Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy (original) (raw)
. Author manuscript; available in PMC: 2012 Nov 1.
SUMMARY
Autophagy, the primary recycling pathway of cells, plays a critical role in mitochondrial quality control under normal growth conditions and in the response to cellular stress. The Hsp90-Cdc37 chaperone complex coordinately regulates the activity of select kinases to orchestrate many facets of the stress response. Although both maintain mitochondrial integrity, the relationship between Hsp90-Cdc37 and autophagy has not been well characterized. Ulk1, one of the mammalian homologues of yeast Atg1, is a serine-threonine kinase required for mitophagy. Here we show that the interaction between Ulk1 and Hsp90-Cdc37 stabilizes and activates Ulk1, which in turn is required for the phosphorylation and release of Atg13 from Ulk1, and for the recruitment of Atg13 to damaged mitochondria. Hsp90-Cdc37, Ulk1 and Atg13 phosphorylation are all required for efficient mitochondrial clearance. These findings establish a direct pathway that integrates Ulk1- and Atg13- directed mitophagy with the stress response coordinated by Hsp90 and Cdc37.
INTRODUCTION
Hsp90 is an abundant chaperone that directs the maturation and activation of a restricted group of metastable proteins, typically kinases and signaling molecules, to orchestrate responses to cellular stress (Li et al., 2009). Most Hsp90 clients adopt their final configuration only once they are post-translationally activated (e.g., by ligand binding and/or phosphorylation) in a manner that is facilitated by their interaction with Hsp90. The half-life and thus the activity of most Hsp90 clients relies on their association with Hsp90 and its co-chaperones, as they are rapidly degraded by the proteasome following release from the chaperone complex. The expression and activity of heat shock proteins is dramatically induced in response to heat shock and other proteotoxic stressors. This response, coupled with post-translational modifications of client proteins in complex with Hsp90, maintains cellular homeostasis by coordinately regulating changes in signal transduction pathways and transcriptional responses that promote cell survival and proliferation.
Maintenance of healthy mitochondria is essential for cellular homeostasis, as this organelle produces ATP and other essential metabolites as well as the building blocks for protein, nucleic acid and lipid biosynthesis. In addition, mitochondria harbor pools of intracellular calcium and are the principal target and relay center for cell death cascades (de Moura et al., 2010). Hsp90 also appears to be involved in mitochondrial homeostasis, specifically by regulating ubiquitin proteasome-mediated turnover of mitochondrial proteins (Margineantu et al., 2007) and the maintenance of mitochondrial membrane potential (Kang et al., 2007).
Autophagy also has important roles in controlling mitochondrial homeostasis (Bhatia-Kissova and Camougrand, 2010). Autophagy functions as the primary recycling pathway of the cell, where it directs lysosome-mediated destruction of its cellular cargo, including damaged or dysfunctional mitochondria (Kundu and Thompson, 2008). Flux through the autophagy pathway markedly increases when cells are faced with metabolic or proteotoxic stress that ensues following exposure to noxious environmental cues, for example starvation, hypoxia or heat (Amaravadi and Thompson, 2007; Liu et al., 2010). Indeed, increased turnover of mitochondria is manifest under all of these conditions (Gamboa and Andrade, 2010; Kim et al., 2007; Oberley et al., 2008; Zhang et al., 2008) and dysregulation of this process is linked to disease, including diabetes, neurodegeneration and cancer (de Moura et al., 2010; Gottlieb and Carreira, 2010). Despite the importance of Hsp90 and autophagy in maintaining mitochondrial integrity and cellular homeostasis, the interplay of the Hsp90 chaperone complex and autophagy in mitochondrial clearance has not been explored.
In yeast, the serine-threonine kinase Atg1 directs the autophagy machinery to appropriate cargo in response to changes in the availability of carbon and nitrogen (Mizushima, 2010). Ulk1, one of the mammalian homologues of Atg1, is required for starvation-induced autophagy (Chan et al., 2007) and for clearance of mitochondria in terminally differentiating erythroid cells (Kundu et al., 2008). Here, we report that Ulk1 function requires its physical interaction with Hsp90 and the kinase-specific co-chaperone Cdc37. This interaction promotes Ulk1 stability and activation, and is necessary for Ulk1-directed phosphorylation of its interacting partner Atg13 at serine 318. Further, Atg13 phosphorylation promotes its release from Ulk1 and its localization to damaged mitochondria. Accordingly, Hsp90, Cdc37, Ulk1 kinase activity and Atg13 phosphorylation are all required for efficient mitochondrial clearance. These findings define an Ulk1- and Atg13-dependent pathway that integrates autophagy into the Hsp90-coordinated stress response to govern mitochondrial homeostasis.
RESULTS
Ulk1 Interacts with the Hsp90-Cdc37 Chaperone Complex
Ulk1 plays a critical role in the autophagy-mediated clearance of mitochondria during erythroid maturation (Kundu et al., 2008). To gain insight into Ulk1 regulation, we used an unbiased proteomics approach to identify Ulk1-interacting proteins. Hsp90 and Cdc37 were identified as Ulk1-interacting partners by LC/MS and immunoblot analyses following affinity purification of Flag-tagged Ulk1 from K562 cells (Figure 1A) and NTAP-tagged Ulk1 from 293T cells (data not shown). Cdc37 was also linked to Ulk1 in a large-scale proteomics-based screen of autophagy networks (Behrends et al., 2010). The observed interactions between Ulk1 and Hsp90-Cdc37 were not an artifact of Ulk1 overexpression, as endogenous anti-Ulk1 immunoprecipitates from wild type mouse embryo fibroblasts (MEFs) contained endogenous Hsp90 and Cdc37 (Figure 1B). Similarly, endogenous Ulk1 was detected in endogenous anti-Cdc37 immunoprecipitates from wild-type MEFs (Figure S1A).
Figure 1. Ulk1 Kinase Activity and Stability are regulated by the Hsp90-Cdc37 Chaperone Complex.

(A) Hsp90 and Cdc37 were identified as Ulk1 interacting proteins using an unbiased proteomics approach. K562 cells were transfected with the indicated expression constructs. Ulk1-K46R is a ULK1 mutant with mildly impaired kinase activity (Dorsey et al., 2009a). The anti-Flag immunoprecipitates were resolved by SDS-PAGE and the three bands excised from the silver stained gel (top panel, marked by arrows) were identified as Ulk1, HSP90β and CDC37 by liquid chromatography-mass spectrometry (LC-MS) and Mascot prediction analysis. Bottom panel, protein identities were confirmed by immunoblot analysis.
(B) Wild type MEFs were treated with 17AAG or were transiently transfected with pooled control (non-targeting, NT) or cdc37 siRNA. Endogenous Ulk1 and interacting proteins were immunoprecipitated using an anti-Ulk1 antibody. Ulk1, Hsp90 and Cdc37 were detected by immunoblot analysis.
(C) Flag-Ulk1 was immunoprecipitated from K562 cells treated with the indicated amount of 17AAG for 1 hr. Ulk1 protein levels were assessed by immunoblot analysis. Ulk1 kinase activity was determined by _in vitr_o kinase assays. Reactions were analyzed by SDS-PAGE and 32P-labeled Ulk1 and MBP were detected by autoradiography.
(D) 293T cells were transfected with the indicated NTAP vector and treated with either vehicle (−) or 2.5 μM 17AAG (+) for 5 hr prior to pulse labeling the cells with 35S-Translabel for 15 minutes. 35S-labeled NTAP-Ulk1 was affinity purified and assessed by autoradiography.
(E) Wild type (ulk1+/+) MEFs were transfected twice with pooled control (non-targeting) or cdc37 siRNA. Top panel, expression of Ulk1, Cdc37 and Actin was assessed by immunoblot analysis 48 hr after the second transfection. Bottom panel, expression of mRNA was analyzed by RT-qPCR (TaqMan) analyses using primer/probe combinations specific for ulk1, cdc37, and 18S RNA. Relative expression (log2) was calculated using the Pffafl comparative Ct method. Expression of ulk1 and cdc37 in cdc37 siRNA treated samples was normalized to 18S and calibrated to control samples (i.e., those transfected with non-targeting siRNA).
