Population Genetic Inference from Personal Genome Data: Impact of Ancestry and Admixture on Human Genomic Variation (original) (raw)
Abstract
Full sequencing of individual human genomes has greatly expanded our understanding of human genetic variation and population history. Here, we present a systematic analysis of 50 human genomes from 11 diverse global populations sequenced at high coverage. Our sample includes 12 individuals who have admixed ancestry and who have varying degrees of recent (within the last 500 years) African, Native American, and European ancestry. We found over 21 million single-nucleotide variants that contribute to a 1.75-fold range in nucleotide heterozygosity across diverse human genomes. This heterozygosity ranged from a high of one heterozygous site per kilobase in west African genomes to a low of 0.57 heterozygous sites per kilobase in segments inferred to have diploid Native American ancestry from the genomes of Mexican and Puerto Rican individuals. We show evidence of all three continental ancestries in the genomes of Mexican, Puerto Rican, and African American populations, and the genome-wide statistics are highly consistent across individuals from a population once ancestry proportions have been accounted for. Using a generalized linear model, we identified subtle variations across populations in the proportion of neutral versus deleterious variation and found that genome-wide statistics vary in admixed populations even once ancestry proportions have been factored in. We further infer that multiple periods of gene flow shaped the diversity of admixed populations in the Americas—70% of the European ancestry in today’s African Americans dates back to European gene flow happening only 7–8 generations ago.
Introduction
Understanding the relative effects of different population genetic forces on the apportionment of human genomic variation is a central focus of medical and population genomics.1–5 Much of what we know comes from analyzing patterns of common, and, therefore, ancient genetic polymorphisms via genotyping across diverse human populations.6–8 Recent studies have used sequencing approaches to reveal a more complete and genome-wide picture of variation, including lower-frequency variants with a more recent evolutionary origin.6,9 In particular, exome sequencing across thousands of participants in medical genetic studies of complex disease now provides strong evidence that rare alleles in the human genome are enriched with mutations that have functional (and presumably deleterious) consequences.10 However, little is known empirically about whether genomes from different populations harbor similar levels of neutral and deleterious variation and whether population bottlenecks and recent population growth play critical roles.
To understand the impact of demographic history and natural selection on genomic variation, we analyzed data from 50 human genomes sequenced to high coverage (at least 18–20×). All the genomes analyzed here come from participants who are part of the 1000 Genomes Project or HapMap Project and who have given consent for public release of genomic data. Two key questions we address are how to analyze genomes from admixed populations and how to interpret individual-level patterns of variation in light of inferred demographic history.
Intercontinental travel and the processes of colonialism have dramatically reshaped the historical distribution of human genetic variation. In particular, much of the world’s population now traces its genetic ancestry to multiple continental source populations. Understanding how to analyze and interpret admixed genomes will be critical for enabling transethnic and multiethnic medical genetic studies2–4 and for ensuring that the findings of genetics research are broadly applicable.5
Interpreting admixed genomes is easiest when the source populations are highly diverged; the ancestry of specific genomic segments can then be inferred on the basis of panels of genotyped SNPs from populations related to the source populations.11–17 In this study, we combined such a method of local-genomic-ancestry assignment with the analysis of full-genome sequence data from 12 individuals with recent ancestry from distinct continental populations. Our analyses allow us to refine our understanding of the effects of population history on patterns of variation both among global populations and within the genomes of individuals who have admixed ancestry.
A hypothesis raised by sequencing exomes from African American and European American individuals is that the former contain proportionally fewer damaging mutations than the latter as a result of a larger effective population size.4 This hypothesis has not been tested in additional populations and has potentially profound implications for how to interpret personal genomic variation, particularly for American genomes that contain segments of European, African, and Native American ancestry. Understanding these dynamics is key to properly controlling for ancestry in transethnic and multiethnic medical sequencing studies, in which segments might vary in the background rate of neutral versus deleterious variation.
Given the stochastic nature of the evolutionary process, we might imagine that individual genomes might harbor so much stochastic noise that small population-level differences might be obscured. However, methods have recently been proposed for inferring demographic history from singly sequenced genomes18 by the exploitation of variation in the depth of the time to the most recent ancestor (TMRCA) for the paternal and maternal chromosomes among the thousands of unlinked genomic regions in a given genome. In this manuscript, we show that individual admixed genomes contain a wealth of information about the admixture history and about the ancient demography of the source populations, and we propose a diversity of tools for leveraging this information at the individual and population levels.
Material and Methods
Genotype Phasing and Reference Panels
Phased haplotypes required for local-ancestry inference were obtained from genotyped trios with the use of BEAGLE.19 For the YRI (Yoruba in Ibadan, Nigeria) and CEU (Utah residents with ancestry from northern and western Europe from the CEPH collection) reference panels, as well as for ASW (African ancestry in Southwest USA) and MXL (Mexican ancestry in Los Angeles, California) individuals, we used SNP genotypes reported by the Phase 3 HapMap Project.6 For sample NA19730, we used genotypes from HapMap 3 release 1. For NA19648 and the PUR (Puerto Rican in Puerto Rico) trio, we used genotypes obtained from Complete Genomics sequencing at HapMap 3 positions. Given that currently available Native American reference panels do not include trio samples, we generated Affymetrix 6.0 SNP array data for ten trios of Native American populations from Mexico, which capture the diversity of Native American ancestry present in the samples analyzed (Figure S2, available online). Analysis utilized the four unique haplotypes (transmitted and nontransmitted) represented by each trio.
Local-Ancestry Assignment
We performed local-ancestry assignment by using PCAdmix.20,21 This approach relies on phased data from reference panels and the admixed individuals. Each chromosome is analyzed independently, and local-ancestry assignment is based on loadings from principal-component (PC) analysis of the three putative ancestral population panels. The scores from the first two PCs are calculated in windows of 100 SNPs for each panel individual (after the removal of SNPs with linkage disequilibrium [LD] > 0.80 for a total of 288,112 SNPs and a median window length of 847 kb). For each window, the distribution of individual scores within a population is modeled to fit a multivariate normal distribution. Given an admixed chromosome, these distributions are used for computing likelihoods of belonging to each panel. These scores are then analyzed in a hidden Markov model (HMM) with transition probabilities as in Bryc et al.22 The g (generations) parameter in the HMM transition model was determined iteratively for maximizing the total likelihood of each analyzed population. Local-ancestry assignments were determined with a 0.9 posterior probability threshold for each window with the use of the forward-backward algorithm. In analyses that required estimating the length of continuous ancestry tracts, the Viterbi algorithm was used. An assessment of the accuracy of this approach is given in the Appendix SA.
