Targeted transcriptional activation of silent oct4 pluripotency gene by combining designer TALEs and inhibition of epigenetic modifiers (original) (raw)
Abstract
Specific control of gene activity is a valuable tool to study and engineer cellular functions. Recent studies uncovered the potential of transcription activator-like effector (TALE) proteins that can be tailored to activate user-defined target genes. It remains however unclear whether and how epigenetic modifications interfere with TALE-mediated transcriptional activation. We studied the activity of five designer TALEs (dTALEs) targeting the oct4 pluripotency gene. In vitro assays showed that the five dTALEs that target distinct sites in the oct4 promoter had the expected DNA specificity and comparable affinities to their corresponding DNA targets. In contrast to their similar in vitro properties, transcriptional activation of oct4 by these distinct dTALEs varied up to 25-fold. While dTALEs efficiently upregulated transcription of the active oct4 promoter in embryonic stem cells (ESCs) they failed to activate the silenced oct4 promoter in ESC-derived neural stem cells (NSCs), indicating that as for endogenous transcription factors also dTALE activity is limited by repressive epigenetic mechanisms. We therefore targeted the activity of epigenetic modulators and found that chemical inhibition of histone deacetylases by valproic acid or DNA methyltransferases by 5-aza-2′-deoxycytidine facilitated dTALE-mediated activation of the epigenetically silenced oct4 promoter in NSCs. Notably, demethylation of the oct4 promoter occurred only if chemical inhibitors and dTALEs were applied together but not upon treatment with inhibitors or dTALEs only. These results show that dTALEs in combination with chemical manipulation of epigenetic modifiers facilitate targeted transcriptional activation of epigenetically silenced target genes.
INTRODUCTION
The ability to specifically manipulate the expression of endogenous genes by engineered designer transcription factors has wide-ranging applications in basic and applied biology (1–4). Availability of suitable DNA-binding scaffolds that can be tailored to bind user-defined target sequences has been the major limitation in the generation and application of designer transcription factors. Recent studies however demonstrated that transcription activator-like effector proteins (TALEs) from the plant pathogenic bacterial genus Xanthomonas contain a DNA-binding domain that can be adjusted to bind any desired target sequence with high specificity (5–9). The TALE DNA-binding domain is composed of tandem arranged 33–35 amino acid repeats, with each repeat binding to one base (10,11). Base preferences of repeats are specified by residues 12 and 13, known as the repeat variable diresidues (RVDs), that determine preferential pairing with A (NI), C (HD), G (NK) and T (NG) nucleotides, respectively. The use of this TALE code facilitates the assembly of TALE repeat arrays that bind any desired DNA sequence (12).
A recent study investigated a large number of dTALEs and found that most, but not all, activated the desired target promoters (5). Notably, the epigenetically controlled oct4 and c-myc gene could not be upregulated by their matching dTALEs, suggesting that epigenetic modifications affect dTALE-mediated gene activation.
We systematically investigated the application of dTALEs to the murine pluripotency gene oct4 to clarify how epigenetic modifications affect their performance. The inspection of five dTALEs that bind to distinct DNA sequences within the oct4 promoter revealed similar affinities to their DNA targets but up to 25-fold differences in their efficiency as transcriptional activators. Further studies revealed that dTALE-mediated activation of a silent oct4 promoter in neural stem cells (NSCs) can be drastically improved by treatment with the histone deacetylase (HDAC) inhibitor valproic acid (VPA) and the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine (5azadC). These data suggest that chromatin modifications that are involved in transcriptional gene silencing, hinder dTALE-mediated gene activation and that simultaneous inhibition of HDACs and DNA methyltransferases may overcome this limitation of dTALE technology.
MATERIALS AND METHODS
Construction of plasmids
A Gateway cassette from pGWB5 (13) was amplified (forward primer: 5′-GGGGCGATCGCACAAGTTTGTACAAAAAAGCTGAACGAG-3′; reverse primer: 5′- GGGGCGGCCGCAACCACTTTGTACAAGAAAGCTGAACG-3′), thereby adding AsiSI and NotI restriction sites. This fragment was cloned via AsiSI and NotI into pCAG_mCh (14) generating pCAG_mCh_GW. The VP16AD was amplified from RSV
E2F1-VP16 (15) (forward primer: 5′GGGGGTCTCTCACCATGGATCCTGCCCCCCCGACCGATGTCAGC-3′; reverse primer:5′-GGGGGTCTCCCTTCTACCCACCGTACTCGTCAATTCCAAGG-3′), thereby adding a BamHI restriction site to the 5′ end and cloned into pENTR-D- TOPO (Invitrogen) generating pENTR-D-BamHI_VP16AD. TALE repeat arrays were generated via multi-fragment cut-ligation using golden gate cloning (16) and ligated either into pENTR-D-TALE-Δrep-_Bpi_I-A or pENTR-D-TALE-Δrep-_Bpi_I-AC-VP16AD. All entry clones were transferred by LR recombination (Invitrogen) into the expression vector pCAG_mCh_GW.
The oct4 reporter construct (p_oct4_-GFP) was generated by inserting the XhoI/AvrII fragment of GOF-18 (17) which includes the basepairs −1 to −4716 upstream of the transcriptional start site of oct4 together with a linker oligo (5′-CCTAGGTGAGCCGTCTTTCCACCAGGCCCCCGGCTCGGGGTGCGATCGCCGCCCATGG-3′) into pGL-3 basic (Promega) cut with XhoI/NcoI. Subsequently, the Luciferase ORF was removed by cutting with KasI/FseI and the eGFP ORF (amplified with: forward primer: 5′-AAAGGCGCCAGTGAGCAAGGGCG-3′; reverse primer: 5′-AAAGGCCGGCCTTACTTGTACAGCTCGTCC-3′) was inserted.
