Inflammatory Mediators Alter the Astrocyte Transcriptome and Calcium Signaling Elicited by Multiple G-Protein-Coupled Receptors (original) (raw)
Abstract
Inflammation features in CNS disorders such as stroke, trauma, neurodegeneration, infection, and autoimmunity in which astrocytes play critical roles. To elucidate how inflammatory mediators alter astrocyte functions, we examined effects of transforming growth factor-β1 (TGF-β1), lipopolysaccharide (LPS), and interferon-gamma (IFNγ), alone and in combination, on purified, mouse primary cortical astrocyte cultures. We used microarrays to conduct whole-genome expression profiling, and measured calcium signaling, which is implicated in mediating dynamic astrocyte functions. Combinatorial exposure to TGF-β1, LPS, and IFNγ significantly modulated astrocyte expression of >6800 gene probes, including >380 synergistic changes not predicted by summing individual treatment effects. Bioinformatic analyses revealed significantly and markedly upregulated molecular networks and pathways associated in particular with immune signaling and regulation of cell injury, death, growth, and proliferation. Highly regulated genes included chemokines, growth factors, enzymes, channels, transporters, and intercellular and intracellular signal transducers. Notably, numerous genes for G-protein-coupled receptors (GPCRs) and G-protein effectors involved in calcium signaling were significantly regulated, mostly down (for example, Cxcr4, Adra2a, Ednra, P2ry1, Gnao1, Gng7), but some up (for example, P2ry14, P2ry6, Ccrl2, Gnb4). We tested selected cases and found that changes in GPCR gene expression were accompanied by significant, parallel changes in astrocyte calcium signaling evoked by corresponding GPCR-specific ligands. These findings identify pronounced changes in the astrocyte transcriptome induced by TGF-β1, LPS, and IFNγ, and show that these inflammatory stimuli upregulate astrocyte molecular networks associated with immune- and injury-related functions and significantly alter astrocyte calcium signaling stimulated by multiple GPCRs.
Introduction
Both inflammation and reactive astrogliosis are features of many CNS disorders including stroke, trauma, neurodegeneration, infection, and autoimmunity (Lucas et al., 2006; Barres, 2008; Sofroniew and Vinters, 2010), yet how their interactions influence one another is not well understood. Astrocytes produce and respond to numerous molecules involved in immune and inflammatory signaling, including chemokines, cytokines, growth factors, and many small molecules (Eddleston and Mucke, 1993; John et al., 2003; Lovatt et al., 2007; Cahoy et al., 2008; Sofroniew, 2009; Zamanian et al., 2012). Nevertheless, the roles that astrocytes play during CNS immune and inflammatory responses, and the effects of inflammatory mediators on astrocyte functions, are not well defined.
During CNS inflammation, multiple cell types can produce multiple inflammatory mediators. Combinatorial interactions of inflammatory signaling molecules are becoming recognized as able to provide unique instructions to specific cells, as demonstrated for certain immune cells (Korn et al., 2009). In contrast, combinatorial effects of multiple inflammatory mediators on astrocytes are unclear. In this study, we (1) determined the combinatorial effects of several representative inflammatory mediators on astrocyte gene expression profiles in vitro, (2) used bioinformatic data analyses to look for changes that might impact on specific astrocyte functions, and (3) examined the functional consequences of certain gene expression changes.
As representative inflammatory mediators we examined transforming growth factor-β1 (TGF-β1), a cytokine produced during CNS inflammation in many disorders (Lindholm et al., 1992; Krupinski et al., 1996; Ata et al., 1997; Swardfager et al., 2010; Friedman and Dingledine, 2011), and lipopolysaccharide (LPS) and interferon-gamma (IFNγ), which are canonical innate inflammatory mediators used extensively to study cellular responses to inflammation (Raetz and Whitfield, 2002; Schroder et al., 2004; Daginakatte et al., 2008). Microglial production of factors like IFNγ upregulate expression of toll-like receptors in astrocytes (Holm et al., 2012). There are numerous contexts in vivo during CNS disorders in which astrocytes are exposed to combinations of TGF-β1 and innate inflammatory mediators such as LPS and IFNγ. We have shown previously that TGF-β1 enhances the LPS and IFNγ-induced expression of inducible nitric oxide synthase (iNOS) or cyclooxygenase-2 in astrocytes (Hamby et al., 2006a, 2008, 2010).
Here, we asked how these inflammatory mediators, alone and in combination, might alter astrocyte functions by modulating entire gene expression profiles. We conducted genome-wide microarray profiling and bioinformatic analyses (Coppola et al., 2009; Coppola, 2011) on well characterized preparations of purified primary astrocyte cultures (Hamby et al., 2006b) exposed to TGF-β1, LPS, and IFNγ alone and in combination. Expression levels of many genes, molecular networks, and functional pathways were significantly altered. Our attention was drawn particularly to changes in numerous G-protein-coupled receptors (GPCRs) and their intracellular effectors because of the potential impact on astrocyte functions. Changes in GPCRs are implicated in various inflammatory conditions (Lattin et al., 2007) and GPCRs play central roles in astrocyte calcium signaling. Astrocytes display spontaneous and ligand-evoked intracellular calcium concentration ([Ca2+]i) increases that represent a form of astrocyte excitability, and signaling via changes in astrocyte [Ca2+]i is under investigation as a means of mediating dynamic astrocyte functions, including interactions with synapses and regulation of blood flow (Verkhratsky et al., 1998; Iadecola and Nedergaard, 2007; Barres, 2008; Attwell et al., 2010; Halassa and Haydon, 2010). We therefore evaluated astrocyte calcium signaling evoked by ligands of various GPCRs and found that changes in gene expression induced by combinatorial inflammatory treatment were accompanied by parallel changes in ligand-evoked [Ca2+]i increases.
Materials and Methods
Astrocyte culture.
Primary astrocyte cultures were prepared from cerebral cortices of postnatal (1–3-d-old), male and female C57BL/6 mice as previously described (Hamby et al., 2006a,b). In brief, plating media consisted of l-glutamine-free DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Hyclone), 10% calf serum (CS; Hyclone), 2 mm l-glutamine, 50 IU/ml penicillin, 50 μg/ml streptomycin, and 10 ng/ml epidermal growth factor (R&D Systems). Upon confluence, astrocyte cultures were purified of microglia by treatment with 8 μm cytosine β-d-arabinofuranoside (Sigma) for 5–6 d to substantially reduce microglia followed by treatment with 75 mm l-leucine methyl ester (Sigma, 60–90 min) 1 d before experimentation to completely eradicate any residual microglia (Hamby et al., 2006a,b). Cells were maintained in medium consisting of l-glutamine-free DMEM, 10% CS, 2 mm l-glutamine, 50 IU/ml penicillin, and 50 μg/ml streptomycin. For imaging experiments, astrocytes were passaged onto glass coverslips. All cultures were maintained at 37°C in a humidified atmosphere of 6% CO2 and used after 14–31 d in vitro.
Cytokine and endotoxin exposure.
Purified astrocyte cultures were exposed to four different experimental stimulation conditions in stimulation medium (SM), which consisted of l-glutamine-free DMEM supplemented with 5% CS, 2 mm l-glutamine, 50 IU/ml penicillin, and 50 μg/ml streptomycin. (1) Basal (B): 32 or ∼48 h in SM; (2) TGF-β1 (T): 32 or ∼48 h in SM with TGF-β1 (3 ng/ml; human recombinant, R&D Systems); (3) LPS + IFNγ (LG ); 24 h in SM before addition of LPS (Escherichia coli 0127:B8, 2 μg/ml; Sigma) plus IFNγ (recombinant mouse, 3 ng/ml; R&D Systems) for a further 8 or ∼24 h; (4) TGF-β1 + LPS + IFNγ (TLG): 24 h in SM with TGF-β1 before addition of LPS + IFNγ for a further 8 or ∼24 h, for mRNA expression analyses, or [Ca2+]i imaging, respectively. The 24 h duration of TGF-β1 treatment before addition of SM alone or that with LPS plus IFNγ was chosen on the basis of previous experiments showing a maximal and stable effect of TGF-β1 on induction of iNOS and enhancement of nitric oxide production in purified astrocyte cultures over this time period (Hamby et al., 2006a). In agreement with previous studies, no cytopathological changes were observed in the astrocytes following treatment with these agents at the doses used in the present studies.
Microarray profiling and analysis.
Microarray profiling was performed as previously described (Coppola et al., 2009; Sarafian et al., 2010; Coppola, 2011). In brief, total RNA was isolated from astrocytes using the Qiagen RNeasy Kit. RNA quantity and quality was assessed using a Nanodrop (Nanodrop Technologies) and the Agilent Bioanalyzer (Agilent Technologies), respectively. Total RNA (200 ng) was amplified, biotinylated, and hybridized on Illumina Mouse ref-8 v2.0 BeadChip arrays, querying the expression of ∼25,600 well annotated Refseq transcripts, as per manufacturer's protocol. Slides were scanned using Illumina BeadStation and signal extracted using Illumina BeadStudio software (Illumina).
Raw data were analyzed using Bioconductor packages (www.bioconductor.org). Quality-control analysis was performed using several indices including interarray Pearson correlation, clustering based on variance, and the mean absolute deviation (MAD) using the top 1000 most variant probes (Coppola, 2011). Data were normalized using quantile normalization. Analysis of differential expression was performed using linear model fitting (LIMMA package) (Smyth, 2004). After linear model fitting, a Bayesian estimate of differential expression was calculated using a false discovery rate (FDR) of 5%. Nonsignificant (NS) results indicate no change in expression levels relative to the basal or treated cells as indicated. NS does not indicate whether the cells actually express the gene or not. Raw and normalized data have been deposited in NCBI's Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE36089 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36089). Pathway and network analyses were performed using Ingenuity Pathway Analysis (Ingenuity Systems; www.ingenuity.com). Venn diagram was made using SmartDraw software.
Quantitative real time PCR.
RNA was extracted using RNeasy Mini Kit (Qiagen) following the manufacturer's directions. cDNA was synthesized from 0.5 μg RNA using MMLV reverse transcriptase primed with oligo(dT)(12–18), as previously described (Hamby et al., 2006a). Quantitative real-time PCR (qRT-PCR) was performed using a LightCycler 480 (Roche) and LightCycler 480 SW 1.5 software (Roche) using Taqman Probes (Applied Biosystems). Twenty microliter reactions (in H2O) consisted of 2.5 μl cDNA, 10 μl Taqman Universal Gene Expression Master Mix (Applied Biosystems), and 2 μl of a specific Taqman Probe. Taqman probes (Assay ID; Applied Biosytems) were used to detect expression of mouse Gjb2 (Mm00433643_s1), Lif (Mm00434762_g1), Il6 (Mm00446190_m1), Tlr2 (Mm00442346_m1), Ccl5 (Mm01302427_m1), Cxcl1 (Mm04207460_m1), Nos2 (Mm00440502_m1), Sema4a (Mm00443140_m1), Cxcl10 (Mm00445235_m1), Klf2 (Mm01244979_g1), Myd88 (Mm00440338_m1), Gp130 (Mm00439665_m1), S1pr3 (Mm04229896_m1), Ednrb (Mm00432989_m1), Ednra (Mm01243722_m1), Cxcr4 (Mm01292123_m1), Adra2a (Mm00845383_s1), Adora1 (Mm01308023_m1), F2rl1 (Mm00433160_m1), P2ry1 (Mm00435471_m1), or the housekeeping gene Gapdh (Mm99999915_g1) for normalization of total cDNA/sample. Cycling conditions were 94°C for 10 min, followed by 45 cycles at 95°C for 15 s and 60°C for 15 s with all ramp up/down rates at 1.6°C/s. Relative quantification of cDNA was calculated using the comparative cycle threshold (_C_T) method, wherein target _C_T levels were normalized to _Gapdh C_T values from the same sample (separate wells). The normalized _C_T values were then compared with a calibrator value (untreated astrocytes) to determine the relative fold or log2-fold changes in target mRNA levels over the calibrator.
Intracellular calcium imaging.
Astrocytes were imaged as described previously (Shigetomi and Khakh, 2009). Briefly, astrocytes were loaded with 2.5 μm Fluo-4 AM (Invitrogen) in the presence of 0.05% Pluronic F-127 (20% solution in dimethylsulfoxide) in physiological buffer for 30 min, then transferred to dye-free buffer for 30 min before experimentation to allow cleavage of the AM ester group. Physiological buffer consisted of 110 mm NaCl, 5.4 mm KCl, 1.8 mm CaCl2, 0.8 mm MgCl2, 10 mm d-glucose, and 10 mm HEPES, adjusted to pH 7.40 using NaOH, in deionized ultrapure water. Fluorescence was captured using an Olympus BX51 or BX61WI microscope with an Olympus 40× 0.8 NA water-immersion objective lens and a cooled CCD camera (Imago). Excitation was provided by a Polychrome V or Polychrome IV monochromater (TILL Photonics). The hardware was controlled by a personal computer, an appropriate frame grabber (TILL Photonics), and macros driven by TILLvisION software. Appropriate filters (Chroma Technology) were chosen for Fluo-4 and Fura2 imaging (Glen Spectra). Imaged cells were continually perfused with either physiological buffer alone or containing various GPCR agonists using a custom-made fast solution switcher that was controlled manually or via a digital interface controlling a bank of solenoid valves (Lee Company), both allowing for fast exchange of solutions in seconds (Shigetomi et al., 2012). GPCR agonists used were as follows: uridine 5′-diphosphoglucose disodium salt (UDP-Glc; Sigma), Uridine 5′-diphosphogalactose disodium salt (UDP-Gal; Sigma), 2-chloro-N6-cyclopentyladenosine (CCPA; Tocris Bioscience), guanfacine HCl (Tocris Bioscience), B-HT 933 (Tocris Bioscience), (−)-epinephrine (Sigma), recombinant mouse CXCL12/SDF-1a (R&D Systems), IRL1620 (Tocris Bioscience), BQ-3020 (Tocris Bioscience), endothelin-1 (Tocris Bioscience), adenosine (Tocris Bioscience), ADPβS (Calbiochem), ADP (Sigma), or ATP (Sigma). Data were analyzed using National Institutes of Health ImageJ, Microsoft Excel, and Graphpad Prism software.
siRNA transfection.