Admixture Characterization from Ancestry Tract-Length Distributions
The length distribution of continuous ancestry tracts obtained with the Viterbi analysis in PCAdmix was compared to predictions with the use of an extended space Markov model of ancestry along the genome. States of this Markov model are labeled by both population of origin and time of migration. Transition rates between states are functions of the time-dependent gene flow.23 Because the number of time-dependent gene-flow models is very large, we chose simple parameterizations of the models; for African Americans, we considered a pulse of European and Native American migration; this was followed by a pulse of African migration and another pulse of European migration. Timing and magnitude of each migration pulse was allowed to vary and was fitted to the data in a maximum-likelihood framework.23 For the Mexican Americans, we considered a model with continuous gene flow for both Native and European ancestries and a pulse of gene flow for African ancestry.
Genome Sequence Data
We analyzed full-genome sequence data from 50 individuals (Table S1), including 48 samples (coverage of 46–80×) from 11 populations sequenced by Complete Genomics and two samples (NA19836 and NA19730; coverages of 21× and 18×, respectively) sequenced with SOLiD technology. This study utilized genomic data generated from cell lines of deidentified individuals from the HapMap Project and 1000 Genomes Project.
SOLiD Data Processing
We used the Applied Biosystems (ABI) SOLiD Corona Lite pipeline to align reads to the reference human sequence (NCBI build h36, UCSC hg18). The program guarantees finding all alignments between a read and the reference sequence with up to M mismatches.24 We allowed up to two mismatches for 25 bp reads and up to five mismatches for 50 bp reads. After alignment, a pairing step was performed for identifying mate pairs that are in the proper order and orientation and that lie at distances within the empirical distribution of distances for the libraries created (target insert size of 1,500 bp). Pairs of reads only aligning in one location in the color-space reference with up to the given number of mismatches were referred to as uniquely aligned and were used for the subsequent analysis steps (beads with a missing pair were not used for SNP detection). The total average haploid coverage of uniquely mapping paired reads achieved was 21× for sample NA19836 and 18× for NA19730. The ABI SOLiD diBayes algorithm was used for SNP detection from the color-space alignments.24 diBayes is a Bayesian algorithm that includes color-space error detection—an error model that uses probe and positional errors, as well as color quality values—and the prior probability of population heterozygosity in a framework similar to that of PolyBayes.25 The raw diBayes output for chr1–chr22 and chrX consisted of 3,824,140 variant positions for NA19730 and 4,681,232 variant positions for NA19836.
We applied an additional set of filters before beginning our analysis. First, we removed variants with a depth of coverage greater than or equal to 100. From the remaining variants, we established a maximum coverage threshold of 3 standard deviations above the mean coverage, which corresponded to a coverage threshold of 36 for NA19730 and of 38 for NA19836. Next, in order to limit spurious SNPs caused by mapping artifacts or indels, we removed all variants located in regions annotated as simple repeats (by the tandem repeats finder program26) as reported on the UCSC Genome Browser, as well as positions located within 50 bp of a small indel identified by the SOLiD small indel tool. For analyses in which correct assignment of heterozygous genotypes is critical, we further restricted SNPs to a set with coverage of at least six reads. We identified genomic regions in which a SNP call could have been made by the application of the same read depth, small indel, and simple-repeat filters. Additionally, we identified uncalled regions of questionable sequence quality by using the calling failure flags reported by the diBayes consensus call file. Combining these callable regions with identified SNPs permitted us to identify positions for which an individual was homozygous for the reference allele.
Complete Genomics Data Processing
Genome data for individuals sequenced by Complete Genomics are available at the website provided in the Web Resources. These data were generated and analyzed with Complete’s local de novo assembly-based pipeline.27 Specifically, we used the alignments against NCBI build 36 and data processed with pipeline version 1.10. We selected sites with single-nucleotide variants (SNVs) called at the default thresholds as included in the CG “var” files, and we considered “no-call” regions separately from those confidently called homozygous reference in each genome. Because our analysis focused on SNVs, positions containing called insertions, deletions, substitutions, or partial (half) calls were excluded from this analysis. Copy-number- and structural-variation predictions from the CG pipeline were not considered.
TMRCA Estimation from Sequence Data
Estimated TMRCA for two haplotypes was calculated in nonoverlapping 10 kb windows for each pair of homologous chromosomes in each individual. For SOLiD data, analysis was limited to callable positions that contained a read depth of at least 6. Complete Genomics data were filtered as described above. To be included in the analysis, a 10 kb window had to contain at least 5 kb of “callable” sequence, contain 5 kb of sequence that aligned to a primate out group, and be entirely contained within a single ancestry assignment class. We additionally removed all windows that overlapped with regions annotated as segmental duplications in the UCSC Genome Browser.
On the basis of the observed number of heterozygous positions, TMRCA was estimated as described by Tavare et al.28 under a constant population-size assumption. The conditional expected time for a pair of chromosomes to coalesce given k nucleotide differences in a specified nonrecombining region is
E(T2|knucleotidedifferences)=(1+k)(1+θ),
where θ is the population genetic mutation rate (θ = 4 N e μ), N e is the effective population size, and μ is the per-generation mutation rate for this genomic region. This estimator calibrates SNP diversity by an estimate of the local mutation rate. Mutation rates were normalized on the basis of comparisons to chimpanzees with the use of genome alignments to the human hg18 assembly obtained from the UCSC Genome browser. Following Glazko and Nei,29 we fixed the human-chimp separation to be 240,000 generations and expressed times in years by assuming 25 years per generation and an effective population size of 10,000.
PSMC Analysis from Sequence Data
Pairwise sequentially Markovian coalescent (PSMC) analysis was performed as described with the use of default parameters.18 In brief, the input to the PSMC method is an encoding of 100 bp windows along the genome and indicates whether each window contains at least one heterozygous position or no heterozygous positions or is not callable. For the ancestry-specific analysis of admixed genomes, we labeled the ancestry segments considered callable and split the data into separate segments for analysis whenever a stretch of 15 kb of noncallable data was encountered.