The promoter mutants TB83, TB68, TB60 and TB31 were generated by site-directed mutagenesis using a _Asi_SI/_Aat_II derived sub-cloned poct4-GFP fragment as template with either forward primer: 5′-TCTCCCACCCCCACAGCTCTGCTCCTTTGGGGAGGGAGAGGTGAAAC-3′, 5′-GCTCTGCTCCTCCACCCACCCAGGGGTTGGGGAGGGAGAGGTGAAACCG-3′,′′ 5′-CCTCCACCCACCCAGGGGGCGGGGCCTTGGGGAGGGAGAGGTGAAACCG-3′ or 5′-GGTCAAGGCTAGAGGGTGGGATTGGGTTGGGGAGGGAGAGGTGAAACCG-3′ together with reverse primer: 5′-GAAACTGAGGCGAGCGCTATCTG-3′, thereby deleting TB83, TB68; TB60 and TB31 and inserting them individually at the position of TB31.
Immunofluorescence staining
For immunostaining, ogNSCs were grown on cover slips and transiently transfected with the T-83VP16 construct for Oct4 stainings or untransfected for Pax6, Nestin and Olig2 stainings. Cells were fixed with 2.0% or 3.7% formaldehyde in phosphate-buffered salie (PBS) and permeabilized in PBS containing 0.2% Triton X-100. The Oct4 staining was performed using a goat primary antibody against the murine Oct4 (goat; 1:1000, Santa Cruz) and a secondary anti-goat antibody coupled to Alexa Fluor 647 (1:2000, Molecular Probes). The neural stem cell markers Pax6 (rabbit; 1:1000, Millipore), Nestin (mouse monoclonal, Rat-401; 1:10, Developmental Studies Hybridoma Bank, University of Iowa) and Olig2 (rabbit; 1:500, Millipore) were detected with secondary antibodies conjugated to Alexa Fluor 488 (Molecular Probes). The antibodies were diluted in PBS containing 0.02% Tween 20 and 2% bovine serum albumin (BSA). Cells were counterstained with DAPI and mounted in Vectashield (Vector Laboratories). Images were acquired with a Zeiss Axioplan 2 fluorescence microscope equipped with a Plan-NEOFLUAR 40×/1.3 oil objective (Zeiss).
Cell culture, transfection and fluorescence-activated cell sorting
HEK293T cells (18) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 50 µg/ml gentamicin and 10% fetal bovine serum (FBS). For expression of fusion proteins, HEK293T cells were transfected with polyethylenimine (Sigma). ogESCs were cultured as described (19). ogNCSs were cultured in N2B27 medium supplemented with 20 ng/ml FGF-2 and EGF. NSCs and ESCs were transfected using Lipofectamin 2000 (Invitrogen) according to the manufacturer's instructions and sorted with a fluorescence-activated cell sorting (FACS) Aria II instrument (Becton Dickinson).
Generation of transgenic cell lines
ogESCs were generated by transfecting wt J1 ESCs (20) with the p_oct4_-GFP reporter construct and repeated sorting for eGFP expression. Finally, single cell sorting was used to obtain a clonal transgenic cell line.
Derivation of NSCs from ESCs
ogESCs were differentiated into ogNSCs as previously described (21–23). In brief, 3.5 × 105 cells were plated in a 25 cm2 culture flask with N2B27 medium containing 1000 U/ml of LIF (ESGRO, Millipore). The next day the medium was exchanged against N2B27 without LIF to initiate differentiation into the neural lineage. After 7 days cells were plated in Euromed-N (Euroclone) supplemented with 20 ng/ml EGF and FGF2 (Peprotech). After 5 days, neurospheres were collected and plated in gelatin-coated flasks in N2B27 medium containing 20 ng/ml EGF and FGF2 to allow outgrowth of NSCs.
Treatment of ogNSCs with epigenetic inhibitors
VPA sodium salt (Sigma-Aldrich) was dissolved in PBS at a concentration of 250 mM and sterile filtered. 5-aza-2′-deoxycytidine (Sigma-Aldrich) was dissolved in PBS at a concentration of 30 mM. Trichostatin A (TSA; Sigma-Aldrich) was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 5 mM. Cells were transfected with the T-83 construct as described above. Medium was changed after 12 h against medium containing dilutions of the respective inhibitor or combination thereof as indicated in Figure 4 and respective legend. Cells were cultured for additional 36 h followed by FACS and quantitative real-time-polymerase chain reaction (qRT-PCR).
Figure 4.

Activation of the endogenous oct4 gene in NSCs requires inhibition of repressive epigenetic mechanisms. (A) Relative eGFP intensities as measured by flow cytometry of mCherry-positive ogNSCs transfected with the VP16 T-83 dTALE construct (T-83). Cells transfected with control plasmid (blue) or T-83 (red) were untreated or treated with TSA (30 nM), VPA (620 µM), 5azadC (10 nM) or a combination of VPA (310 µM) and five azadC (5 nM). (B) Relative levels of endogenous oct4 mRNA measured by quantitative real-time PCR of transfected, mCherry-positive ogNSCs from (A) as well as untransfected ogNSCs and ogESCs as a reference. (C) DNA methylation levels of the oct4 promoter in samples from (A) and of ogESCs as well as ogNSCs as reference. Percentage of methylation represents the average of five CpG sites in the proximal part of the oct4 promoter. (D, E) Relative mRNA levels of tet1 and nanog as determined by quantitative real-time PCR of samples from (A) and of ogESCs as well as ogNSCs as reference. (F) Fluorescence microscopy images of ogNSCs transfected with the T-83 construct in combination with 5azadC treatment (10 nM) or no drug. Samples were stained for Oct4 protein (A647) and counterstained with DAPI. mCherry channel shows cells transfected with T-83. eGFP channel shows expression of the oct4 reporter transgene. Scale bar represents 25 µm. For images of samples treated with the other inhibitors, see Supplementary Figure S4. Error bars represent standard deviation from two to three independent experiments. Asterisks indicate samples where no mRNA was detectable by quantitative real-time PCR.
