), and MRS2179 (12 events, 5 cells; P > 0.23). C, apyrase (20 U ml−1) did not inhibit the LiGluR-evoked Ca2+ transients in the LiGluR(−) cells. Sample traces, and summary plots of the evoked Ca2+ transients in control (15 events, 6 cells), and in apyrase (Apy, n = 16 events, 5 cells; P > 0.26). D, LiGluR-evoked Ca2+ transients in the LiGluR(−) cells in control (n = 18 events, 6 cells; grey traces) were reduced in amplitude, duration, and frequency by the group I/II mGluR antagonist MCPG (500 μm, 9 events, 6 cells; red traces) and the mGluR5 antagonist MPEP (100 μm, 9 events, 7 cells; green traces).
ATP, a gliotransmitter candidate, induces type-2 purinergic receptor (P2R)-mediated Ca2+ rises in astrocytes (Fam et al. 2000). To test the possibility of LiGluR-evoked ATP release, we studied the effect of P2R antagonists on the LiGluR-evoked Ca2+ transients in LiGluR(−) astrocytes. As the P2R antagonist PPADS interfered with the LiGluR-evoked Ca2+ rises in LiGluR(+) astrocytes (Supplemental Table 2), we tested another P2R antagonist, suramin (100 μm), and a P2Y1 receptor antagonist, MRS2179 (25 μm), which both reduced the ATP-evoked Ca2+ rises (Supplemental Fig. 3_A_), without having an effect on LiGluR-evoked Ca2+ signals in LiGluR(+) astrocytes (Supplemental Table 2). Both antagonists had no effect on the LiGluR-evoked Ca2+ transients in LiGluR(−) astrocytes (Fig. 5_B_). We also tested the effect of the ectonucleotidase apyrase, which degrades extracellular ATP and inhibits purinergic signalling in astrocytes (Stout et al. 2002; Bowser & Khakh, 2007). Control experiments showed that apyrase had no effect on the LiGluR-evoked Ca2+ rise in LiGluR(+) cells (Supplemental Table 2). We found that apyrase did not alter the Ca2+ transients in LiGluR(−) astrocytes (Fig. 5_C_). Together, these results indicate that LiGluR-evoked astrocyte-to-astrocyte communication is not due to ATP release.
Glutamate, another gliotransmitter candidate, activates metabotropic glutamate receptors (mGluR) and mediates Ca2+ rise in astrocytes (Gordon et al. 2009; Sasaki et al. 2011). The group I mGluR5 mediates astrocytic Ca2+ elevation via the phospholipase C (PLC) IP3 pathway (Cai et al. 2000; Fiacco et al. 2009). To test the possibility of LiGluR-evoked glutamate release, we used a wide-spectrum antagonist, MCPG, to block the group I/II mGluRs, and MPEP, a mGluR5 antagonist. Neither MCPG nor MPEP had an effect on LiGluR-evoked Ca2+ signals in LiGluR(+) astrocytes (Supplemental Table 2). Both, applied separately, reduced LiGluR-evoked transient Ca2+ rises in LiGluR(−) astrocytes (Fig. 5_D_). Altogether, these results suggest that LiGluR-evoked Ca2+ transients in LiGluR(−) astrocytes are mediated by glutamate release from LiGluR(+) astrocytes, and they establish LiGluR as a potent tool to study the mechanisms of intercellular communication between electrically silent cells such as the protoplasmic astrocytes.
Exocytosis is not a major route for LiGluR-evoked glutamate release by astrocyte
We next decided to investigate possible pathways of LiGluR-evoked glutamate release, such as reverse transport (Rossi et al. 2000), vesicular release (Hamilton & Attwell, 2010), hemichannels (Ye et al. 2003), and anion channels (Takano et al. 2005; Kimelberg et al. 2006; Park et al. 2009).
The possible involvement of a glutamate transporter (Rossi et al. 2000) is unlikely since we found no effect of the transporter blocker TBOA on LiGluR-evoked Ca2+ transients in LiGluR(−) astrocytes (Supplemental Fig. 3_B_; Supplemental Table 2).
We next investigated the possibility of Ca2+-regulated exocytosis (Hamilton & Attwell, 2010). We previously showed that astrocytic lysosomes undergo asynchronous Ca2+-regulated exocytosis (Li et al. 2008), and recent findings suggest that the lysosomal H+/sialic acid cotransporter, sialin, is a vesicular aspartate/glutamate transporter (Miyaji et al. 2008), raising the possibility of LiGluR-evoked glutamate release by lysosomal exocytosis. To test this possibility, we used the styryl dye FM4–64 to monitor lysosomal exocytosis (Zhang et al. 2007; Li et al. 2008) in LiGluR-transfected astrocytes. Control experiments showed that LiGluR photoactivation evoked similar Ca2+ rises in LiGluR(+) astrocytes in control and after FM4–64 loading (Δ_F_/F_0= 47.6 ± 5.1%, FWHM = 65.3 ± 6.1 s, vs. control, Δ_F/F_0= 49.4 ± 6.8%, FWHM = 50.3 ± 9.5 s, n = 6 trials from 3 cells, P > 0.14; Fig. 6_Aa_). We found that the LiGluR-evoked Ca2+ had no effect on the fluorescence of FM4–64-labelled lysosomes (Δ_F/_F_0, −1.1 ± 4.5%, n = 23 lysosomes from 5 LiGluR(+) cells, vs. control, 0.9 ± 2.6%, n = 12 lysosomes from 3 LiGluR(−) cells, P = 0.6; Fig. 6_A_), indicating that lysosomal exocytosis is not a likely pathway for LiGluR-evoked glutamate release.
Figure 6. Exocytosis is not the pathway for LiGluR-evoked glutamate release by astrocytes.