PolyPhen Analysis
Autosomal SNPs for all individuals were run through an in-house SNP annotation pipeline restricted to those protein-coding variants with a UniProtKB annotation and scored with the PolyPhen-2 algorithm with thresholds set with the use of the HumDiv data set, which is the version recommended for predicting the damaging effect of nonsynonymous SNPs obtained from sequence data.30 For SNPs with a prediction by PolyPhen-2, a probability of being deleterious was reported. These probabilities were partitioned with cutoffs of 0.2 and 0.8 for assigning categories of “benign,” “possibly damaging,” and “probably damaging” as per PolyPhen-2 documentation. A generalized linear model of the counts in each PolyPhen category was fitted in R under the assumption of a Poisson distribution of counts in each category.
Results
Sequence Variation among Individuals
The genomic data we analyzed were collected by Complete Genomics27 and Applied Biosystems SOLiD24 sequencing from 50 individuals across 11 populations sampled by the 1,000 Genomes and International HapMap Projects. These individuals included those with African (YRI, MKK [Maasai from Kenya], and LWK [Luhya from Webuye, Kenya]), South Asian (GIH [Gujurati Indian from Houston, TX]), European (CEU and TSI [Tuscans from Italy]), and East Asian (CHB [Han Chinese from Beijing] and JPT [Japanese from Tokyo]) ancestry, as well as African Americans (ASW), Mexican Americans (MXL), and Puerto Ricans (PUR). After applying a series of filters, we assessed the quality of the SNP calls made from short-read sequence data by comparing them to genotypes inferred from high-density arrays on a subset of individuals, and we found an average of greater than 99.5% concordance across millions of common genetic variants (Table S1). As a first means of comparing these genomes, we estimated nucleotide diversity across an average of 2.5 billion callable sites by using two metrics: the average number of SNPs per kilobase relative to the genome reference sequence and the average nucleotide heterozygosity for each genome (Figure 1 and Table S2). Consistent with expectations from studies of haplotype and sequence diversity,6,31 we found striking variation in genome-wide nucleotide diversity across individuals sequenced with the same technologies. For example, African genomes harbored, on average, 18% more genetic variation than non-African genomes. As expected,32 regions of the genome with higher levels of selective constraint as measured by the GERP algorithm33 showed fewer polymorphisms and proportionally more heterozygous than homozygous alternative sites per genome (Figure S1). However, there are systematic differences across genomes of varying ethnic ancestry in this ratio.
Figure 1.
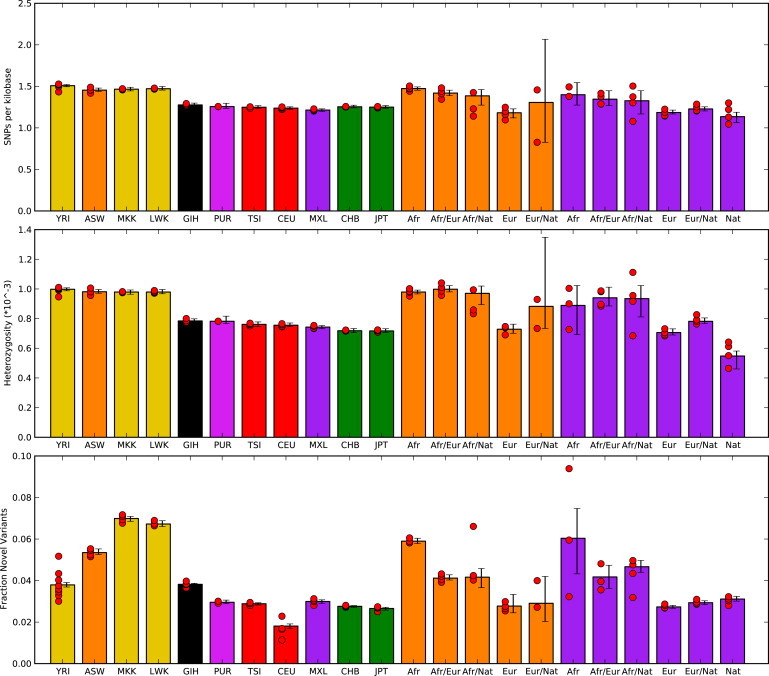
Summary Statistics from Individual Sequenced Genome
Individual diversity statistics are given on the basis of sequence data from Complete Genomics. In addition to mean values from each population, results partitioned by inferred local genomic ancestry are given for the ASW (African ancestry in Southwest USA) (orange bars) and MXL (Mexican ancestry in Los Angeles, California) (purple bars) populations. Only individuals with at least 1 MB of each assigned ancestry are included. Novel SNPs were determined relative to variants discovered by the 1000 Genomes low-coverage sequencing pilot and were limited to genomic positions interrogated by the project. Red circles represent mean values for each sample, and error bars represent 95% confidence intervals found by bootstrap resampling across all chromosomes from samples for each population.
Sequence Variation within Individuals with Recent Admixed Ancestry
Several aspects of sequence variation, including the density of SNPs, the degree of nucleotide heterozygosity, the proportion of novel SNPs relative to other sequencing efforts, and the ratio of synonymous to nonsynonymous polymorphisms, vary in admixed individuals in a manner correlated with global ancestry (Figure 1 and Table S2). However, these measures of diversity are not simple weighted averages over values of the different ancestries; for example, we observed more low-frequency alleles in the five sequenced MXL individuals than in other populations, such as CEU or CHB.
Limiting the analysis to genomic segments from the MXL population inferred to have European ancestry removes this effect (see Figure 2 and the next section). The presence of more rare variants than expected in a neutral, constant-size panmictic population (standard neutral model [SNM]) has been demonstrated in human populations as a result of nonneutral evolution and nonconstant population sizes; Figure 2 shows that admixture also leads to more rare variants than expected in the SNM.
Figure 2.
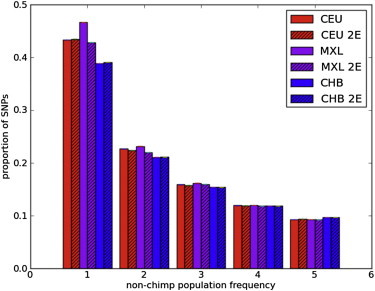
Impact of Admixture on the Site Frequency Spectrum
The MXL population shows more rare variants than the CEU (Utah residents with ancestry from northern and western Europe from the CEPH collection) or CHB (Han Chinese from Beijing) populations. Limiting consideration to MXL segments with inferred European ancestry (MXL 2E) removes this effect.
