Phosphorothioate DNA as an antioxidant in bacteria (original) (raw)
Abstract
Diverse bacteria contain DNA with sulfur incorporated stereo-specifically into their DNA backbone at specific sequences (phosphorothioation). We found that in vitro oxidation of phosphorothioate (PT) DNA by hydrogen peroxide (H2O2) or peracetic acid has two possible outcomes: DNA backbone cleavage or sulfur removal resulting in restoration of normal DNA backbone. The physiological relevance of this redox reaction was investigated by challenging PT DNA hosting Salmonella enterica cells using H2O2. DNA phosphorothioation was found to correlate with increasing resistance to the growth inhibition by H2O2. Resistance to H2O2 was abolished when each of the three dnd genes, required for phosphorothioation, was inactivated. In vivo, PT DNA is more resistant to the double-strand break damage caused by H2O2 than PT-free DNA. Furthermore, sulfur on the modified DNA was consumed and the DNA was converted to PT-free state when the bacteria were incubated with H2O2. These findings are consistent with a hypothesis that phosphorothioation modification endows DNA with reducing chemical property, which protects the hosting bacteria against peroxide, explaining why this modification is maintained by diverse bacteria.
INTRODUCTION
Many bacteria contain DNA that is stereo-specifically phosphorothioate (PT) modified at specific sequences (1–6). DNA phosphorothioation is controlled by five clustered genes, dndA–E, which are located on genetic islands (7,8). Four of the genes, dndA and dndC–E, are essential for the PT modification, while inactivation of dndB, resulted in increased phosphorothioation and altered sequence preference (1,3 and personal communication). The biological function(s) of DNA phosphorothioation remained elusive until the recent discoveries of a Streptomyces coelicolor gene that specifically restricts PT DNA from Streptomyces lividans, and a restriction system in Salmonella enterica serovar Cerro 87 against PT-free heterologous DNA (9,10).
The extent of phosphorothioation varies between different host strains, ranging from 300 to 3000 modifications per 106 nt (6). In the case of Escherichia coli B7A and S. enterica 87, the amount is around 750 per 106 nt, and in S. lividans 474 per 106 nt. The study of 63 Vibrio strains revealed that seven of them possess from six per 104 nt to as high as two to three PT modifications per 103 nt (6).
PTs modification are also sequence specific (1,6). A high frequency of GA was found in Bermanella marisrubri RED65 and Hahella chejuensis KCTC2396, using high pressure (or high performance) liquid chromatography (HPLC)/mass method. While the dinucleotide sequence of GG was found in Pseudomonas fluorescens Pf0-1, S. lividans 1326 and Geobacter uraniumreducens Rf4. In the case of E. coli B7A and S. enterica 87, the most commonly PT modified dinucleotide sequences are GA and GT. A high frequency of PT modifications at GA, GT, GG and CC were found in the seven Vibrio strains. Using cleavage and ligation methods, a conserved modification sequence of -cGGCCgccg- (including a highly conserved 4-bp central core -GGCC-) was identified in S. lividans, indicating that the modification is double stranded (ds) (1).
Phosphorothioation across the bacterial genome DNA leads to the often-observed Dnd phenotype: DNA degradation during electrophoresis (4). It was proposed that the DNA degradation comes from specific double-strand cleavage at the site of the modification by Tris-peracetic acid, which is generated at the anode during gel electrophoresis (11,12).
We asked what chemical property of phosphorothioation leads to the DNA ds breakage (Dnd phenotype), and whether this susceptibility to ds DNA cleavage makes the host bacterial cell sensitive or insensitive to oxidative environment.
MATERIALS AND METHODS
Reagents
PT-containing dinucleotides d(GPSA) (PS indicates the phosphorus–sulfur bond) and dinucleotide d(GA) were synthesized by Sangon Biotech (Shanghai) Co., Ltd. Per-acetic acid (PAA) was purchased from Shanghai Habo Company or obtained by mixing 1 M acetic acid with 1 M hydrogen peroxide (H2O2) (1 M Stock), and keeping the mixture dark at room temperature for 72 h. PAA was then kept in the dark and stored at 4°C until use. PAA–TAE was made by mixing 1% 1 M Stock PAA with TAE buffer (40 mM tris base adjusted to pH 7.5 using acetic acid and 0.8 mM ethylenediaminetetraacetic acid (EDTA)).
H–phosphonate modified d(GHA) dinucleotide was synthesized by Sangon Biotech (Shanghai) Co., Ltd. and released from resin using Ammonium hydroxide.
Oxidation of PT modified DNA
Plasmid DNA was linearized using XhoI or EcoRI according to protocols provided by NEB Co., Ltd. Genomic or linearized plasmid DNAs was dissolved in 1× TAE to a concentration of 300 ng/μl. A total amount of 30 ng/μl of DNA were incubated in PAA–TAE or electrophoresis buffer (sampled at the anode after running for 2 h) for 20 min. DNA was then precipitated using propanol according to Sambrook (13). The precipitated DNA was resuspended in TAE buffer for agarose gel electrophoresis analysis.