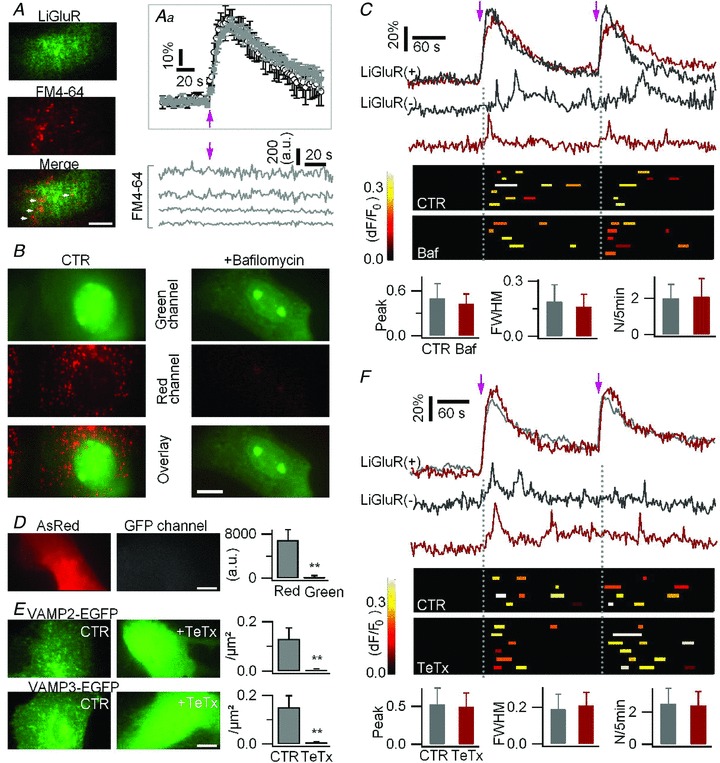
A, astrocytes were transfected with LiGluR-GFP and loaded with the red-fluorescent lysosomal marker FM4–64. Inset (Aa) average LiGluR-evoked Ca2+ rises were similar in control (black trace) and FM4–64-loaded cells (grey trace; n = 6 trials, 3 cells for each). Bottom right, fluorescence intensity of the FM6–64 loaded lysosomal vesicles was unchanged upon LiGluR photoactivation. B, inhibition of V-ATPase activity by bafilomycin A1 (100 nm, 1 h) abolished the vesicular accumulation (red channel, epifluorescence) of acridine orange (15 μm, 15 min). C, LiGluR-evoked Ca2+ rises in LiGluR(−) astrocytes were unchanged in cells treated with bafilomycin (Baf) for 1 h. Astrocytes were co-transfected with LiGluR–RFP and AsRed, and loaded with Fluo-4 AM. Top, sample traces (grey, control; red, bafilomycin). Middle, summary plots of the LiGluR-evoked Ca2+ events in LiGluR(−) cells. Bottom, the relative peak amplitude, FWHM, and frequency (N/5 min) of the Ca2+ transients (16 events, 6 cells in control; 14 events, 5 cells in bafilomycin, P > 0.39). D, transfection of TeTx in astrocytes did not affect the GFP imaging channel. Right, fluorescence counts in red and green channels, respectively (n = 10 cells). AsRed was co-transfected to localize the transfected cell. E, transfection of TeTx in astrocytes abolished the vesicular targeting of VAMP2-EGFP and VAMP3-EGFP. Right, puncta density (n = 16–19 cells for each). F, LiGluR-evoked near-membrane Ca2+ transients in LiGluR(−) cells were unchanged in astrocytes co-transfected with TeTx (grey, control, 15 events, 4 cells; red, TeTx, 19 events, 6 cells; P > 0.32). Bars, 10 μm.
Astrocytes contain small synaptic-like vesicles which are suggested to carry glutamate vesicular transporters and undergo Ca2+-dependent exocytosis (Bezzi et al. 2004; Hamilton & Attwell, 2010). Both lysosomes and small synaptic-like vesicles are acidic compartments. To test a possible involvement of all these acidic vesicles in the LiGluR-evoked glutamate release, we used the V–ATPase inhibitor bafilomycin A1 to disrupt the vesicular proton gradient (Bowman et al. 1988; Zhou et al. 2000; Angulo et al. 2004; Fiacco et al. 2007). Since long-term bafilomycin treatment can cause cell death (Nakashima et al. 2003), we made control experiments measuring mitochondrial fragmentation, a sign of cell death (Frank et al. 2001) (Supplemental Fig. 4_A_). We found that a limited (1 h) treatment, which affects minimally the mitochondria, is sufficient to abolish the vesicular pH gradient, as shown by the disruption of vesicular accumulation of the fluorescent weak base acridine orange (Fig. 6_B_). This bafilomycin treatment affected neither the LiGluR-evoked Ca2+ rise in LiGluR(+) astrocytes (Supplemental Table 2), nor the LiGluR-evoked Ca2+ transients in LiGluR(−) astrocytes (Fig. 6_C_), arguing against an involvement of acidic compartments in LiGluR-evoked glutamate release.
To further investigate a possible involvement of the synaptic-like small vesicles in LiGluR-evoked glutamate release, we used tetanus toxin (TeTx), an exocytosis blocker which cleaves VAMP2 (synaptobrevin II) and VAMP3 (cellubrevin) (McMahon et al. 1993) v-SNARE proteins suggested to be expressed by small astrocytic vesicles (Hamilton & Attwell 2010). Since bath application of TeTx did not cleave effectively the v-SNARE proteins as shown by the residual vesicular targeting of VAMP2–enhanced green fluorescent protein (EGFP) and VAMP3-EGFP (Supplemental Fig. 4_B_), we transfected astrocytes with the TeTx light chain to abolish the v-SNARE vesicular targeting (Fig. 6_D_–E). Astrocytes co-transfected with TeTx and LiGluR responded normally to light activation (Supplemental Table 2), and LiGluR-evoked Ca2+ signals in LiGluR(−) astrocytes were unchanged (Fig. 6_F_), suggesting that VAMP2/3-dependant exocytosis is not involved in LiGluR-evoked glutamate release from astrocytes. Altogether these results indicate that the LiGluR-evoked glutamate release in astrocytes does not involve the exocytosis of either lysosomes or of small synaptic-like vesicles.