In vitro methylation and reporter gene assay
In vitro methylation of p_oct4_-GFP was performed using M.SssI (New England Biolabs). Forty-five units of enzyme were incubated with 45 µg of plasmid DNA in the presence of 160 µM SAM overnight. After 3 h of incubation, fresh SAM (160 µM) was added to ensure complete methylation. Methylation status of the plasmid after in vitro methylation was tested by digestion with MspI and HpaII (Fermentas). For the reporter gene assay HEK293T cells were plated in six-well plates and grown to 70% confluence. Subsequently, cells were co-transfected with the reporter plasmid and the respective dTALE construct. Forty-eight hours after transfection cells were lysed in PBS containing 0.5% NP40 and mammalian protease inhibitors. The lysate was cleared by centrifugation and eGFP and mCherry fluorescence was measured with a Tecan Infinite M1000 plate reader.
RNA Isolation, cDNA synthesis and qRT-PCR
Isolation of RNA and reverse transcription was carried out as described previously (19). Real-time PCR analysis was performed on the 7500 Fast Real-Time PCR System (Applied Biosystems) at standard reaction conditions using either the TaqMan Gene Expression Master Mix (Applied Biosystems) or the Power SYBR Green PCR Master Mix (Applied Biosystems). The following TaqMan Gene expression assays were used: gapdh (Assay ID: Mm99999915_g1), oct4 (Assay ID: Mm00658129_gH) and nanog (Assay ID: Mm01617761_g1). Primer sequences for SYBR Green PCRs: gapdh (For 5′catggccttccgtgttccta 3′ Rev 5′cttcaccaccttcttgatgtcatc 3′); tet1 (for 5′ccaggaagaggcgactacgtt 3′ Rev 5′ ttagtgttgtgtgaacctgatttattgt 3′) and hnf4a (for 5′ caagaggtccatggtgtttaagg 3′, Rev 5′ cggctcatctccgctagct 3′). Relative mRNA levels were normalized to gapdh and calculated with the comparative CT Method (ΔΔCT Method).
In vitro DNA-binding assays
In vitro DNA-binding assays were performed as described previously (24,25) with the following modifications. Two differentially fluorescently labeled DNA substrates corresponding to position − 39 to +18 (A) and −88 to −31 (B) relative to the TSS of the oct4 promoter (Figure 3) were used in direct competition. Substrates were prepared by annealing 5′ ATTO550 or ATTO647N labeled lower strand with the respective unlabeled upper strand oligonucleotide. For competition assays, 200-nM ATTO647N-labeled substrate A and ATTO550-labeled substrate B were added and incubated at room temperature (RT) for 1 h with constant mixing. Fluorescence intensities were measured with a Tecan Infinite M1000 plate reader using the following excitation/emission wavelengths: 490 ± 10 nm / 511 ± 10 nm for eGFP, 550 ± 15 nm / 580 ± 15 nm for ATTO550 and 650 ± 10 nm / 670 ± 10 nm for ATTO647N. The measurements were normalized using standard curves from purified eGFP and ATTO-dye-labeled oligonucleotides. Moreover, a control set of each substrate with distinct fluorophores was used for normalization.
Figure 3.

(A)DNA-binding properties of oct4 eGFP-dTALE fusion proteins in vitro. Schematic representation of the 102-bp upstream of the transcriptional start site (TSS) of the oct4 promoter, including the binding site of the Sp1/Sp3 transcription factors, the hormone responsive element (HRE) and two CpG sites (open circles). oct4 eGFP–dTALE fusion proteins are depicted at the position of their target sequence and numbered according to the distance between the 5′ end of their target sequence and the TSS of the oct4 gene. Binding assays were performed using fluorescently labeled double-stranded DNA substrates corresponding to position −39 to +18 [substrate A (SA)] and −88 to −31 [substrate B (SB)] relative to the TSS of the oct4 gene. Note that substrate A includes the targeting sequences of dTALEs T-11 and T-31 and substrate B includes the targeting sequences of dTALEs T-60, T-68 and T-83. (B) DNA binding of eGFP-dTALE fusions to the specific substrate in competition with the respective unspecific substrate. Shown are fluorescent intensity ratios of bound labeled DNA substrate/eGFP-dTALE fusions. eGFP was used as negative control. Values represent means and ±SEM from three independent experiments. (C) DNA affinity measurements of the five dTALEs as measured by fluorescence polarization. Upper panel shows the data points acquired for each dTALE and the corresponding fitted curves. The table contains the _K_d values for each dTALE calculated from the fittings using gnuplot and the function 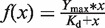 .
.
Fluorescence polarization measurements
DNA affinity was determined by fluorescence polarization measurements. eGFP-dTALE fusion proteins were purified as described above and eluted from the Sepharose beads by addition of 250 mM imidazol. Different concentrations of GFP-dTALE fusion proteins were incubated with their specific ATTO647N-labeled substrates (1 nM). After incubation for 30 min, at RT fluorescence polarization was measured with a Tecan Infinite M1000 plate reader using 635 nm for excitation and 670 ± 10 nm for emission. The data of fluorescence polarization over protein concentration were fitted with 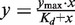 using gnuplot (http://www.gnuplot.info).
using gnuplot (http://www.gnuplot.info).
DNA methylation analysis
For the analysis of DNA methylation levels at the oct4 promoter genomic DNA was isolated using the NucleoSpin Triprep Kit (Macherey-Nagel). Bisulfite treatment was performed using the EZ DNA Methylation-Gold™ Kit (Zymo Research Corporation) according to the manufacturer's protocol. Subsequently, the oct4 promoter sequence was amplified in a semi-nested PCR using the primers:
- F1: 5′-ATGGGTTGAAATATTGGGTTTATTTA-3′
- F2: 5′-GTAAGAATTGAGGAGTGGTTTTAG-3′
- R1: 5′ACCCTCTAACCTTAACCTCTAAC 3′
- R2 = R1 with 5′biotin
The biotinylated PCR products of the second PCR were analyzed by pyrosequencing (Varionostic GmbH, Ulm, Germany). The pyrosequencing covered five CpG sites of which the average methylation level was calculated. DNA methylation levels of major satellite repeats and H19 promoter was performed as previously described (25).