A similar protocol was used for the H2O2 and PAA–TAE oxidation of the dinucleotide. The dinucleotides d(GA) and d(GPSA) were dissolved to a concentration of 400 μM in water (stock solution). An amount of 20 μM of dinucleotides were incubated in PAA–TAE, 10 mM PAA or H2O2 for 30 min. The reaction products were analyzed directly using LC–MS or lyophilized and kept at −20°C until use.
For the generation of the hydrogen–phosphonate dinucleotide d(GHA), 20 μM d(GPSA) was incubated in 30 mM PAA supplemented with 40 mM acetic acid at room temperature for 30 min. The reaction products were then lyophilized and kept at −20°C until use.
Liquid chromatography mass spectrometry analysis
Liquid chromatography–mass spectrometry (HPLC/MS) analysis was routinely carried out on an Agilent 1100 series LC/MSD Trap system. Ten microliters of final reaction sample was loaded on Agilent’s C18 reversed phase column (250 × 4.6 mm, 5 μm) and the absorbance was monitored at 254 and 210 nm. HPLC was carried out at a flow rate of 0.3 ml/min at room temperature. Eluent A was 0.1% acetic acid in water, and eluent B was 0.1% acetic acid in acetonitrile. HPLC conditions used were 1–13% buffer B for 35.5 min, 13–30% buffer B for another 20 min and then 30–1% buffer B for 5 min. Drying gas flow: 10 l/min; nebulizer pressure: 30 psi; drying gas temperature: 325°C; capillary voltage: 3200 V (using the positive detection mode and scanning between 50 and 900 m/z for mass spectrometer analysis).
The LC-electrospray (ES)-quadrupole time of flight (QTOF) (Agilent 1200 system, Agilent Co., CA) equipped with a 2.1- by 30-mm C18 reverse-phase column (Agilent Co., CA) was used for high resolution analysis of the reaction products. The flow rate was set at 0.2 ml/min. HPLC was carried out using a two-step program starting with 98% solvent A (0.1% formic acid in water) and 2% solvent B (0.1% formic acid in acetonitrile) to 20% solvent B over 20 min and with 20 solvent B to 95% solvent B for 5 min. Tandem MS (MS/MS) was performed in the target-dependent mode under these conditions: drying gas flow: 8 ml/min; nebulizer pressure: 35 psi; drying gas temperature: 300°C; sheath gas temperature: 300°C; sheath gas flow: 7 ml/min; capillary, 3500 V; skimmer, 65 V; OCT RF V, 750 V; fragmentor, 170 V; ion polarity: positive, and the mass scanning range was set at m/z 50–1500.
The Agilent 1200 series LC system equipped with YMC ODS-AQ reversed phase column (250 × 4.6 mm, 5 μm) was used for quantification of H2O2 reduced by d(GPSA). Absorbance was measured at 254 and 210 nm. Eluent A contained 0.1% acetic acid in water, and eluent B contained 0.1% acetic acid in acetonitrile under this condition: 1–12% buffer B for 10 min, 12–40% buffer B for 5 min and 40% of 1% buffer B for 5 min.
H2O2 calibration curve was prepared from UV254nm absorption peak areas and H2O2 concentrations from 10 to 100 mM. Under this condition, the UV254 absorption peak area shows collinearity with H2O2 concentration. The calibration curves for d(GPSA) and other analytes were prepared similarly.
Bacterial growth curves and H2O2 treatment
Salmonella enterica serovar Cerro 87 wild-type and the dptB-E gene mutants were cultured in Luria Broth medium at 37°C overnight. The cultures were then diluted using LB medium to 1 × 108 cfu in a volume of 100 μl in Greiner 96-well plates. H2O2 was then added to the final concentration of 0, 2.2, 4.4 and 8.8 mM. The growth curves were monitored using Gen5 (Biotek) from Gene Co. Ltd. The cultures were kept shaking at 37°C. OD600 was monitored for 12 h at 10-min intervals.
For the analysis of DNA ds cleavage caused by H2O2 oxidization, S. enterica serovar Cerro 87 strains were cultured in LB at 37°C overnight. The cultures were then diluted by LB medium to OD600 1.5, and H2O2 was added to a final concentration of 176, 528, 704 and 880 mM. The cultures were kept at 37°C for 2 h shaking at 220 rpm. Total DNA was then extracted and analyzed using 1% agarose gel electrophoresis. Software Quantity One from Bio-Rad company was used for the gel densitometry measurements.
RESULTS
The cleavage by PAA–TAE of natural and synthetic PT DNA
Distinctive features are associated with PT modification: DNA degradation during normal or pulse-field gel electrophoresis (the Dnd phenotype) which is not inhibited by formaldehyde and proteinase K treatment; the Dnd phenotype can be repressed if the electrophoretic buffer is supplemented with a small amount of reducing agents, especially those containing sulfur, like thiourea, or if Tris is replaced by HEPES in the electrophoresis buffer. The DNA degradation is due to the DNA cleavage specifically at the site of PT modification (1,11,12).