LiGluR-evoked glutamate release from astrocyte via anion channels
Previous experiments suggest that glutamate release from astrocytes can also be mediated by non-selective hemichannels (Ye et al. 2003) and anion channels (Takano et al. 2005; Kimelberg et al. 2006; Park et al. 2009).
We first studied the effect of LiGluR activation on the fluorescence of calcein, a hemichannel-permeant dye (Stout et al. 2002; Thompson et al. 2006). Control experiments showed that mechanical stimulation without membrane rupture (Li et al. 2008) induced significant calcein release as expected from hemichannel opening (Stout et al. 2002) (−26.3 ± 7.9% in 20 s, n = 5; Supplemental Fig. 5_A_), and that LiGluR-evoked Ca2+ elevations in LiGluR(+) astrocytes were similar in control and in calcein-loaded astrocytes (Fig. 7_Aa_). We showed that the calcein fluorescence in LiGluR(+) astrocytes was unchanged by the LiGluR-evoked Ca2+ elevation (Δ_F_/_F_0=−2.2 ± 3.6%, vs. −0.6 ± 1.2% in control cells without LiGluR, n = 7, P = 0.33; Fig. 7_A_). These results, together with the lack of effect of carbenoxolone (Fig. 5_A_), argue against the idea of LiGluR-evoked hemichannel-mediated glutamate release in astrocytes.
Figure 7. An anion channel mediates LiGluR-evoked glutamate release by astrocytes.
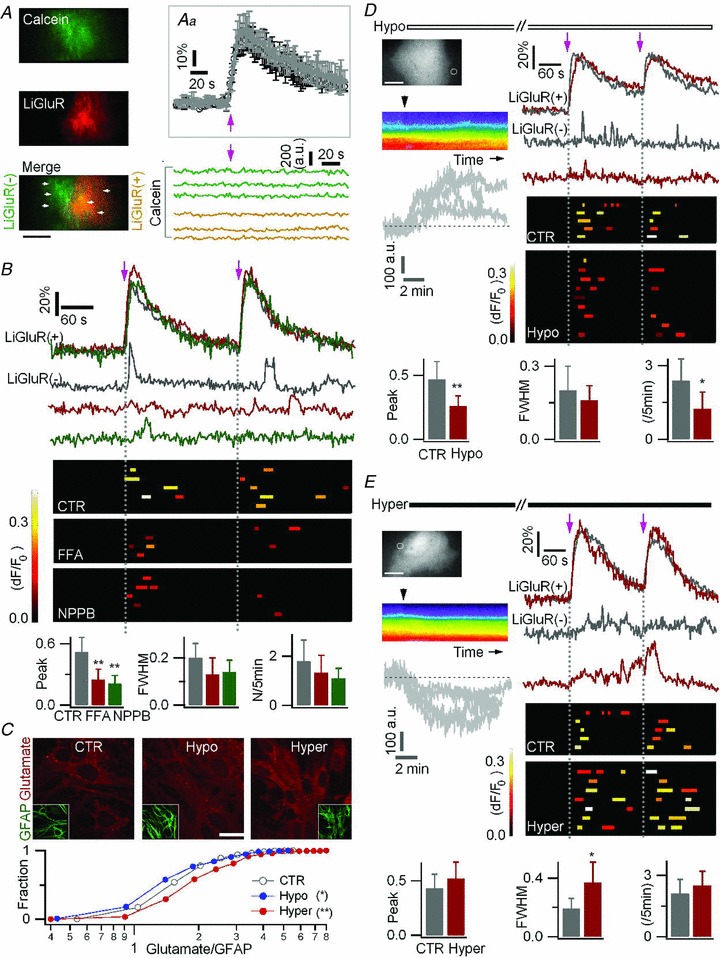
A, astrocytes were co-transfected with LiGluR-RFP and AsRed, and loaded with calcein AM. Inset (Aa), similar LiGluR-evoked Ca2+ rises in LiGluR(+) astrocytes in the absence (average black trace, n = 6) and in the presence of calcein (average grey trace, n = 6). Bottom, LiGluR photoactivation (arrow) left the calcein fluorescence stable in the ROIs (left panel, arrowhead, 2 μm × 2 μm) in LiGluR(+) cells (green traces) and LiGluR(−) cells (yellow traces). Bar, 5 μm. B, astrocytes were co-transfected with LiGluR-RFP and AsRed, and loaded with Fluo-4 AM. The LiGluR-evoked Ca2+ transients in LiGluR(−) astrocytes were reduced by the anion channel blockers, flufenamic acid (FFA, 50 μm, red) and 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB, 100 μm, green). Control (CTR, grey traces, 12 events, 5 cells). FFA (red traces, 7 events, 4 cells). NPPB (green traces, 7 events, 5 cells. C, astrocytes were immunostained with glutamate (red) and GFAP (green) antibodies, and the glutamate/GFAP fluorescence intensity ratio was plotted as a cumulative histogram. Intracellular glutamate immunostaining was altered by 20 min treatment of hypo- (blue) or hyper-tonic solutions (red) (n = 244–606 regions from 3 trials for each condition, KS test). Bar, 10 μm. D–E, effect of hypo-osmotic (−25 mm NaCl) and hyper-osmotic (+50 mm sucrose; 15–20 min) solutions on LiGluR-evoked Ca2+ transients in LiGluR(−) astrocytes co-transfected with LiGluR-RFP and AsRed, and loaded with Fluo-4 AM. Left, pseudocolour kymographs show the transient cell swelling or shrinking, respectively, following the change of osmotic pressure (black arrowhead, application time) on GFP-expressing astrocytes. The same result is obtained when plotting the fluorescence change in ROIs lining the outer/inner side of the cell border (grey traces, n = 4 cells each). Right, sample traces and summary plots of LiGluR-evoked Ca2+ transients in LiGluR(−) astrocytes. Bottom, statistical comparison of LiGluR-evoked Ca2+ transients in control (CTR, 14 events, 5 cells), hypotonic (Hypo, 16 events, 9 cells), and hypertonic (Hyper, 23 events, 7 cells) solutions. Bars, 10 μm.