(F) Pulse-chase analyses were performed on 293T cells expressing the indicated NTAP vector and treated with either vehicle (control) or 2.5 μM 17AAG.
(G) Wild type MEFs were pretreated with the H+ATPase inhibitor bafilomycin-A1 (10 nM) or with the proteasome inhibitor MG132 (10 nM) for 1 hr prior to the addition of 2.5 μM 17AAG for 5 hr, as indicated. The levels of Ulk1 and Hsp90 were determined by immunoblotting. Lysates from _ulk1_−/− MEFs served as a control.
Activation of specific kinase clients by the Hsp90-Cdc37 chaperone complex involves the assembly of a salt-stable heterocomplex of Hsp90, Cdc37 and the kinase client (Hartson et al., 2000). The formation of these meta-stable complexes is blocked by Hsp90 antagonists such as 17-Allylamino-17-Demethoxygeldanamycin (17AAG), a synthetic derivative of geldanamycin that binds to the _N_-terminal ATP binding pocket of Hsp90, inhibiting ATP binding and hydrolysis, which are required for chaperone function (Hartson et al., 2000; Pearl and Prodromou, 2006). Consistent with the notion that Ulk1 is a client of the Hsp90-Cdc37 chaperone complex, treatment with 2.5μM 17AAG for 1 hr disrupted the interaction between Ulk1, Hsp90 and Cdc37 (Figure 1B, left panels and Figure S1B). Similarly, silencing cdc37 expression by siRNA disrupted the interaction between endogenous Ulk1 and Hsp90 (Figure 1B, right panels).
The Hsp90-Cdc37 Chaperone Complex Regulates Ulk1 Kinase Activity
Agents that disrupt the interaction of Hsp90 with its kinase clients inactivate kinase activity and/or lead to their destruction by the ubiquitin-proteasome pathway (Caplan et al., 2007). To determine if the Hsp90-Cdc37 complex regulates Ulk1 kinase activity, anti-Flag immunoprecipitates isolated from Flag-Ulk1-expressing K562 cells treated with increasing doses of 17AAG for 1 hr were incubated with 32P-γATP and the general kinase substrate myelin basic protein (MBP). 17AAG treatment inhibited both Ulk1 autophosphorylation and phosphorylation of MBP in vitro (Figure 1C). Pulse-chase studies demonstrated that newly synthesized Ulk1 migrates faster in an SDS-PAGE gel and rapidly shifts up, suggesting that Ulk1 is phosphorylated shortly after synthesis (Figure 1F, top panel). To test if Hsp90 plays a role in these early phosphorylation events, Ulk1 was purified from transfected 293T cells that were pre-treated with 17AAG for 5 hr and then pulse-labeled with 35S-methionine and 35S-cysteine for 15 minutes. Newly synthesized Ulk1 purified from 17AAG-treated cells migrated faster than Ulk1 from vehicle-treated cells, suggesting that disrupting Hsp90 interactions with newly synthesized Ulk1 inhibits Ulk1 phosphorylation (Figure 1D). Ulk1 isolated from cells under normal growth conditions is hyperphosphorylated, at least in part due to autophosphorylation (Chan et al., 2009; Dorsey et al., 2009a), and treatment with lambda phosphatase in vitro leads to electrophoretic migration similar to kinase-dead Ulk1 mutants (Dorsey et al., 2009a). Treatment of Ulk1-expressing 293T cells with 17AAG for 5 hr led to the appearance of a faster migrating form of Ulk1 that co-migrated with a kinase-dead hypophosphorylated Ulk1 mutant (Ulk1-K46A) (Figure S1C), suggesting that Hsp90 inhibition impaired Ulk1 phosphorylation. Finally, silencing c_dc37_ also led to increased Ulk1 electrophoretic mobility, consistent with a requirement for Cdc37 for Ulk1 activation (Figure 1E).
Stable isotope labeling with amino acids in cell culture (SILAC) followed by high resolution tandem mass spectrometry confirmed that the change in Ulk1 phosphorylation following disruption of Hsp90 function was due to a decrease in Ulk1 kinase activity. To quantify Ulk1 phosphorylation following Hsp90 inhibition we employed SILAC, where one population of cells is grown in normal (light) media and another is grown in the presence of media containing 13C6-labeled lysine and 13C6 15N4-labeled arginine (heavy) (Amanchy et al., 2005). Ulk1 kinase activity is required for phosphorylation of S1047 in the Ulk1 _C_-terminus (Dorsey et al., 2009a). Indeed, S1047 phosphorylation was 147-fold more abundant in wild type Ulk1 versus in kinase-dead Ulk1-K46A (after normalizing to unmodified Ulk1 peptides, Figure S1D, top panels). Treatment with 17AAG (5hr) resulted in a 3-fold decrease (after normalization to unmodified peptides) in S1047 phosphorylation (Figure S1D, bottom panels), consistent with the observation that Hsp90 inhibition impairs phosphorylation of newly synthesized Ulk1 (Figure 1D). Collectively, these findings indicate that the interaction of Ulk1 with the Hsp90-Cdc37 complex is an early event that stabilizes the mature phosphorylated conformation of Ulk1, which includes autophosphorylation of S1047.
The Hsp90-Cdc37 Chaperone Complex Regulates Ulk1 Stability
Pulse chase analyses have demonstrated that, under normal growth conditions, Ulk1 is a long-lived protein with a half-life close to 24 hr (Dorsey et al., 2009a). We therefore assessed the effects of disrupting the Hsp90-Cdc37-Ulk1 interaction on Ulk1 turnover. Notably, 17AAG treatment triggered rapid turnover of Ulk1 in NTAP-Ulk1-expressing 293T cells compared to vehicle treated cells (Figure 1F). The steady state levels of endogenous Ulk1 protein were also significantly reduced in MEFs treated with 2.5μM 17AAG for 5 or 21 hr, without effects on ulk1 mRNA levels (Figure S1E). Similarly, silencing cdc37 expression triggered reductions in the steady state levels of Ulk1 protein, without affecting ulk1 mRNA levels (Figure 1E). The reduction in the steady state levels of Ulk1 was proportional to the degree of Cdc37 knockdown, and was most apparent with combined shRNA/siRNA-mediated knockdown of Cdc37 (Figure S1F). In addition, cdc37 knockdown markedly increased the sensitivity of Ulk1 to the effects of 17AAG (data not shown), as shown for other bona fide kinase clients of Hsp90 (Smith et al., 2009). Finally, the destabilizing effects of 17AAG on endogenous Ulk1 were abolished by co-treatment with the proteasome inhibitor MG132 (Figure 1G), but not the H+ATPase inhibitor, bafilomycin-A1, which inhibits lysosome-mediated degradation of autophagic cargo (Yoshimori et al., 1991). Therefore, the interaction between Ulk1 and the Hsp90-Cdc37 chaperone complex stabilizes Ulk1 and disrupting this complex triggers Ulk1 degradation by the proteasome.
The Hsp90-Cdc37 complex regulates the stability and activity of numerous kinases by direct binding to the catalytic kinase domain (Caplan et al., 2007). Using Ulk1 deletion constructs we demonstrated that the _N_-terminal kinase domain of Ulk1 (residues 1–279) was necessary and sufficient for mediating the interaction of Ulk1 with Hsp90 and Cdc37 (Figure S2A) and for regulating its sensitivity to degradation following 17AAG treatment (Figure S2C and D). Notably, although Ulk1 and the related mammalian Atg1 homologue Ulk2 share significant homology within their kinase domains, we did not detect Cdc37 or Hsp90 in Ulk2 immunoprecipitates (Figure S2B), nor were there decreases in the steady state levels of Ulk2 following treatment with 17AAG (Figure S2C). Furthermore, the stability of yeast Atg1 was not altered in temperature sensitive mutants that disable Cdc37 and Hsp82 (yeast Hsp90) (personal communication, Usha Nair and Daniel Klionsky). Together, these data provide evidence of the specificity of the Ulk1-Hsp90-Cdc37 interaction, and suggest an evolutionary divergence in Hsp90 regulation of Ulk1 and other Atg1 homologues.