RESULTS
Design and construction of five dTALEs targeting the murine oct4 promoter
We generated five dTALEs each targeting a distinct 19-bp sequence of the murine pluripotency gene oct4 to test whether the position of the target sequence influences the efficiency of dTALE-mediated promoter activation. These five dTALEs targeted sequences upstream or downstream of the Sp1/Sp3/hormone responsive element (HRE) box (Figure 1A, Supplementary Figure S1). DNA-binding TALE repeat arrays were generated by Golden Gate cloning as described previously (6) and transferred to mammalian expression vectors by Gateway recombination (Supplementary Figure S1A). To monitor transfection efficiency and expression levels, mCherry (26) was fused to the N-terminus of the dTALEs. Furthermore, we replaced the transcriptional activation domain (AD) of the Xanthomonas wild-type TALE protein (wtdTALEs) with the VP16 AD from the herpes simplex virus (VP16dTALEs) and compared the activity of these two distinct dTALE architectures.
Figure 1.

Activation of a transgenic oct4 reporter construct by dTALEs in HEK293T cells. (A) Schematic representation of the 102-bp fragment upstream of the transcriptional start site (TSS) of the oct4 promoter, including the binding site of the Sp1/Sp3 transcription factors, the hormone responsive element (HRE) and two CpG sites (open circles). oct4_-specific dTALEs are depicted in correspondence of the location of their target sequence and designated according to the distance between the 5′ end of their target sequence and the TSS. (B) Fluorescence microscopy images of HEK293T cells co-transfected with the p_oct4_- GFP reporter construct and the T-83 dTALE constructs. Left panel shows cells transfected with the T-83 dTALE fused to the wild-type AD (wt AD). Right panel shows cells transfected with the T-83 dTALE fused to the VP16 AD. Scale bar = 200 µm. (C) Transcriptional activation of the unmethylated p_oct4-GFP reporter construct by oct4_-specific dTALEs. eGFP expression was normalized to cells co-transfected with a control plasmid encoding the fluorescent protein mCherry (mCh) and poct4-GFP reporter construct. (D) Transcriptional activation of the in vitro methylated p_oct4-GFP reporter construct by _oct4_-specific dTALEs. eGFP expression was normalized to cells co-transfected with a control plasmid (mCh) and poct4-GFP reporter construct. To allow for a direct comparison of expression levels in (C) and (D) the data observed on the methylated promoter were normalized to the mCherry values observed with the unmethylated promoter (C). Error bars in (C) and (D) represent standard deviation from three independent experiments.
dTALEs targeting distinct sites in the oct4 promoter have similar affinities in vitro but differ strongly in their in vivo performance as transcriptional activators
The activity of the different dTALEs was first analyzed in a transient reporter gene assay. HEK293T cells were co-transfected with an oct4 promoter-driven eGFP reporter (p_oct4-eGFP_) and a constitutively expressed dTALE construct. Expression was analyzed by fluorescence measurement 48 h after transfection. Notably, the VP16dTALEs activated the oct4 promoter to significantly higher levels than the corresponding wtdTALEs (Figure 1C), despite the fact that the latter were expressed at slightly higher levels (Supplementary Figure S2). The most distal dTALE (T-83) yielded the strongest transcriptional activation with both, the wtdTALE and VP16dTALE architecture (Figure 1B and C). To test whether the variable efficiency of the dTALEs with distinct repeat arrays is caused by the location of the target sites within the promoter, we replaced base pairs −31 to −102 of the oct4 reporter construct which contain the target sites of four of the five dTALEs, with a shorter sequence containing one dTALE target site only. The resulting four reporter constructs have the respective dTALE target site at the same position (Figure 2A). Transcriptional activation of these four mutated reporter constructs by the corresponding VP16dTALEs was greatly reduced as compared to the activation level of the wild-type oct4 reporter. Three of the four VP16dTALEs induced similar eGFP expression levels (Figure 2B) while T-60 exhibited a slightly stronger activation in the mutated promoter. The enhanced activity of T-60 is possibly due to the overlap of its target site with the SP1 site in the wild-type promoter, which also results in a relatively higher background in cells transfected only with the mutated reporter construct containing the T-60 binding site (Figure 2C). The dTALEs used in this study were designed to target different sequences within the promoter region of oct4. The distinct RVD compositions of these dTALE repeat arrays might result in different binding affinity, causing the observed difference in transcriptional activation. We therefore determined the affinity and specificity of our dTALEs in vitro using fluorescently labeled DNA substrates and eGFP–dTALE fusion proteins. We found specific binding of all five dTALEs to their respective DNA substrates (Figure 3A and B). Dissociation constants were determined by fluorescence polarization and all dTALEs tested yielded affinities for their specific substrates, with _K_d values in the low nanomolar to high picomolar range (Figure 3C). Notably, the dTALE T-83, which was the strongest transcriptional activator in vivo, had a comparably low affinity in vitro. Together, these data strongly suggest that the observed variations in dTALE-mediated activation of the oct4 promoter are not due to inherent differences in their binding affinity.
Figure 2.
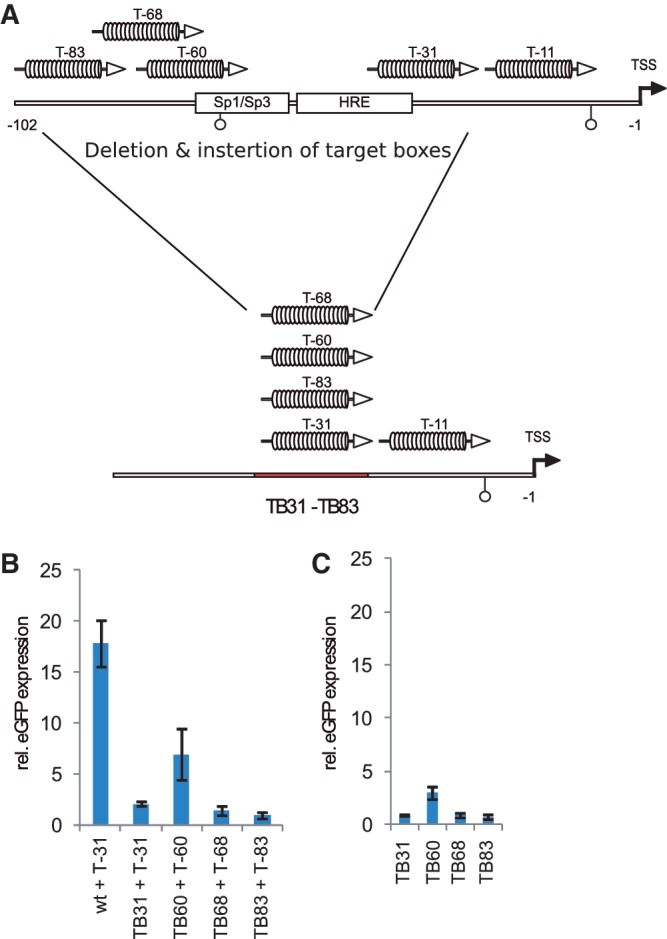
The location of a dTALE target sequence within the oct4 promoters can affect its functionality. (A) Schematic representation of an oct4 promoter deletion construct in which base pairs −31 to −102 relative to the TSS were deleted and the target sequences of the four dTALEs were inserted yielding the reporter constructs TB31, TB60, TB68 and TB83. (B) Transcriptional activation of the reporter constructs TB31, TB60, TB68 and TB83 by corresponding dTALEs. (C) Background activity of the mutated reporter constructs in cells co-transfected with respective reporter and mCherry control.