We next turned to anion channel blockers, flufenamic acid (FFA) and 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB), which did not significantly affect LiGluR-evoked Ca2+ rise in LiGluR(+) astrocytes (Supplemental Table 2). However, each blocker caused a reduction of the LiGluR-evoked Ca2+ signalling in LiGluR(−) astrocytes (Δ_F_/F_0= 0.25 ± 0.1 for FFA, 0.21 ± 0.11 for NPPB, vs. control, 0.52 ± 0.14, n = 7–12 events, P < 0.01; Fig. 7_B_). Hypotonic solutions induce cell swelling, anion channel opening, and enhance glutamate release, the hypertonic solutions having opposite effects (Takano et al. 2005; Kimelberg et al. 2006; Duran et al. 2010). As shown previously (Benfenati et al. 2011), hypotonic (−17%) solution induced Ca2+ rises in astrocytes (Supplemental Fig. 5_B), and therefore we could not perform competition experiments and submit simultaneously the astrocytes to osmotic shock and LiGluR photoactivation. However, we argued that if LiGluR-evoked glutamate release involves a channel, osmotic shocks should change the amount of glutamate available for release. Using immunostaining (Gorg et al. 2010), we first confirmed that a 20 min treatment with hypotonic (−17%) solution led to a reduction of intracellular glutamate content, an opposite effect being induced by hypertonic (+23%) solution (Fig. 7_C_). We estimated the LiGluR-evoked astrocyte-to-astrocyte communication in cells challenged by a change of osmotic pressure, when osmotically altered Ca2+ levels had stabilized, and the LiGluR-evoked Ca2+ rise in LiGluR(+) cells were normal (Supplemental Table 2). Interestingly, the LiGluR-evoked Ca2+ transients in LiGluR(−) astrocytes were reduced by the hypotonic solution treatment (Fig. 7_D_), and enhanced by the hypertonic solution treatment (Fig. 7_E_), as expected if release occurs by efflux of glutamate from the cytosol. Altogether, these results suggest that LiGluR-evoked glutamate release involves a glutamate-permeable anion channel.
Discussion
Here, we show that photoactivation of the light-gated Ca2+-permeable channels, LiGluR and CatCh, unlike ChR2, evokes Ca2+ signals in cultured cortical astrocytes. Using evanescent-field excitation for near-membrane Ca2+ imaging and epifluorescence for LiGluR activation and inactivation, we could precisely and reliably control the amplitude and duration of the LiGluR-evoked Ca2+ rise. We also show that the photoactivation of LiGluR triggered astrocyte-to-astrocyte communication due to anion channel-mediated glutamate release. These results demonstrate the power of LiGluR, in association with all-optical tools, to control Ca2+ activity and to monitor downstream events in electrically silent cells. Finally, the light activation with genetically encoded proteins holds the promise for a specific remote control of astrocyte activity in situ.
Low-intensity (0.3 mW mm-2 for tens of milliseconds) light pulses reliably trigger Ca2+ rise in astrocytes expressing LiGluR. The CatCh photoactivation could also induce Ca2+ rise, although it required an order of magnitude more power and longer illumination (15.1 mW mm-2, 1 s), and unlike the stable LiGluR-evoked responses, the CatCh-evoked responses faded in amplitude upon repeated stimulation. Unlike LiGluR and CatCh, ChR2(H134R), even using pulses of higher intensity (27.3 mW mm-2, up to 1 s), which activate neurons, failed to evoke substantial Ca2+ rise in astrocytes. In line with a previous study (Zhang & Oertner, 2007), we found that in neurons, the ChR2-evoked Ca2+ transients are mostly due to the secondary activation of VGCCs. In astrocytes we found that ChR2 photoactivation is unable to generate secondary Ca2+ since astrocytes have low input resistance and express little VGCCs (Verkhratsky & Steinhäuser, 2000). Hence, the inability of ChR2 to evoke reliable Ca2+ signals in astrocytes results probably from its relatively weak Ca2+ permeability. Previous work in oocytes showed that ChR2 behaves as a Ca2+ permeable channel; however the experiments were conducted in elevated 80 mm Ca2+, and the current measured in the presence of BAPTA to inactivate a Ca2+-sensitive chloride current was much smaller than the current measured in 110 mm Na+ (Nagel et al. 2003). Moreover, lowering the external Ca2+ concentration from 80 mm to 20 mm or 2 mm made the ChR2-gated Ca2+ rises in HEK cells much reduced and less reliable (Lin et al. 2009). Finally, the relative Ca2+ permeability of ChR2 (_P_Ca/_P_Na∼0.12) estimated in 20 mm Ca2+ (Lin et al. 2009) is relatively weak, in comparison with LiGluR (_P_Ca/_P_mono∼1.2) (Egebjerg & Heinemann, 1993) and CatCh (_P_Ca/_P_Na∼0.24) (Kleinlogel et al. 2011). The ChR2 is mainly permeable to H+ (_P_H/_P_Na∼1.06 × 106) (Nagel et al. 2003; Lin et al. 2009), as confirmed in our H+-GFP quenching experiments. The LiGluR-mediated Ca2+ rises we have seen in cultured cortical astrocytes are reliable and temporally well controlled. This contrasts with the variable ChR2-gated Ca2+ signals seen in brainstem astrocytes, which require long-lasting (tens of seconds) illumination (Gourine et al. 2010). The differences between our observations and those of Gourine et al. could be attributed to their longer illumination and to specialized features of their brainstem astrocytes that responded with a Ca2+ rise to a gentle acidification (pH 7.4 to 7.2), a feature not found for cortical astrocytes (Gourine et al. 2010). Interestingly, CatCh-evoked Ca2+ signals, like near-membrane spontaneous Ca2+ events (Shigetomi et al. 2010), depend only on the Ca2+ influx. In contrast, LiGluR-evoked signals, like near-membrane ATP-evoked events (Shigetomi et al. 2010), involve both Ca2+ influx and Ca2+ stores. Our results indicate LiGluR and CatCh as useful tools to investigate Ca2+-regulated cellular responses in astrocytes and possibly in other non-excitable cells.