Astrocytes plated on 12 mm coverslips in 24-well plates were transfected with experimental or negative control siRNA using Lipofectamine 2000 (Invitrogen) per manufacturer's instructions. Several hours before transfection, astrocytes were washed with and fed penicillin and streptomycin-free Optimem (Invitrogen). Before transfection, culture medium was removed and 400 μl Optimem medium was added. After making the siRNA (40pmol/well) plus Lipofectamine 2000 (1 μl/well) in Optimem mixture, 100 μl was added into each well. Experimental siRNAs used were as follows: Ednrb, EdnrbMSS203783; P2ry1, P2ry1MSS207363; and P2ry14, P2ry14MSS204176 (Stealth Select RNAi siRNA; Invitrogen). Negative control siRNA used was Stealth Select RNAi siRNA Negative Control LO GC (catalog #12935–200; Invitrogen). Astrocytes were fed with maintenance medium the following day and imaged for calcium measurements 4 d post-transfection.
In vivo injections of cytokine and endotoxin.
All in vivo experiments were conducted using wild-type, male and female C57BL/6 mice from an in-house breeding colony. Mice were housed in a 12 h light/dark cycle in a specific pathogen-free facility with controlled temperature and humidity and allowed free access to food and water, and all surgical procedures and experiments were conducted according to protocols approved by the Chancellor's Animal Research Committee of the Office for Protection of Research Subjects at University of California Los Angeles. All surgical procedures were performed under sterile conditions with isoflurane in oxygen-enriched air as the general anesthesia and using an operating microscope (Zeiss) and rodent stereotaxic apparatus (David Kopf) as described previously (Myer et al., 2006). The skull was exposed and a burr hole was drilled with a high-speed dental drill. Solutions of 1 μl of sterile PBS or TGFβ+LPS+IFNγ prepared in PBS were injected stereotaxically into the frontal, sensorimotor cortex using the target coordinates of 0.0 mm anterior to bregma, 1.5 mm lateral to bregma, and a depth of 0.75 mm below the cortical surface. Injections were made at a speed of 0.2 μl per minute using glass micropipettes (pulled and ground to 25–50 μm tips) connected via specialized connectors and high-pressure tubing (Kopf) to a 10 μl syringe (Hamilton) under the control of a microinfusion pump (Harvard Instruments). TGFβ+LPS+IFNγ PBS consisted of TGF-β1 (0.3 μg/μl; human recombinant, R&D Systems) and LPS (2 μg/μl; E. coli 0127:B8, Sigma) plus IFNγ (0.3 μg/μl; recombinant mouse, R&D Systems). Analgesic was given before wound closure and every 12 h for 48 h postinjury. No adverse effects were observed.
Immunohistochemistry.
After survival times of 1 d, 3 d, or 5 d after forebrain injections, mice received terminal anesthesia by barbiturate overdose and were perfused transcardially with PBS followed by 10% formalin in PBS. Brains were removed, postfixed overnight, and cryoprotected in buffered 30% sucrose for at least 2 d. Coronal frozen sections (40 μm) were prepared using a cryostat microtome (Leica) and processed for fluorescence immunohistochemistry as described previously (Faulkner et al., 2004; Voskuhl et al., 2009). The following primary antibodies were used: mouse anti-GFAP (1:500; Sigma), rabbit anti-CXCL1 (1:100; Lifespan BioSciences), rabbit anti-CCL7 (1:100; Lifespan BioSciences), goat anti-CXCL10 (1:200; Santa Cruz Biotechnology), rabbit anti-IL-6 (1:100; Abcam), and rabbit anti-ADRA2A (1:200; Lifespan BioSciences. Fluorescence immunohistochemistry was performed using secondary antibodies with Alexa 488 (green) (Invitrogen) and Cy5 (red) (Vector Laboratories). The nuclear stain, 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI; 2 ng/ml, Invitrogen), was used to label nuclei. Stained sections were examined and photographed using fluorescence and laser scanning confocal microscopy (Zeiss).
Results
Characterization of genomic analyses
To identify genome-wide changes in astrocyte gene expression induced by inflammatory mediators, we used well characterized cell cultures of purified astrocyte that are >99% astrocytes and essentially free of microglia and other cell types (Hamby et al., 2006b), together with a previously established model of combinatorial stimulation with TGF-β1 and LPS+IFNγ (Hamby et al., 2006a, 2008, 2010). We used Illumina BeadChip-based microarray profiling to compare genome-wide effects of treating these cultures with TGF-β1 alone, LPS+IFNγ, or all three stimuli (TGFβ+LPS+IFNγ) combined, relative to basal (untreated) astrocytes. Raw and normalized data have been deposited in NCBI's Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE36089 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36089). Hierarchical clustering (Fig. 1A) highlighted a high degree of similarity among n = 4 independent samples from distinct astrocyte cultures within each given treatment group, thereby demonstrating that the changes in gene expression were due to the specific treatment conditions and not due to culture-to-culture variability. In addition, we assessed the purity of the astrocyte cultures used for gene array analysis by examining eight probes from the microarrays for three genes selectively associated with microglia—Iba1 (three probes, 1212938, 1218123, and 2804487), Emr1/F4/80 (three probes, 1216880, 2756353, and 2847787), and Csf1r/c-fms (two probes, 1230485 and 2710819)—and found expression levels compatible with low or no expression, as well as no significant differences among any of the probes in any of the three treatment conditions TGF-β1, LPS+IFNγ, or TGFβ+LPS+IFNγ. We further conducted RT-PCR for the microglial marker, Iba1, and found no detectable bands in any of the three treatment conditions in contrast with bands obtained from positive control samples (data not shown). Together these findings confirm both the integrity of our basic experimental protocol and the absence of microglial contamination from the purified astrocyte cultures examined for the present study.
Figure 1.
Effects on astrocyte genomic profiles of treatment with TGF-β1, LPS+IFNγ, or all three stimuli combined. A, Gene array clustering of the top 1000 most variable genes shown as a colored heatmap depicting the MAD value for each interarray comparison for samples 1–4 for each treatment in astrocytes as indicated. B, basal; T, TGF-β1; LG, LPS+IFNγ; TLG, TGF-β1+LPS+IFNγ. The color map key (top left) represents a low MAD value indicating very little variability (yellow) between comparisons whereas red represents a high MAD value indicating greater differences between comparisons. Dendrogram indicates clustering of samples and treatments based on similarity. B, Heatmap indicating significant changes (FDR p value ≤ 0.05) in gene expression (black, no change; red, increase; green, decrease) due to TGF-β1 (T vs B), LPS+IFNγ (LG vs B), and TGF-β1+LPS+IFNγ (TLG vs B), compared with basal astrocytes, as well as TLG vs T and TLG versus LG. Genes are clustered by similarity. Boxed region denoted by asterisks indicates examples of genes that behaved differently across different treatment conditions. C, Total number of genes significantly (FDR p ≤ 0.05) altered (up, red; down, green) by each treatment comparison. D, Venn diagram depicts the number of genes changed in a treatment-specific and nonspecific manner. Within each segment, upper red numerical values = number of genes upregulated. Lower green numerical values = number of genes downregulated. Total numbers of genes changed are labeled outside of the Venn diagram beside its respective circle's treatment condition. E, Flow chart depicting filtering strategy conducted using statistical cutoffs to generate subsequent datasets for analysis. Numbers indicate number of probes within each filtered dataset and a–d demarcate the four new dataset categories for subsequent analysis. S, significant; NS, nonsignificant. F, Categories of additive and synergistic treatment interactions emerging from filtering strategy in E. All categories a–d had a further change from either T (c) or LG (b) or both (a, d) due to TLG treatment. Numbers of gene probes are shown indicating additive or synergistic gene regulation by TLG treatment per category. A–E, n = 4 independent samples from distinct astrocyte cultures.
Treatment-dependent global changes in gene expression profiles
We began our data evaluations by characterizing the global changes in gene expression induced by the different treatment conditions—TGF-β1 alone, LPS+IFNγ, or TGFβ+LPS+IFNγ—each compared with untreated (basal) astrocytes, using a stringent criterion of significance that corrects for multiple testing using a FDR of < 0.05. These analyses revealed that large-scale changes in gene expression occurred as a result of treatment with either LPS+IFNγ (6149 differentially expressed probes) or TGFβ+LPS+IFNγ (6864) relative to basal (untreated) astrocytes, whereas treatment with TGF-β1 alone (1420) induced relatively fewer changes (Fig. 1B–E). All three stimulation conditions elicited approximately proportionate changes with respect to the number of upregulated genes (TGF-β1, 736; LPS+IFNγ, 2582; TGFβ+LPS+IFNγ, 2934) versus downregulated genes (TGF-β1, 684; LPS+IFNγ, 3567; TGFβ+LPS+IFNγ, 3930) (Fig. 1C). Importantly, while some genes were altered (up or down) by only one treatment in a treatment-specific manner, others were consistently altered across two or all three of the treatments (Fig. 1B,D).
Greatest fold changes in specific genes
To identify the most highly regulated genes, we sorted the list of gene probes according to the greatest log2-fold changes in gene expression (FDR p ≤ 0.05) induced by each of the three treatment conditions relative to basal. Table 1 shows the top 10 genes with the greatest log2-fold increases and decreases in expression for each treatment condition, TGF-β1, LPS+IFNγ, or TGFβ+LPS+IFNγ, relative to basal. It is noteworthy that 7 of the top 10 genes that were most highly upregulated by combinatorial treatment with TGFβ+LPS+IFNγ encode molecules involved in immune and inflammatory processes, namely the chemokines, Cxcl1, Cxcl9, Cxcl10, Ccl2, Ccl5, and Ccl7 and the toll-like receptor, Tlr2 (Table 1).
Table 1.
Top upregulated and downregulated genes
| Top upregulated genes | Top downregulated genes | ||||||
|---|---|---|---|---|---|---|---|
| Probe ID | Gene | Gene description | log2 ratio | Probe ID | Gene | Gene description | log2 ratio |
| TGF-β1+LPS+IFNγ | TGF-β1+LPS+IFNγ | ||||||
| 2763245 | Cxcl1 | Chemokine (C-X-C motif) ligand 1 | 7.77 | 2602581 | Gmpr | Guanosine monophosphate reductase | −3.49 |
| 1215862 | Cxcl9 | Chemokine (C-X-C motif) ligand 9 | 7.77 | 2958067 | RIKEN cDNA 6030405A18 gene | −3.44 | |
| 3142803 | Cxcl10 | Chemokine (C-X-C motif) ligand 10 | 7.34 | 2741063 | Aif1l | Allograft inflammatory factor 1-like | −3.42 |
| 2835117 | Ccl7 | Chemokine (C-C motif) ligand 7 | 7.26 | 2736471 | Ccno | Cyclin O | −3.21 |
| 1231814 | Ccl5 | Chemokine (C-C motif) ligand 5 | 7.25 | 2768972 | Fam107a | Family with sequence similarity 107, member A | −2.93 |
| 3160750 | Expressed sequence AA467197 | 6.67 | 2678366 | Psd2 | Pleckstrin and Sec7 domain containing 2 | −2.90 | |
| 1225204 | Rsad2 | Radical _S_-adenosyl methionine domain containing 2 | 6.55 | 2967750 | Fam123a | Family with sequence similarity 123, member A | −2.74 |
| 2733733 | Tlr2 | Toll-like receptor 2 | 6.50 | 2628174 | Acas2l | Acyl-CoA synthetase short-chain family member 1 | −2.73 |
| 1245710 | Ccl2 | Chemokine (C-C motif) ligand 2 | 6.48 | 2954824 | Ppp1r1b | Protein phosphatase 1, regulatory (inhibitor) subunit 1B | −2.62 |
| 2662802 | Ptx3 | Pentraxin related gene | 6.47 | 3062267 | Acot1 | Acyl-CoA thioesterase 1 | −2.61 |
| LPS+IFNγ | LPS+IFNγ | ||||||
| 1215862 | Cxcl9 | Chemokine (C-X-C motif) ligand 9 | 7.84 | 2741063 | Aif1l | Allograft inflammatory factor 1-like | −3.00 |
| 2763245 | Cxcl1 | Chemokine (C-X-C motif) ligand 1 | 7.77 | 2768972 | Fam107a | Family with sequence similarity 107, member A | −2.98 |
| 1231814 | Ccl5 | Chemokine (C-C motif) ligand 5 | 7.40 | 2958067 | RIKEN cDNA 6030405A18 gene | −2.80 | |
| 3142803 | Cxcl10 | Chemokine (C-X-C motif) ligand 10 | 7.39 | 2602581 | Gmpr | Guanosine monophosphate reductase | −2.56 |
| 2835117 | Ccl7 | Chemokine (C-C motif) ligand 7 | 6.89 | 1242170 | Adra2a | Adrenergic receptor, α 2a | −2.49 |
| 2733733 | Tlr2 | Toll-like receptor 2 | 6.50 | 2652414 | Ncald | Neurocalcin delta | −2.49 |
| 2662802 | Ptx3 | Pentraxin related gene | 6.48 | 1214880 | Dmn | Desmuslin (Dmn), transcript variant 2 | −2.48 |
| 1225204 | Rsad2 | Radical _S_-adenosyl methionine domain containing 2 | 6.47 | 2736471 | Ccno | Cyclin O | −2.41 |
| 2652857 | Ifi47 | IFNγ inducible protein 47 | 6.37 | 1246821 | RIKEN cDNA E130203B14 gene | −2.39 | |
| 2426853 | Ubd | Ubiquitin D | 6.35 | 2954824 | Ppp1r1b | Protein phosphatase 1, regulatory (inhibitor) subunit 1B | −2.35 |
| TGF-β1 | TGF-β1 | ||||||
| 2633350 | Mfap4 | Microfibrillar-associated protein 4 | 3.51 | 2958067 | RIKEN cDNA 6030405A18 gene | −2.13 | |
| 3160638 | Ripply1 | ripply1 | 3.08 | 1230586 | Traf1 | Tnf receptor-associated factor 1 | −1.54 |
| 2742042 | Nupr1 | Nuclear protein 1 | 2.90 | 2862026 | RIKEN cDNA 1700084C01 gene | −1.48 | |
| 2793062 | Rasl11b | RAS-like, family 11, member B | 2.62 | 1231274 | Cklfsf5 | CKLF-like MARVEL Transmembrane domain containing 5 | −1.48 |
| 2706269 | Hspb1 | Heat shock protein 1 | 2.20 | 2702266 | Crygs | Crystallin, gamma S | −1.42 |
| 2966104 | Htra1 | HtrA serine peptidase 1 | 2.14 | 2742426 | Selenbp1 | Selenium binding protein 1 | −1.33 |
| 1255871 | Loxl1 | Lysyl oxidase-like 1 | 2.11 | 2700408 | Mgll | Monoglyceride lipase | −1.31 |
| 2675697 | Kdelr3 | KDEL Endoplasmic reticulum protein retention receptor 3 | 2.04 | 1249886 | Rln1 | relaxin 1 | −1.26 |
| 2636424 | Itgbl1 | Integrin, β-like 1 | 1.99 | 1226356 | Vat1l | Vesicle amine transport protein 1 homolog-like (T. californica) | −1.23 |
| 3160292 | Akr1c19 | Aldo-keto reductase family 1, member C19 | 1.99 | 2798402 | Lcat | Lecithin cholesterol acetyltransferase | −1.23 |
Synergistic effects of combinatorial treatment
We next asked whether gene regulatory changes in the combinatorial treatment group, TGFβ+LPS+IFNγ, were simply the sums of the effects of TGF-β1 treatment and LPS+IFNγ treatment, or whether there were additional effects due to synergistic combinatorial interactions. To examine the gross extent of change, we examined heat maps that represented the number of genes changed by TGF-β1 versus basal, LPS+IFNγ versus basal, TGFβ+LPS+IFNγ versus basal, TGFβ+LPS+IFNγ versus TGF-β1, and TGFβ+LPS+IFNγ versus LPS+IFNγ (Fig. 1B). This comparison indicated at a gross level that there were subsets of genes that behaved differently across conditions (Fig. 1B, boxed region denoted by asterisks). Further analyses of these comparisons using an FDR p ≤ 0.05 revealed that substantial numbers of genes were altered by TGFβ+LPS+IFNγ versus TGF-β1 (5574 total) or by TGFβ+LPS+IFNγ versus LPS+IFNγ (1038 total), in comparison with genes regulated by TGFβ+LPS+IFNγ versus basal (6864) or LPS+IFNγ versus basal (6149) or TGF-β1 versus basal (1420) (Fig. 1E). These findings suggested the potential for combinatorial treatment interactions in which treatment with TGFβ+LPS+IFNγ might lead to substantial changes in gene expression relative to treatment with either TGF-β1 or LPS+IFNγ alone.