Hsp90 Regulates Starvation-Induced Autophagy
Ulk1 has been implicated in starvation-induced autophagy (Chan et al., 2007). Thus, we hypothesized that Hsp90 controlled Ulk1-directed, starvation-induced autophagy. Ulk1-deficient MEFs have impaired flux through the autophagy pathway following amino acid starvation, although given the high rate of turnover of lipidated LC3 (LC3-II) in these cells it is necessary to inhibit lysosomal degradation of LC3 using the H+-ATPase inhibitor Bafilomycin A to appreciate the defect (Jung et al., 2009). To quantify autophagic flux in MEFs, we developed a highly reproducible firefly luciferase-based assay that exploits the autophagy-dependent turnover of LC3. LC3 is conjugated to phosphatidylethanolamine (PE) following cleavage of the pro-form at glycine 120 and both cleavage and modification are required for fusion of isolation membranes to form autophagosomes. Notably, PE-conjugated LC3 decorates both the inner and outer membranes of autophagosomes, and is degraded following fusion of autophagosomes with lysosomes (Dorsey et al., 2009b); thus, rates of LC3 turnover are an accurate measure of flux through the autophagy pathway. Firefly luciferase was fused in frame with the _N_-terminus of LC3 to generate the Luc-LC3 reporter. We generated MSCV-based vectors that express either luciferase-LC3 (Luc-LC3) or a fusion point mutant (Luc-LC3G120A) that abolishes PE modification, thus uncoupling LC3 degradation from the autophagic machinery (Figure S3A). Using this reporter assay, ulk1−/− MEFs exhibited marked defects in rates of Luc-LC3 degradation (relative to Luc-LC3G120A) following amino acid starvation (Figure S3B). Indeed, defects in LC3 turnover in ulk1−/− MEFs were comparable to those observed in MEFs lacking Atg7, an E1-like enzyme required for the autophagy pathway (Figure S3B). The defect in LC3 turnover manifest in ulk1−/− MEFs was rescued following reconstitution with wild type Ulk1 but not with the kinase-dead Ulk1-K46A mutant (Figure S3C). Thus, Ulk1 kinase activity is required for starvation-induced autophagy. Finally, this assay was used to assess the role of Hsp90 in amino acid starvation-induced autophagy. Pretreatment with 17AAG impaired starvation-induced autophagy in ulk1+/+ MEFs to an extent similar to defects in LC3 turnover manifest in ulk1−/− cells (Figure S3D). 17AAG also impaired accumulation of lipidated LC3 in Bafilomycin A-treated, amino acid-starved MEFs (data not shown), and inhibited LC3 punctae formation in a screen for drugs inhibiting starvation-induced autophagy (Criollo et al., 2010). Collectively, these data indicate that Hsp90 and Ulk1 kinase activity are required for starvation-induced autophagy.
Autophagy is a cell survival mechanism engaged by metabolic stress, including acute starvation (Stipanuk, 2009). We therefore assessed the viability of ulk1+/+ and ulk1−/− MEFs by trypan blue dye exclusion following amino acid deprivation. Notably, starvation triggered cell death of ulk1−/− but not ulk1+/+ MEFs (Figure S3E) and this response was mitigated by reconstituting ulk1−/− MEFs with wild type Ulk1 but not with kinase-dead Ulk1-K46A (Figure S3F). Finally, Hsp90 inhibition also augmented cell death following amino acid deprivation (Figure S3G), consistent with a role for Hsp90 in promoting survival in response to metabolic stress.
Hsp90 is Necessary for Autophagy-Mediated Clearance of Mitochondria during Erythroid Differentiation
Mitochondrial clearance occurs during the final steps of erythroid maturation, and this response requires Ulk1 (Kundu et al., 2008). Since the Hsp90-Cdc37 complex was necessary for the stability and activity of Ulk1 and 17AAG impairs autophagy, we tested the effects of Hsp90 inhibition on mitochondrial clearance during terminal erythroid maturation. Treatment of differentiating erythroid cells with 2.5μM 17AAG triggered marked reductions in Ulk1 protein levels in two independent erythroid cultures (Figure 2A and Figure S4A, left panels) without affecting ulk1 mRNA levels (Figure S4A, right panel). 17AAG treatment did not impair reticulocyte development (Figure 2B), yet it significantly reduced the number of reticulocytes harboring autophagosomes, especially those containing mitochondria (Figure 2C–2D), and led to corresponding increases in the levels of mitochondrial proteins and overall mitochondrial mass (Figure S4A–C). Thus, Ulk1 stabilization by the Hsp90-Cdc37 complex is required for efficient autophagy-mediated clearance of mitochondria during erythroid differentiation.
Figure 2. Hsp90 is Required for Autophagy-Mediated Clearance of Mitochondria in Erythroid Cells.

(A) Erythroid progenitors isolated from day E12.5 ulk1+/+ fetal livers were cultured with erythropoietin and harvested at the indicated intervals. Vehicle (Control) or 2.5 μM 17AAG was added (as indicated) during the final 24 hr of culture. Immunoblot analyses assessed levels of Ulk1, Atg13 and Hsp90 (loading control). The asterisks (*) denote non-specific bands detected with the Ulk1 or Atg13 antibodies.
(B–D) Erythroblast cultures derived from the spleens of wild type Balb/c mice infected with the anemia-inducing strain of Friend leukemia virus (FVA) were treated with vehicle (Control) or 2.5 μM 17AAG during the final 24 hr of culture. Samples were prepared for light (benzidine-stained cytospins, the scale bars represent 100μM) and electron microscopy (the scale bars represent 500 nm) 48 hr after the initiation of the culture and representative images are shown in (B) and (C), respectively. Arrows highlight reticulocytes in (B), which are enucleated and express hemoglobin (benzidine-positive). (C,D) Reticulocytes were identified on electron micrographs and assessed for the presence of autophagosomes containing single mitochondria (Am), autophagosomes containing multiple mitochondria (Am*) and autolysosomes (AL). The percentage of reticulocytes (mean from each of two independent experiments, _n_>20 cells per experiment) with autophagosomes containing mitochondria is plotted in (D). *_p_=0.007 (Student’s _t_-test).
The Hsp90-Cdc37 Chaperone Complex and Ulk1 are Essential for Clearance of Depolarized Mitochondria
Disease associated mutations in PARK2 (Abbas et al., 1999; Shimura et al., 2000), the gene encoding the E3 ligase Parkin, impair the elimination of damaged mitochondria (Geisler et al., 2010; Lee et al., 2010; Narendra et al., 2010; Vives-Bauza et al., 2010). Investigations of Parkin’s role in targeting mitochondria for degradation by autophagy have established a highly reproducible, quantitative and genetically tractable cell based assay for identifying and characterizing genes involved in mitochondrial clearance (Narendra et al., 2008). The assay involves treatment of Parkin-expressing cells with the proton ionophore carbonyl cyanide m-chlorophenyl hydrazone (CCCP), which triggers loss of mitochondrial membrane potential and induces the selective clearance of depolarized mitochondria (Narendra et al., 2008). We used this assay to thoroughly assess the roles of Ulk1 kinase activity and Hsp90-Cdc37 in mitophagy.
As expected (Narendra et al., 2008), we observed complete clearance of mitochondria in most Parkin-expressing wild type MEFs following treatment with CCCP (Figure 3), but not following treatment with vehicle or in untransfected MEFs with or without CCCP (data not shown). Inhibition of Hsp90-Cdc37 function in wild type MEFs by 17AAG or cdc37 knockdown significantly reduced the number of Parkin-expressing MEFs that cleared mitochondria (Figure 3A–3B and Figure S5B–S5C). Similar defects in mitochondrial clearance were observed in ulk1−/− MEFs (Figure 3A and Figure S5B). Mitochondria at various stages of clearance were scored based on Parkin and Tom20 staining (Figure S5A). As observed in ulk1−/− MEFs, a significant proportion of the ulk1+/+ MEFS treated with 17AAG or following silencing of Cdc37 contained mitochondria in small clusters that co-localized with Parkin (Figure 3A, Figure S5B–S5D); thus, the primary defect in clearance occurs after Parkin recruitment to depolarized mitochondria. By contrast, ulk1−/− MEFs treated with 17AAG showed diffuse localization of Parkin (Figure S5B). Finally, the ability to efficiently clear mitochondria following CCCP treatment was restored in Parkin-expressing ulk1−/− MEFs reconstituted with wild type Ulk1, but not in those expressing kinase-dead Ulk1-K46A (Figure 3D) despite comparable protein levels (Figure 4A). Thus, Ulk1 kinase activity is essential for Parkin-dependent clearance of damaged mitochondria.