Astrocytes in culture release gliotransmitters in a Ca2+-dependent manner which shares similarities with astrocyte-to-neuron communication in situ (Halassa & Haydon, 2010). The first demonstration of exocytosis by small synaptic-like vesicle carrying a vesicular glutamate transporter came from cultured astrocytes (Bezzi et al. 2004), and therefore, although astrocytes in culture differ from their in situ counterparts (Cahoy et al. 2008), they stand as a good model to establish new tools and to study the cellular and molecular mechanisms of gliotransmitter release. Here we show that the LiGluR-evoked Ca2+ rises evoke Ca2+ transients in adjacent LiGluR(−) astrocytes which are consistent with a chemical transmission between LiGluR(+) and LiGluR(−) astrocytes. Unlike the stereotyped light-gated Ca2+ rises in LiGluR(+) astrocytes, the Ca2+ transients in LiGluR(−) cells have a small amplitude, relatively short duration, and, unlike the glutamate-evoked Ref-Ca responses, a variable latency probably due to a sluggish coupling between the Ca2+ rise and the activation of the release pathway in LiGluR(+) cells. Combining pharmacological dissections with the all-optical investigation, we show that LiGluR photoactivation induces glutamate release to activate mGluR receptor in neighbouring astrocytes.
Among the mechanisms of glutamate release by astrocytes, a fast Ca2+-regulated exocytosis which shares features with neurotransmitter release was proposed as a major pathway (Bezzi et al. 2004; Hamilton & Attwell, 2010; Parpura et al. 2011). Nonetheless, we showed that astrocytes handle the exo/endo-cytotic marker FM4–64 differently from neurons (Li et al. 2009), and that mechanical stimulation triggers asynchronous Ca2+-dependant exocytosis of sialin-positive lysosomes (Li et al. 2008). Here we show that LiGluR photoactivation triggers astrocyte-to-astrocyte communication which operates on a slow time base, is insensitive to blockers of vesicular release, bafilomycin and TeTx (Hamilton & Attwell, 2010), and does not induce FM4–64-labelled lysosomal exocytosis. These results indicate that the exocytosis of neither small synaptic-like vesicles nor lysosomes is involved in LiGluR-evoked glutamate release from astrocytes in culture.
Using hemichannel and glutamate transporter blockers, we also exclude hemichannels and glutamate transporters as possible pathways for LiGluR-evoked glutamate release. Our experiments using anion channel blockers and treatments with hypo/hypertonic solutions suggest anion channels as the pathway for the LiGluR-gated glutamate release. Glutamate-permeable channels in astrocytes include volume-regulated anion channels and Ca2+-activated chloride channels (Takano et al. 2005; Kimelberg et al. 2006; Park et al. 2009). Identifying among the cloned channels (Duran et al. 2010) the channel that mediates the LiGluR-evoked glutamate release will require new experiments.
The LiGluR-evoked astrocyte-to-astrocyte communication needs relatively long lasting Ca2+ rises, raising the question of the functional significance of our results. Similar Ca2+ signals were observed in situ in conditions such as Alzheimer's disease (Kuchibhotla et al. 2009), epilepsy (Ding et al. 2007; Gomez-Gonzalo et al. 2010), and NMDA receptor-mediated slow inward currents (SICs) (Fellin et al. 2004; Shigetomi et al. 2008). LiGluR-evoked Ca2+ rises that mimic the response to a physiological application of glutamate failed to evoke glutamate-mediated astrocyte-to-astrocyte communication. More experiments in situ will be needed to clarify whether astrocyte glutamate release is relevant mainly to pathological conditions, or whether smaller Ca2+ transients evoked by LiGluR in the very thin astrocytic processes present in situ will be sufficient to mediate astrocyte-to-astrocyte and astrocyte-to-neuron communications in physiological conditions.
The opsins offer the advantage of working without introduction of the photochrome. This makes them easier to use but the lack of efficacy and reproducibility to activate astrocytes can be a problem since it might require toxic levels of light. The light intensity used to induce Ca2+ rise in astrocytes with ChR2 (Gourine et al. 2010) might induce a local change in temperature of the order of ∼5°C according to the quantitative estimation made by Yizhar et al. (2011). Since the MAG photoswitch can be introduced to LiGluR in vivo as shown in the zebrafish spinal cord and the mouse eye (Szobota et al. 2007; Wyart et al. 2009; Caporale et al. 2011), and the LiGuR-mediated Ca2+ elevation requires low light intensity, LiGluR appears as a better tool than the ChR2 to use for the astrocytes in situ.