The enteropathogenic S. enterica serovar Cerro 87 and the soil-dwelling S. lividans, both with heavily PT modified DNA, were chosen to investigate the DNA cleavage reaction. Dinucleotide PT d(GPSA) was synthesized to analyze the reaction products. Cleaved genomic DNA was monitored using agarose gel electrophoresis, while the reaction products of d(GPSA) were analyzed using HPLC/MS. We treated purified genomic DNA and synthetic PT dinucleotide using PAA–TAE buffer (PAA, peracetic; TAE (Tris, Acetic acid, EDTA); PAA–TAE can be generated either subjecting TAE to 2 h of electrophoresis or mixing PAA with TAE) (1,11,12). Exposure of a linearized 5.4-kb plasmid pHZ209 from S. lividans to either PAA–TAE or PAA–TAE mixture causes extensive DNA cleavage (see Supplementary Figure 1SA for the site-specific cleavage of a 9.7 kb linearized plasmid carrying the Salmonella dpt gene cluster), while plasmid treated with TAE buffer alone remains uncut (Figure 1A). Interestingly, the banding patterns caused by PAA–TAE buffer and PAA–TAE mixture are very similar (Figure 1A). Figure 1B and Supplementary Figure S1B show the cleavage of the synthesized PT d(GPSA) using PAA–TAE. Dinucleotide d(GPSA) (peak 1) was completely degraded by PAA–TAE, producing six new UV254 absorbing peaks (2–7, Figure 1B). The cleavage of R or S configuration of d(GPSA) generated the same six reaction products (Figure 1B).
Figure 1.

Cleavage reaction of PT DNA. (A) Agarose gel showing the effect of treating a linearized plasmid pHZ209 isolated from S. lividans with PAA–TAE buffer or by mixing PAA with TAE buffer (normal 40 mM TAE buffer pH 7.5 containing 5 mM per-acetic acid). First lane, PAA and TAE mixture. Second lane, pre-run TAE buffer. Third lane, untreated TAE buffer. (B) HPLC traces showing R or S configuration form of synthesized PT dinucleotide d(GPSA) ([M+H]+ m/z 597, 1) oxidized by PAA–TAE. Six new reaction products were identified by MS, with m/z 565 (2), 581 (3), 332 (4), 252 (5), 316 (6) and 268 (7), respectively. H2O2 peak can also be observed if excess of H2O2 was added to the reaction mixture.
The reaction products
PAA–TAE is a mixture of chemicals, including H2O2, PAA, acetic acid, Tris and EDTA. To clarify which chemical is involved in the cleavage reaction, reactivity of d(GPSA) with H2O2 was analyzed using HPLC/MS. Figure 2A shows that the same six UV254 absorbing peaks were generated (peak 2–7) with H2O2 treatment, although the HPLC peak pattern is different. One main reaction product of H2O2 oxidation was d(GA) (peak 3, ∼60% of reaction products under the reaction condition), with the PT restored to a normal DNA phosphodiester bond.
Figure 2.


PT dinucleotide is oxidized by H2O2 and PAA. (A) Conversion of d(GPSA) to d(GA) by H2O2. d(GPSA) was incubated with (110 mM) H2O2, generating reaction products 2–7 (a). The main product is 3. d(GA) (b) and d(GPSA) (c) were run as control. Note the remaining H2O2 peak can be observed. (B) HPLC traces showing the conversion of PT to H–phosphonate linkage by increasing concentrations of per-acetic acid (PAA). Note the amount of reaction product 2, H–phosphonate dinucleotide [d(GHA)], increased corresponding to increasing concentration of PAA. At 30 mM concentration of PAA, d(GPSA) is converted mainly to d(GHA) (∼67.5% of reaction products under the reaction condition). (C) High resolution mass spectrum analysis of H–phosphonate dinucleotides. Mass fragmentations of d(GPSA) (1), d(GHA) (2) and d(GA) (3). Characteristic mass peaks m/z = 348, 316 and 332, indicating the linkages of PT, H–phosphonate and phosphodiester were marked. Fragmentation peaks corresponding to G (m/z = 152) and A (m/z = 136) were also indicated. (D) HPLC traces showing the hydrolyzed products of synthesized d(GHA) standard and the lyophilized d(GHA) generated by oxidizing d(GPSA) using PAA. Note, after release from the synthesizing column by ammonium hydroxide, all d(GHA) was hydrolyzed, generating products 4–7 mainly. Peak 3 was barely detectable. d(GPSA) oxidized by PAA generated d(GA) (peak 3) as a main product. For mass fragmentation of peak 4–7 (see Supplementary Figure 2S2).
In order to determine what influences the HPLC peak pattern and how is the PT bond cleaved in the reaction, we incubated d(GPSA) using increasing amount of H2O2 or PAA and analyzed the reaction products using HPLC/MS. We found that an increment in concentrations of H2O2 (from 3.4 to 880 mM, Supplementary Figure 2S1) or PAA (from 0.14 to 30 mM, Figure 2B), causes peak 3 (corresponding to d(GA)), to gradually become dominant followed by an increase of peak 2.