Figure 3. Hsp90-Cdc37 and Ulk1 Kinase Activity are Required for Parkin-Mediated Clearance of Depolarized Mitochondria.

(A, B) Mitochondrial clearance in wild type (ulk1+/+) and ulk1−/− MEFs transiently transfected with YFP-Parkin and pretreated with vehicle (control) or 2.5 μM 17AAG for 5 hr prior to treatment with 20 μM CCCP for 18 hr. Cells were stained with anti-Tom20 (an outer mitochondrial membrane protein) antibody and scored for the percentage of YFP-Parkin+ cells lacking mitochondria (B); representative images are shown in (A); the scale bars represent 20 μm.
(C) Mitochondrial clearance in wild type MEFs transiently transfected with either non-targeting (NT) (pool) or cdc37 siRNA (pool or individual) and YFP-Parkin and then treated with CCCP for 18 hr. Cells were stained with anti-Tom20 antibody and scored for the percentage of YFP-Parkin+ cells lacking mitochondria.
(D) Clearance of mitochondria in YFP-Parkin+ cells was similarly assessed in ulk1+/+, ulk1−/− and ulk1−/− MEFs stably expressing either Ulk1 or Ulk1-K46A. The percentage of YFP-Parkin+ cells with complete absence of Tom20 signal (mean + s.e.m., >100 cells from 3 independent experiments) is shown in (B–D) (detailed scoring shown in Figure S5B–D). *p<0.001 (one way ANOVA analysis followed by Holm-Sidak post-hoc analysis).
Figure 4. Hsp90 Regulates Ulk1-Directed Phosphorylation of Atg13 at Serine 318.

(A) Immunoblot analysis of whole cell lysates of ulk1+/+, ulk1−/− and ulk1−/− MEFs stably expressing either Ulk1 or Ulk1-K46A.
(B) Immunoblot analysis of whole cell lysates of 293T cells transiently transfected with the indicated NTAP-tagged expression constructs, which were treated with vehicle (Control) or 2.5 μM 17AAG for 5 hr.
(C) Immunoblot analysis of whole cell lysates of ulk1−/− or ulk1−/− MEFs stably expressing Ulk1 transfected with either pooled non-targeting (NT), cdc37 or atg13 siRNA.
(D) Quantitative SILAC LC-MS/MS analyses of purified Atg13 from 293T cells expressing either Ulk1 or kinase-dead Ulk1-K46A (top panels); or from Ulk1-expressing μM 17AAG for 5 hr (bottom panels). Top left panel, extracted ion chromatograms showing the relative abundance of the phosphorylated Atg13 S318 peptide (~40%) from Ulk1-expressing cells, and the predominance of the unmodified Atg13 peptide from Ulk1-K46A expressing cells. Top middle panel, full MS scan of the Atg13 S318 phosphorylated peptide demonstrating a quantitative ~150-fold increase (149±26.4) in S318 phosphorylation of Atg13 when co-expressed with Ulk1 versus Ulk1-K46A (values are the mean ± s.d., _n_=2). Bottom left panel, extracted ion chromatogram demonstrating a reduction in the relative abundance of Atg13 S318 phosphorylation from 17AAG-treated cells. Bottom middle panel, full MS scan of the phosphorylated S318 Atg13 peptide demonstrating a ~3 fold reduction (2.9±0.1) in phosphorylation of Atg13 S318 following 17AAG treatment (values are the mean ± s.d., _n_=2). Right panels, full MS scans of one of the several unmodified Atg13 peptides that were used to normalize protein levels, which demonstrate that purified “light” (left peaks) and “heavy” (right peaks) Atg13 samples were mixed at a ratio very close to 1:1.
(E) 293T cells were transiently transfected with NTAP-Ulk1, NTAP-Ulk1-K46A and/or Atg13 as indicated, and treated with vehicle (−) or 17AAG (+) for 2 hr. Ulk1, p318S-Atg13, total Atg13 and GAPDH were detected by immunoblot analyses.
Ulk1 phosphorylates Atg13 in an Hsp90-Cdc37-dependent manner
Atg13 is a known interacting partner of Ulk1 and a suspected Ulk1 substrate (Chan et al., 2009; Hosokawa et al., 2009; Jung et al., 2009). Consistent with this notion, a faster migrating form of endogenous Atg13 was observed in ulk1−/− versus ulk1+/+ MEFs (Figure 4A). Further, reconstituting ulk1−/− MEFs with wild type Ulk1 but not kinase-dead Ulk1-K46A restored the migration of endogenous Atg13 migration to that seen in wild type MEFs (Figure 4A). Similarly, the migration of overexpressed Atg13 in 293T cells was altered by co-expressing wild type Ulk1but not by kinase-dead Ulk1-K46A (Figure 4B, left panels), and the shift in Atg13 migration was abolished by 17AAG (Figure 4B, right panels). Finally, a faster migrating form of endogenous Atg13 was evident in 17AAG-treated ulk1+/+ erythroid cultures (Figure 2A) and following cdc37 knockdown in _ulk1_−/− MEFs engineered to express wild type Ulk1 (Figure 4C and 5D, bottom).
Figure 5. Hsp90-Cdc37 and Ulk1 Kinase Activity Regulate Release of Atg13.

(A) Immunoblot analysis of anti-Ulk1 immunoprecipitates from whole cell lysates of ulk1+/+, ulk1−/− and ulk1−/− MEFs stably expressing either Ulk1 or Ulk1-K46A.
(B) Immunoblot analysis of NTAP affinity-purified precipitates from 293T cells transfected with the indicated vector and then treated with 2.5 μM 17AAG (+) or vehicle (−) for 5 hr.
(C) Immunoblot analysis of anti-Ulk1 immunoprecipitates from ulk1−/− MEFs stably expressing Ulk1 and treated with 2.5 μM 17AAG for the indicated times.
(D) Immunoblot analysis of anti-Ulk1 immunoprecipitates from ulk1−/− MEFs stably expressing Ulk1 and transfected twice with pooled non-targeting or cdc37 siRNA.
(E) Representative images of HeLa cells co-transfected with HA-Atg13 and either NTAP-Ulk1 or NTAP-Ulk1-K46A and treated with vehicle (Control) or 2.5 μM 17AAG for 5 hr. Cells were stained with anti-HA and anti-Ulk1 antibodies. Scale bar represents 10 μM.
Collectively, these findings suggested that Atg13 is a substrate of Ulk1, and that the Hsp90-Cdc37 complex regulates Ulk1-mediated phosphorylation of Atg13. To directly assess the phosphorylation of Atg13 by Ulk1, a SILAC-based mass spectrometric approach was used to identify phosphorylation sites. Ulk1 and Atg13 were co-expressed in heavy labeled 293T cells, and Ulk1-K46A and Atg13 were co-expressed in cells grown in normal (light) media. These analyses established Atg13 S318 as a site of Ulk1 phosphorylation (Figure 4D, top panels). Moreover, a 3-fold decrease in Atg13 S318 phosphorylation (after normalization to unmodified Atg13 peptides) was evident in cells treated with 17AAG (Figure 4D, bottom panels), a response almost identical to the effect of 17AAG on pS1047 in Ulk1 (Figure S1D, bottom panels).