In conclusion, we show that the Ca2+-permeable LiGluR and CatCh extend our repertoire of tools to control astrocyte activity and to study downstream cellular events regulated by Ca2+ elevation. Opsin-derived proteins and LiGluR-based approaches control very efficiently neuronal activity in vitro and in situ (Janovjak et al. 2010; Szobota & Isacoff, 2010; Fenno et al. 2011). Our results in the present study show that LiGluR allows us not only to efficiently shape the Ca2+ elevations, but also to image the Ca2+ activity of electrically silent cells using highly sensitive TIRFM, allowing a better understanding of astrocytic signalling.
Acknowledgments
We thank Patrice Jegouzo for technical support, Florent Jean for administrative assistance, Ernst Bamberg (MPI für Biophysik, Frankfurt, Germany) for the CatCh-YFP plasmid. We also thank Etienne Audinat for discussion and comments of an earlier version of the manuscript. Confocal imaging was performed at the Paris Descartes imaging plateform. The work was supported by the European Union (FP6-STRP no. 037897-AUTOSCREEN, FP7-ERA-NET no. 006-03-NANOSYN), and the Agence Nationale de la Recherche (ANR PNANO 05-051, ANR P3N 09-044-02). D.L. acknowledges post-doctoral funding from Ecole des Neurosciences de Paris Ile-de-France (ENP), EU and ANR. E.Y.I. received funding from the National Institutes of Health Nanomedicine Development Center for the Optical Control of Biological Function (PN2EY018241) and the ENP sabbatical programme.
Glossary
Abbreviations
CatCh
Ca2+ translocating channelrhodopsin
ChR2
channelrhodopsin 2
EPI
epifluorescence
ER
endoplasmic reticulum
FWHM
full-width at half-maximum
GFAP
glial fibrillary acidic protein
LiGluR
light-gated glutamate receptor 6
MAG
maleimide-azobenzene-glutamate
ROI
region of interest
SIC
slow inward current
TeTx
tetanus toxin
TIRF(M)
total internal reflection fluorescence (microscopy)
VGCC
voltage-gated calcium channel
Author contributions
D.L., E.Y.I., M.O. and N.R. designed the research; K.H. prepared cell culture and performed the fluorescence immunostaining; D.L. performed the experiments and analysed the data; D.L., E.Y.I., M.O. and N.R. wrote the manuscript. Experiments were done at Paris Descartes university. All authors have approved the final version for publication.
References
- Agulhon C, Fiacco TA, McCarthy KD. Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signaling. Science. 2010;327:1250–1254. doi: 10.1126/science.1184821. [DOI] [PubMed] [Google Scholar]
- Agulhon C, Petravicz J, McMullen AB, Sweger EJ, Minton SK, Taves SR, Casper KB, Fiacco TA, McCarthy KD. What is the role of astrocyte calcium in neurophysiology? Neuron. 2008;59:932–946. doi: 10.1016/j.neuron.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson M, Hanse E. Astrocytes impose postburst depression of release probability at hippocampal glutamate synapses. J Neurosci. 2010;30:5776–5780. doi: 10.1523/JNEUROSCI.3957-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci. 2004;24:6920–6927. doi: 10.1523/JNEUROSCI.0473-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati V, Caprini M, Dovizio M, Mylonakou MN, Ferroni S, Ottersen OP, Amiry-Moghaddam M. An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc Natl Acad Sci U S A. 2011;108:2563–2568. doi: 10.1073/pnas.1012867108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi P, Gundersen V, Galbete JL, Seifert G, Steinhauser C, Pilati E, Volterra A. Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat Neurosci. 2004;7:613–620. doi: 10.1038/nn1246. [DOI] [PubMed] [Google Scholar]
- Bowman EJ, Siebers A, Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci U S A. 1988;85:7972–7976. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowser DN, Khakh BS. Vesicular ATP is the predominant cause of intercellular calcium waves in astrocytes. J Gen Physiol. 2007;129:485–491. doi: 10.1085/jgp.200709780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Schools GP, Kimelberg HK. Metabotropic glutamate receptors in acutely isolated hippocampal astrocytes: developmental changes of mGluR5 mRNA and functional expression. Glia. 2000;29:70–80. doi: 10.1002/(sici)1098-1136(20000101)29:1<70::aid-glia7>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Caporale N, Kolstad KD, Lee T, Tochitsky I, Dalkara D, Trauner D, Kramer R, Dan Y, Isacoff EY, Flannery JG. LiGluR restores visual responses in rodent models of inherited blindness. Mol Ther. 2011;19:1212–1219. doi: 10.1038/mt.2011.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro A, Parker I. “Optical patch-clamping”: single-channel recording by imaging Ca2+ flux through individual muscle acetylcholine receptor channels. J Gen Physiol. 2005;126:179–192. doi: 10.1085/jgp.200509331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, Coulter DA, Carmignoto G, Haydon PG. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J Neurosci. 2007;27:10674–10684. doi: 10.1523/JNEUROSCI.2001-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran C, Thompson CH, Xiao Q, Hartzell HC. Chloride channels: often enigmatic, rarely predictable. Annu Rev Physiol. 2010;72:95–121. doi: 10.1146/annurev-physiol-021909-135811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzubay JA, Jahr CE. The concentration of synaptically released glutamate outside of the climbing fiber-Purkinje cell synaptic cleft. J Neurosci. 1999;19:5265–5274. doi: 10.1523/JNEUROSCI.19-13-05265.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egebjerg J, Heinemann SF. Ca2+ permeability of unedited and edited versions of the kainate selective glutamate receptor GluR6. Proc Natl Acad Sci U S A. 1993;90:755–759. doi: 10.1073/pnas.90.2.