Structural mass spectrum analysis revealed that the substance in peak 2 contained a hydrogen-phosphonate bond dinucleotide d(GHA) (Figure 2C).
The structure of d(GHA) was further confirmed by the synthesized of a standard compound. Release of synthesized d(GHA) from the resin using ammonium hydroxide resulted in its hydrolysis, generating peak 3–7, identical to the PAA degradation products of d(GPSA) (Figure 2D).
The H–phosphonate compound d(GHA) is stable under acidic conditions and is the likely intermediate in the cleavage of d(GPSA). High resolution Mass analysis of the reaction products contained in peak 4–7 revealed that d(GHA) was hydrolyzed at either the 3′- or 5′-side of H–phosphonate bond, but not at both sides, generating the four hydrolyzed products (Figure 2C and D and Supplementary Figure 2S2).
d(GPSA) as a reductant
The above data established that the PT bond could be oxidized by H2O2 and peracetic acid, and that there are two outcomes of the oxidative reaction: sulfur exchange with oxygen and restoration of a phosphodiester bond, or removal of sulfur generating H–phosphonate bond that is susceptible to hydrolysis. These results suggest that the reaction is a reduction–oxidation reaction.
The possibility that the PT linkage can function as a reductant and consumption of H2O2 was further investigated. The reduction of H2O2 by d(GPSA) was monitored using HPLC. Figure 3 shows that, in sharp contrast to the d(GA) control, which has no effect on H2O2, increasing the concentration of d(GPSA) directly correlated with a decrease in H2O2 levels.
Figure 3.
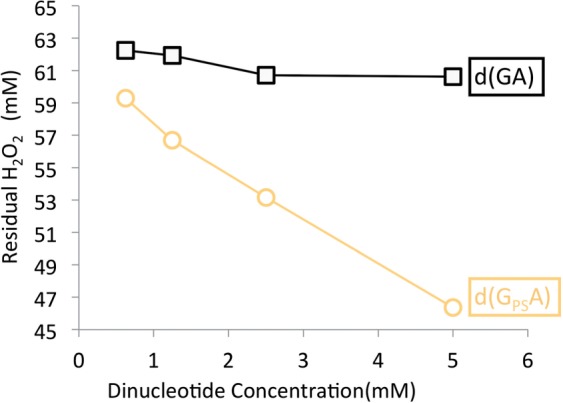
Consumption of H2O2 by d(GPSA). H2O2 incubated with increasing amounts of d(GPSA) or d(GA) (0.625, 1.25, 2.5 and 5 mM, respectively). The amount of H2O2 reduced was monitored using HPLC peak area.
H2O2 resistance of S. enterica
As shown above, oxidization of PT DNA backbone by H2O2 or peracetic acid will result in either the restoration to a normal phosphodiester linkage, or the cleavage of the backbone. In the former case, when the modification-hosting bacteria are in the presence of peroxides, like H2O2, PT bonds can function as a reductant: specifically under the condition of H2O2. The restoration to a normal DNA backbone is innocuous. In the later case, although PT modified bonds can still function as a reductant, the cleavage of the DNA backbone might be lethal, since in vivo repair of double-strand DNA breaks could be difficult—even a single non-repaired break might result in the death of the bacterium. We asked what is the actual response of the modification-hosting bacterial cell to a H2O2 containing environment.
Salmonella enterica serovar Cerro 87 contains the four clustered dptB-E genes which are orthologous to the dndB–E genes shared by many PT hosting bacteria. Hypothetically, DptB could be a regulator whose absence increases the amount of DNA phosphorothioation, and DptC, D and E are required for replacing a non-bridging O with S in the DNA backbone (3,5).
Growth curves showed that the S. enterica serovar Cerro 87 wild-type and dptB mutant that contains PT DNA are quite resistant to H2O2, and started growth quickly at higher H2O2 concentrations (Figure 4A and B). Inactivating dptC-E in S. enterica individually by non-polar in-frame deletions showed that loss of phosphorothioation correlated with an increasing lag phase after the addition of H2O2, and cessation of growth at 4.4 mM H2O2. (Note that at the highest H2O2 concentration (4.4 mM), the OD decreased slightly, possibly due to bleaching of the medium.) These results suggested that PT modification increased the H2O2 resistance of S. enterica.
Figure 4.

PT DNA hosting bacteria are resistant to H2O2. H2O2 treatment of S. enterica serovar 87 and its dpt mutant derivatives. The growth rates of the strains were monitored at OD600. The genotypes are indicated under the corresponding growth curves.
Resistance to DNA double-strand break caused by H2O2
The in vivo susceptibility of PT genomic DNA to double-strand break caused by peroxide was further investigated.
Salmonella enterica WT and dptB-E gene mutant cells were treated using increasing amount of H2O2. Chromosomal DNAs were then extracted and DNA double-strand breaks were monitored by gel electrophoresis. The gels were run in TAE buffer supplemented with thiourea to protect the modified DNA from degradation during electrophoresis. Figure 5 shows more degradation can be observed in unmodified compared to PT modified DNA, indicating that the phosphorothioation protected DNA from oxidative double-strand breakage in vivo.