We also generated a polyclonal antibody specific for Atg13pS318 by immunizing rabbits with a peptide containing pS318. Using 293T cells engineered to co-express Ulk1 and Atg13, western blot analyses with this antibody confirmed that phosphorylation of Atg13 S318 occurred in an Ulk1-kinase dependent manner, and that this was inhibited by 17AAG (Figure 4E). Thus, Atg13 is phosphorylated by Ulk1 at S318, and this modification is facilitated by the interaction of Ulk1 and Hsp90-Cdc37.
Hsp90-Cdc37 and Ulk1 kinase activity control release of Atg13 from Ulk1
Given these findings, we more thoroughly characterized the relationship between UIk1, Atg13, Hsp90 and Cdc37. First, the steady state levels of endogenous Atg13 did not decrease as dramatically as endogenous Ulk1 following 17AAG treatment (Figure S6A), suggesting that Ulk1 is not the only determinant of Atg13 stability in MEFs. Indeed, ulk1−/− MEFs showed only a slight decrease in steady state levels of Atg13 (Figure 4A and S6B). However, ulk1−/− MEFs reconstituted with either wild type or kinase-dead Ulk1 (K46A) showed increased steady state levels of Atg13, suggesting that Ulk1 can stabilize Atg13 and that this occurs in a kinase-independent manner (Figure 4A and S6B). In addition, silencing of ulk2 in the ulk1−/− MEFs (Egan et al., 2010) significantly decreased steady state levels of endogenous Atg13 (Figure S6C), indicating that in MEFs both Ulk1 and Ulk2 contribute to stabilization of Atg13. By contrast, in erythroid cells, where ulk2 mRNA expression is minimal (Kundu et al., 2008), loss of Ulk1 is sufficient to dramatically decrease steady state levels of endogenous Atg13 (Figure S6D). Collectively, these data and those showing that Ulk2 is not a client of Hsp90 (Figure S2) suggest that Atg13 stability is maintained in MEFs following 17AAG treatment as a result of its interaction with Ulk2.
Interestingly, immunoprecipitation analyses of ulk1+/+ MEFs, and ulk1−/− MEFs reconstituted with either wild type or kinase-dead Ulk1-K46A demonstrated an inverse correlation between Ulk1 kinase activity and the relative amount of Atg13 present in anti-Ulk1 immunoprecipitates. Although the amount of Hsp90 and Cdc37 immunoprecipitated with Ulk1 was proportional to the level of Ulk1, higher levels of Atg13 were co-immunoprecipitated from ulk1−/− MEFs reconstituted with kinase-dead Ulk1-K46A (Figure 5A). Similar findings were observed from precipitation of NTAP-Ulk1 or NTAP-Ulk1-K46A overexpressed in 293T cells (Figure 5B). Furthermore, treatment with 17AAG or knockdown of cdc37, which impaired Ulk1 stability and kinase activity (Figure 1), triggered increases in the amount of Atg13 that co-immunoprecipitated with overexpressed Ulk1 (Figure 5B–5D). 17AAG treatment also increased Atg13 immunoprecipitation with endogenous Ulk1 (Figure S6A). Given these findings, we also examined the subcellular distribution of Ulk1 and Atg13 by confocal microscopy. Although Ulk1 and Atg13 were diffusely cytoplasmic when co-expressed in HeLa cells, inhibition of Ulk1 kinase activity by treatment with 17AAG or by expressing kinase-dead Ulk1-K46A triggered the formation of discrete Ulk1+/Atg13+ foci (Figure 5E) that were not LC3+ (data not shown). Collectively, these observations suggest that though the interaction of Ulk1 with Hsp90-Cdc37 is necessary for Atg13 phosphorylation, the activation of Ulk1 kinase by Hsp90-Cdc37 promotes release of Atg13 from the Ulk1-Hsp90-Cdc37 complex.
Atg13 localizes to damaged mitochondria and is necessary for mitochondrial clearance
There are conflicting results regarding the role of orthologous yeast Atg1-Atg13 complex in mitophagy (Kanki and Klionsky, 2010; Kanki et al., 2009; Okamoto et al., 2009). Therefore, we tested if Atg13 contributes to mitophagy in mammalian cells. Notably, atg13 knockdown significantly impaired mitochondrial clearance in Parkin-expressing MEFs treated with CCCP (Figure 6A–6B). Silencing of ATG13 in Parkin-expressing HeLa cells also impaired clearance of depolarized mitochondria (Figure S7B). Strikingly, assessment of the subcellular distribution of Ulk1 and Atg13 in HeLa cells engineered to express Parkin and treated with CCCP demonstrated that Atg13, but not Ulk1, co-localized with Tom20+ mitochondrial clusters in most Parkin-expressing cells (Figures 6C and S7A). Atg13 did not localize to mitochondria in the absence of CCCP and Parkin (data not shown). Moreover, overexpressing the kinase-dead Ulk1 mutant, which inhibits Atg13 phosphorylation (Figure 4B) and mitochondrial clearance (Figure S7C) prevented Atg13 localization to damaged mitochondria (Figure 6C and S7A). Finally, Hsp90 inhibition impaired localization of Atg13 to damaged mitochondria (Figure 6C and S7A). Collectively, these findings suggest that mitochondrial damage triggers Ulk1-dependent phosphorylation and release of Atg13 to mitochondria.
Figure 6. Atg13 is Required for Mitochondrial Clearance and Localizes to Damaged Mitochondria in an Ulk1- and Hsp90-dependent manner.

(A, B) Wild type (ulk1+/+) MEFs were transiently transfected with either pooled non-targeting or atg13 siRNA and YFP-Parkin, and were then treated with 20μM CCCP for 18 hr. Cells were then stained with anti-Tom20 antibody and scored for the percentage of YFP-Parkin+ cells lacking mitochondria. Representative images are shown in (A) and the percentage of YFP-Parkin+ cells with complete absence of Tom20 signal (mean + s.e.m., >100 cells from three independent experiments) is shown in (B). *p<0.001 (Student’s _t_-test). Scale bar represents 20μM. Atg13 silencing was confirmed by immunoblot analysis (B).
(C) HeLa cells were transfected with YFP-Parkin together with Ulk1, Ulk1-K46A and/or Atg13 expression constructs (as indicated). Cells were treated with 20μM CCCP for 8 hr. As indicated, cells were treated for 5 hr with 2.5 μM 17AAG prior to addition of CCCP. Cells were then stained with anti-HA (green) and anti-Tom20 (red) antibodies. Representative merged images of Parkin+ CCCP-treated cells are shown (top panels). The corresponding single-channel pseudocolored images are shown in Figure S7A. Line scans (bottom panels) indicate the degree of co-localization between Atg13 (green) and Tom20+ mitochondria (red) in Parkin+ cells, and correlate to the lines drawn in the magnified images (middle panels). Intensity profiles (in arbitrary units) were obtained using NIS elements AR 3.10 software from Nikon. Scale bar represents 10μM.
Atg13 S318 Phosphorylation is Required for Parkin-Mediated Mitochondrial Clearance
To assess if Atg13 phosphorylation on S318 plays roles in Parkin-mediated mitophagy, we generated a serine-to-alanine (non-phosphorylatable) substitution mutation of Atg13 (Atg13-S318A). Although Atg13 overexpression had little effect on Parkin-dependent clearance of depolarized mitochondria, similar levels of Atg13-S318A exerted a dominant negative effect on mitophagy (Figures 7A–7B). Interestingly, although Ulk1-K46A dominantly inhibited both starvation-induced autophagy (data not shown and (Chan et al., 2009)) and mitochondrial clearance (Figure S7C), overexpression of Atg13-S318A did not impair starvation-induced autophagy, but resulted in increased LC3 turnover under normal growth conditions and following nutrient deprivation (Figure S7D). Thus, phosphorylation of Atg13 at S318 is required for mitophagy, but not basal or starvation-induced autophagy, implying that differential phosphorylation of Atg13 by Ulk1 may influence downstream functions of Atg13.
Figure 7. Ulk1-Mediated Phosphorylation of Atg13 at S318 is Required for Efficient Clearance of Depolarized Mitochondria.