To dissect the nature of these potential combinatorial treatment interactions, we first determined how many gene probes were modulated significantly (FDR ≤0.05) by combinatorial treatment in either the up or down direction relative to the individual treatments, regardless of whether or not the combinatorial effects were additive or synergistic. To simplify this analysis, we grouped gene probes into four categories (a–d) for evaluation based on whether individual treatments had (FDR ≤ 0.05) or had not (FDR > 0.05) caused significant changes in gene expression (Fig. 1Ea–d,Fa–d). In this manner we identified (a) 295 gene probes in which TGF-β1 and LPS+IFNγ had both caused significant changes versus basal (Fig. 1Ea); (b) 322 gene probes in which TGF-β1 had not, and LPS+IFNγ had, caused significant changes versus basal (Fig. 1Eb); (c) 125 gene probes in which TGF-β1 had but LPS+IFNγ had not caused significant changes versus basal (Fig. 1Ec); and (d) 90 gene probes in which neither TGF-β1 nor LPS+IFNγ had caused significant changes versus basal (Fig. 1Ed); where in all of these cases (a–d) the combinatorial treatment had caused significant changes relative to the individual treatments (Fig. 1Ea–d; Fig. 1E, filtering criteria). To determine which of these combinatorial treatment interactions reflected changes that were additive or synergistic, we further dissected the subsets of genes in categories a–d (Fig. 1F). Within the four categories, we identified substantial numbers of gene probes that exhibited either additive or synergistic combinatorial treatment interactions, with a conservative estimate of a total of >380 genes that were synergistically modulated in either the up or down direction by combinatorial treatment with TGFβ+LPS+IFNγ relative to individual treatments alone (Fig. 1Fa–d). It is noteworthy that these synergistic interactions consisted of different types of gene expression regulation that could not be predicted on the basis of summing the effects of individual stimulators alone. For example, in 90 cases (category d), significant changes in gene expression occurred due to combinatorial treatment with TGFβ+LPS+IFNγ, even though treatment with TGF-β1 alone and LPS+IFNγ alone caused no significant change (Fig. 1Fd). In other cases combinational stimulation with TGFβ+LPS+IFNγ either significantly augmented or attenuated, or in some cases reversed, changes induced by individual treatments (Table 2). Prototypical genes within each category are listed in Table 2.
Table 2.
Types of synergistic interactions and example genes
| Synergistic category* | T | LG | TLG | Example genes |
|---|---|---|---|---|
| d | No change | No change | Up | Itga5, Bmp1, Tgfb1, Homer1, Rims3, Pi4k2b |
| d | No change | No change | Down | Dtx4, Tmem53 |
| b | No change | Up | Further up | Il6, Lif, Gdnf, Nos2, Sod3, Sema4a, Egr2, Ccl8, Ccl12, Gpr83, Gjb2, Slc16a9, Casp8 |
| b | No change | Up | Down (relative to LG) | Calb2, Klf2, Brip1, Ptges, |
| b | No change | Down | Further down | Itpk1, Gpr23 |
| b | No change | Down | Up (relative to LG) | Adora1, Gpr156, Fgf12, Ctgf, Slc4a2, |
| c | Up | No change | Further up | Ccl17, Col12a1, Eln, Lox |
| c | Up | No change | Down (relative to T) | Igf1, Cav1, Sema7a, Cd28, Slc7a5, |
| c | Down | No change | Up (relative to T) | Thbs1, Foxp1 |
| a | Up | Up | Further up | Il11, Cxcl16, P2ry6, P2ry14, Pdgfb, Dok1 |
qRT-PCR validation of microarray genomic profiling
To test the validity of the large-scale changes in gene expression found using the Illumina BeadChip-based microarray profiling, we generated new samples of astrocytes for all treatment conditions and used qRT-PCR to evaluate several highly regulated genes, Cxcl1, Ccl5, Cxcl10, and Tlr2 (Table 1). We found similar changes in mRNA expression to those found via microarray analysis for all genes examined (Fig. 2). To validate the synergistic effects of combinatorial treatment revealed by genome-wide microarray profiling, we used qRT-PCR to evaluate various genes synergistically induced by treatment with TGFβ+LPS+IFNγ compared with individual treatments, Il6, Lif, Gjb2, Nos2, Sema4a, Klf2, and Myd88 (Table 2) and found similar changes in mRNA expression to those found via microarray analysis for all genes examined (Fig. 2). In addition, we examined a gene whose expression was not significantly altered in any treatment condition in the microarray profiling, Gp130, and found that qRT-PCR results confirmed that there was no change in mRNA expression in any treatment condition relative to basal (Fig. 2).
Figure 2.

qRT-PCR validation of microarray data. qRT-PCR was performed on untreated or treated (T, TGF-β1; LG, LPS+IFNγ; TLG, TGF-β1+LPS+IFNγ) primary mouse astrocytes. Fold change in mRNA expression was calculated relative to basal after normalization of expression to GAPDH. Data were expressed as mean relative fold change in mRNA ± SEM (n = 3 from samples independently generated from that in Fig. 1). *, Indicates a significant fold increase compared with basal, whereas # indicates a significant difference in TLG-treated astrocytes compared with LG (p ≤ 0.05) as assessed via repeated-measures ANOVA after normalizing data followed by Newman–Keuls multiple-comparison test. Significantly altered (FDR ≤ 0.05) microarray log2 ratios are provided below treatment-labeled bars.
Bioinformatic analyses of molecular networks, functions, and pathways
We next used the Ingenuity Pathway Analysis software (Ingenuity Systems) to identify functional molecular groupings within our datasets of differentially regulated genes, based on their statistical association with known Associated Network Functions, Molecular and Cellular Functions, and Canonical Pathways in the Ingenuity knowledge base. For this analysis, we used a conservative threshold and selected genes with an absolute log2 fold change of at least 0.20, and an FDR corrected p value ≤0.05. Table 3 shows the top five Associated Network Functions, Table 4 the top five Molecular and Cellular Functions, and Table 5 the top 20 Canonical Pathways, that were significantly altered in each of the three treatment conditions. Comparisons across the different treatment groups indicated pronounced differences, particularly between combinatorial treatment with TGFβ+LPS+IFNγ and treatment with TGF-β1 alone. Recurrent themes among the Associated Network Functions and Molecular and Cellular Functions that were highly modulated by all three treatment conditions included cell death, cell compromise, and cell injury, as well as regulation of cell morphology, cell organization, cell maintenance, cell growth, and cell proliferation (Tables 4, 5). In addition, it is noteworthy that in the combinatorial treatment group, TGFβ+LPS+IFNγ, the most highly regulated network function was antigen presentation, and that all of the top 6, and 17 of the top 20 Canonical Pathways were related directly to immune or inflammatory signaling (Table 5).
Table 3.
Top 5 Associated Networks
| TGF-β1+LPS+IFNγ | LPS+IFNγ | TGF-β1 | ||||||
|---|---|---|---|---|---|---|---|---|
| Top networks | Score | Focus mol. | Top networks | Score | Focus mol. | Top networks | Score | Focus mol. |
| Antigen presentation, cell death | 18 | 35 | Cell death, cell cycle, organismal injury and abnormalities | 20 | 35 | Cellular development, cellular growth and proliferation | 30 | 31 |
| Cell morphology, cellular compromise, DNA replication, recombination, and repair | 18 | 35 | Lipid metabolism, small molecule biochemistry, vitamin and mineral metabolism | 20 | 35 | Lipid metabolism, small molecule biochemistry, vitamin and mineral metabolism | 30 | 31 |
| Genetic disorder, neurological disease | 18 | 35 | Cell-to-cell signaling and interaction, nervous system development and function, cellular development | 20 | 35 | Lipid metabolism, small molecule biochemistry | 30 | 31 |
| Post-translational modification, cell morphology, cellular assembly and organization | 18 | 35 | Organismal injury and abnormalities, cell morphology, cellular assembly and organization | 20 | 35 | Cell death, genetic disorder | 28 | 30 |
| Gastrointestinal disease, hepatic disease | 16 | 34 | Cell death, gene expression, cellular compromise | 18 | 34 | Cellular growth and proliferation, cancer | 25 | 28 |
Table 4.
Top five IPA Molecular and Cellular Functions
| TGF-β1+LPS+IFNγ | LPS+IFNγ | TGF-β1 | |||
|---|---|---|---|---|---|
| Category | p value | Category | p value | Category | p value |
| Cell death | 7.49 × 10−13 | Cell death | 3.57 × 10−9 | Cellular development | 1.07 × 10−6 |
| Cellular assembly and organization | 1.26 × 10−9 | Post-translational modification | 7.21 × 10−9 | Lipid metabolism | 2.18 × 10−5 |
| Cellular function and maintenance | 1.26 × 10−9 | Cellular assembly and organization | 8.40 × 10−9 | Small molecule biochemistry | 2.18 × 10−5 |
| Post-translational modification | 1.79 × 10−9 | Cellular function and maintenance | 8.40 × 10−9 | Vitamin and mineral metabolism | 2.18 × 10−5 |
| Cellular growth and proliferation | 1.04 × 10−8 | Cellular growth and proliferation | 4.44 × 10−9 | Cellular movement | 2.78 × 10−5 |
Table 5.