Local-Genomic-Ancestry Assignment with SNP Haplotypes
To explore the impact of continental ancestry on genetic variation within individuals, we focused on 12 genomes from individuals who have recent (<500 years) ancestry from different continental populations. One individual, sequenced with the SOLiD system, was estimated to have over 88% Native American ancestry (Figure 3A). Because of the relative demographic isolation of European, African, and Native American groups from 20,000 years ago to the dawn of intercontinental travel, individual haplotypes in current populations can often be traced back to a specific continental group over this period. Using the PCAdmix algorithm,20–22 we partitioned each chromosome into segments of inferred African, European, or Native American ancestry. We obtained phased haplotypes for variants genotyped in parent-offspring trios, including a set of Mexican Native American trios genotyped for this project (Figure S2). We analyzed each chromosome independently and yielded tracts of ancestry along each chromosome (Figure 3B). We assessed the accuracy of our assignments by using simulations that construct simulated admixed genomes of known ancestry on the basis of copying phased haplotypes from a reference panel (see Appendix SA). Our simulations indicate that a cutoff of 0.9 applied to the posterior probability for the ancestry of each window (calculated with the forward-backward algorithm) yields an accuracy rate of 80%–99% for local-genomic-ancestry assignment (see Appendix SA). With these criteria, we assigned an inferred ancestry to 79%–95% of each genome (Table S3).
Figure 3.
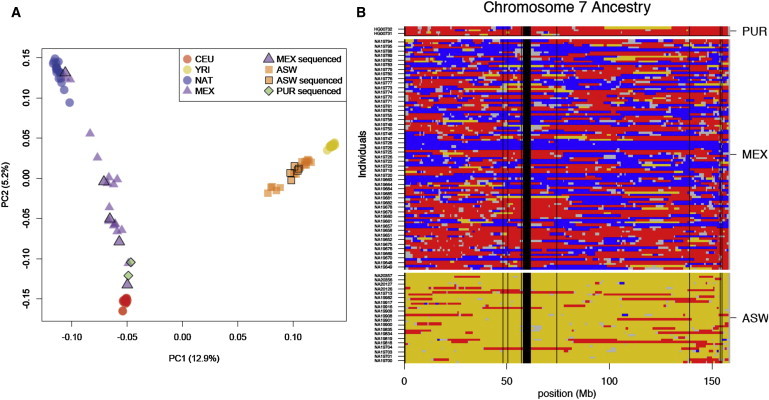
Local-Ancestry Inference
Local genomic ancestry was inferred for the genomes of admixed individuals with the use of PCAdmix.
(A) The first two principal components of variation for admixed individuals are shown relative to European, African, and Native American source populations. The markers outlined in black represent 12 admixed individuals who have been sequenced.
(B) Ancestry assignment for chromosome 7. The use of phased haplotypes obtained from trios permit assignment of ancestry for each transmitted and nontransmitted chromosome separately. The following colors are used: red, inferred European ancestry; yellow, inferred African ancestry; blue, inferred Native American ancestry; gray, regions not assigned; and black, centromere and genome assembly gaps.
Inference of Population History with Ancestry Tract Lengths
To explore the recent history of admixed populations, we considered the length distribution of continuous tracts of inferred ancestry provided by Viterbi decoding.34 Broadly speaking, we expect that the average length of ancestry tracts decreases with time after admixture and that the overall shape of the distribution gives hints about the actual migratory process. Using models including repeated or continuous gene flow, we modeled tract-length distributions obtained from phased SNP genotype information6 for 100 haploid MXL genomes from 25 trios and for 40 haploid ASW genomes from 10 trios.
We observed departures from the predictions of the commonly used “pulse” admixture model, in which each population contributes migrants at a discrete period in time. Specifically, we observed an excess of long European tracts in both populations and an excess of long Native American tracts in the Mexican population, suggesting more recent gene flow from these groups. Indeed, allowing for continuous or repeated migration results in very good agreement with the data (Figure 4) and gives admixture timing estimates somewhat older than those previously obtained from genetic data.35,36 Both models involve continuous gene flow from a European source, and the Mexican American model also includes continuous Native American gene flow. The best-fit model for African Americans involves admixture, starting 15 generations ago, between individuals of African ancestry and a population of mixed European and Native American ancestry. This model suggests that 70% of the European ancestry in today’s African Americans dates back to European gene flow 7–8 generations ago.
Figure 4.
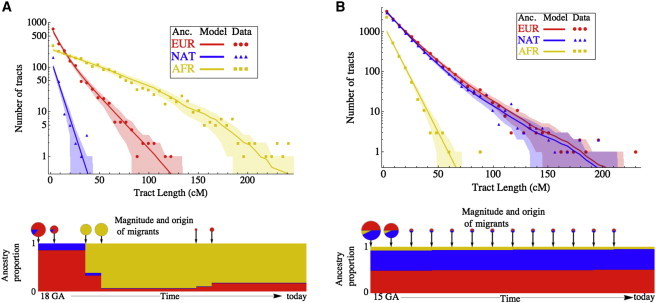
Inference from Admixture Tract-Length Distributions
The distribution of lengths of European, African, and Native American ancestry tracts are shown for the (A) MXL and (B) ASW populations. Analysis considered parents of genotyped HapMap 3 trios. The dots indicate observed data obtained from the Viterbi local-ancestry assignment from PCAdmix. The lines and shading represent predictions and 95% confidence intervals, respectively, obtained from the models indicated. The amount and origin of gene flow are indicated by pie-chart size and coloring, and the ancestry proportion over time in the model population is illustrated below.
Such models are simplifications of the historical processes, and we have limited power to detect some aspects of the migration history, such as the duration of the putative second pulse of European migration; such finer analyses would most likely require the integration of genetic and historical data. Nevertheless, the continuous-gene-flow model in Mexican Americans resolves an apparent inconsistency: although the historical record suggests that the contact between Europeans and Native American predated the onset of the slave trade, the existence of relatively long Native American and short west African tracts suggests that European and west African admixture must have predated contact with Native Americans. Johnson et al.37 hypothesized that this might be due to significant European and west African admixture prior to the development of the slave trade. However, our results show that such a hypothesis is not necessary and that more detailed quantitative models of recent, continuous gene flow can account for the data. We finally point out that even though the continuous-gene-flow models presented here allow for improved agreement with both historical and genetic data, they are still simplifications of the historical process. Distinguishing finer patterns of time-dependent gene flow will most likely require the integration of genetic and historical data.