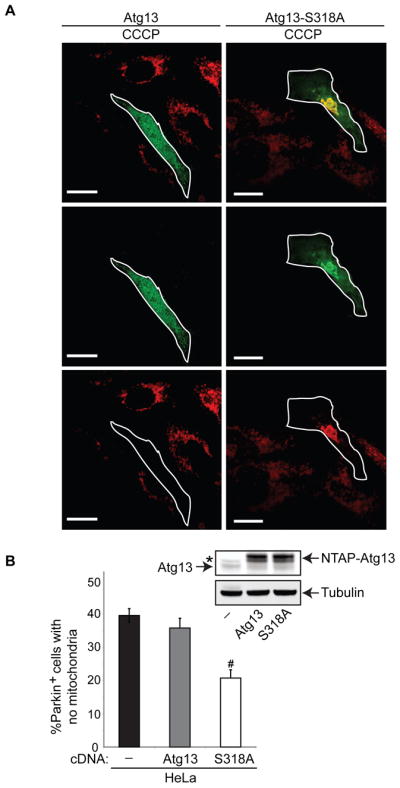
(A, B) HeLa cells were transiently transfected with YFP-Parkin alone, or together with either NTAP-tagged Atg13 or Atg13-S318A. The cells were treated with vehicle (Control) or 20 μM CCCP for 18 hr. Cells were stained with anti-Tom20 antibody and YFP-Parkin+ cells were scored based on the presence or absence of Tom20 staining. Representative images of CCCP-treated cells are shown in (A); the scale bars represent 20 μm. The percentage of YFP-Parkin+ cells lacking Tom20 signal (mean + s.e.m., >100 cells from three independent experiments) is shown in panel (B). #p<0.001 (Student’s _t_-test). Expression of Atg13 and Atg13-S318A were confirmed by immunoblot analysis (arrows highlight endogenous and NTAP-tagged Atg13 and Atg13-S318A as indicated); tubulin was used as a loading control. The asterisk (*) denotes a non-specific band observed with the anti-Atg13 antibody.
DISCUSSION
The data presented herein establish that the Hsp90-Cdc37 chaperone complex regulates mitophagy by modulating the stability and function of Ulk1 and one of its downstream targets, Atg13. Specifically, the interaction with the Hsp90-Cdc37 complex stabilizes Ulk1 by preventing proteasome-mediated degradation and this interaction is required for Ulk1 autophosphorylation and phosphorylation of Atg13 at S318. Strikingly, Ulk1 activation promotes the release of Atg13 from the Hsp90-Cdc37-Ulk1 complex and the localization of Atg13 to depolarized mitochondria, where it plays an essential role in Parkin-dependent mitophagy. Although Hsp90, Cdc37 and Ulk1 regulate the phosphorylation of Atg13 at S318, this modification alone does not appear to be sufficient for promoting Atg13 release, as increased amounts of Ulk1 were not detected in immunoprecipitates of Atg13-S318A versus wild type Atg13, nor did we observe enhanced co-localization of Ulk1 and Atg13-S318A (data not shown). Rather, the release of Atg13 from Ulk1 may depend on a kinase-dependent change in conformation of Ulk1 and/or phosphorylation of Atg13 at sites other than S318. Regardless, the finding that the non-phosphorylatable Atg13-S318A mutant dominantly inhibits mitochondrial clearance highlights the functional significance of Atg13 phosphorylation by Ulk1. Since Hsp90, Cdc37, and Ulk1 together regulate phosphorylation of Atg13 at S318, and all are required for efficient autophagy-mediated clearance of mitochondria, these findings define a new pathway linking mitochondrial homeostasis with the cellular stress response coordinated by Hsp90-Cdc37.
In contrast to the prevailing view that Ulk1 and Atg13 function as a complex, our data indicate that Ulk1 kinase activity promotes the release of Atg13 from Ulk1, and that released Atg13 then localizes to ring-like structures around damaged mitochondria, and thereby contributes to their degradation. Indeed silencing of Atg13 or enforced expression of the non-phosphorylatable Atg13-S318A mutant impairs mitochondrial clearance. Curiously, phosphorylation of Atg13 at S318 is not required for efficient LC3 conversion and degradation (under basal conditions or following amino acid deprivation), suggesting S318 phosphorylation of Atg13 may influence cargo selection during autophagy.
The Hsp90-Cdc37 complex is involved in two distinct paradigms of autophagy-mediated mitochondrial clearance: Parkin-mediated clearance of depolarized mitochondria, and the BNIP3L-dependent developmental clearance of mitochondria in erythroid cells. Although these pathways differ in the mechanism by which mitochondria are targeted for degradation (Parkin versus BNIP3L), both rely on Ulk1-mediated activation of the autophagy pathway, which is regulated by Hsp90 and Cdc37. It is interesting to note that Pink1, a serine-threonine kinase that recruits Parkin to depolarized mitochondria and promotes clearance (Geisler et al., 2010; Narendra et al., 2010; Vives-Bauza et al., 2010), was identified as a client of the Hsp90-Cdc37 chaperone complex (Lin and Kang, 2008; Moriwaki et al., 2008; Weihofen et al., 2008). While the primary defect in mitochondrial clearance occurs after the recruitment of Parkin to mitochondria in ulk1−/− MEFs, and in ulk1+/+ MEFS treated with 17AAG or having knockdown of cdc37, we cannot exclude the possibility that Pink1 function is also affected. In fact, the diffuse localization of Parkin in ulk1−/− MEFs treated with 17AAG suggests that another client of Hsp90, perhaps Pink1, recruits Parkin to mitochondria and maintains the minimal levels of mitochondrial clearance observed the absence of Ulk1. That serine-threonine kinases involved in autophagy (Ulk1) and the mitochondrial targeting pathway (Pink1) are both clients of the Hsp90-Cdc37 chaperone complex suggests that coordinated regulation of mitochondrial turnover is an important homeostatic response that must be preserved, even under adverse conditions.
It has been suggested that Hsp90 inhibition promotes autophagy (Qing et al., 2006; Siegelin et al., 2010). Indeed, under normal growth conditions Hsp90 inhibition triggers an initial Ulk1-dependent increase in autophagy-dependent LC3 degradation (Dorsey and Kundu, unpublished observations), a response consistent with the Hsp90-dependent nature of kinases in the PI3K-mTOR pathway (Basso et al., 2002; Gray et al., 2007; Ohji et al., 2006) that inhibit autophagy (Ravikumar et al., 2004). However, this initial increase in autophagy is self-limiting and is rapidly cancelled by the inactivation of Hsp90-dependent Ulk1 activity, the primary autophagy-related target of mTOR (Hosokawa et al., 2009; Jung et al., 2009). In turn, inactivation of Ulk1 cripples the cells ability to respond to stimuli that induce autophagy, such as starvation or mitochondrial damage.
The exquisite dependence of Ulk1-mediated autophagy and mitochondrial clearance on Hsp90 function has important implications for cancer pathogenesis and treatment. First, it has been proposed that the destabilization of proteins associated with growth in the hypoxic, nutrient-deprived tumor milieu, is compensated by increased expression of Hsp90 and its co-chaperones (Whitesell and Lindquist, 2005). Since autophagy promotes survival under these adverse conditions, the stabilization and activation of Ulk1 by Hsp90 may be an important component of the survival response coordinated by Hsp90 and exploited by certain tumors. Indeed, since many tumors rely on autophagy for survival under stressed conditions (Amaravadi et al., 2007; Livesey et al., 2009; Maclean et al., 2008), the efficacy of Hsp90 inhibitors such as 17AAG may rely on their effects on Ulk1-mediated autophagy.