Figure 5.

In vivo resistance of PT DNA to double-strand breakage caused by H2O2 Salmonella cells were incubated with increasing concentration of H2O2 for 2 h. DNA ds break damage was monitored using agarose gel electrophoresis in TAE buffer containing thiourea. W: wild-type; B–E: dptB–E mutant, respectively. DNA from dpt mutant dptC–E did not contain PT modification, while DNA from dptB mutant contained increased PT modification. The experiment was repeated at least five times. DNA smear is quantified by densitometry and estimations of the percentage are indicated below the gel panel.
The function of sulfur on DNA as a reducing reagent
We suspect that sulfur on the modified DNA could function as a biological reductant in vivo. A prediction of this model is that, when bacterial cells are facing H2O2 containing environments, the sulfur on the modified DNA should be consumed to neutralize the oxidant.
In order to test this hypothesis, S. enterica and its dptB mutant cells were again incubated with H2O2 and the sulfur content on chromosomal DNA was monitored using Dnd phenotype analysis. Figure 6 shows that phosphorothioation on chromosomal DNA, both in the wild-type strain and its dptB mutant, decreased with the addition of increasing amounts of H2O2, suggesting that sulfur on the modified DNA was indeed used to neutralize H2O2.
Figure 6.

In vivo consumption of sulfur on the modified DNA. Salmonella (A) and its dptB mutant (B) were treated using increasing concentration of H2O2 for 2 h (0, 176, 528, 704 and 880 mM, respectively). Sulfur on genomic DNA was monitored by Dnd phenotype analysis (per-acetic acid treatment followed by electrophoresis using buffer contained thiourea). Upper panel: DNA samples not treated with per-acetic acid as a control.
DISCUSSION
By performing in vitro experiments we discovered, that the peroxides, including H2O2 and peracetic acid, react with and are consumed by PT modified DNA. The reduction–oxidation reaction, originally identified as Dnd phenotype, is manifested in every DNA degradation gel from diverse bacteria (8,14–31). In our hands, this reaction observed in TAE buffer, Tris buffer, glycine buffer (11,12,32) and water. None of the reaction conditions is greatly deviate from physiological conditions. It is predictable that when PT DNA meets H2O2 and possibly other peroxides, the redox reaction observed here will happen, suggesting that PT DNA could function as a peroxide reducing reagent in vivo.
Growth curve experiments showed that S. enterica containing naturally phosphorothioated DNA were more resistant to H2O2 than their mutant derivatives that lack the modification, suggesting that PT modification has anti-oxidative physiological function in the hosting bacteria. It is possible that anti-oxidative function could come directly from its reducing chemical properties. Consistent with this view is, sulfur on the modified DNA is consumed upon incubation of the bacterial cells with H2O2 (Figure 6).
We used high concentration of H2O2 (3.4–880 mM) and peracetic acid (0.14–30 mM) to test their reactivity with PT DNA (Figure 2B and Supplementary Figure 2S1). However, the double bonds of DNA, unsaturated membrane lipids and the cysteine SH groups of proteins are prone to oxidation and growth is inhibited when cellular levels of H2O2 reach 2 μM (33). If PT DNA functions as a reducing thiol in vivo, one concern is whether the PT DNA react with physiological scale peroxide concentration. The lower limit of PAA reaction that can be detected by fluorescence is 0.14 mM, while as low as a 10 μM PAA reaction can still be detected to react with the dinucleotide using mass monitoring (Supplementary Figure S3).
Another concern is whether the in vivo consumption of H2O2 by PT DNA is comparable to the consumption by catalases, peroxidases, peroxiredoxins, plus the thiols on proteins and small molecules such as glutathione and Bacillithiol (34–36), and is therefore physiologically significant? There are around 6000 PT sulfur molecules per S. enterica cell (6), correspond to a 6–10 μM of concentration, which is substantially lower than the concentration of glutathione, estimated to be 3–7 mM in bacteria (36). However, the concentration of PT’s is comparable to that of the protein antioxidant thioredoxin, which is estimated to be 10 μM (37). Glutathione is present in most of the gram-negative bacteria, but absent in most of the gram-positive bacteria. Glutathione is involved in multiple physiological functions, including protecting the bacteria against antibiotics, oxidative stress and transition-metal homeostasis (38–40). Thioredoxins are found in nearly all organisms, acting as antioxidants by facilitating the reduction of other proteins by cysteine thio-disulfide exchange (41). It seems unlikely that PT DNA can substitute the known anti-oxidizing mechanisms, such as thioredoxin. Indeed, the growth curve experiment presented here clearly demonstrate that dnd mutants survive treatment with 2 mM H2O2 (Figure 4), which might be explained by the activity of catalase, peroxidase, thioredoxin or a combination of them. In consistent with the idea that most of the added H2O2 is consumed by catalase and other peroxidases, Imlay and colleagues have shown that, by using hydroperoxidase minus (Hpx−) strains of E. coli, simply shifting cells from anaerobic to aerobic growth conditions leads to sufficient H2O2 production to impede growth, due mainly to DNA damage (42,43).