dTALEs activate methylated reporter plasmids
In addition to positional effects, we tested whether the epigenetic state of the promoter might influence the efficiency of dTALE-mediated transcriptional activation. We methylated the p_oct4_-GFP plasmid in vitro (Supplementary Figure S3A) and determined its inducibility by dTALEs. All dTALEs induced eGFP expression from the methylated oct4 promoter, yet to lower levels as compared to the unmethylated reporter (Figure 1D). Notably, the relative differences in the activity of the highly potent T-83 and the other dTALEs were up to 25-fold and thus more pronounced with the methylated as compared to the unmethylated reporter construct (Figure 1D). These results indicate that dTALEs can activate heavily methylated promoter sequences, albeit to a reduced extent, and suggest that the lack of correlation between in vitro binding affinity and in vivo activity of dTALES may reflect their different ability to overcome other repressive epigenetic marks at the target locus.
dTALEs hyperactivate endogenous oct4 expression in embryonic stem cells
To test the ability of dTALEs to activate the endogenous oct4 gene we generated mouse embryonic stem cells (ESCs) stably carrying the p_oct4-GFP_ reporter construct (ogESCs). ogESCs were tested with T-83 fused to the VP16 AD (VP16 T83), the most efficient dTALE, and compared with mCherry control vector. Transfected mCherry-positive cells were selected using FACS and total RNA was isolated followed by reverse transcription and qRT-PCR. FACS analysis showed that ogESCs transfected with the VP16 T-83 had a 3–4-fold higher mean eGFP fluorescence intensity compared to control transfected cells (Supplementary Figure S4A). Transcription of the endogenous oct4 was induced about 2-fold as determined by qRT-PCR (Supplementary Figure S4B). The relatively low induction rate is likely due to the high basal level of oct4 transcription in ESCs and the negative feedback of Oct4 on its own promoter (27).
Activation of oct4 in neural stem cells depends on inhibition of repressive epigenetic mechanisms
To test whether dTALEs can also activate a transcriptionally silent endogenous oct4 promoter, we differentiated ogESCs into NSCs. During this differentiation process the oct4 locus is epigenetically silenced and NSCs no longer express oct4 (28). Analysis by immunofluorescence showed that all cells were positive for the NSC markers (21) Pax6, Nestin and Olig2 (Supplementary Figure S5), indicating successful in vitro differentiation from ogESCs to ogNSCs. The ogNSCs were transfected with the vector encoding the dTALE VP16 T-83 or a control vector encoding mCherry. Forty-eight hours after transfection cells were analyzed by flow cytometry. In contrast to the experiments with ogESCs, the dTALE VP16 T-83 activated neither the transgenic p_oct4-eGFP_ reporter nor the endogenous oct4 promoter in ogNSCs (Figure 4A and B). This could be due to the different epigenetic states of the oct4 promoter in ESCs and NSCs. Whereas the oct4 promoter in ESCs is active and apparently accessible to dTALEs, oct4 is not expressed in NSCs and the promoter might be less prone to dTALE-mediated activation. Therefore, we envisaged that inhibiting the repressive epigenetic modifiers that prevent activation of the oct4 promoter in NSCs could allow dTALE-mediated activation of oct4. To test this hypothesis, we used the HDAC inhibitors TSA (29) or VPA (30) as well as the DNA methyltransferase (Dnmt) inhibitor 5-aza-2'-deoxycytidine (5azadC) (31) to interfere with two major epigenetic mechanisms by which transcriptional silencing of genes is achieved in mammals. Twelve hours after transfection with VP16 T-83, ogNSCs were treated with the respective inhibitor for additional 36 h. Treatment with 5azadC or VPA but not TSA significantly increased relative eGFP expression in cells transfected with VP16 T-83 (Figure 4A and Supplementary Figure S6A–C). Similarly, endogenous oct4 transcript levels were induced up to 60% as compared to the levels in ogESCs. However, a combination of 5azadC and VPA did not show additive nor synergistic effects (Figure 4A and B). Treatment with the inhibitors alone did not result in transcriptional activation of the reporter nor the endogenous oct4 gene (Figure 4A and B), demonstrating that the observed activation was due to the synergistic action of the dTALE and the inhibitors. Cells transfected with the dTALE VP16 T-83 and treated with VPA, 5azadC or combinations of both showed not only increased oct4 transcript levels but also Oct4 protein (Figure 4F). Moreover, treatment of VP16 T-83 transfected cells with VPA, 5azadC or combinations of both exhibited up-regulation of the Oct4 target genes nanog and tet1 (Figure 4C and D) (32–34). By contrast, genes that are not part of the Oct4 regulatory network were not influenced by treatment with inhibitors and/or expression of dTALE VP16 T-83 on transcript (Supplementary Figure S7A).