Top 20 IPA Canonical Pathways
| TGF-β1+LPS+IFNγ | LPS+IFNγ | TGF-β1 | ||||||
|---|---|---|---|---|---|---|---|---|
| Ingenuity canonical pathways | −log (p value) | Ratio | Ingenuity canonical pathways | −log (p value) | Ratio | Ingenuity canonical pathways | −log (p value) | Ratio |
| Activation of IRF by cytosolic PRRs | 9.05 | 0.56 | Molecular mechanisms of cancer | 8.33 | 0.38 | RhoA signaling | 4.18 | 0.18 |
| CD40 signaling | 8.83 | 0.62 | TNFR1 signaling | 7.99 | 0.60 | Wnt/β-catenin signaling | 2.99 | 0.14 |
| Role of IL-17A in arthritis | 8.63 | 0.62 | 4–1BB signaling in T-lymphocytes | 7.91 | 0.70 | Human embryonic stem cell Pluripotency | 2.96 | 0.14 |
| IL-1 signaling | 8.62 | 0.55 | TNFR2 signaling | 7.91 | 0.70 | biosynthesis of steroids | 2.84 | 0.29 |
| Toll-like receptor signaling | 8.37 | 0.67 | LPS-stimulated MAPK signaling | 7.77 | 0.54 | Caveolar-mediated endocytosis signaling | 2.49 | 0.15 |
| CD27 Signaling in lymphocytes | 8.32 | 0.63 | Activation of IRF by cytosolic PRRs | 7.30 | 0.50 | Sulfur metabolism | 2.46 | 0.25 |
| Type I diabetes mellitus signaling | 8.23 | 0.50 | CD27 signaling in lymphocytes | 7.27 | 0.57 | ILK signaling | 2.34 | 0.12 |
| LPS-stimulated MAPK signaling | 8.16 | 0.58 | Toll-like receptor signaling | 7.24 | 0.61 | Factors promoting cardiogenesis in vertebrates | 2.25 | 0.15 |
| IL-17 signaling | 8.15 | 0.61 | Induction of apoptosis by HIV1 | 7.11 | 0.55 | Integrin signaling | 2.07 | 0.11 |
| IL-17A signaling in fibroblasts | 8.09 | 0.69 | Type I diabetes mellitus signaling | 6.87 | 0.45 | Mitotic roles of polo-like kinase | 1.97 | 0.15 |
| 4-1BB signaling in T-lymphocytes | 8.03 | 0.73 | Apoptosis signaling | 6.82 | 0.50 | Actin cytoskeleton signaling | 1.89 | 0.10 |
| Role of MØs, fibroblasts and endo. cells in RA | 7.79 | 0.41 | Type II diabetes mellitus signaling | 6.63 | 0.43 | PPARα/RXRα activation | 1.80 | 0.11 |
| HMGB1 signaling | 7.77 | 0.54 | Interferon signaling | 6.51 | 0.62 | Endothelin-1 signaling | 1.80 | 0.12 |
| IL-6 signaling | 7.52 | 0.54 | NF-κB signaling | 6.51 | 0.42 | Propanoate metabolism | 1.79 | 0.16 |
| NF-κB signaling | 7.27 | 0.46 | TWEAK signaling | 6.44 | 0.61 | Agrin interactions at neuromuscular junction | 1.76 | 0.15 |
| ATM signaling | 7.17 | 0.65 | IL-17A signaling in fibroblasts | 6.42 | 0.62 | Role of MAPK signaling in the pathogenesis of influenza | 1.76 | 0.16 |
| Induction of apoptosis by HIV1 | 7.16 | 0.59 | Acute phase response signaling | 6.16 | 0.43 | Cdc42 signaling | 1.73 | 0.09 |
| Dendritic cell maturation | 7.06 | 0.39 | Death receptor signaling | 6.10 | 0.52 | Selenoamino acid metabolism | 1.68 | 0.19 |
| TNFR2 signaling | 7.02 | 0.70 | Role of RIG1-like Rs in antiviral inn. immunity | 6.03 | 0.46 | Regulation of actin-based motility by Rho | 1.62 | 0.13 |
| TNFR1 signaling | 6.88 | 0.60 | IL-1 signaling | 6.00 | 0.47 | Circadian rhythm signaling | 1.61 | 0.18 |
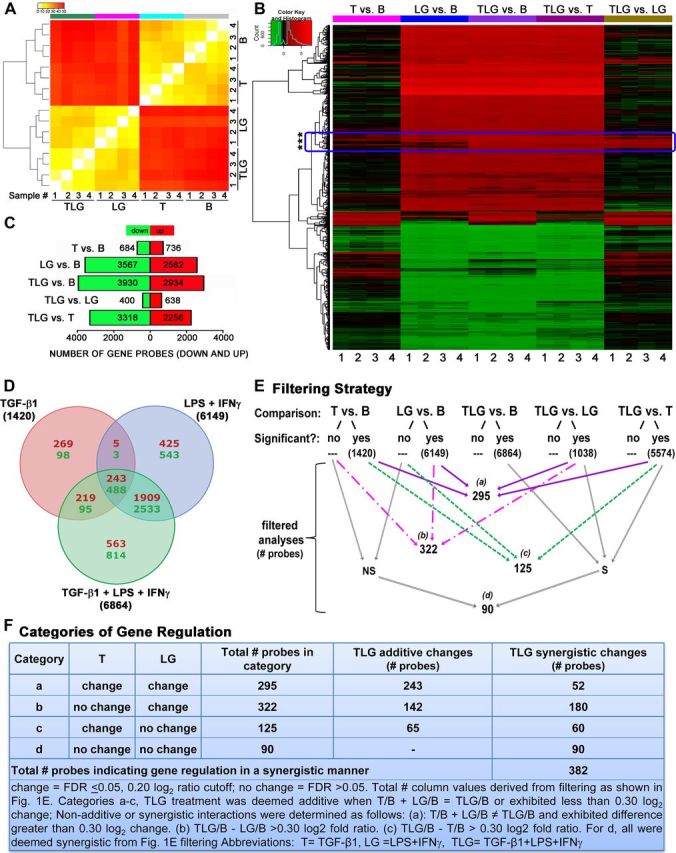
To gain an appreciation of individual genes that were highly regulated in the up or down direction by combinatorial treatment with TGFβ+LPS+IFNγ, we next examined the specific gene sets that comprised certain Associated Network Functions and Canonical Pathways. Due to space limitations, only selected examples are shown. We focused particularly on networks and pathways associated with immune and inflammatory signaling, regulation of cell compromise, death, growth, and proliferation that had been identified above as the main general themes represented in our analysis. These themes are heavily represented among the top three most regulated Associated Network Functions, which include Antigen Presentation and Cell Death (Fig. 3A); Cell Morphology, Compromise, and DNA Replication, Recombination, and Repair (Fig. 3B); and Genetic Disorder and Neurological Disease (Fig. 3C). The themes of immune and inflammatory signaling overwhelmingly dominated the most highly regulated Canonical Pathways (Table 5), illustrative examples of which include CD40 Signaling (Fig. 4A), IL-1 Signaling (Fig. 4B), and Chemokine Signaling (Fig. 4C). Inspection of these networks and pathways indicated that individual genes highly regulated by our combinatorial treatment with TGFβ+LPS+IFNγ included a wide variety of chemokines, cytokines, growth factors, and intercellular and intracellular signaling molecules, as well as diverse structural and regulatory molecules (Figs. 3, 4).
Figure 3.

Top three networks of significantly associated molecules in TGF-β1+LPS+IFNγ-treated astrocytes. A–C, Schematic diagrams of the top three predesignated Associated Molecular Networks identified using Ingenuity Pathway Analysis software (IPA; Ingenuity) as being affected by TGF-β1+LPS+IFNγ treatment of astrocytes. As defined in Table 3, the top three networks are Antigen Presentation and Cell Death (A); Cell Morphology, Cellular Compromise, and DNA Replication, Recombination, and Repair (B); and Genetic Disorder and Neurological Disease (C). Molecules are indicated by standard abbreviations. Molecular categories are indicated by icons defined in the icon key. Relative changes in gene expression are depicted by gradated shades of color coding: red, up; green, down. Direct and indirect interactions between molecules are depicted by solid and dotted lines, respectively.
Figure 4.
Three IPA Canonical Signaling Pathways: CD40, IL-1, and Chemokine Signaling in TGF-β1+LPS+IFNγ-treated astrocytes. A–C, Schematic diagrams of three of the top significantly altered predesignated Canonical Signaling identified using Ingenuity Pathway Analysis software (IPA; Ingenuity) as being affected by TGF-β1+LPS+IFNγ-treatment of astrocytes. As selected from Table 5, these pathways are CD40 Signaling (A), IL-1 Signaling (B), and Chemokine Signaling (C). Molecules are indicated by standard abbreviations. Relative changes in gene expression are depicted by gradated shades of color coding: red, up; green, down; white, no change or not applicable. Direct and indirect interactions between molecules are depicted by solid and dotted lines, respectively.
Regulation of specific genes in recognized functional categories
To further our understanding of the effects of all of our treatment conditions on specific genes, we next identified specific gene members according to commonly recognized functional molecular categories. We focused on categories broadly related to the main functional themes identified by the bioinformatic analysis described above and on categories relevant to astrocyte biology, and included also certain genes that exhibited little or no regulation (Table 6). These evaluations illustrated that a large number chemokines and cytokines exhibited a very high degree of regulation, as well as many growth factors; extracellular matrix molecules; transmembrane receptors; inflammation-related enzymes that produce small effector molecules; and intracellular signaling molecules including transcription factors, connexins, amino acid transporters, ion channels, and other astrocyte-related molecules (Table 6). It is noteworthy that many of these genes (demarcated by asterisks) exhibited synergistic regulation by combinatorial treatment with TGFβ+LPS+IFNγ relative to individual treatments with either TGF-β1 or LPS+IFNγ alone.
Table 6.
Individual genes
| Probe ID | Gene | TLG versus B | LG versus B | T versus B |
|---|---|---|---|---|
| Cytokines, chemokines | log2 ratio | log2 ratio | log2 ratio | |
| 2777498 | Il1b | 2.40 | 1.84 | NS |
| 1243601 | Il6 *(b) | 3.84 | 2.03 | NS |
| 2630852 | Il7 | 1.18 | 1.18 | NS |
| 1243862 | Il11 *(b) | 4.30 | 0.67 | NS |
| 2610234 | Il15 | 2.79 | 2.96 | NS |
| 2792502 | IL17d | −1.05 | −0.66 | −0.62 |
| 2711461 | Tgfb1 | 0.49 | NS | NS |
| 2899863 | Tnf | 1.29 | NS | NS |
| 2690603 | Spp1 | 2.94 | NS | 1.30 |
| 1245710 | Ccl2 (Mcp1) | 6.48 | 6.23 | NS |
| 1231814 | Ccl5 | 7.25 | 7.40 | NS |
| 2835117 | Ccl7 | 7.26 | 6.89 | NS |
| 1238886 | Ccl8 *(b) | 2.51 | 1.83 | NS |
| 2771766 | Ccl12 *(b) | 2.53 | 1.60 | NS |
| 2717015 | Ccl17 *(d) | 0.84 | NS | NS |
| 1238886 | Ccl8 | 2.51 | 1.83 | NS |
| 2763245 | Cxcl1 | 7.77 | 7.77 | NS |
| 1215862 | Cxcl9 | 7.77 | 7.84 | NS |
| 3142803 | Cxcl10 | 7.34 | 7.39 | 0.50 |
| 2658908 | Cxcl12(SDF-1) | 2.69 | 3.05 | NS |
| 2687586 | Cxcl16 | 4.47 | 3.82 | NS |
| Growth factors and related | ||||
| 3137287 | Lif * (b) | 2.29 | 0.53 | NS |
| 3105417 | Bdnf | 0.87 | −0.49 | 1.05 |
| 2659994 | Gdnf *(d) | 0.99 | NS | NS |
| 2701233 | Fgf1 | −1.84 | −1.87 | NS |
| 1239151 | Fgf2 | 0.84 | 0.53 | NS |
| 1225565 | Igf1 *(c) | 0.92 | NS | 1.45 |
| 2937596 | Ngfb | 0.81 | 0.35 | 0.51 |
| 2618714 | Pdgfb *(d) | 2.28 | NS | NS |
| 2989955 | Bmp1 *(d) | 0.72 | NS | NS |
| 2659151 | Thbs1 *(d) | 1.23 | NS | NS |
| 2484527 | Vegfa | 0.41 | NS | NS |
| Immune and inflammation-related receptors | ||||