Inference of Population History from Sequence Data
The observed patterns of sequence variation among the genomes of individuals with recent admixed ancestry reflect population-history differences—e.g., periods of growth and bottleneck leading to changes in effective population size—experienced by the ancestral populations that contribute to the genomic repertoire of present-day admixed populations. Because of recombination, the diploid genome of a single individual represents samplings of hundreds of thousands of independent lineages that coalesced sometime in the past. We explored the distribution of the age of these coalescence events (the TMRCA) by calculating sequence divergence between pairs of homologous chromosomes in each individual for nonoverlapping 10 kb segments. On the basis of divergence from chimpanzees and assumptions about chimp-human speciation time and long-term effective population sizes,29 we estimated the TMRCA for each genomic window.28 This approach shows clear differences among major population groups. For example, the genomes of European populations have a higher fraction of more recent TMRCA than do those of African populations (Figure 5A), and Asian populations are more recent than European populations, consistent with stronger bottlenecks in these groups.38 We applied this approach to the genomes of the admixed individuals and analyzed each category of ancestry separately. In this manner, we could study multiple ancestral populations by sequencing single individuals. This analysis largely recapitulated expected patterns of history; Native American portions of the genome had the highest rate of recent coalescence times and were followed by Europeans (Figure 5B and Figure S3). Because local-ancestry inference can be performed with proxy populations, this demonstrates how whole-genome demographic analysis can be performed in the absence of data from the actual source populations. Furthermore, only genotype data from the panel samples are required for performing such analyses on sequencing data from the admixed population.
Figure 5.
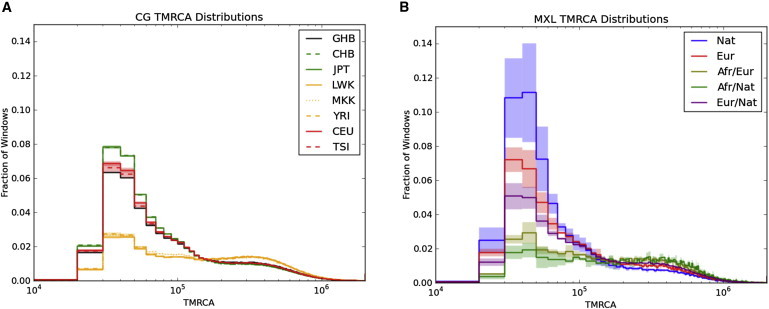
Distribution of Inferred TMRCA
The distribution of inferred TMRCA is calculated in 10 kb windows scaled with chimpanzee divergence and is shown for (A) eight populations and (B) local ancestry in MXL. The lines indicate means, and the shading represents 95% confidence intervals for each bin determined from the samples depicted in Figure 1.
The proportion of lineages that coalesce in a time period is informative about the effective population size at that time. We used these data to explore changes in effective population sizes over time by using the recently described PSMC model,18,39 which also considers sequence differences between homologous chromosomes within a single individual. Using a larger set of genomes, we recapitulated the basic picture of human population history previously reported,18 but our larger sample size allowed for a finer analysis. Using this method on 50 individual genomes, we were able to clearly observe population-size change associated with the out-of-Africa migration (Figure 6A). We also saw long term (>100,000 years ago) changes in inferred effective population size, which might indicate the formation and dissolution of population structure among ancestral human groups.18 We found that individuals from a given population sequenced with the same technology show very consistent results but that differences between Complete Genomics and SOLiD sequencing platforms result in differences in inferred population sizes, emphasizing that the PSMC model might be highly sensitive to technical biases in whole-genome sequencing. Further study of systematic technical biases in genome sequencing associated with a variety of technologies is warranted.40,41 Because the results from the Complete Genomics platform are in better agreement to those of Li and Durbin (when they are applied to samples from related populations) and because Table S1 shows that Complete Genomics data have a higher concordance rate with genotype data than do the Solid Technology data analyzed in this study (perhaps as a result of higher coverage), we propose that inaccuracies in the genomes sequenced with Solid Technology might explain the bulk of the difference.
Figure 6.
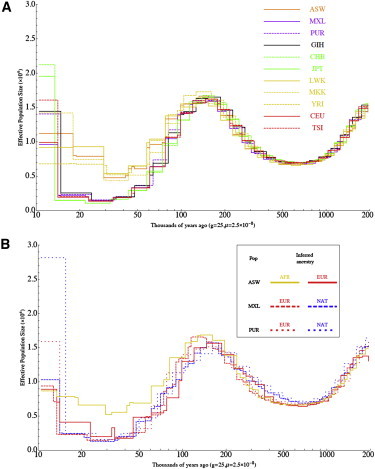
Demographic History Inferred with PSMC
Estimates of effective population size over time are shown for (A) 11 populations and (B) three ancestries on the basis of local-ancestry inference in three populations with the use of PSMC, which estimates effective population sizes at different time intervals on the basis of the distribution of TMRCA estimates across the genome. Lines represent mean values obtained from separate analysis of each sample.
In the hope of learning about the demography of Native American groups, we applied this approach to partitioned admixed genomes, including Native American genomic segments inferred from the MXL samples and African segments from the ASW samples (Figure 6B and Figure S4). To avoid biases due to errors in phasing variants discovered by sequencing, we limited our analysis to segments of double African, double Native American, or double European ancestries, and it is therefore noisier than results based on whole genomes. On the basis of individual admixed African American genomes, we were able to recover the histories inferred from African and European individuals. Applying the same method to segments of inferred Native American ancestry, we found in the Native American ancestral population evidence of a strong bottleneck of a magnitude comparable to that observed in European and Asian groups. The PSMC method does not allow for accurate inferences over the last ∼25,000 years, and recent differences in demography are therefore not distinguished by this method. The development of methods that can handle many individuals is therefore highly desirable for resolving many questions about recent human demography, including past populations whose descendents have had complex admixture histories.