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fam SR, Gallagher CJ, Salter MW. P2Y1 purinoceptor-mediated Ca2+ signaling and Ca2+ wave propagation in dorsal spinal cord astrocytes. J Neurosci. 2000;20:2800–2808. doi: 10.1523/JNEUROSCI.20-08-02800.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–743. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Fenno L, Yizhar O, Deisseroth K. The development and application of optogenetics. Annu Rev Neurosci. 2011;34:389–412. doi: 10.1146/annurev-neuro-061010-113817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiacco TA, Agulhon C, McCarthy KD. Sorting out astrocyte physiology from pharmacology. Annu Rev Pharmacol Toxicol. 2009;49:151–174. doi: 10.1146/annurev.pharmtox.011008.145602. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, Agulhon C, Taves SR, Petravicz J, Casper KB, Dong X, Chen J, McCarthy KD. Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron. 2007;54:611–626. doi: 10.1016/j.neuron.2007.04.032. [DOI] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- Gomez-Gonzalo M, Losi G, Chiavegato A, Zonta M, Cammarota M, Brondi M, Vetri F, Uva L, Pozzan T, de Curtis M, Ratto GM, Carmignoto G. An excitatory loop with astrocytes contributes to drive neurons to seizure threshold. PLoS Biol. 2010;8:e1000352. doi: 10.1371/journal.pbio.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GR, Iremonger KJ, Kantevari S, Ellis-Davies GC, MacVicar BA, Bains JS. Astrocyte-mediated distributed plasticity at hypothalamic glutamate synapses. Neuron. 2009;64:391–403. doi: 10.1016/j.neuron.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorg B, Morwinsky A, Keitel V, Qvartskhava N, Schror K, Haussinger D. Ammonia triggers exocytotic release of L-glutamate from cultured rat astrocytes. Glia. 2010;58:691–705. doi: 10.1002/glia.20955. [DOI] [PubMed] [Google Scholar]
- Gorostiza P, Volgraf M, Numano R, Szobota S, Trauner D, Isacoff EY. Mechanisms of photoswitch conjugation and light activation of an ionotropic glutamate receptor. Proc Natl Acad Sci U S A. 2007;104:10865–10870. doi: 10.1073/pnas.0701274104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Kasymov V, Marina N, Tang F, Figueiredo MF, Lane S, Teschemacher AG, Spyer KM, Deisseroth K, Kasparov S. Astrocytes control breathing through pH-dependent release of ATP. Science. 2010;329:571–575. doi: 10.1126/science.1190721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science. 2009;324:354–359. doi: 10.1126/science.1167093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol. 2010;72:335–355. doi: 10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton NB, Attwell D. Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci. 2010;11:227–238. doi: 10.1038/nrn2803. [DOI] [PubMed] [Google Scholar]
- Janovjak H, Szobota S, Wyart C, Trauner D, Isacoff EY. A light-gated, potassium-selective glutamate receptor for the optical inhibition of neuronal firing. Nat Neurosci. 2010;13:1027–1032. doi: 10.1038/nn.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelberg HK, Macvicar BA, Sontheimer H. Anion channels in astrocytes: biophysics, pharmacology, and function. Glia. 2006;54:747–757. doi: 10.1002/glia.20423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinlogel S, Feldbauer K, Dempski RE, Fotis H, Wood PG, Bamann C, Bamberg E. Ultra light-sensitive and fast neuronal activation with the Ca2+-permeable channelrhodopsin CatCh. Nat Neurosci. 2011;14:513–518. doi: 10.1038/nn.2776. [DOI] [PubMed] [Google Scholar]
- Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science. 2009;323:1211–1215. doi: 10.1126/science.1169096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Hérault K, Oheim M, Ropert N. FM dyes enter via a store-operated calcium channel and modify calcium signaling of cultured astrocytes. Proc Natl Acad Sci U S A. 2009;106:21960–21965. doi: 10.1073/pnas.0909109106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Ropert N, Koulakoff A, Giaume C, Oheim M. Lysosomes are the major vesicular compartment undergoing Ca2+-regulated exocytosis from cortical astrocytes. J Neurosci. 2008;28:7648–7658. doi: 10.1523/JNEUROSCI.0744-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY, Lin MZ, Steinbach P, Tsien RY. Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys J. 2009;96:1803–1814. doi: 10.1016/j.bpj.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, Ushkaryov YA, Edelmann L, Link E, Binz T, Niemann H, Jahn R, Sudhof TC. Cellubrevin is a ubiquitous tetanus-toxin substrate homologous to a putative synaptic vesicle fusion protein. Nature. 1993;364:346–349. doi: 10.1038/364346a0. [DOI] [PubMed] [Google Scholar]
- Miyaji T, Echigo N, Hiasa M, Senoh S, Omote H, Moriyama Y. Identification of a vesicular aspartate transporter. Proc Natl Acad Sci U S A. 2008;105:11720–11724. doi: 10.1073/pnas.0804015105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadrigny F, Li D, Kemnitz K, Ropert N, Koulakoff A, Rudolph S, Vitali M, Giaume C, Kirchhoff F, Oheim M. Systematic colocalization errors between acridine orange and EGFP in astrocyte vesicular organelles. Biophys J. 2007;93:969–980. doi: 10.1529/biophysj.106.102673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadrigny F, Rivals I, Hirrlinger PG, Koulakoff A, Personnaz L, Vernet M, Allioux M, Chaumeil M, Ropert N, Giaume C, Kirchhoff F, Oheim M. Detecting fluorescent protein expression and co-localisation on single secretory vesicles with linear spectral unmixing. Eur Biophys J. 2006;35:533–547. doi: 10.1007/s00249-005-0040-8. [DOI] [PubMed] [Google Scholar]