It seems likely that, complementary to the known anti-oxidizing mechanisms, PT modification, enhance the innate anti-oxidation capability of the hosting bacteria by protecting DNA against peroxide. It is also possible that the PT modifications may be quickly replaced when consumed by oxidation, providing for a high turnover rate that increases the overall reducing capacity for the cell. Figure 5 clearly shows that PT modified genomic DNA, both from the wild-type strain and the dndB mutant, is very quite resistant to DNA ds break damage caused by H2O2. This viewpoint was further tested by introducing dpt gene cluster into a DNA phosphorothioation non proficient strain E. coli DH10B. Figure 4S shows that introducing DNA PT modification enhances the anti-oxidation capability of the bacteria. This explains why the dpt gene cluster resides in genetic mobile elements and is kept by diverse bacteria while the modification is not essential.
In vitro oxidation of the PT dinucleotide by PAA or H2O2 results in a mixture of DNA backbone cleavage and just removal of S from the DNA without backbone cleavage. Contrary to the expectation, PT DNA in vivo is more resistant to oxidative ds DNA breakage caused by H2O2 than unmodified DNA (Figure 5). The oxidative reaction is directed toward the exchange of sulfur with oxygen rather than to backbone cleavage. An explanation for this apparent paradox is that d(GHA), the intermediate of cleavage reaction pathway, is preferentially produced under high peroxide concentration (Figure 2B), which is less likely inside a bacterial cell. However even a single ds DNA breakage is lethal (or harmful), which should make the PT DNA hosting bacteria sensitive to oxidative environments [especially considering the fact that there are approximately 6000 modifications per cell (6)]. An alternatively explanation, is that the cell can choose the PT oxidation pathway. This possibility is currently under our intensive investigation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1–4.
FUNDING
The National Science Foundation of China, the Ministry of Science and Technology [973 (2012CB721004) and 863 programs]; Ministry of Education of China, the Shanghai Municipal Council of Science and Technology and Shanghai Leading Academic Discipline Project B203; State Key Laboratory of Bio-organic and Natural Products Chemistry (CAS); National Program of Development of Transgenic New Species of China. Funding for open access charge: the Ministry of Science and Technology [973 (2012CB721004)].
Conflict of interest statement. None declared.
Supplementary Material
Supplementary Data
ACKNOWLEDGEMENTS
The authors are grateful to Dr Daniel Blanchard from UC Berkeley and Dr Tobias Kieser for editing this article.
REFERENCES
- 1.Liang JD, Wang ZJ, He XY, Li JL, Zhou XF, Deng ZX. DNA modification by sulfur: analysis of the sequence recognition specificity surrounding the modification sites. Nucleic Acids Res. 2007;35:2944–2954. doi: 10.1093/nar/gkm176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang L, Chen S, Xu T, Taghizadeh K, Wishnok JS, Zhou X, You D, Deng Z, Dedon PC. Phosphorothioation of DNA in bacteria by dnd genes. Nat. Chem. Biol. 2007;3:709–710. doi: 10.1038/nchembio.2007.39. [DOI] [PubMed] [Google Scholar]
- 3.Xu TG, Liang JD, Chen S, Wang LR, He XY, You DL, Wang ZJ, Li AY, Xu ZL, Zhou XF, et al. DNA phosphorothioation in Streptomyces lividans: mutational analysis of the dnd locus. BMC Microbiol. 2009;9:41. doi: 10.1186/1471-2180-9-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou XF, Deng ZX, Firmin JL, Hopwood DA, Kieser T. Site-specific degradation of Streptomyces lividans DNA during electrophoresis in buffers contaminated with ferrous iron. Nucleic Acids Res. 1988;16:4341–4352. doi: 10.1093/nar/16.10.4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou X, He X, Liang J, Li A, Xu T, Kieser T, Helmann JD, Deng Z. A novel DNA modification by sulphur. Mol. Microbiol. 2005;57:1428–1438. doi: 10.1111/j.1365-2958.2005.04764.x. [DOI] [PubMed] [Google Scholar]
- 6.Wang L, Chen S, Vergin KL, Giovannoni SJ, Chan SW, DeMott MS, Taghizadeh K, Cordero OX, Cutler M, Timberlake S, et al. DNA phosphorothioation is widespread and quantized in bacterial genomes. Proc. Natl Acad. Sci. USA. 2011;108:2963–2968. doi: 10.1073/pnas.1017261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He X, Ou HY, Yu Q, Zhou X, Wu J, Liang J, Zhang W, Rajakumar K, Deng Z. Analysis of a genomic island housing genes for DNA S-modification system in Streptomyces lividans 66 and its counterparts in other distantly related bacteria. Mol. Microbiol. 2007;65:1034–1048. doi: 10.1111/j.1365-2958.2007.05846.x. [DOI] [PubMed] [Google Scholar]