Impact of Ancestry on Patterns of Deleterious Variation
Demography impacts the relative allele-frequency distribution of neutral and functional alleles. Populations that have undergone bottlenecks followed by growth, for example, are expected to have fewer segregating sites, proportionally more segregating deleterious variants,4 and a higher proportion of rare genetic variants than a constant-sized population. To facilitate comparison across all panel populations, we focused on the distribution of heterozygous and homozygous nonreference alleles within individuals (i.e., the two-chromosome frequency spectrum) and its variation across ancestry and functional class. Focusing on amino-acid-changing variants, we obtained counts Counts AZP of variants in bins defined by ancestry A (e.g., ASW, CEU, CHB, etc.), zygosity Z (homozygous nonreference or heterozygous), and PolyPhen2-predicted functional impact P (benign, possibly damaging, or probably damaging30). Obtaining a mechanistic model accounting for the joint effects of population size fluctuations, selection, and gene flow is challenging. As a simple alternative, we considered different generalized linear models for Counts AZP. The basic, fully saturated model has the form
Log(CountsAZP)=β+βA+βZ+βP+βAZ+βAP+βZP+βAZP,
where β represents fitting parameters (see Appendix SB for model details and additional motivation). A first observation is that the linear and pairwise interaction terms do not provide a satisfactory description of the data and that β AZP terms are highly significant: when compared to the ASW population, the out-of-Africa panels CEU, CHB, GIH, JPT, and TSI are expected to exhibit increased homozygosity and increased PolyPhen deleterious alleles (Figure 7). In addition, we found that the increase in probably damaging variants is more pronounced among homozygous sites (all p < 0.00036, see Supplemental Data and Appendix SB). Thus, the expected proportion of recessive variants among out-of-Africa populations not only is higher than what would be predicted by the increase of homozygosity alone and by the larger number of deleterious variants alone but is also larger than what would be predicted on the basis of a product of the two effects. This effect also holds if we consider the inferred diploid local ancestries in admixed genomes; segments of either double Native American, double European, or joint Native American and European ancestries show excesses of homozygous probably damaging variants (all p < 7.2 × 10−6, see Supplemental Data).
Figure 7.
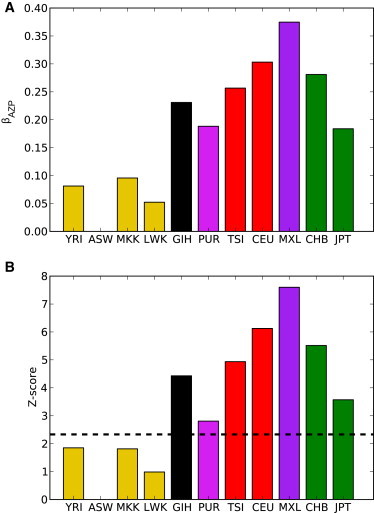
Interaction Coefficients Inferred for a Model of Deleterious Variant Counts
Estimated values for β AZP, the interaction term representing joint effects of population ancestry, zygosity, and PolyPhen status on the counts of variants, are shown for Z = “homozygous” and p = “probably damaging” for each population. Coefficient values (A) are given relative to counts in the ASW population. Z scores of coefficient significance are given in (B). The dashed line corresponds to p = 0.01.
We can also use the GLM model to test whether the inferred local ancestries provide a complete description of the observed patterns of diversity in the admixed genomes from different populations. Interestingly, we found that an additional term, β POP(I)Z, describing the effect of the population-of-origin pop(I) on the Counts IAZP per individual in the admixed populations, leads to an AIC (Akaike information criterion) improvement of 57. We thus found that African Americans have the highest degree of heterozygosity and are followed by the PUR and the MXL groups even after the effects of global ancestry proportions have been taken into account (Table S7). This can indeed be observed in Figure 1: the heterozygosity in African Americans is higher than that of the MXL samples for all inferred local-ancestry categories. Importantly, these effects are consistent across individuals from a given population: adding individual-level terms β IZ does not provide significant improvement to the fit, and indeed, the inferred β IZ terms are very similar for different individuals in the same ancestry. (Table S8)
Discussion
Human-population history, including prehistoric migrations and population-size changes, as well as more recent migrations induced by exogenous settlement of territories in the Americas, Australia, Africa, and Asia, have profoundly affected the distribution of genetic variation around the world. An open question is the degree to which individual_-_level data can be used for drawing inferences regarding these population_-_level processes. By analyzing full-genome sequence data from 50 individuals across 14 populations, we demonstrate that personal genomic data recapitulates key aspects of the population history from which the genomes were drawn. This includes ancient demographic events (such as the out-of-Africa bottleneck), as well as the much more recent “personal genetic history” of admixed genomes with regard to the proportion of ancestry drawn from different ancestral populations and a broad outline of the history of how admixture occurred. Simulations suggest that the inference strategy we have used is accurate for the Mexican, Puerto Rican, and African American populations studied here.
Many of our analyses of individual human genomes recapitulated what has been learned through decades of sequencing population-based samples. For example, we found that human genomes vary by nearly 50% in the degree of nucleotide heterozygosity and that west African genomes are vastly more diverse than non-African genomes. We also found, surprisingly, that the proportion of homozygous versus heterozygous benign, possibly, and probably damaging alleles annotated per genome is highly consistent for genomes from the same population and differs dramatically across genomes from different populations. Furthermore, the generalized linear model we introduce emphasizes subtle differences in heterozygosity between segments of identical continental ancestry in different populations; ASW samples showed the highest overall heterozygosity after continental ancestry was factored in. This observation is consistent with proportionally less inbreeding in the ASW population. It is also possible that the European, African, and Native American source populations for the ASW samples happened to have higher diversity than the corresponding MXL source populations. Finally, at this level of accuracy, we might also be detecting small levels of misassigned ancestry, which would tend to increase diversity across all ancestry categories in populations with more west African ancestry.
We have found that we can reliably reconstruct the continental ancestry along genomes of individuals with admixed ancestry as evidenced both by simulations that show an accuracy rate of 80%–99% for local-ancestry assignment and by the consistency of nucleotide diversity among inferred genomic segments (Figure 1). For example, the amount of genetic diversity in segments of African ancestry in African Americans resembles that of sequenced west African populations (∼0.1% nucleotide heterozygosity, ∼5% novel alleles relative to the 1000 Genomes Pilot Project, and 1.5 SNPs per kilobase), and similarly, the segments of inferred European ancestry in African Americans and Mexicans resemble those of sequenced west European populations (∼0.07% nucleotide heterozygosity, 2% novelty rate relative to the 1000 Genomes Pilot Project, and one SNP per kilobase). This gives us confidence that the results we present for Native American ancestry provide useful information about the genomic diversity of this ancestry, for which sequence data were not available. Such an approach will be useful in the study of the numerous historical populations whose descendents share ancestors with diverged populations.