As both, 5azadC and VPA, have been reported to induce DNA demethylation (31,35) we investigated the effects of these inhibitors on the DNA methylation levels of the oct4 promoter. Interestingly, in all samples that were treated with the inhibitors only and/or transfected with the control plasmid no change in DNA methylation levels was observed. However, expression of the dTALE VP16 T-83 together with VPA and/or 5azadC treatment caused a reduction of DNA methylation at the oct4 promoter by ∼30% (Figure 4E). Treatment with inhibitors alone or in combination with the dTALEs did not influence methylation levels at the h19 locus and major satellite repeats, showing that the observed effect is specific for the oct4 promoter (Supplementary Figure S7B and S7C). These results suggest a synergistic effect of dTALEs and epigenetic inhibitors in mammalian cells.
DISCUSSION
Variable efficiency of different dTALEs in transcriptional activation
In eukaryotic cells, transcriptional activation involves the concerted action of multiple factors recognizing target sites at different positions of gene promoters. The possibility to generate dTALEs that bind different sites within the promoter of target genes opens new possibilities to probe and optimize conditions for targeted transcriptional activation. We designed a panel of dTALEs targeting distinct sites in the murine oct4 promoter and compared their performance in vitro and in vivo. Expression of dTALE T-83 resulted in a 2-fold increase of the oct4 mRNA in ESCs. Previous studies reported a number of dTALEs targeting distinct promoters (5–7,36,37). However, none of these studies has systematically investigated whether the relative position of a dTALE target site within a promoter affects its functionality. We engineered five dTALEs, each targeting a distinct 19-bp sequence within the oct4 promoter. All dTALEs yielded _K_d values in the low nanomolar to high picomolar range and were expressed at similar levels but differed largely in their efficiency in vivo. Remarkably T-83, the dTALE with one of the lowest binding affinity, showed the highest efficiency in oct4 promoter activation. Our data obtained with recombinant oct4 promoter constructs showed that deletions in the native oct4 promoter severely affected dTALE performance, indicating that the presence of a specific binding site is not sufficient for efficient activation of transcription. These results suggest that the different capacity of dTALEs to activate transcription is less determined by their intrinsic DNA-binding properties but rather by their interactions at target promoters. Studies with the viral transactivator VP16 had previously indicated position-dependent interactions of the VP16 activation domain, possibly with basal transcription factors (38). Therefore, it is likely that also dTALEs are involved in complex interactions at the promoter of target genes that may either hinder or promote transcriptional activation.
As multiple _cis_- and _trans_-acting factors and epigenetic modifications are involved in the regulation of promoter activity, it will be difficult to predict the efficiency of a dTALE in silico. Hence, it seems important to construct and test multiple dTALEs for a given target promoter to obtain the most effective transcriptional activator. In the past, the assembly of genes that encode custom-designed repeat arrays was challenging and thus construction of multiple dTALEs targeting one promoter was not a realistic task. However, this is no longer a bottleneck since the recently established hierarchical ligation-based ‘Golden Gate’ cloning approaches facilitate rapid generation of genes encoding TALE repeat arrays (5,6,9,36,37,39,40).
Another potential bottleneck in the selection of efficient dTALEs is the analysis of promoter activation by RT-PCR or comparable assays. By contrast, promoter–reporter fusions constructs facilitate rapid quantitative comparison of multiple dTALEs but may not adequately reflect the transcriptional regulation of the corresponding endogenous genes. In this context, it should be noted that the dTALE (T-83), performing best on plasmid reporters, also most efficiently activated the chromosomal oct4 promoter (Supplementary Figure S6E). Thus, promoter–reporter fusions may greatly facilitate the screening of different dTALE repeat arrays and experimental conditions that can then be verified and optimized in a second step by monitoring transcription of the endogenous genes.
Transcriptional activation by dTALEs is facilitated by epigenetic inhibitors
In a recent study, dTALEs were shown to activate an episomal oct4 reporter but not the endogenous oct4 promoter (5). Similarly, we observed a lack of dTALE-mediated oct4 activation in NSCs, where the promoter is silent. In ESCs, however, where the oct4 promoter is active, our dTALE clearly increased oct4 transcription, suggesting that dTALE activity depends on the epigenetic state of the promoter. These results are consistent with the reported multistep inactivation of the oct4 promoter that occurs during cellular differentiation after implantation and involves H3K9 as well as DNA methylation. This tight epigenetic control apparently safeguards against inappropriate reactivation of the oct4 gene and thus prevents uncontrolled proliferation and cancer (41–43).
We found that chemical inhibition of repressive epigenetic modifiers like Dnmts and HDACs enabled dTALE-mediated transcriptional activation of silent oct4 in NSCs. Interestingly, of the two HDAC inhibitors tested only VPA but not TSA treatment allowed efficient transcriptional activation of oct4 by dTALEs. A similar difference between the two inhibitors was previously reported for cellular reprogramming and oct4 promoter activation (44). One possible explanation for the different efficacy of the two inhibitors could be their different target specificities (45). Interestingly, VPA was shown to specifically affect the proximal region of the oct4 promoter (46) where also the T-83 dTALE binds. This might also explain why inhibitor treatment of cells transfected with T-31, the dTALE with the next greatest activity in the reporter assays, did not facilitate the activation of oct4 (Supplementary Figure S6E).
Previous studies reported that high concentrations of VPA and/or 5azadC induce demethylation and reactivation of silent genes (35,47,44,46,38). Under our experimental conditions, however, these inhibitors induced DNA demethylation of the oct4 promoter only in combination with dTALEs, indicating a synergistic effect. A possible explanation could be that binding of the dTALE interferes with maintenance of DNA methylation and, thus, in combination with the epigenetic inhibitors leads to reduction of methylation levels. Such a synergistic effect would be consistent with the recent realization that DNA methylation is rather dynamic and functionally linked to other epigenetic pathways (48). The synergy between low concentrations of epigenetic inhibitors and dTALEs suggests that silent target genes could be activated without genome-wide demethylation and thus avoid unwanted side effects.