Collectively, the findings presented herein support a model (see Graphical Abstract) whereby interactions of Ulk1 with the Hsp90-Cdc37 complex stabilizes an active form of Ulk1 that promotes flux through specific arms of the autophagy pathway in response to metabolic or proteotoxic cues. Starvation, mitochondrial depolarization and other stimuli that trigger autophagy activate Ulk1, promoting Ulk1-mediated phosphorylation and release of its essential interacting partner, Atg13, and increased flux through the autophagy pathway. Phosphorylation of Atg13 at specific sites, for example S318, may favor selective degradation of mitochondria. As Ulk1 stability and activity rely on the Hsp90-Cdc37 complex, the function of both Ulk1 and downstream targets, including Atg13, is also subject to regulation by environmental and intracellular cues that alter Hsp90 activity. The Hsp90-Cdc37-to-Ulk1-to-Atg13 pathway thus coordinates and integrates autophagy and mitochondrial homeostasis with the cellular stress response.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
Flag-tagged Ulk1 and deletion constructs in the pME18S vector (gift of Dr. Toshifumi Tomoda) (Yan et al., 1998), HA-Atg13 (Jung et al., 2009), HA-Ulk1 (Jung et al., 2009) and NTAP-Ulk1 (Dorsey et al., 2009a) were previously described. The YFP-Parkin construct was also described (Narendra et al., 2008). An insert containing the coding sequences for mCherry-EGFP-LC3b (from pDEST mCherry-EGFPLC3b vector, a gift of Dr. Terje Johannsen) was subcloned into the LPC retroviral vector, and used to generate stably expressing MEFs as described (Tresse et al., 2010). Methods used for generating the following plasmid constructs are described in Supplemental Experimental Procedures: Flag- and NTAP- tagged Ulk1 kinase-dead (K46A) mutants, MSCV-luciferase-LC3B and MSCV-luciferase-G120ALC3B; and NTAP-Atg13 and NTAP-Atg13-S318A.
Proteomics
Ulk1-interacting proteins were visualized by silver staining (Invitrogen) according to the manufacturer’s protocol. Gel slices containing bands of interest were digested with trypsin as described (Strader et al., 2006). Peptides were separated using a 10-cm C18 column; samples were run at 200nl/min for 45min on a NanoLC (Eksigent). Online nanospray was used to spray the separated peptides into LTQ (Thermo Electron). Xcalibur was used to acquire the raw data and databases including NCBI and Swissprot were searched using Mascot (Perkins et al., 1999). Criteria used for confident protein identification were: Peptide score ≥ 30; _P_-value of peptide < 0.05; Protein score ≥70; Number of unique peptides ≥ 2. The bands shown in Figure 1A were identified as Ulk1 (Mascot score 2274, 85 queries matched, 32 unique peptides), HSP90β (Mascot score 1507, 69 queries matched, 28 unique peptides), and CDC37 (Mascot score 430, 15 queries matched, 8 unique peptides). Details of methods used to quantify protein phosphorylation by SILAC are described in Supplemental Experimental Procedures.
Cell Culture, Transfection and Drug Treatment; Generation of MEFs; Immunoprecipitation; Immunoblot Analyses and Antibodies; Pulse-Chase Analyses, Gene Silencing; Microscopy; Erythroid Cultures; Flow Cytometry; Luc-LC3 Luciferase Reporter Assay; and Cell Viability Assays
Please see Supplemental Experimental Procedures.
In Vitro Kinase Assays
Flag-Ulk1 was immunoprecipitated from K562 cells and eluted with Flag peptide (Sigma Aldrich) as described above. In vitro kinase reactions were performed as follows. Eluted Flag-Ulk1 was incubated in kinase buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 10 mM MgCl2, 0.5 mM DTT) supplemented with 1 mg/ml myelin basic protein fragments (Sigma Aldrich), 25 μM nonradioactive ATP, phosphatase inhibitors (Pierce), and 2.5 μCi γ-32P-labeled ATP at 37 °C for 15 minutes. Kinase reactions were stopped by adding an equal volume of 2x sample buffer, and samples were immediately boiled and separated on SDS-PAGE gels, which were then fixed in 30% methanol 10% acetic acid, stained with Coomassie blue, dried, and exposed to Kodak MR resolution film.
Quantitative Real-Time PCR
Total RNA was isolated from cells using TRIzol Reagent (Invitrogen). The reverse transcription (RT) reaction was carried out using the iScript cDNA™ synthesis kit (Bio-Rad) according to the manufacturer’s instructions. TaqMan® Gene Expression Assays containing FAM-labeled primer/probe sets specific for ulk1, ulk2, akt1, cdc37, and 18S were obtained from Applied Biosystems. The real-time PCR reactions were performed in a total reaction volume of 25 μL using FastStart TaqMan® Probe Master (Roche) reagent and results were analyzed with the iCycler IQ™ real-time PCR detection system (Bio-Rad). Relative expression (log2) was calculated using the Pfaff quantification method (Pfaffl, 2001) after normalization of cycle thresholds to 18S RNA and calibration to respective controls.
Statistical Analyses
Co-localization of Parkin and mitochondria or mitochondrial clearance was assessed by visually scoring ≥ 100 Parkin-positive cells per condition from at least three independent experiments and is represented as the mean ± s.e.m. Statistical analysis was performed using SigmaPlot; significance was assessed by two-tailed paired Student’s _t_-test, or one- or two-factor ANOVA analysis followed by Holm-Sidak post-hoc analysis.
Supplementary Material
01
Highlights.
- Hsp90-Cdc37 interacts with Ulk1, regulating its stability and activation
- Ulk1 phosphorylates Atg13 on S318 and promotes its release to damaged mitochondria
- Ulk1-directed Atg13 S318 phosphorylation is regulated by Hsp90-Cdc37 interactions
- Atg13 S318 phosphorylation is selectively required for mitophagy
Acknowledgments
We are grateful to: Charles J. Sherr (St. Jude Children’s Research Hospital [SJCRH], Memphis, TN) for providing Cdc37 antibody; Emily Tresse (SJCRH) for the LPC-mCherry-EGFP-LC3b construct; Toshifumi Tomoda (Beckman Research Institute of City of Hope, Duarte, CA) for Flag-Ulk1 deletion constructs; Sharon Tooze (London Research Institute, Cancer Research UK, London, UK) for providing an Atg13 antibody; Mark Hall (The Scripps Research Institute [TSRI], Jupiter, Florida) for providing the MSCV-Gateway-IRES-GFP vector; Reuben Shaw (The Salk Institute for Biological Studies, La Jolla, CA) for providing ulk1−/− MEFs stably expressing ulk2 shRNA; Jennifer Moore (SJCRH) for assistance with generation of MEFs; Aaron Poole (SJCRH), Valerie Cavett (TSRI), Chunying Yang (TSRI), and Laura Alsina (TSRI) for technical support; the Proteomics Core facilities of Scripps Florida (TSRI) and the University of Pennsylvania; the Biomedical Imaging Core facilities at SJCRH (Samuel Connell, Jennifer Peters and Sharon Frase) and University of Pennsylvania (Neelima Shaw and Ray Meade); and Robert Matts (Oklahoma State University) for helpful discussions. This research was partially supported by grants from the National Institutes of Health to M.K. (HL084199), F.C.D. (CA123777), J.L.C. (CA076379), P.A.N. (DK074519), and C.B.T. (CA099179), the NINDS intramural program to R.J.Y, the Burroughs Welcome Fund to M.K., the American Society of Hematology to M.K., by monies from the State of Florida to J.L.C. at Scripps Florida, and by monies from the American Lebanese Syrian Associated Charities (ALSAC) to P.A.N. and M.K.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbas N, Lucking CB, Ricard S, Durr A, Bonifati V, De Michele G, Bouley S, Vaughan JR, Gasser T, Marconi R, et al. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson’s Disease. Hum Mol Genet. 1999;8:567–574. doi: 10.1093/hmg/8.4.567. [DOI] [PubMed] [Google Scholar]
- Amanchy R, Kalume DE, Pandey A. Stable isotope labeling with amino acids in cell culture (SILAC) for studying dynamics of protein abundance and posttranslational modifications. Sci STKE. 2005;2005:pl2. doi: 10.1126/stke.2672005pl2. [DOI] [PubMed] [Google Scholar]
- Amaravadi RK, Thompson CB. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res. 2007;13:7271–7279. doi: 10.1158/1078-0432.CCR-07-1595. [DOI] [PubMed] [Google Scholar]
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem. 2002;277:39858–39866. doi: 10.1074/jbc.M206322200. [DOI] [PubMed] [Google Scholar]