Using a generalized linear model to study the distribution of predicted deleterious or benign alleles, we found that demographic history and natural selection have had a complex interplay to pattern genomes from diverse human populations. Our results from 50 sequenced human genomes are consistent with deep exome data10 and genome-wide population-genetics analyses42,43 that suggest that negative selection is the predominant mode of selection in functional regions of the human genome.
Aside from characteristics of the source populations, we have examined the genomic diversity of the admixed population themselves. Genome-wide statistics of the admixed individuals are consistent with models of their genomes as mosaics of segments from the diverged populations. An important consequence for population-genomics analysis is the observation that the allele-frequency distribution had more rare variants than expected under a uniform population. Additionally, we show that the mosaic ancestry patterns themselves provide information specific to recent gene-flow intensities. Using detailed gene-flow models, we have obtained evidence for continuous gene flow in both African Americans and Mexicans, resolving an apparent inconsistency observed in previous studies assuming a discrete gene-flow event.
The analysis we have presented here integrated trio-phased genotype data from source-population proxies and the admixed populations, as well as full sequence data from admixed individuals. One limitation of the current analysis was the difficulty of accurately phasing rare variants obtained in the sequence data. We expect that the availability of such phased data (through trio sequencing or new experimental approaches44,45) will considerably increase the power of inferences for events both prior and posterior to the gene flow.
As the medical community continues to expand its efforts to increase the representation of diverse populations and as the availability of whole-genome sequence data increases, the need for detailed and quantitative models for interpreting and modeling admixed genomes will increase. Many of the standard population-genetics tools do not apply directly to such populations, but we have shown that the application of local-ancestry inference can provide a simple basis for developing tractable extensions of standard methods for learning about complex demographic events in recently admixed populations.
Acknowledgments
We thank Melisa Barker, Fiona Hyland, Clarence Lee, Kirk Lohmueller, Kevin McKernan and Koni Wright for advice about this project. J.M.K was supported by National Institutes of Health training grant T32HG000044. Additional support was provided by grants R01ES015794 and R01HL088133 (E.G.B.) and 1R44HG3667 (M.G.R). Vrunda Sheth, Stephen F. McLaughlin, Heather E. Peckham, Christina A. Bormann Chung, Sarah Stanley, Kevin Pearlstein, Elizabeth Levandowsky, and Francisco M. De La Vega are or were employees of Life Technologies during this study. Archie Russell and Martin Reese are employees of Omicia. The Varimed database has been licensed to Personalis. Atul Butte is a founder and consults for Personalis. Carlos D. Bustamante also consults for Personalis, as well as Ancestry.com, Locus Development, and the 23andMe.com project “Roots into the Future.” Stephen E. Lincoln was an employee of Complete Genomics during this study. Jake Byrnes is an employee of Ancestry.com.
Contributor Information
Francisco M. De La Vega, Email: francisco.dlv@gmail.com.
Carlos D. Bustamante, Email: cdbustam@stanford.edu.
Supplemental Data
Document S1. Figures S1–S6, Tables S1–S8, and supplemental appendices giving sensitivity and specificity results based on simulations.
Web Resources
The URLs for data presented herein are as follows:
- Complete Genomics public FTP site, ftp://ftp2.completegenomics.com/
- PCAdmix Software, http://sites.google.com/site/pcadmix/
Accession Numbers
Data for the two genomes sequenced with SOLiD technology are available at the Sequence Read Archive under accession number SRA056457.
References
- 1.International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chakraborty R., Weiss K.M. Admixture as a tool for finding linked genes and detecting that difference from allelic association between loci. Proc. Natl. Acad. Sci. USA. 1988;85:9119–9123. doi: 10.1073/pnas.85.23.9119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg N.A., Huang L., Jewett E.M., Szpiech Z.A., Jankovic I., Boehnke M. Genome-wide association studies in diverse populations. Nat. Rev. Genet. 2010;11:356–366. doi: 10.1038/nrg2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lohmueller K.E., Indap A.R., Schmidt S., Boyko A.R., Hernandez R.D., Hubisz M.J., Sninsky J.J., White T.J., Sunyaev S.R., Nielsen R. Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451:994–997. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bustamante C.D., Burchard E.G., De la Vega F.M. Genomics for the world. Nature. 2011;475:163–165. doi: 10.1038/475163a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altshuler D.M., Gibbs R.A., Peltonen L., Altshuler D.M., Gibbs R.A., Peltonen L., Dermitzakis E., Schaffner S.F., Yu F., Peltonen L., International HapMap 3 Consortium Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L., Myers R.M. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 8.Auton A., Bryc K., Boyko A.R., Lohmueller K.E., Novembre J., Reynolds A., Indap A., Wright M.H., Degenhardt J.D., Gutenkunst R.N. Global distribution of genomic diversity underscores rich complex history of continental human populations. Genome Res. 2009;19:795–803. doi: 10.1101/gr.088898.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., Broad GO. Seattle GO. NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bercovici S., Geiger D. Inferring ancestries efficiently in admixed populations with linkage disequilibrium. J. Comput. Biol. 2009;16:1141–1150. doi: 10.1089/cmb.2009.0105. [DOI] [PubMed] [Google Scholar]
- 12.Patterson N., Hattangadi N., Lane B., Lohmueller K.E., Hafler D.A., Oksenberg J.R., Hauser S.L., Smith M.W., O’Brien S.J., Altshuler D. Methods for high-density admixture mapping of disease genes. Am. J. Hum. Genet. 2004;74:979–1000. doi: 10.1086/420871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falush D., Stephens M., Pritchard J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang H., Coram M., Wang P., Zhu X., Risch N. Reconstructing genetic ancestry blocks in admixed individuals. Am. J. Hum. Genet. 2006;79:1–12. doi: 10.1086/504302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Price A.L., Tandon A., Patterson N., Barnes K.C., Rafaels N., Ruczinski I., Beaty T.H., Mathias R., Reich D., Myers S. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet. 2009;5:e1000519. doi: 10.1371/journal.pgen.1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pasaniuc B., Sankararaman S., Kimmel G., Halperin E. Inference of locus-specific ancestry in closely related populations. Bioinformatics. 2009;25:i213–i221. doi: 10.1093/bioinformatics/btp197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sankararaman S., Sridhar S., Kimmel G., Halperin E. Estimating local ancestry in admixed populations. Am. J. Hum. Genet. 2008;82:290–303. doi: 10.1016/j.ajhg.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H., Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Browning S.R., Browning B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brisbin A. Cornell University; Ithaca, New York: 2010. Linkage Analysis for Categorical Traits and Ancestry Assignment in Admixed Individuals. [Google Scholar]