| 2829008 | Lifr | 0.25 | 0.26 | NS |
| 3138904 | Ntrk2 (TrkB) | −0.80 | −0.66 | NS |
| 2602711 | Tgfbr1 | −0.23 | −0.28 | NS |
| 2760979 | Tgfbr2 | NS | 0.73 | −0.61 |
| 2789239 | Tgfbr3 | −0.66 | −0.51 | −0.43 |
| 2733733 | Tlr2 | 6.50 | 6.50 | NS |
| 2697002 | Tlr3 | 2.48 | 2.53 | NS |
| 2752966 | Tlr4 | 0.47 | 0.60 | NS |
| 2742075 | CD14 *(b) | 3.51 | 2.97 | NS |
| 2740852 | F2r (Par1) | NS | −0.28 | NS |
| 2513826 | Tnfrsf11b | 3.38 | 2.12 | 1.52 |
| 1226099 | Tnfsf13b *(b) | 2.30 | 1.29 | NS |
| 3115796 | CD40 | 6.07 | 5.60 | NS |
| 2589865 | CD28 *(c) | NS | NS | 0.47 |
| Transcription factors and intracellular signaling molecules | ||||
| 1245579 | Jak2 | 1.63 | 1.44 | NS |
| 2655721 | Stat1 | 4.15 | 4.23 | NS |
| 2657822 | Stat2 | 2.71 | 2.87 | NS |
| 3096144 | Stat3 | 1.33 | 1.38 | NS |
| 2618176 | Soc3 | 1.70 | 1.14 | 0.67 |
| 2938893 | Smad3 | 1.30 | 1.41 | NS |
| 2706631 | Smad4 | −0.59 | −0.55 | NS |
| 2971767 | MyD88 *(b) | 2.72 | 3.02 | NS |
| 2937735 | Irak2 | 0.86 | NS | 0.98 |
| 2592476 | Nfkb1 | 2.21 | 1.91 | NS |
| 2714534 | Map2k1 | 1.07 | 0.71 | NS |
| 1245924 | Map3k8 | 2.73 | 2.63 | NS |
| 2599782 | Irf1 | 4.82 | 5.09 | −0.35 |
| 2664319 | Irf3 | −0.21 | NS | NS |
| log2 ratio | log2 ratio | log2 ratio | ||
| 2621752 | Irf5 *(b) | 2.04 | 1.66 | NS |
| 2652857 | Ifi47 | 6.37 | 6.37 | NS |
| 2604029 | Klf2 *(b) | 0.57 | 1.53 | NS |
| 1252078 | Foxp1 *(d) | 0.31 | NS | NS |
| Extracellular matrix and cell–cell contact related | ||||
| 2699488 | Lamb2 | −0.71 | −0.72 | NS |
| 2974798 | Lamc1 | 0.95 | 1.08 | NS |
| 2775307 | Lamc2 | 1.12 | 0.74 | NS |
| 2670486 | Bcan | −1.04 | −0.59 | NS |
| 2993652 | Ncan | NS | −0.63 | NS |
| 1254975 | Has2 | 1.67 | 1.31 | NS |
| 2864172 | Chst3 | 0.67 | NS | 0.47 |
| 1216374 | Chst7 | 0.92 | 0.97 | NS |
| 2643049 | Chst8 | −0.80 | −1.34 | 1.14 |
| 1215528 | Chst11 | 1.56 | 0.49 | 0.83 |
| 2862538 | Col12a1 | 1.19 | NS | NS |
| 2599692 | Colm (Gliomedin) *(a) | 1.54 | 0.31 | 0.41 |
| 2697380 | Notch3 *(a) | 0.49 | −0.35 | 1.36 |
| 2753809 | Mmp3 | 2.74 | 1.85 | NS |
| 1215120 | Sema4A *(b) | 1.72 | 0.33 | NS |
| 2790357 | Sema7a *(c) | 0.47 | NS | 1.07 |
| 2670172 | Itga5 *(d) | 0.77 | NS | NS |
| Inflammatory-related enzymes | ||||
| 1223499 | Nos2 *(b) | 2.53 | 1.28 | NS |
| 2650280 | Sod2 | 1.42 | 1.57 | NS |
| 1241892 | Sod3 | 1.06 | 0.66 | NS |
| 2786442 | Ptges | 1.60 | 1.97 | NS |
| 2997494 | Lox *(c) | 2.42 | NS | 1.32 |
| Channels and connexins | ||||
| 2980661 | Aqp1 | 0.21 | NS | NS |
| 2757232 | Aqp4 | NS | NS | NS |
| 1214634 | Aqp9 *(d) | −0.65 | NS | NS |
| 1225901 | Aqp11 | −0.73 | −0.67 | −0.45 |
| 1244291 | Gja1 (Cx43) | −0.97 | −1.11 | NS |
| 2691641 | Gja5 (Cx40) *(b) | 0.64 | 0.27 | NS |
| 2999627 | Gjb2 (Cx26) *(b) | 2.91 | 1.32 | NS |
| 2946088 | Panx1 | 0.76 | 0.46 | NS |
| 2621548 | TrpA1 | NS | NS | NS |
| Transporters | ||||
| 3123195 | Slc1a2 (Glt1) | NS | 0.61 | NS |
| 2905589 | Slc6a1 (Gat1) | −0.26 | −0.26 | NS |
| 1212645 | Slc6a13 (Gat3) | NS | NS | NS |
| 2948143 | Slc7a11 (xCT) | 2.49 | 2.98 | NS |
| Astrocyte-related molecules | ||||
| 2706906 | Fthfd (Aldh1l1) | −0.65 | −0.39 | NS |
| 2644496 | Glul (Gln Synthetase) | −0.97 | −0.70 | NS |
| 1214715 | Gfap | NS | NS | NS |
| 2634742 | S100b | NS | NS | NS |
| 2840213 | Cryab | NS | NS | NS |
| 2712075 | Lcn2 (lipocalin2) | 4.98 | 4.98 | 0.82 |
| 2697304 | Eln (elastin) *(c) | 1.81 | NS | 0.95 |
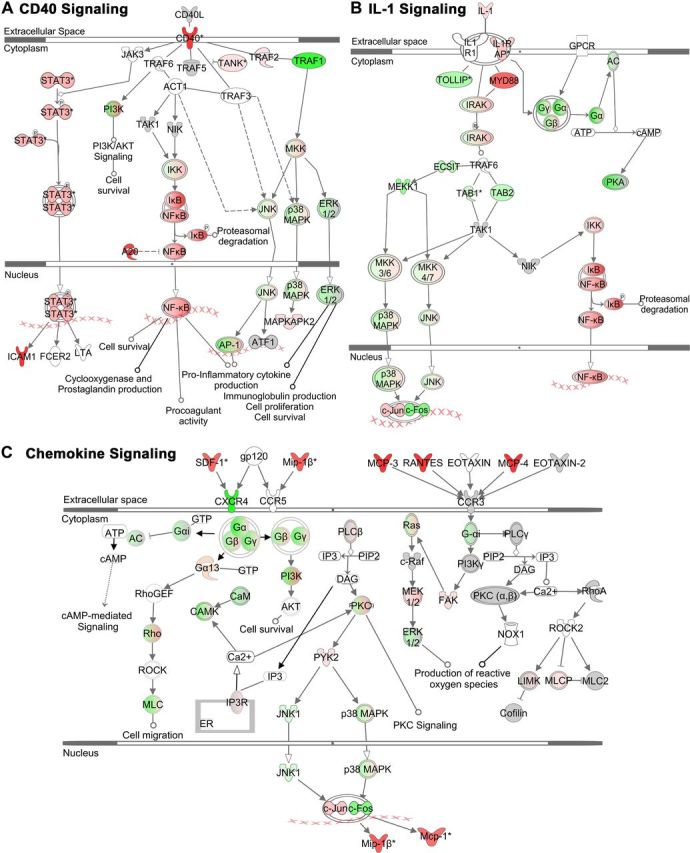
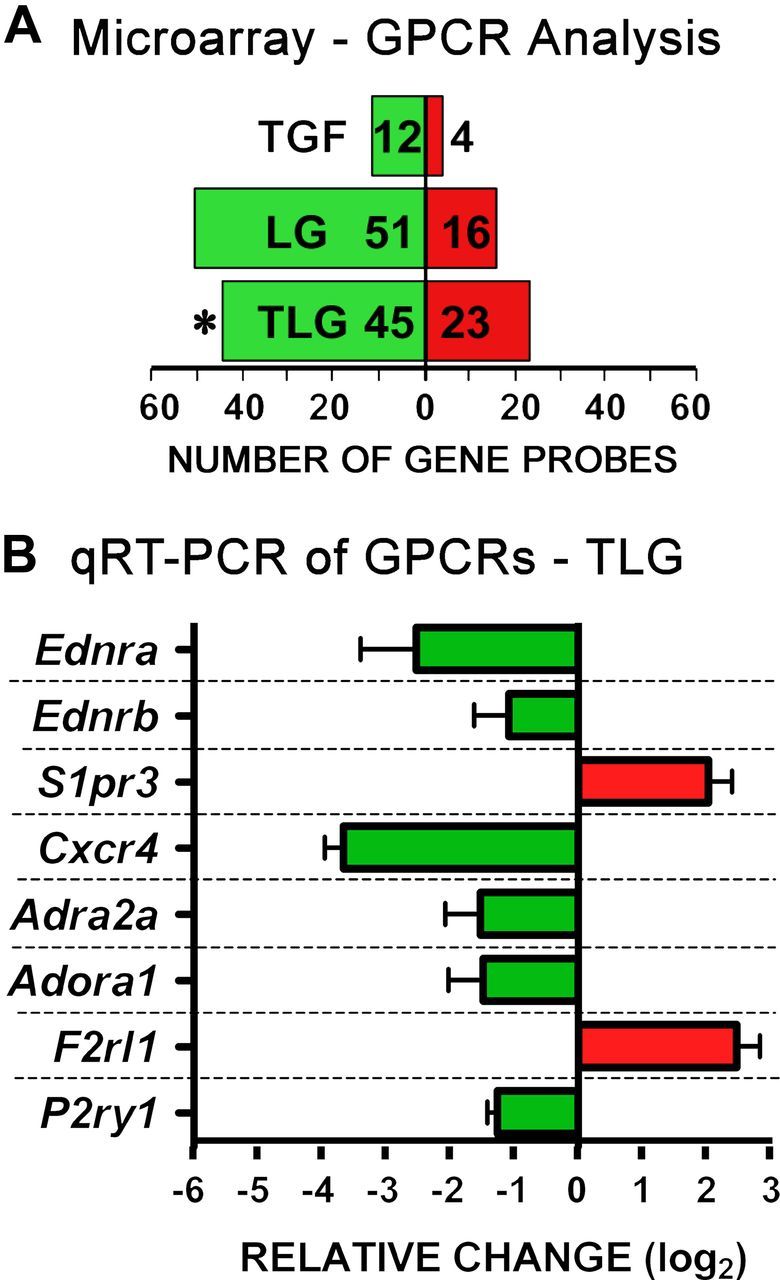
Regulation of genes encoding GPCRs and their ligands
We next examined the effects of combinatorial treatment with TGFβ+LPS+IFNγ and noted a high degree of upregulation or downregulation of many genes encoding molecules associated with calcium signaling, including the GPCRs Cxcr4 (Fig. 4C), Adra2a (Table 1), P2ry6 (Fig. 3A; Table 2), P2ry14 (Table 2), Adora1 (Table 2), Gpr23 and Gpr156 (Table 2), and G-protein subunits, Gα,β,γ (Fig. 4B,C). Moreover, in certain cases there were multiple differentially expressed genes at all three signaling levels (extracellular trigger, transmembrane receptor, and intracellular effector), for example, the chemokine signaling pathway of ligand SDF-1α/Cxcl12, receptor Cxcr4, and intracellular effectors (Gα,β,γ), as shown in Figure 4C. These observations suggested the potential for inflammatory stimuli to substantially modulate astrocyte calcium signaling. We found these changes of particular interest because astrocytes display GPCR-mediated [Ca2+]i increases that are under extensive investigation as potential means of mediating various dynamic astrocyte functions (Verkhratsky et al., 1998; Attwell et al., 2010; Halassa and Haydon, 2010). We therefore performed a systematic analysis of all genes encoding known GPCRs [i.e., those annotated on the International Union of Basic and Clinical Pharmacology (IUPHAR) database] (Sharman et al., 2011) to look for highly regulated genes among GPCRs, and G-protein effectors. This analysis revealed a large number of significantly regulated genes (Fig. 5A), and also indicated a bias toward downregulation among those genes that were significantly modulated, as reflected in the significantly greater number of genes that were downregulated versus upregulated (assessed via Fisher's exact test, p ≤ 0.05; Fig. 5A), as well as in the magnitude of the changes among individual genes (Tables 7, 8). To validate the changes revealed by genomic microarray profiling, we performed qRT-PCR and found similar changes in mRNA expression of selected GPCR genes, Cxcr4, Adra2a, Ednra, Ednrb, P2ry1, Adora1, S1pr3, and F2rl1 (Fig. 5B). Canonical Pathway (Ingenuity) analysis further revealed that, as in the case of CXCR4 (Fig. 4C), various other pathways exhibited highly regulated genes at the levels of both receptor and intracellular effectors, including the type 2A α-adrenergic receptor (Fig. 6A), endothelin receptors type A and B (Fig. 6B), and various P2Y receptors (Fig. 6C). These findings pointed toward the potential for diverse effects of inflammatory stimuli on different forms of ligand-evoked astrocyte calcium signaling. We also noted that certain other GPCRs implicated in astrocyte calcium signaling were not significantly regulated by the inflammatory stimuli used in this study, including various metabotropic glutamate receptors (mGluR) subunits, mGlur (probe #s Grm1, ILMN_2665238; Grm3, ILMN_1238513; Grm4, ILMN_3162152; Grm6, ILMN_3162125; Grm7, ILMN_2872782; and Grm8, ILMN_2711523).
Figure 5.
Genome-wide analysis of differentially expressed genes encoding GPCRs and qRT-PCR validation. A, All genes designated to encode GPCRs according to the IUPHAR database were cross-referenced with our microarray dataset (Fig. 1, n = 4 from Fig. 1) and total number of gene probes showing differential expression after treatment of astrocytes with T, LG, or TLG versus B was calculated. *, A significant skew in genes differentially expressed, down versus up (p ≤ 0.05 Fisher's exact test). B, basal; T, TGF-β1; LG, LPS+IFNγ; TLG, TGF-β1+LPS+ IFNγ. B, qRT-PCR validation of a cross section of differentially expressed genes encoding GPCRs in astrocytes exposed to TLG. Data are expressed as mean fold change (log2) relative to basal + SEM (n = 3 from 3 independently generated samples from those used for gene arrays).
Table 7.
Top upregulated and downregulated GPCRs by TGF-β1+LPS+IFNγ
| Gene | Gene description | log2 Ratio |
|---|---|---|
| Top 15 upregulated GPCRs | ||
| P2ry6 | Purinergic receptor P2Y, G-protein coupled 6 | 2.11 |
| S1pr3 | Endothelial differentiation, sphingolipid | 1.13 |
| GPCR 3 | ||
| Gpr158 | GPCR 158 | 1.04 |
| Gpr83 | GPCR 83 | 1.01 |
| Ccrl2 | Chemokine (C-C motif) receptor-like 2 | 0.85 |
| Fzd1 | Frizzled homolog 1 (Drosophila) | 0.83 |
| P2ry14 | Purinergic receptor P2Y, G-protein coupled 14 | 0.78 |
| Gpr123 | GPCR | 0.73 |
| Gpr172b | GPCR172B | 0.58 |
| Gpr108 | GPCR 108 | 0.42 |
| Vipr1 | Vasoactive intestinal peptide receptor 1 | 0.35 |
| Niacr1 | GPCR 109A/niacin receptor 1 | 0.34 |
| F2rl1 | Coagulation factor II (thrombin) receptor-like 1 | 0.30 |
| Opn3 | Opsin 3 | 0.27 |
| Gpr64 | GPCR 64 | 0.27 |
| Top 15 downregulated GPCRs | ||
| Adra2a | α-2A-adrenergic receptor | −2.55 |
| Cxcr4 | Chemokine (C-X-C motif) receptor 4 | −1.85 |
| Ednrb | Endothelin receptor type B | −1.67 |
| Gpr146 | GPCR 146, transcript variant 2 | −1.57 |
| Fzd2 | Frizzled homolog 2 (Drosophila) | −1.31 |
| P2ry1 | Purinergic receptor P2Y, G-protein coupled 1 | −1.29 |
| Ednra | Endothelin receptor type A | −1.17 |
| Gpr19 | GPCR 19 | −1.16 |
| Lpar4 | GPCR 23 | −1.15 |
| Gpr30 | GPCR 30 | −1.06 |
| Gpr12 | GPCR 12 | −1.00 |
| Gpr85 | GPCR 85 | −0.94 |
| Celsr2 | Cadherin, EGF LAG seven-pass G-type receptor 2 | −0.93 |
| Gprc5b | GPCR, family C, group 5, member B | −0.92 |
| Adora1 | Adenosine A1 receptor | −0.87 |
Table 8.