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia-Kissova I, Camougrand N. Mitophagy in yeast: actors and physiological roles. FEMS Yeast Res. 2010;10:1023–1034. doi: 10.1111/j.1567-1364.2010.00659.x. [DOI] [PubMed] [Google Scholar]
- Caplan AJ, Mandal AK, Theodoraki MA. Molecular chaperones and protein kinase quality control. Trends Cell Biol. 2007;17:87–92. doi: 10.1016/j.tcb.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Chan EY, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem. 2007;282:25464–25474. doi: 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- Chan EY, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol. 2009;29:157–171. doi: 10.1128/MCB.01082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criollo A, Senovilla L, Authier H, Maiuri MC, Morselli E, Vitale I, Kepp O, Tasdemir E, Galluzzi L, Shen S, et al. The IKK complex contributes to the induction of autophagy. EMBO J. 2010;29:619–631. doi: 10.1038/emboj.2009.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Moura MB, dos Santos LS, Van Houten B. Mitochondrial dysfunction in neurodegenerative diseases and cancer. Environ Mol Mutagen. 2010;51:391–405. doi: 10.1002/em.20575. [DOI] [PubMed] [Google Scholar]
- Dorsey FC, Rose KL, Coenen S, Prater SM, Cavett V, Cleveland JL, Caldwell-Busby J. Mapping the phosphorylation sites of Ulk1. J Proteome Res. 2009a;8:5253–5263. doi: 10.1021/pr900583m. [DOI] [PubMed] [Google Scholar]
- Dorsey FC, Steeves MA, Prater SM, Schroter T, Cleveland JL. Monitoring the autophagy pathway in cancer. Methods Enzymol. 2009b;453:251–271. doi: 10.1016/S0076-6879(08)04012-3. [DOI] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2010;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamboa JL, Andrade FH. Mitochondrial content and distribution changes specific to mouse diaphragm after chronic normobaric hypoxia. Am J Physiol Regul Integr Comp Physiol. 2010;298:R575–583. doi: 10.1152/ajpregu.00320.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Gottlieb RA, Carreira RS. Autophagy in health and disease. 5. Mitophagy as a way of life. Am J Physiol Cell Physiol. 2010;299:C203–210. doi: 10.1152/ajpcell.00097.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray PJ, Jr, Stevenson MA, Calderwood SK. Targeting Cdc37 inhibits multiple signaling pathways and induces growth arrest in prostate cancer cells. Cancer Res. 2007;67:11942–11950. doi: 10.1158/0008-5472.CAN-07-3162. [DOI] [PubMed] [Google Scholar]
- Hartson SD, Irwin AD, Shao J, Scroggins BT, Volk L, Huang W, Matts RL. p50(cdc37) is a nonexclusive Hsp90 cohort which participates intimately in Hsp90-mediated folding of immature kinase molecules. Biochemistry. 2000;39:7631–7644. doi: 10.1021/bi000315r. [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007;131:257–270. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- Kanki T, Klionsky DJ. The molecular mechanism of mitochondria autophagy in yeast. Mol Microbiol. 2010;75:795–800. doi: 10.1111/j.1365-2958.2009.07035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T, Wang K, Baba M, Bartholomew CR, Lynch-Day MA, Du Z, Geng J, Mao K, Yang Z, Yen WL, Klionsky DJ. A genomic screen for yeast mutants defective in selective mitochondria autophagy. Mol Biol Cell. 2009;20:4730–4738. doi: 10.1091/mbc.E09-03-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112:1493–1502. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol. 2008;3:427–455. doi: 10.1146/annurev.pathmechdis.2.010506.091842. [DOI] [PubMed] [Google Scholar]
- Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol. 2010;189:671–679. doi: 10.1083/jcb.201001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang T, Schwartz SJ, Sun D. New developments in Hsp90 inhibitors as anti-cancer therapeutics: mechanisms, clinical perspective and more potential. Drug Resist Updat. 2009;12:17–27. doi: 10.1016/j.drup.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Kang UJ. Characterization of PINK1 processing, stability, and subcellular localization. J Neurochem. 2008;106:464–474. doi: 10.1111/j.1471-4159.2008.05398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TT, Hu CH, Tsai CD, Li CW, Lin YF, Wang JY. Heat Stroke Induces Autophagy as a Protection Mechanism against Neurodegeneration in the Brain. Shock. 2010 doi: 10.1097/SHK.0b013e3181e761c1. [DOI] [PubMed] [Google Scholar]
- Livesey KM, Tang D, Zeh HJ, Lotze MT. Autophagy inhibition in combination cancer treatment. Curr Opin Investig Drugs. 2009;10:1269–1279. [PubMed] [Google Scholar]
- Maclean KH, Dorsey FC, Cleveland JL, Kastan MB. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Invest. 2008;118:79–88. doi: 10.1172/JCI33700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margineantu DH, Emerson CB, Diaz D, Hockenbery DM. Hsp90 inhibition decreases mitochondrial protein turnover. PLoS One. 2007;2:e1066. doi: 10.1371/journal.pone.0001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–139. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Moriwaki Y, Kim YJ, Ido Y, Misawa H, Kawashima K, Endo S, Takahashi R. L347P PINK1 mutant that fails to bind to Hsp90/Cdc37 chaperones is rapidly degraded in a proteasome-dependent manner. Neurosci Res. 2008;61:43–48. doi: 10.1016/j.neures.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberley TD, Swanlund JM, Zhang HJ, Kregel KC. Aging results in increased autophagy of mitochondria and protein nitration in rat hepatocytes following heat stress. J Histochem Cytochem. 2008;56:615–627. doi: 10.1369/jhc.2008.950873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohji G, Hidayat S, Nakashima A, Tokunaga C, Oshiro N, Yoshino K, Yokono K, Kikkawa U, Yonezawa K. Suppression of the mTOR-raptor signaling pathway by the inhibitor of heat shock protein 90 geldanamycin. J Biochem. 2006;139:129–135. doi: 10.1093/jb/mvj008. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009;17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem. 2006;75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Qing G, Yan P, Xiao G. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of IkappaB kinase (IKK) Cell Res. 2006;16:895–901. doi: 10.1038/sj.cr.7310109. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Siegelin MD, Plescia J, Raskett CM, Gilbert CA, Ross AH, Altieri DC. Global Targeting of Subcellular Heat Shock Protein-90 Networks for Therapy of Glioblastoma. Mol Cancer Ther. 2010 doi: 10.1158/1535-7163.MCT-10-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JR, Clarke PA, de Billy E, Workman P. Silencing the cochaperone CDC37 destabilizes kinase clients and sensitizes cancer cells to HSP90 inhibitors. Oncogene. 2009;28:157–169. doi: 10.1038/onc.2008.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipanuk MH. Macroautophagy and its role in nutrient homeostasis. Nutr Rev. 2009;67:677–689. doi: 10.1111/j.1753-4887.2009.00252.x. [DOI] [PubMed] [Google Scholar]
- Strader MB, Tabb DL, Hervey WJ, Pan C, Hurst GB. Efficient and specific trypsin digestion of microgram to nanogram quantities of proteins in organic-aqueous solvent systems. Anal Chem. 2006;78:125–134. doi: 10.1021/ac051348l. [DOI] [PubMed] [Google Scholar]
- Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao TP, Dantuma NP, Taylor JP. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. 2010;6:217–227. doi: 10.4161/auto.6.2.11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihofen A, Ostaszewski B, Minami Y, Selkoe DJ. Pink1 Parkinson mutations, the Cdc37/Hsp90 chaperones and Parkin all influence the maturation or subcellular distribution of Pink1. Hum Mol Genet. 2008;17:602–616. doi: 10.1093/hmg/ddm334. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- Yan J, Kuroyanagi H, Kuroiwa A, Matsuda Y, Tokumitsu H, Tomoda T, Shirasawa T, Muramatsu M. Identification of mouse ULK1, a novel protein kinase structurally related to C. elegans UNC-51. Biochem Biophys Res Commun. 1998;246:222–227. doi: 10.1006/bbrc.1998.8546. [DOI] [PubMed] [Google Scholar]
- Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem. 1991;266:17707–17712. [PubMed] [Google Scholar]
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01