- 21.Henn B.M., Botigué L.R., Gravel S., Wang W., Brisbin A., Byrnes J.K., Fadhlaoui-Zid K., Zalloua P.A., Moreno-Estrada A., Bertranpetit J. Genomic ancestry of North Africans supports back-to-Africa migrations. PLoS Genet. 2012;8:e1002397. doi: 10.1371/journal.pgen.1002397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bryc K., Auton A., Nelson M.R., Oksenberg J.R., Hauser S.L., Williams S., Froment A., Bodo J.M., Wambebe C., Tishkoff S.A., Bustamante C.D. Genome-wide patterns of population structure and admixture in West Africans and African Americans. Proc. Natl. Acad. Sci. USA. 2010;107:786–791. doi: 10.1073/pnas.0909559107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gravel S. Population genetics models of local ancestry. Genetics. 2012;191:607–619. doi: 10.1534/genetics.112.139808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKernan K.J., Peckham H.E., Costa G.L., McLaughlin S.F., Fu Y., Tsung E.F., Clouser C.R., Duncan C., Ichikawa J.K., Lee C.C. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–1541. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marth G.T., Korf I., Yandell M.D., Yeh R.T., Gu Z., Zakeri H., Stitziel N.O., Hillier L., Kwok P.Y., Gish W.R. A general approach to single-nucleotide polymorphism discovery. Nat. Genet. 1999;23:452–456. doi: 10.1038/70570. [DOI] [PubMed] [Google Scholar]
- 26.Benson G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drmanac R., Sparks A.B., Callow M.J., Halpern A.L., Burns N.L., Kermani B.G., Carnevali P., Nazarenko I., Nilsen G.B., Yeung G. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327:78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- 28.Tavaré S., Balding D.J., Griffiths R.C., Donnelly P. Inferring coalescence times from DNA sequence data. Genetics. 1997;145:505–518. doi: 10.1093/genetics/145.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glazko G.V., Nei M. Estimation of divergence times for major lineages of primate species. Mol. Biol. Evol. 2003;20:424–434. doi: 10.1093/molbev/msg050. [DOI] [PubMed] [Google Scholar]
- 30.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conrad D.F., Jakobsson M., Coop G., Wen X., Wall J.D., Rosenberg N.A., Pritchard J.K. A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat. Genet. 2006;38:1251–1260. doi: 10.1038/ng1911. [DOI] [PubMed] [Google Scholar]
- 32.Goode D.L., Cooper G.M., Schmutz J., Dickson M., Gonzales E., Tsai M., Karra K., Davydov E., Batzoglou S., Myers R.M., Sidow A. Evolutionary constraint facilitates interpretation of genetic variation in resequenced human genomes. Genome Res. 2010;20:301–310. doi: 10.1101/gr.102210.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper G.M., Stone E.A., Asimenos G., Green E.D., Batzoglou S., Sidow A., NISC Comparative Sequencing Program Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pool J.E., Nielsen R. Inference of historical changes in migration rate from the lengths of migrant tracts. Genetics. 2009;181:711–719. doi: 10.1534/genetics.108.098095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith M.W., Patterson N., Lautenberger J.A., Truelove A.L., McDonald G.J., Waliszewska A., Kessing B.D., Malasky M.J., Scafe C., Le E. A high-density admixture map for disease gene discovery in african americans. Am. J. Hum. Genet. 2004;74:1001–1013. doi: 10.1086/420856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Price A.L., Patterson N., Yu F., Cox D.R., Waliszewska A., McDonald G.J., Tandon A., Schirmer C., Neubauer J., Bedoya G. A genomewide admixture map for Latino populations. Am. J. Hum. Genet. 2007;80:1024–1036. doi: 10.1086/518313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson N.A., Coram M.A., Shriver M.D., Romieu I., Barsh G.S., London S.J., Tang H. Ancestral components of admixed genomes in a Mexican cohort. PLoS Genet. 2011;7:e1002410. doi: 10.1371/journal.pgen.1002410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keinan A., Mullikin J.C., Patterson N., Reich D. Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans. Nat. Genet. 2007;39:1251–1255. doi: 10.1038/ng2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McVean G.A., Cardin N.J. Approximating the coalescent with recombination. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005;360:1387–1393. doi: 10.1098/rstb.2005.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harismendy O., Ng P.C., Strausberg R.L., Wang X., Stockwell T.B., Beeson K.Y., Schork N.J., Murray S.S., Topol E.J., Levy S., Frazer K.A. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009;10:R32. doi: 10.1186/gb-2009-10-3-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lam H.Y., Clark M.J., Chen R., Chen R., Natsoulis G., O’Huallachain M., Dewey F.E., Habegger L., Ashley E.A., Gerstein M.B. Performance comparison of whole-genome sequencing platforms. Nat. Biotechnol. 2012;30:78–82. doi: 10.1038/nbt.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McVicker G., Gordon D., Davis C., Green P. Widespread genomic signatures of natural selection in hominid evolution. PLoS Genet. 2009;5:e1000471. doi: 10.1371/journal.pgen.1000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hernandez R.D., Kelley J.L., Elyashiv E., Melton S.C., Auton A., McVean G., Sella G., Przeworski M., 1000 Genomes Project Classic selective sweeps were rare in recent human evolution. Science. 2011;331:920–924. doi: 10.1126/science.1198878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitzman J.O., Mackenzie A.P., Adey A., Hiatt J.B., Patwardhan R.P., Sudmant P.H., Ng S.B., Alkan C., Qiu R., Eichler E.E., Shendure J. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat. Biotechnol. 2011;29:59–63. doi: 10.1038/nbt.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fan H.C., Wang J., Potanina A., Quake S.R. Whole-genome molecular haplotyping of single cells. Nat. Biotechnol. 2011;29:51–57. doi: 10.1038/nbt.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Figures S1–S6, Tables S1–S8, and supplemental appendices giving sensitivity and specificity results based on simulations.