- Nagel G, Brauner M, Liewald JF, Adeishvili N, Bamberg E, Gottschalk A. Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr Biol. 2005;15:2279–2284. doi: 10.1016/j.cub.2005.11.032. [DOI] [PubMed] [Google Scholar]
- Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P, Bamberg E. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A. 2003;100:13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima S, Hiraku Y, Tada-Oikawa S, Hishita T, Gabazza EC, Tamaki S, Imoto I, Adachi Y, Kawanishi S. Vacuolar H+-ATPase inhibitor induces apoptosis via lysosomal dysfunction in the human gastric cancer cell line MKN-1. J Biochem. 2003;134:359–364. doi: 10.1093/jb/mvg153. [DOI] [PubMed] [Google Scholar]
- Numano R, Szobota S, Lau AY, Gorostiza P, Volgraf M, Roux B, Trauner D, Isacoff EY. Nanosculpting reversed wavelength sensitivity into a photoswitchable iGluR. Proc Natl Acad Sci U S A. 2009;106:6814–6819. doi: 10.1073/pnas.0811899106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Oh SJ, Han KS, Woo DH, Mannaioni G, Traynelis SF, Lee CJ. Bestrophin-1 encodes for the Ca2+-activated anion channel in hippocampal astrocytes. J Neurosci. 2009;29:13063–13073. doi: 10.1523/JNEUROSCI.3193-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpura V, Grubisic V, Verkhratsky A. Ca2+ sources for the exocytotic release of glutamate from astrocytes. Biochim Biophys Acta. 2011;1813:984–991. doi: 10.1016/j.bbamcr.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Perea G, Araque A. Astrocytes potentiate transmitter release at single hippocampal synapses. Science. 2007;317:1083–1086. doi: 10.1126/science.1144640. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403:316–321. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Kuga N, Namiki S, Matsuki N, Ikegaya Y. Locally synchronized astrocytes. Cereb Cortex. 2011;21:1889–1900. doi: 10.1093/cercor/bhq256. [DOI] [PubMed] [Google Scholar]
- Scemes E, Giaume C. Astrocyte calcium waves: what they are and what they do. Glia. 2006;54:716–725. doi: 10.1002/glia.20374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Bowser DN, Sofroniew MV, Khakh BS. Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J Neurosci. 2008;28:6659–6663. doi: 10.1523/JNEUROSCI.1717-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Kracun S, Sofroniew MV, Khakh BS. A genetically targeted optical sensor to monitor calcium signals in astrocyte processes. Nat Neurosci. 2010;13:759–766. doi: 10.1038/nn.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. 2002;277:10482–10488. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- Szobota S, Isacoff EY. Optical control of neuronal activity. Annu Rev Biophys. 2010;39:329–348. doi: 10.1146/annurev.biophys.093008.131400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szobota S, Gorostiza P, Del Bene F, Wyart C, Fortin DL, Kolstad KD, Tulyathan O, Volgraf M, Numano R, Aaron HL, Scott EK, Kramer RH, Flannery J, Baier H, Trauner D, Isacoff EY. Remote control of neuronal activity with a light-gated glutamate receptor. Neuron. 2007;54:535–545. doi: 10.1016/j.neuron.2007.05.010. [DOI] [PubMed] [Google Scholar]
- Takano T, Kang J, Jaiswal JK, Simon SM, Lin JH, Yu Y, Li Y, Yang J, Dienel G, Zielke HR, Nedergaard M. Receptor-mediated glutamate release from volume sensitive channels in astrocytes. Proc Natl Acad Sci U S A. 2005;102:16466–16471. doi: 10.1073/pnas.0506382102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RJ, Zhou N, MacVicar BA. Ischemia opens neuronal gap junction hemichannels. Science. 2006;312:924–927. doi: 10.1126/science.1126241. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Steinhäuser C. Ion channels in glial cells. Brain Res Brain Res Rev. 2000;32:380–412. doi: 10.1016/s0165-0173(99)00093-4. [DOI] [PubMed] [Google Scholar]
- Volgraf M, Gorostiza P, Numano R, Kramer RH, Isacoff EY, Trauner D. Allosteric control of an ionotropic glutamate receptor with an optical switch. Nat Chem Biol. 2006;2:47–52. doi: 10.1038/nchembio756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lou N, Xu Q, Tian GF, Peng WG, Han X, Kang J, Takano T, Nedergaard M. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat Neurosci. 2006;9:816–823. doi: 10.1038/nn1703. [DOI] [PubMed] [Google Scholar]
- Wyart C, Del Bene F, Warp E, Scott EK, Trauner D, Baier H, Isacoff EY. Optogenetic dissection of a behavioural module in the vertebrate spinal cord. Nature. 2009;461:407–410. doi: 10.1038/nature08323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Davidson TJ, Mogri M, Deisseroth K. Optogenetics in neural systems. Neuron. 2011;71:9–34. doi: 10.1016/j.neuron.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci. 2003;23:3588–3596. doi: 10.1523/JNEUROSCI.23-09-03588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YP, Oertner TG. Optical induction of synaptic plasticity using a light-sensitive channel. Nat Methods. 2007;4:139–141. doi: 10.1038/nmeth988. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Chen G, Zhou W, Song A, Xu T, Luo Q, Wang W, Gu XS, Duan S. Regulated ATP release from astrocytes through lysosome exocytosis. Nat Cell Biol. 2007;9:945–953. doi: 10.1038/ncb1620. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Petersen CC, Nicoll RA. Effects of reduced vesicular filling on synaptic transmission in rat hippocampal neurones. J Physiol. 2000;525:195–206. doi: 10.1111/j.1469-7793.2000.t01-1-00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]