- 8.Ou HY, He X, Shao Y, Tai C, Rajakumar K, Deng Z. dndDB: a database focused on phosphorothioation of the DNA backbone. PLoS One. 2009;4:e5132. doi: 10.1371/journal.pone.0005132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu GA, Ou HY, Wang T, Li L, Tan HR, Zhou XF, Rajakumar K, Deng ZX, He XY. Cleavage of phosphorothioated DNA and methylated DNA by the type IV restriction endonuclease ScoMcrA. PLos Genet. 2010;6:e1001253. doi: 10.1371/journal.pgen.1001253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu TG, Yao F, Zhou XF, Deng ZX, You DL. A novel host-specific restriction system associated with DNA backbone S-modification in Salmonella. Nucleic Acids Res. 2010;38:7133–7141. doi: 10.1093/nar/gkq610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ray T, Weaden J, Dyson P. Tris-dependent site-specific cleavage of Streptomyces lividans DNA. FEMS Microbiol. Lett. 1992;96:247–252. doi: 10.1016/0378-1097(92)90412-h. [DOI] [PubMed] [Google Scholar]
- 12.Ray T, Mills A, Dyson P. Tris-dependent oxidative DNA strand scission during electrophoresis. Electrophoresis. 1995;16:888–894. doi: 10.1002/elps.11501601149. [DOI] [PubMed] [Google Scholar]
- Sambrook,J. (2001) Molecular Cloning: A Laboratory Manual. 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- 14.O’Reilly LC. A method for overcoming DNA degradation during PFGE for Serratia marcescens. J. Microbiol. Methods. 2011;85:173–174. doi: 10.1016/j.mimet.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 15.Miled-Bennour R, Ells TC, Pagotto FJ, Farber JM, Kerouanton A, Meheut T, Colin P, Joosten H, Leclercq A, Besse NG. Genotypic and phenotypic characterisation of a collection of Cronobacter (Enterobacter sakazakii) isolates. Int. J. Food Microbiol. 2010;139:116–125. doi: 10.1016/j.ijfoodmicro.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 16.Ribeiro RL, Machry L, Brazil JM, Ramos TM, Avelar KE, Pereira MM. Technical improvement to prevent DNA degradation of Leptospira spp. in pulsed field gel electrophoresis. Lett. Appl. Microbiol. 2009;49:289–291. doi: 10.1111/j.1472-765X.2009.02641.x. [DOI] [PubMed] [Google Scholar]
- 17.Bosilevac JM, Guerini MN, Kalchayanand N, Koohmaraie M. Prevalence and characterization of salmonellae in commercial ground beef in the United States. Appl. Environ. Microbiol. 2009;75:1892–1900. doi: 10.1128/AEM.02530-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banerjee SK, Farber JM. Susceptibility of Vibrio parahaemolyticus to tris-dependent DNA degradation during pulsed-field gel electrophoresis. J. Clin. Microbiol. 2009;47:870–871. doi: 10.1128/JCM.02306-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leclair D, Pacyotto F, Farber JM, Cadieux B, Austin JW. Comparison of DNA fingerprinting methods for use in investigation of type E botulism outbreaks in the Canadian arctic. J. Clin. Microbiol. 2006;44:1635–1644. doi: 10.1128/JCM.44.5.1635-1644.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciofu O, Riis B, Pressler T, Poulsen HE, Hoiby N. Occurrence of hypermutable Pseudomonas aeruginosa in cystic fibrosis patients is associated with the oxidative stress caused by chronic lung inflammation. Antimicrobial Agents Chemother. 2005;49:2276–2282. doi: 10.1128/AAC.49.6.2276-2282.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang YS, Yakrus MA, Graviss EA, Williams-Bouyer N, Turenne C, Kabani A, Wallace RJ. Pulsed-field gel electrophoresis study of Mycobacterium abscessus isolates previously affected by DNA degradation. J. Clin. Microbiol. 2004;42:5582–5587. doi: 10.1128/JCM.42.12.5582-5587.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murase T, Nagato M, Shirota K, Katoh H, Otsuki K. Pulsed-field gel electrophoresis-based subtyping of DNA degradation-sensitive Salmonella enterica subsp enterica serovar Livingstone and serovar Cerro isolates obtained from a chicken layer farm. Vet. Microbiol. 2004;99:139–143. doi: 10.1016/j.vetmic.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 23.Silbert S, Boyken L, Hollis RJ, Pfaller MA. Improving typeability of multiple bacterial species using pulsed-field gel electrophoresis and thiourea. Diagn. Micr. Infec. Dis. 2003;47:619–621. doi: 10.1016/s0732-8893(03)00164-0. [DOI] [PubMed] [Google Scholar]
- 24.Hata M, Suzuki M, Matsumoto M, Takahashi M, Yamazaki M, Hiramatsu R, Matsui H, Sakae K, Suzuki Y, Miyazaki Y. A new clonal line of Salmonella Saintpaul having emerged and prevailed since 1999 in Aichi, Japan. Jpn J. Infect. Dis. 2003;56:77–79. [PubMed] [Google Scholar]