Top upregulated and downregulated GPCR effectors by TGF-β1+LPS+IFNγ
| Gene | Gene description | log2 ratio |
|---|---|---|
| Top 5 upregulated G-proteins | ||
| Gnb4 | Guanine nucleotide binding protein (Gnbp; G-protein), β 4 | 0.91 |
| Gnl1 | Gnbp (G-protein)-like 1 (Gna-rs1) | 0.60 |
| Gng2 | Gnbp (G-protein), γ 2 transcript variant 1 | 0.55 |
| Gna13 | Gnbp (G-protein), α 13 | 0.40 |
| Gnb2l1 | Gnbp (G-protein), β polypeptide 2 like 1 (Gnb2-rs1) | 0.33 |
| Top 5 downregulated G-proteins | ||
| Gnao1 | Gnbp (G-protein), α O transcript variant A | −1.68 |
| Gng7 | Gnbp (G-protein), γ 7 subunit transcript variant 2 | −1.29 |
| Gna14 | Gnbp (G-protein), α 14 | −1.12 |
| Gnb5 | Gnbp (G-protein), β 5 transcript variant 2 | −0.75 |
| Gnai2 | Gnbp (G-protein), α inhibiting 2 | −0.58 |
Figure 6.
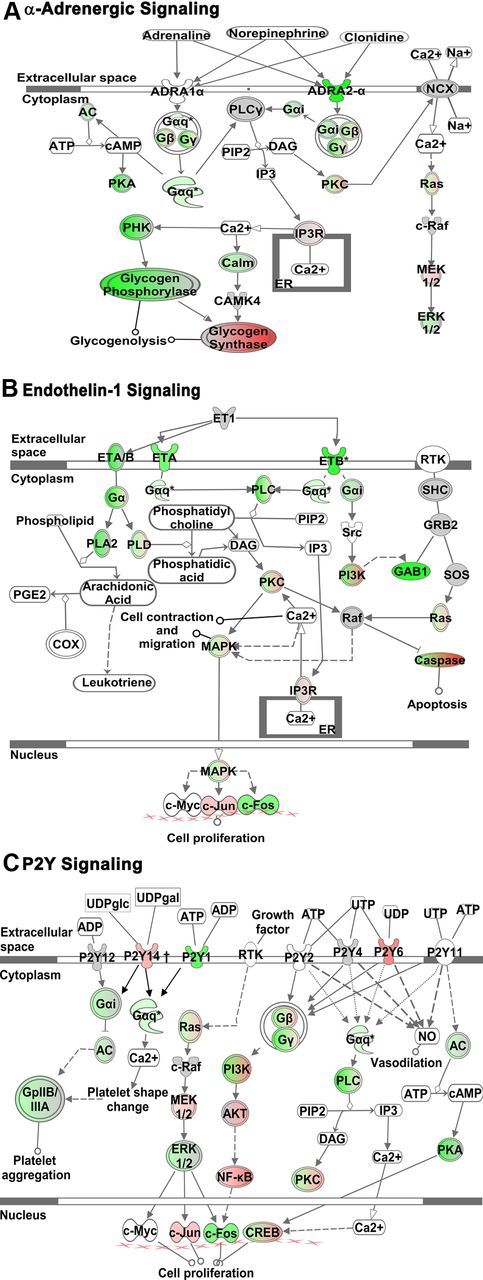
GPCR Canonical Signaling Pathways. A–C, Schematic diagrams of three significantly altered GPCR Canonical Signaling Pathways identified using Ingenuity Pathway Analysis software (IPA; Ingenuity) as being affected by TGF-β1+LPS+IFNγ-treatment of astrocytes. As selected from Table 7, these pathways are α-Adrenergic Signaling (A), Endothelin-1 Signaling (B), and P2Y Signaling (C). Molecules are indicated by standard abbreviations. Relative changes in gene expression are depicted by gradated shades of color coding: red, up; green, down; white, no change. Direct and indirect interactions between molecules are depicted by solid and dotted lines, respectively.
Effects of TGFβ+LPS+IFNγ on astrocyte GPCR-evoked calcium signaling
Given that stimulation with combinatorial inflammatory mediators substantially altered the gene expression of many GPCRs and G-protein effectors in astrocytes, we next evaluated the effects of treatment with TGFβ+LPS+IFNγ on GPCR-mediated astrocyte calcium signaling. We examined GPCRs whose expression levels had been significantly modulated down or up by TGFβ+LPS+IFNγ treatment (Table 7), and focused on a cross section of GPCRs activated by different molecular classes of ligands, including cytokines, growth factors, catecholamines, and purine nucleotides.
Before examining GPCR regulated changes in [Ca2+]i, we asked whether treatment with TGFβ+LPS+IFNγ altered the resting [Ca2+]i in astrocytes. Findings from Fura2 bulk-loaded astrocytes indicated that resting [Ca2+]i levels did not differ significantly (p > 0.05, un-paired t test) between untreated and TGFβ+LPS+IFNγ-treated astrocytes, with values approximating 90 nm in both cases (basal, 84 ± 6 nm; TGFβ+LPS+IFNγ, 89 ± 4 nm, mean ± SEM; n = 84–98 from 5 to 6 separate experiments), a value similar to that reported previously (Shigetomi et al., 2012). This result is consistent with our microarray data showing that expression of the TrpA1 ion channel, a contributor to astrocyte resting calcium levels (Shigetomi et al., 2012), is not altered by treatment with TGFβ+LPS+IFNγ compared with basal (Table 6). These resting calcium measurements also indicate that astrocyte cell membranes were intact. Second we examined whether spontaneous (nonevoked) increases in [Ca2+]i varied between basal and TGFβ+LPS+IFNγ-treated astrocytes. Interestingly, we found that TGFβ+LPS+IFNγ-treated cells had fewer spontaneous events over time (basal, 0.09 ± 0.02 events/min; TGFβ+LPS+IFNγ, 0.03 ± 0.01 events/min, mean ± SEM, p ≤ 0.05 assessed via unpaired t test; n = 84–111 from 5 to 6 experiments), but the peak amplitude of these spontaneous events did not differ significantly from one another (basal, 0.22 ± 0.05; TGFβ+LPS+IFNγ, 0.41 ± 0.18 mean ± SEM; p > 0.05 as assessed via Mann–Whitney test).
We next determined whether GPCR ligand-evoked increases in astrocyte [Ca2+]i differed among TGFβ+LPS+IFNγ-treated astrocytes compared with basal. We first examined representative receptors whose gene expression levels had declined after TGFβ+LPS+IFNγ treatment, Cxcr4, Adra2a, Ednrb, Adora, and P2ry1 (Table 7; Fig. 5). Stimulation of astrocytes with various selective agonists for these receptors increased [Ca2+]i in control astrocytes, including CXCL12/SDF-1α, the endogenous and selective agonist of chemokine receptor CXCR4; guanfacine HCl, an ADRA2A-selective agonist; IRL1620, an endothelin receptor type B (EDRNB)-selective agonist; CCPA, an ADORA1-selective agonist; and ADPβS, a P2Y1-selective agonist (Fig. 7). For each of these agonists, TGFβ+LPS+IFNγ-treated astrocytes exhibited a significantly diminished increase in [Ca2+]i levels compared with untreated (basal) astrocytes (Fig. 7). For four of these agonists, SDF-1α, guanfacine HCl, CCPA, and IRL1620, the TGFβ+LPS+IFNγ-induced diminution in [Ca2+]i elevations was due at least in part to a significant reduction in the number of astrocytes that responded, whereas for ADPβS, all cells continued to respond (Fig. 7). In addition, application of another adrenergic ADRA2-selective agonist, B-HT 933, to Fluo-4-loaded astrocytes revealed a similar functional response to that of the ADRA2A-selective agonist guanfacine with the peak increase in [Ca2+]i in TGFβ+LPS+IFNγ-treated astrocytes significantly attenuated compared with that of basal (basal, 0.9 ± 0.1 dF/F; TGFβ+LPS+IFNγ 0.2 ± 0.1, mean peak increase in [Ca2+]i ± SEM; n = 85–92 from 3 to 4 experiments; p ≤ 0.05 as assessed via Mann–Whitney test). Additionally, a different EDRNB-selective agonist BQ-3020 was found to mirror that of IRL1620 with the peak increase in [Ca2+]i significantly attenuated in TGFβ+LPS+IFNγ-treated versus basal astrocytes (basal, 4.1 ± 0.2 dF/F; TGFβ+LPS+IFNγ, 1.8 ± 0.2 dF/F mean peak increase in [Ca2+]i, ±SEM; n = 58–111 from three experiments p ≤ 0.05 as assessed via Mann–Whitney test). We then examined a receptor whose gene expression levels had increased after TGFβ+LPS+IFNγ treatment, P2ry14 (Table 7). To stimulate P2Y14, we used the receptor-selective and endogenous ligand, UDP-glucose (Abbracchio et al., 2003). Interestingly, in basal astrocytes, UDP-glucose had no detectable effect on astrocyte [Ca2+]i; however, in astrocytes treated with TGFβ+LPS+IFNγ, UDP-glucose resulted in a significant increase in [Ca2+]i, in ∼50% of the cells (Fig. 7). It is noteworthy that all of the decreases or increases in ligand-evoked [Ca2+]i responses that were induced by treatment of astrocytes with TGFβ+LPS+IFNγ mirrored the relative up or down changes measured in gene expression of the respective receptors: Cxcr4, Adra2a, Ednrb, Adora1, P2ry14, and P2ry1 (Table 7; Fig. 5B).
Figure 7.
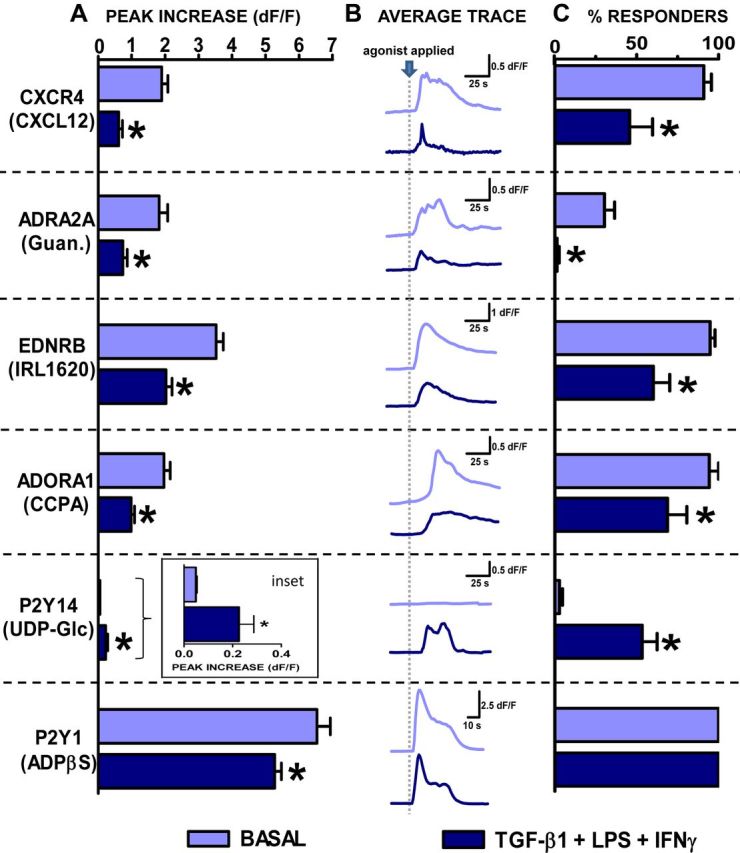
Calcium responses elicited by selective GPCR agonists in untreated and TGF-β1+LPS+IFNγ-treated astrocytes. Untreated and TGF-β1+LPS+IFNγ (TLG)-treated astrocytes were bulk loaded with the calcium indicator dye Fluo-4 and imaged before, during (30 s), and after application of agonists selective for specific GPCRs as indicated: left, CXCL12, 30 ng/ml; guanfacine HCl (Guan.), 10 μm; IRL1620, 100 nm; CCPA, 100 nm; UDP-Glc, 100 μm; and ADPβS, 10 μm. Data are represented as (A) mean peak increase in intracellular calcium levels ([Ca2+]i) (dF/F) + SEM, (B) average trace depicting [Ca2+]i (dF/F) over time (s), and (C) mean percentage cells responding + SEM. Significance (p ≤ 0.05) was assessed via (A) Mann–Whitney test or (C) unpaired t test following normalization of data. *, Indicates significantly different from untreated (basal) control. (CXCL12, n = 86–107 cells from 3 experiments; guanfacine HCl, n = 78–93 from 4 experiments; IRL1620, n = 115–146 from 3–5 experiments; CCPA, n = 83–89 from 4 experiments; UDP-Glc, n = 44–47 from 3 experiments; ADPβS, n = 67–94 from 3 experiments.)
In addition to testing GPCR-selective agonists as just described, we wished to determine whether endogenously produced GPCR agonists would yield similar results. To do so we tested the effects of epinephrine, which activates the adrenoceptor family of receptors; endothelin-1, which activates the endothelin family of receptors; adenosine, which activates the adenosine family of receptors; UDP-galactose, another P2Y14-selective endogenous agonist; and ADP, which activates the purinergic P2Y family receptors. In each case, the endogenously produced ligands elicited approximately similar responses to those noted with the selective agonists, and the effect of TGFβ+ LPS+IFNγ treatment on ligand-evoked responses was essentially the same (compare Figs. 7 and 8). As expected, the response for ligands capable of activating more than one receptor led to a larger rise in [Ca2+]i.
Figure 8.

Calcium responses elicited by endogenous GPCR agonists in untreated and TGF-β1+LPS+IFNγ-treated astrocytes. Untreated and TGF-β1+LPS+IFNγ (TLG)-treated astrocytes were bulk loaded with the calcium indicator dye Fluo-4 and imaged before, during (30 s), and after application of agonists (epinephrine, 100 ng/ml; endothelin-1, 100 nm; adenosine, 1 μm; UDP-Gal, 300 μm; ADP, 1 μm) as indicated (left column). Data are represented as (A) mean peak increase in intracellular calcium levels [Ca2+]i (dF/F) + SEM, (B) average trace depicting [Ca2+]i (dF/F) over time (s), and (C) Mean percentage cells responding + SEM. Significance (p ≤ 0.05) was assessed via (A) Mann–Whitney test or (C) unpaired t test following normalization of data. *, Indicates significantly different from untreated (basal) control. (Epinephrine, n = 66–85 cells from 3 experiments; endothelin-1, n = 130–133 from 3 experiments; adenosine, n = 64 from 3 experiments; UDP-Gal, n = 40–47 from 3 experiments; ADP, n = 33–35 from 3–4 experiments.)