In summary, we demonstrated that combining dTALEs with DNA methylation and/or HDAC inhibitors facilitates selective activation of the endogenous oct4 pluripotency gene. As in turn also Oct4 target genes are reactivated, dTALEs could be used for reprogramming of somatic cells to induced pluripotent stem cells (iPSCs). It remains to be investigated whether single or combinations of several dTALEs are more efficient than present reprogramming strategies involving the Oct4 protein itself. However, in contrast to native transcription factors, dTALEs can be specifically directed against single genes or selected combinations of target genes and thereby allow dissection of complex transcription networks to identify key factors in biological processes like pluripotency and differentiation. The combination with epigenetic inhibitors may, in some cases, facilitate the activation of tightly repressed genes and further expand the utility of dTALEs in basic and applied biosciences.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–7.
FUNDING
The Deutsche Forschungsgemeinschaft (SPP 1356 and SFBs 646/TR5) to H.L.; grants from the 2Blades foundation to T.L.; The Elite Network of Bavaria (International Doctorate Program NanoBioTechnology) to C.S.S.; S.B. is a fellow of the graduate school Life Science Munich (LSM). Funding for open access charge: The Deutsche Forschungsgemeinschaft.
Conflict of interest statement. None declared.
Supplementary Material
Supplementary Data
ACKNOWLEDGEMENTS
We thank Luca Gentile and Hans R. Schöler (Max Planck Institute for Molecular Biomedicine, Münster) for providing the GOF-18 construct, DG Johnson (Howard Hughes Medical Institute, Durham) for providing the E2F1-VP16 fusion and Kerry Tucker (Ruprecht-Karls-University,Heidelberg) for providing wt J1 ESCs. Furthermore, we thank Fabian Köhler and Tobias Anton for help with the reporter gene assays. We thank Alex Buschle for help with the immunofluorescence staining and Carina Frauer for advice on the DNA binding assays. CSS and KT gratefully acknowledge the International Max Planck Research School for Molecular and Cellular Life Sciences (IMPRS-LS). H.L., T.L., S.B. and F.S. conceived the study. H.L., T.L., F.S., S.B. and R.M. designed the experiments. S.B. performed the cell biological experiments and DNA methylation analysis. K.T. performed the DNA binding assays. R.M. and J.E. established a collection of plasmids for assembly of dTALE genes. RM designed and cloned the dTALEs used in this study. CSS isolated RNA samples, performed the expression analysis and DNA methylation analysis. S.B., F.S., T.L. and H.L. wrote the manuscript.
REFERENCES
- 1.Klug A. The discovery of zinc fingers and their development for practical applications in gene regulation and genome manipulation. Q. Rev. Biophys. 2010;43:1–21. doi: 10.1017/S0033583510000089. [DOI] [PubMed] [Google Scholar]
- 2.Segal DJ. Toward controlling gene expression at will: selection and design of zinc finger domains recognizing each of the 5’-GNN-3' DNA target sequences. Proc. Natl. Acad. Sci. USA. 2011;96:2758–2763. doi: 10.1073/pnas.96.6.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cathomen T, Joung JK. Zinc-finger nucleases: the next generation emerges. Mol. Ther. 2008;16:1200–1207. doi: 10.1038/mt.2008.114. [DOI] [PubMed] [Google Scholar]
- 4.De Francesco L. Move over ZFNs. Nat. Biotechnol. 2011;29:681–684. doi: 10.1038/nbt.1935. [DOI] [PubMed] [Google Scholar]
- 5.Zhang F, Cong L, Lodato S, Kosuri S, Church GM, Arlotta P. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat. Biotechnol. 2011;29:149–153. doi: 10.1038/nbt.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morbitzer R, Elsaesser J, Hausner J, Lahaye T. Assembly of custom TALE-type DNA binding domains by modular cloning. Nucleic Acids Res. 2011;39:5790–5709. doi: 10.1093/nar/gkr151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 8.Mahfouz MM, Li L, Shamimuzzaman M, Wibowo A, Fang X, Zhu J-K. De novo-engineered transcription activator-like effector (TALE) hybrid nuclease with novel DNA binding specificity creates double-strand breaks. Proc. Natl. Acad. Sci. USA. 2011;108:2623–2628. doi: 10.1073/pnas.1019533108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 11.Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 12.Bogdanove AJ, Schornack S, Lahaye T. TAL effectors: finding plant genes for disease and defense. Curr. Opin. Plant Biol. 2010;13:394–401. doi: 10.1016/j.pbi.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 13.Nakagawa T, Kurose T, Hino T, Tanaka K, Kawamukai M, Niwa Y, Toyooka K, Matsuoka K, Jinbo T, Kimura T. Development of series of gateway binary vectors, pGWBs, for realizing efficient construction of fusion genes for plant transformation. J. Biosci. Bioeng. 2007;104:34–41. doi: 10.1263/jbb.104.34. [DOI] [PubMed] [Google Scholar]
- 14.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 15.Johnson DG. Oncogenic capacity of the E2F1 Gene. Proc. Natl. Acad. Sci. USA. 1994;91:12823–12827. doi: 10.1073/pnas.91.26.12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engler C, Gruetzner R, Kandzia R, Marillonnet S. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One. 2009;4:e5553. doi: 10.1371/journal.pone.0005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yeom Y, Fuhrmann G, Ovitt C, Brehm A, Ohbo K, Gross M, Hubner K, Scholer H. Germline regulatory element of Oct-4 specific for the totipotent cycle of embryonal cells. Development. 1996;122:881–894. doi: 10.1242/dev.122.3.881. [DOI] [PubMed] [Google Scholar]