- 25.Liesegang A, Tschape H. Modified pulsed-field gel electrophoresis method for DNA degradation-sensitive Salmonella enterica and Escherichia coli strains. IJMM. 2002;291:645–648. doi: 10.1078/1438-4221-00180. [DOI] [PubMed] [Google Scholar]
- 26.Koort JMK, Lukinmaa S, Rantala M, Unkila E, Siitonen A. Technical improvement to prevent DNA degradation of enteric pathogens in pulsed-field gel electrophoresis. J. Clin. Microbiol. 2002;40:3497–3498. doi: 10.1128/JCM.40.9.3497-3498.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klaassen CHW, de Valk-Van Haren HA, Horrevorts AM. Pulsed-field gel electrophoresis can yield DNA fingerprints of degradation-susceptible Clostridium difficile strains—Reply. J. Clin. Microbiol. 2002;40:3547–3547. doi: 10.1128/JCM.40.9.3546-3547.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inglis TJJ, O’Reilly L, Foster N, Clair A, Sampson J. Comparison of rapid, automated ribotyping and DNA macrorestriction analysis of Burkholderia pseudomallei. J. Clin. Microbiol. 2002;40:3198–3203. doi: 10.1128/JCM.40.9.3198-3203.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fawley WN, Wilcox MH. Pulsed-field gel electrophoresis can yield DNA fingerprints of degradation-susceptible Clostridium difficile strains. J. Clin. Microbiol. 2002;40:3546–3547. doi: 10.1128/JCM.40.9.3546-3547.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romling U, Tummler B. Achieving 100% typeability of Pseudomonas aeruginosa by pulsed-field gel electrophoresis. J. Clin. Microbiol. 2000;38:464–465. doi: 10.1128/jcm.38.1.464-465.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corkill JE, Graham R, Hart CA, Stubbs S. Pulsed-field gel electrophoresis of degradation-sensitive DNAs from Clostridium difficile PCR ribotype 1 strains. J. Clin. Microbiol. 2000;38:2791–2792. doi: 10.1128/jcm.38.7.2791-2792.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boybek A, Ray TD, Evans MC, Dyson PJ. Novel site-specific DNA modification in Streptomyces: analysis of preferred intragenic modification sites present in a 5.7 kb amplified DNA sequence. Nucleic Acids Res. 1998;26:3364–3371. doi: 10.1093/nar/26.14.3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mongkolsuk S, Helmann JD. Regulation of inducible peroxide stress responses. Mol. Microbiol. 2002;45:9–15. doi: 10.1046/j.1365-2958.2002.03015.x. [DOI] [PubMed] [Google Scholar]
- 34.Antelmann H, Helmann JD. Thiol-based redox switches and gene regulation. Antioxid. Redox. Sign. 2011;14:1049–1063. doi: 10.1089/ars.2010.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faulkner MJ, Helmann JD. Peroxide stress elicits adaptive changes in bacterial metal ion homeostasis. Antioxid. Redox. Sign. 2011;15:175–189. doi: 10.1089/ars.2010.3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newton GL, Rawat M, La Clair JJ, Jothivasan VK, Budiarto T, Hamilton CJ, Claiborne A, Helmann JD, Fahey RC. Bacillithiol is an antioxidant thiol produced in Bacilli. Nat. Chem. Biol. 2009;5:625–627. doi: 10.1038/nchembio.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holmgren A, Ohlsson I, Grankvist ML. Thiroedoxin from Escherichia coli. Radioimmunological and enzymatic determinations in wild type cells and mutants defective in phage T7 DNA replication. J. Biol. Chem. 1978;253:430–436. [PubMed] [Google Scholar]
- 38.Allocati N, Federici L, Masulli M, Di Ilio C. Glutathione transferases in bacteria. FEBS J. 2009;276:58–75. doi: 10.1111/j.1742-4658.2008.06743.x. [DOI] [PubMed] [Google Scholar]
- 39.Allocati N, Federici L, Masulli M, Di Ilio C. Distribution of glutathione transferases in Gram-positive bacteria and Archaea. Biochimie. 2012;94:588–596. doi: 10.1016/j.biochi.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 40.Masip L, Veeravalli K, Georgiou G. The many faces of glutathione in bacteria. Antioxid. Redox. Sig. 2006;8:753–762. doi: 10.1089/ars.2006.8.753. [DOI] [PubMed] [Google Scholar]
- 41.Nordberg J, Arner ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Rad. Biol. Med. 2001;31:1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 42.Park S, You X, Imlay JA. Substantial DNA damage from submicromolar intracellular hydrogen peroxide detected in Hpx-mutants of Escherichia coli. Proc. Natl Acad. Sci. USA. 2005;102:9317–9322. doi: 10.1073/pnas.0502051102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Imlay JA, Linn S. DNA damage and oxygen radical toxicity. Science. 1988;240:1302–1309. doi: 10.1126/science.3287616. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data