Last, we determined the effects of siRNA knockdown of several representative GPCRs on ligand-evoked increases in [Ca2+]i. Basal or TGFβ+LPS+IFNγ-treated astrocytes were transfected with control siRNA or siRNA specific for the Endrb, P2ry1, or P2ry14 receptors. For P2ry1, selective siRNA resulted in a 43–50% reduction in the peak increases in [Ca2+]i compared with control siRNA (Fig. 9). For Ednrb and P2ry14, selective siRNA resulted in an 85–90% reduction in the peak increases in [Ca2+]i compared with control siRNA, essentially abolishing the responses (Fig. 9). To test whether the siRNA knockdown effect might be due to decrease in cell health or viability, stimulation with ATP was used. These tests showed that ATP responses remained intact despite a decrease in the specific receptor-evoked [Ca2+]i that was due to receptor-specific siRNA (compared with control siRNA). For example, in P2y14 siRNA-transfected astrocytes, ATP (10 μm) elicited a peak increase in [Ca2+]i that was similar to that elicited in negative control siRNA-transfected astrocytes (control siRNA, 5.2 ± 0.3 dF/F; P2ry14 siRNA, 4.8 ± 0.1 dF/F, mean ± SEM; p > 0.05 as assessed via Mann–Whitney test; n = 63–107 from 3 to 4 separate experiments). These observations indicated that the siRNA knockdown effects were receptor specific, and not due to a general inability of these cells to respond to agonists in evoking a rise in [Ca2+]i.
Figure 9.
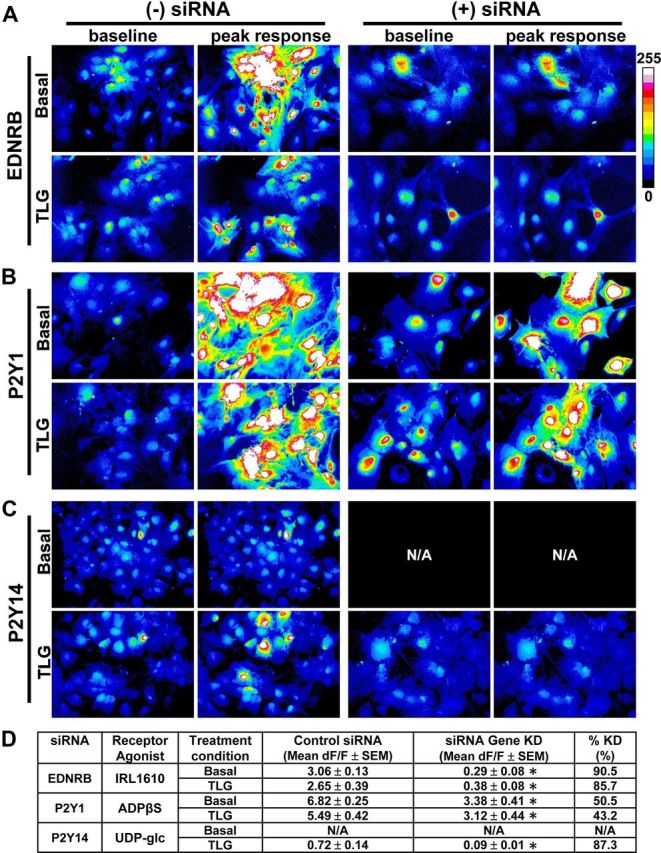
Representative images of peak responses to GPCR receptor-selective agonists with or without receptor-specific siRNA knockdown. Normal nontransfected [(−) siRNA], negative control siRNA-transfected or _Ednrb_-, _P2ry1_-, or _P2ry14_-siRNA transfected [(+) siRNA] astrocytes were exposed to medium alone (basal) or TGF-β1+LPS+IFNγ (TLG). From 22–30 hours following administration of control medium or that containing TLG, astrocytes were bulk loaded with the calcium indicator dye Fluo-4 and imaged before, during (30 s), and after application of receptor-selective agonists: EDNRB (A), 100 nm IRL1620, P2Y1 (B), 10 μm ADPβS, P2Y14 (C), and UDP-Glc, 300 μm. Shown are representative images for baseline and the peak increase in [Ca2+]i (dF intensity), as indicated, in response to the agonist application for both basal and TLG-treated cells with and without siRNA. Each image is representative of at least three experiments. Note: Given that UDP-Glc did not elicit a response in basal astrocytes, siRNA knockdown for P2y14 in basal astrocytes was not performed as indicated via N/A. D, Quantification of functional knockdown (KD) of agonist-triggered response (i.e., KD of measured increase in [Ca2+]i) (from A–C). Results are presented as mean dF/F ± for each treatment condition and as percentage KD. *, Indicates a significant KD compared with respective control siRNA, as assessed by Mann–Whitney test (p ≤ 0.05).
Together, these siRNA findings confirmed that the observed changes in [Ca2+]i were mediated via the specific receptors studied.
Effects of TGFβ+LPS+IFNγ on astrocyte expression of representative molecules in vivo
The findings presented thus far were derived from in vitro experiments using primary cultures of cortical astrocytes. We next sought to test the potential relevance in vivo of our observations. To do so, we examined the effects of our combinatorial inflammatory stimuli on expression by cortical astrocytes in vivo of several representative proteins whose genes had been strongly upregulated or downregulated in vitro. We injected TGFβ+LPS+IFNγ or PBS as a control into the frontal, sensorimotor cortex of wild-type mice. After survival times of 1–5 d, double staining immunohistochemistry was conducted for the canonical marker of reactive astrocytes, glial fibrillary acidic protein (GFAP; Sofroniew, 2009), in combination with various other proteins. In particular, we examined CXCL1, CXCL10, and CCL7 as three representative proteins whose genes had been highly upregulated by exposure to TGFβ+LPS+IFNγ in vitro (Table 1) and IL-6 as a representative protein whose gene was synergistically upregulated by combinatorial interactions of TGFβ+LPS+IFNγ (Fig. 2, Table 2). We also examined ADRA2A as a representative protein whose gene had been strongly downregulated in vitro (Fig. 5, Table 8) and that we had studied functionally (Figs. 7, 8).
In agreement with previous reports (Myer et al., 2006), we found that in the central layers of naive cerebral cortex that was not injected, GFAP expression was low and could be detected by immunohistochemistry only in randomly scattered cells (Fig. 10A). As expected, GFAP expression was upregulated in the immediate vicinity of the injection sites after injection of either PBS (Fig. 10B) or TGFβ+LPS+IFNγ (Fig. 10C). Interestingly, the level of GFAP increase in individual astrocytes did not differ substantively in cortex injected with either PBS or TGFβ+LPS+ IFNγ (Fig. 10B,C), but the cortical area over which the increased GFAP expression occurred was larger in mice injected with TGFβ+LPS+IFNγ.
Figure 10.
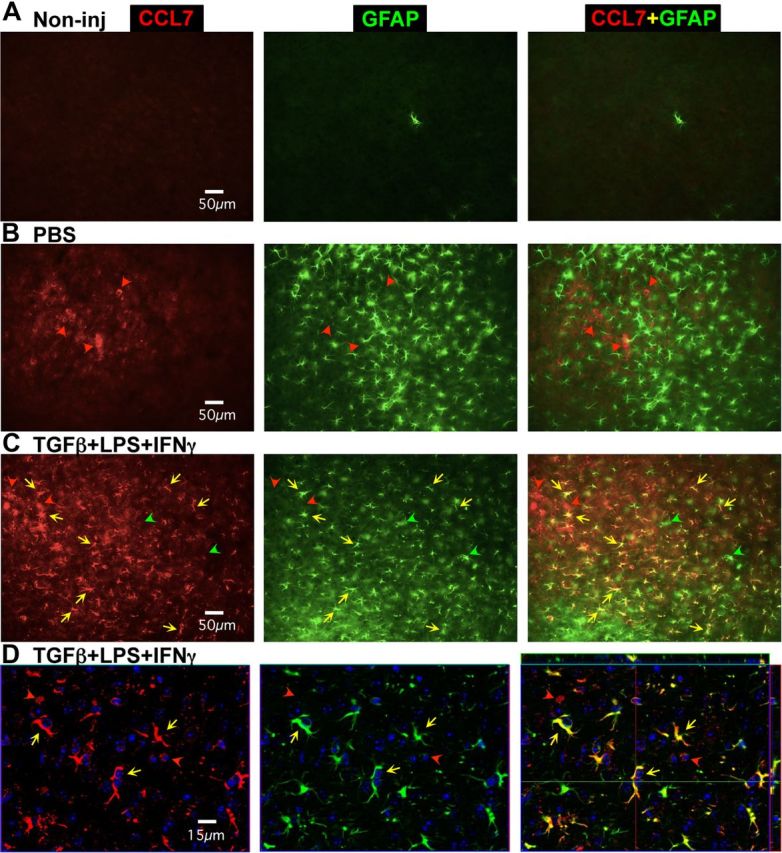
Changes in expression of GFAP and CCL7 induced in vivo by injection of PBS or TGFβ+LPS+IFNγ into cerebral cortex. A–D, Single channel and merged two-color fluorescence survey (A–C) and detail (D) images of immunohistochemical staining for CCL7 (red) and GFAP (green) in mid-layers (3–5) of cerebral cortex of wild-type mice that were either noninjected (A) or injected with PBS (B) or TGFβ+LPS+IFNγ 5 d previously (C, D). D, Nuclei are counterstained with DAPI (blue) and the images were obtained using confocal microscopy. A, In noninjected cortex, CCL7 is not detectably expressed by any cells and GFAP is detectable in only one astrocyte in this frame. B, In PBS-injected cortex, CCL7 is expressed by a few cells that do not express GFAP (red arrowheads), whereas GFAP is detectable in many reactive astrocytes in the immediate vicinity of the injection site. C, D, In TGFβ+LPS+IFNγ-injected cortex, CCL7 is expressed both by cells that do not express GFAP (red arrowheads) as well as by many GFAP-expressing reactive astrocytes (yellow arrows). Some GFAP-expressing reactive astrocytes express little or no detectable CCL7 (green arrowheads).
In noninjected cortex, we found that none of the four molecules examined for potential upregulation, CXCL1, CXCL10, CCL7, and IL-6, could be detected in any cell type (Fig. 10A, representative example of CCL7). In cortex injected with PBS, three of these molecules, CXCL1, CXCL10, and CCL7, could not be detected in any reactive astrocytes even though these astrocytes had upregulated GFAP (Figs. 10B, 11A–C), whereas a few GFAP-expressing reactive astrocytes could be found that expressed small amounts of detectable IL-6 (Fig. 11D). In addition, after PBS injection these molecules were detectable in cells that were clearly not astrocytes as defined by size, shape, and lack of GFAP expression (Figs. 10B, 11A–D).
Figure 11.
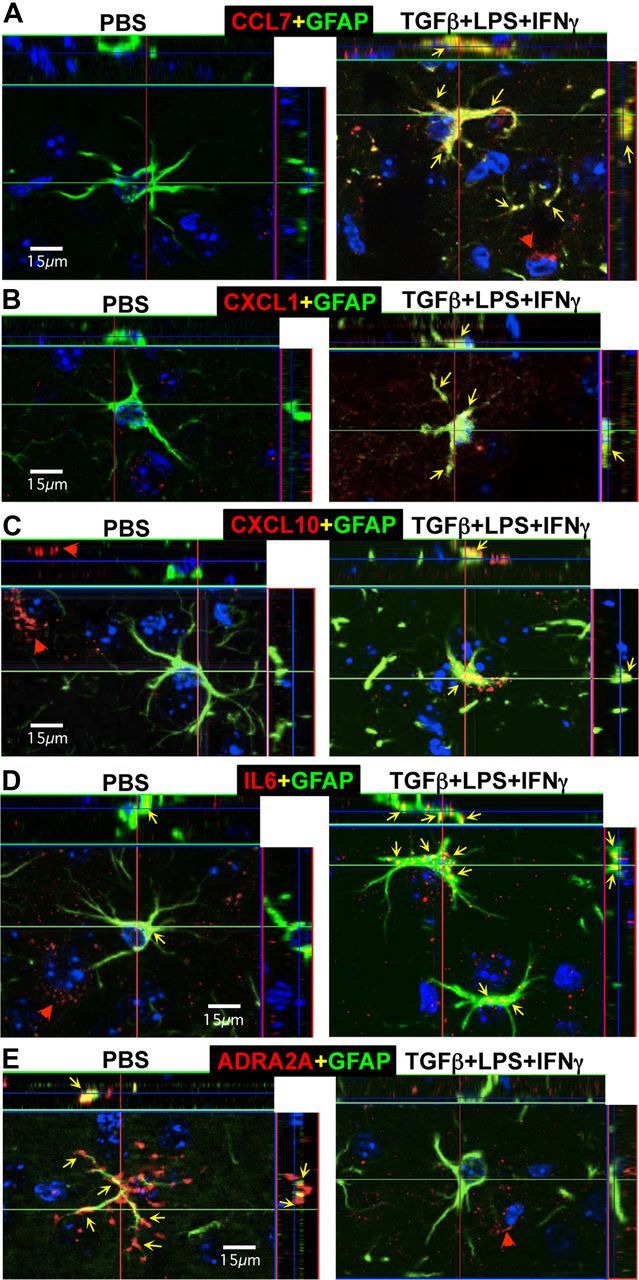
Changes in expression of CCL7, CXCL1, CXCL10, IL-6, and ADRA2A induced in vivo in GFAP-expressing reactive astrocytes by injection of TGFβ+LPS+IFNγ into cerebral cortex. A–E, Multicolor, fluorescence, and confocal microscopic images of immunohistochemical staining for CCL7, CXCL1, CXCL10, IL-6, or ADRA2A (red), in combination with GFAP (green) and the nuclear stain, DAPI (blue), in mid-layers (3–5) of cerebral cortex of wild-type mice injected with PBS or TGFβ+LPS+IFNγ 1 d previously. Red arrowheads denote cells that express only CCL7, CXCL1, CXCL10, IL-6, or ADRA2A and do not express GFAP. Yellow arrows denote sties of coexpression of CCL7, CXCL1, CXCL10, IL-6, or ADRA2A within reactive astrocytes that also express GFAP. Note that after injection of PBS, GFAP-expressing reactive astrocytes do not express detectable levels of CCL7 (A), CXCL1 (B), and CXCL10 (C), whereas some reactive astrocytes do express small amounts of IL-6 (D) and some robustly express ADRA2A (E). After injection of TGFβ+LPS+IFNγ, many reactive astrocytes express detectable levels of CCL7 (A), CXCL1 (B), and CXCL10 (C), and upregulate expression of IL-6 (D) and markedly downregulate expression of ADRA2A (E).