- 18.DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, Calos MP. Analysis of mutation in human cells by using an Epstein–Barr virus shuttle system. Mol. Cell. Biol. 7:379–387. doi: 10.1128/mcb.7.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szwagierczak A, Bultmann S, Schmidt CS, Spada F, Leonhardt H. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2010;38:e181. doi: 10.1093/nar/gkq684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 21.Conti L, Pollard SM, Gorba T, Reitano E, Toselli M, Biella G, Sun Y, Sanzone S, Ying Q-L, Cattaneo E, et al. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol. 3:e283. doi: 10.1371/journal.pbio.0030283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ying Q-L, Stavridis M, Griffiths D, Li M, Smith A. Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat. Biotechnol. 2003;21:183–186. doi: 10.1038/nbt780. [DOI] [PubMed] [Google Scholar]
- 23.Ying QL, Smith AG. Defined conditions for neural commitment and differentiation. Methods Enzymol. 2003;365:327–341. doi: 10.1016/s0076-6879(03)65023-8. [DOI] [PubMed] [Google Scholar]
- 24.Rottach A, CFrauer C, Pichler G, Bonapace IM, Spada F, Leonhardt H. The multi-domain protein Np95 connects DNA methylation and histone modification. Nucleic Acids Res. 2010;38:1796–1804. doi: 10.1093/nar/gkp1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frauer C, Rottach A, Meilinger D, Bultmann S, Fellinger K, Hasenöder S, Wang M, Qin W, Söding J, Spada F, et al. Different binding properties and function of CXXC zinc finger domains in Dnmt1 and Tet1. PLoS One. 2011;6:e16627. doi: 10.1371/journal.pone.0016627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 27.Pan G, Li J, Zhou Y, Zheng H, Pei D. A negative feedback loop of transcription factors that controls stem cell pluripotency and self-renewal. FASEB J. 2006;20:1730–1732. doi: 10.1096/fj.05-5543fje. [DOI] [PubMed] [Google Scholar]
- 28.Kim JB, Zaehres H, Wu G, Gentile L, Ko K, Sebastiano V, Araúzo-Bravo MJ, Ruau D, Han DW, Zenke M, et al. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature. 2008;454:646–650. doi: 10.1038/nature07061. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 30.Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santi DV, Garrett CE, Barr PJ. On the mechanism of inhibition of DNA-cytosine methyltransferases by cytosine analogs. Cell. 1983;33:9–10. doi: 10.1016/0092-8674(83)90327-6. [DOI] [PubMed] [Google Scholar]
- 32.Chen X, Fang F, Liou Y-C, Ng H-H. Zfp143 regulates Nanog through modulation of Oct4 binding. Stem Cells. 2008;26:2759–2767. doi: 10.1634/stemcells.2008-0398. [DOI] [PubMed] [Google Scholar]
- 33.Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, Laiho A, Tahiliani M, Sommer CA, Mostoslavsky G, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodda DJ, Chew J-L, Lim L-H, Loh Y-H, Wang B, Ng H-H, Robson P. Transcriptional regulation of nanog by OCT4 and SOX2. J. Biol. Chem. 2005;280:24731–24737. doi: 10.1074/jbc.M502573200. [DOI] [PubMed] [Google Scholar]
- 35.Dong E, Chen Y, Gavin DP, Grayson DR, Guidotti A. Valproate induces DNA demethylation in nuclear extracts from adult mouse brain. Epigenetics. 2010;5:730–735. doi: 10.4161/epi.5.8.13053. [DOI] [PubMed] [Google Scholar]
- 36.Geiβler R, Scholze H, Hahn S, Streubel J, Bonas U, Behrens S-E, Boch J. Transcriptional activators of human genes with programmable DNA-specificity. PloS One. 2011;6:e19509. doi: 10.1371/journal.pone.0019509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weber E, Gruetzner R, Werner S, Engler C, Marillonnet S. Assembly of designer TAL effectors by Golden Gate cloning. PLoS One. 2011;6:e19722. doi: 10.1371/journal.pone.0019722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hagmann M, Georgiev O, Schaffner W. The VP16 paradox: herpes simplex virus VP16 contains a long-range activation domain but within the natural multiprotein complex activates only from promoter-proximal positions. J. Virol. 1997;71:5952–5962. doi: 10.1128/jvi.71.8.5952-5962.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li T, Huang S, Zhao X, Wright DA, Carpenter S, Spalding MH, Weeks DP, Yang B. Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res. 2011;39:6315–6325. doi: 10.1093/nar/gkr188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scholze H, Boch J. TAL effectors are remote controls for gene activation. Curr. Opin. Microbiol. 2011;14:47–53. doi: 10.1016/j.mib.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 41.Feldman N, Gerson A, Fang J, Li E, Zhang Y, Shinkai Y, Cedar H, Bergman Y. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat. Cell Biol. 2006;8:188–194. doi: 10.1038/ncb1353. [DOI] [PubMed] [Google Scholar]
- 42.Gidekel S, Pizov G, Bergman Y, Pikarsky E. Oct-3/4 is a dose-dependent oncogenic fate determinant. Cancer Cell. 2003;4:361–370. doi: 10.1016/s1535-6108(03)00270-8. [DOI] [PubMed] [Google Scholar]
- 43.Looijenga LHJ, Stoop H, de Leeuw HPJC, de Gouveia Brazao CA, Gillis AJM, van Roozendaal KEP, van Zoelen EJJ, Weber RFA, Wolffenbuttel KP, van Dekken H, et al. POU5F1 (OCT3/4) identifies cells with pluripotent potential in human germ cell tumors. Cancer Res. 2003;63:2244–2250. [PubMed] [Google Scholar]
- 44.Huangfu D, Maehr R, Guo W, Eijkelenboom A, Snitow M, Chen AE, Melton DA. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 2008;26:795–797. doi: 10.1038/nbt1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim T-Y, Bang Y-J, Robertson K. Histone deacetylase inhibitors for cancer therapy. Epigenetics. 2006;1:14–23. doi: 10.4161/epi.1.1.2644. [DOI] [PubMed] [Google Scholar]
- 46.Teng HF, Kuo Y-L, Loo MR, Li CL, Chu TW, Suo H, Liu HS, Lin KH, Chen SL. Valproic acid enhances Oct4 promoter activity in myogenic cells. J. Cell. Biochem. 2011;110:995–1004. doi: 10.1002/jcb.22613. [DOI] [PubMed] [Google Scholar]
- 47.Al-Salihi M, Yu M, Burnett DM, Alexander A, Samlowski W, Fitzpatrick FA. The depletion of DNA methyltransferase-1 and the epigenetic effects of 5-aza-2’deoxycytidine (decitabine) are differentially regulated by cell cycle progression. Epigenetics. 2011;6:1021–1028. doi: 10.4161/epi.6.8.16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011;146:866–872. doi: 10.1016/j.cell.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data