In cortex injected with TGFβ+LPS+IFNγ, all four molecules examined for potential upregulation, CXCL1, CXCL10, CCL7, and IL-6, were immunohistochemically detectable in many cells that expressed GFAP and were clearly reactive astrocytes as defined by size and shape (Figs. 10C,D, 11A–E). Interestingly, the expression levels of all four molecules appeared to be markedly heterogeneous in reactive astrocytes, such that some astrocytes expressed high levels, some low, and some undetectable levels of the molecules (Figs. 10C,D, 11A–E). In addition, the expression patterns and timing differed among the four molecules, such that expression by astrocytes of CXCL1 and CXCL10 peaked at the early time points and had disappeared by 5 d, whereas expression of CCL7 and IL-6 persisted in many astrocytes through 5 d (Fig. 10).
These findings confirm in vivo the upregulation by TGFβ+LPS+IFNγ of several representative molecules whose genes we identified as highly upregulated in vitro. In addition, these in vivo findings support our in vitro observation that treatment of primary cultures of cortical astrocytes with TGFβ+LPS+IFNγ did not further upregulate expression of GFAP (Table 6). Primary cortical astrocyte cultures express high levels of GFAP and have long been thought by some investigators to exhibit certain characteristics of reactive astrocytes, such as elevated levels of GFAP expression. Elegant studies combining large-scale genetic analyses of different types of in vitro and in vivo preparations of astrocytes have now clearly shown that primary cortical astrocyte cultures are more similar to reactive astrocytes than to astrocytes in healthy tissue (Foo et al., 2011; Zamanian et al., 2012). Our observation that GFAP is essentially undetectable in astrocytes in unmanipulated cortical tissue in vivo and is upregulated to the same degree in individual cortical astrocytes after injection in vivo of either PBS or TGFβ+LPS+IFNγ is consistent with the notion that primary cultures of cortical astrocytes are in a reactive state in which GFAP is already upregulated and was not further upregulated by our doses of TGFβ+LPS+IFNγ.
For the molecule being examined for potential downregulation, ADRA2A, we found that in cerebral cortex injected with PBS, ADRA2A could be detected by immunohistochemistry in GFAP-positive astrocytes in cerebral cortex in the immediate vicinity of the PBS injections and that expression levels were heterogeneous among different astrocytes, with some exhibiting high expression levels (Fig. 11E) and others low or undetectable expression levels. In cerebral cortex injected with TGFβ+LPS+IFNγ, despite the large number of GFAP-positive reactive astrocytes, there was an essential absence of ADRA2A-positive astrocytes (Fig. 11E). These findings confirm in vivo the downregulation by TGFβ+LPS+IFNγ of a representative molecule whose gene we identified as markedly downregulated in vitro and are consistent with our functional observations that treatment with TGFβ+LPS+IFNγ markedly reduced both the peak response and the percentage responders of astrocytes stimulated in vitro with the ADRA2A-selective agonist guanfacine (Fig. 7).
Discussion
In this study we found that stimulation of astrocytes with the inflammatory mediators TGF-β1, LPS, and IFNγ alone and in combination significantly altered the expression of many genes, molecular networks, and functional pathways. Combinatorial stimulation with all three mediators led to both additive and synergistic changes, and substantially modulated numerous astrocyte molecular networks and pathways associated with immune- and injury-related functions. Expression levels of many GPCRs and G-protein effectors involved in calcium signaling were significantly altered, and these expression changes were accompanied by parallel changes in astrocyte calcium signaling evoked by corresponding GPCR-specific ligands. Astrocytes play many essential roles in healthy CNS (Barres, 2008; Attwell et al., 2010; Halassa and Haydon, 2010) and respond to CNS insults through a process known as reactive astrogliosis, in which astrocytes modify their gene expression in response to stimulation by numerous factors (Pekny and Nilsson, 2005; Sofroniew, 2009; Hamby and Sofroniew, 2010). The impact of inflammatory mediators on the functions of astrocytes and reactive astrocytes is poorly understood and is of increasing interest with the growing awareness of critical roles of astrocytes in healthy and injured CNS (Barres, 2008; Nedergaard et al., 2010; Sofroniew and Vinters, 2010). The findings of this study shed light on how inflammatory stimuli, alone and in combination, may influence a wide variety of astrocyte functions.
Additive and synergistic effects of TGF-β1, LPS, and IFNγ on astrocyte genomic profiles
There is an extensive and well reviewed literature on the effects of many different individual inflammatory mediators on the expression by astrocytes of numerous individual genes (Eddleston and Mucke, 1993; John et al., 2003; Sofroniew, 2009). Microarray profiling has previously been applied to generate transcriptomic datasets of astrocytes acutely isolated from either healthy CNS (Lovatt et al., 2007; Cahoy et al., 2008) or CNS after stroke or peripheral LPS injections (Zamanian et al., 2012) or hyperammonemia (Lichter-Konecki et al., 2008), or astrocyte cell cultures prepared by a novel immunopanning procedure (Foo et al., 2011), or primary astrocyte cell cultures treated with LPS or IL-1 (Pang et al., 2001; John et al., 2005). Our findings here extend these studies not only by documenting transcriptome changes induced in primary astrocyte cultures by the regulatory cytokine, TGF-β1, and the canonical innate inflammatory mediators, LPS+IFNγ, but also by demonstrating that combinatorial interactions among these inflammatory mediators can lead to synergistic changes. Notably, using highly stringent criteria, we found that combinatorial treatment with TGFβ+LPS+IFNγ resulted in synergistic changes in >380 gene probes that could not be predicted by summing the effects of individual treatments with TGF-β1 or LPS+IFNγ alone. qRT-PCR confirmed synergistic regulation of selected genes. These findings show that synergistic interactions of inflammatory mediators can regulate astrocyte production of molecules with key functions in the CNS response to inflammation (IL-6, SEMA4a, NOS-2, etc.) in ways not predicted by individual effects of those mediators. The importance of combinatorial interactions among different inflammatory stimuli in regulating specific responses of T-cells is well documented (Korn et al., 2009). Our findings show that combinatorial exposure to multiple inflammatory mediators can lead to large-scale synergistic and unpredictable changes in astrocyte gene expression. Further study is warranted to understand the biological implications and intracellular signaling mechanisms.
In vivo confirmation of in vitro observations
We confirmed by immunohistochemistry that several representative proteins whose genes we identified as highly upregulated or downregulated in vitro were upregulated or downregulated in astrocytes in vivo after injection of TGFβ+LPS+IFNγ into cerebral cortex. Our findings in vivo and in vitro are in broad agreement with a recent analysis of genome-wide changes induced in astrocytes in vivo by peripheral treatment with LPS, which showed pronounced upregulation by reactive astrocytes (compared with nonreactive astrocytes) of many genes encoding the same chemokines, cytokines, and other molecules found in our study in vitro (Zamanian et al., 2012). Interestingly, our immunohistochemical analysis at the single cell in vivo revealed that the patterns, levels, and time course of expression of GFAP, chemokine, cytokines, and other molecules was markedly heterogeneous among different individual reactive astrocytes after two different triggers of reactivity, injection of PBS, or TGFβ+LPS+IFNγ. These observations provide further support for the notion that reactive astrogliosis is not a stereotypic all-or-none response as once believed, but rather is a highly varied and heterogeneous process that can involve many different changes in gene expression that can vary over time and can resolve, as regulated by many different specific signaling mechanisms in a context and temporally dependent manner (Sofroniew, 2009). In addition, our present in vitro and in vivo findings extend these ideas by providing direct evidence that exposure to inflammatory mediators can alter the nature of astrocyte reactivity and shift it toward exerting more proinflammatory effects. In this context it is interesting to speculate that peripheral infections that produce such inflammatory mediators may exert pronounced and context-altering effects on astrocyte reactivity, thereby having important and as yet poorly understood repercussions in a wide variety of CNS disorders in which astrocyte reactivity is prominent (Sofroniew and Vinters, 2010).
TGFβ+LPS+IFNγ modulates astrocyte genomic networks toward immune- and injury-related functions
A striking feature of the effects of combinatorial treatment with TGFβ+LPS+IFNγ on astrocytes is that all of the top 5 most upregulated genes were chemokines, and 7 of the top 10 were related to immune or inflammatory functions, all of which exhibited pronounced increases in the range of ∼125- to 250-fold (7–8 log2-fold). In addition, bioinformatic analysis revealed that the top regulated Associated Network Function was antigen presentation, and that 17 of the top 20 Canonical Pathways involved chemokine or cytokine signaling or innate immune regulation. Highly upregulated individual genes included numerous chemokines, cytokines, growth factors, their receptors, and components of their signaling pathways. Together, these findings add to the growing body of evidence that astrocytes play complex roles in CNS immune and inflammatory responses that may involve both pro- and anti-inflammatory effects (Bush et al., 1999; Brambilla et al., 2005; Okada et al., 2006; Herrmann et al., 2008; Sofroniew, 2009; Voskuhl et al., 2009; Li et al., 2011). Here, we both confirm, and newly identify, a large number of specific candidate molecules whose production by astrocytes is regulated by inflammatory stimuli that could potentially participate in or mediate such effects. For example, CXCL12, CCL2, and CD40 have all been shown to regulate T-cell trafficking in the CNS (Mahad and Ransohoff, 2003; Sitati et al., 2007; McCandless et al., 2008; Moll et al., 2009; Patel et al., 2010). IL-11 attenuates inflammation and protects myelin in experimental autoimmune encephalomyelitis (EAE) (Gurfein et al., 2009). IL-15 is implicated in T-, B-, and natural killer (NK) cell activation and proliferation (Steel et al., 2012). CCL8 is implicated in eosinophil recruitment (Islam et al., 2011). IL-6 is implicated as an essential inducer of pathogenic antibody production in neuromyelitis optica (NMO) (Chihara et al., 2011). Combinational interactions between IL-6 and TGF-β1 underlie the conversion of CD4 T-cells into Th17 T-cells that are widely implicated in CNS immune defense and autoimmune disorders including EAE, NMO, and multiple sclerosis (MS) (Korn et al., 2009). We show here that TGFβ+LPS+IFNγ markedly upregulated astrocyte production of all of these molecules. Our findings strongly support the notion that astrocytes are key regulators of CNS immune and inflammatory responses, and identify numerous novel candidate molecules that may mediate astrocyte effects on CNS immune and inflammatory responses. We provide transcriptomic datasets with which to generate hypotheses regarding astrocyte molecules regulated by these inflammatory mediators that may be involved in astrocyte roles in autoimmune diseases like MS, NMO, and EAE, as well as more generally in CNS inflammation associated with diverse disorders including stroke, trauma, neurodegeneration, and infection.
Bioinformatic analysis also revealed that combinatorial stimulation with TGFβ+LPS+IFNγ substantially modulated molecular and cellular networks associated with cell death, cell compromise, and cell injury, as well as regulation of cell morphology, cell maintenance, cell compromise, and cell growth and proliferation. These observations are consistent with the large body of evidence that reactive astrocytes undergo cell hypertrophy and proliferation as well as cell injury and death while subserving critical roles in the response to CNS injury and tissue repair. This notion was supported further by analysis of individual genes (Table 6), which indicated pronounced regulation of a wide variety of molecules associated with such responses, including numerous growth factors, molecules involved in extracellular matrix remodeling, and enzymes that generate small molecule mediators involved in regulation of blood flow. Notably, many of the changes induced by inflammatory stimuli in astrocytes may be adaptive and beneficial in some contexts, such as response to infectious agents, but might be harmful in other contexts where overexpression of certain chemokines and cytokines may contribute to autoimmune disease, or the overproduction of certain molecules such as nitric oxide might contribute to neural dysfunction or degeneration (Sofroniew, 2009; Sofroniew and Vinters, 2010).
TGFβ+LPS+IFNγ modulate astrocyte calcium signaling
Calcium signaling is broadly implicated in regulating many aspects of cell function (Clapham, 2007) and can be modulated by tissue injury in ways that alter cell functions (Sayeed, 2000). Astrocyte calcium signaling is under investigation for a potential role in mediating astrocyte influences on neuronal activity and blood flow in the healthy CNS (Iadecola and Nedergaard, 2007; Gordon et al., 2008; Attwell et al., 2010; Halassa and Haydon, 2010), as well as in responses to ischemia, cell injury, edema/osmotic stress, or inflammation (Duffy and MacVicar, 1996; Neary et al., 2003; Delbro et al., 2009; Thrane et al., 2011). The impact of inflammatory mediators on astrocyte calcium signaling is poorly understood. Our genomic screening analysis revealed that exposure of astrocytes to the combinatorial inflammatory stimuli of TGF-β1, LPS, and IFNγ, led to substantial changes in the expression of many genes among GPCRs and their intracellular effector molecules involved in inositol trisphosphate-mediated calcium signaling. Notably, there was a significant downward trend in the GPCR-encoding genes that were regulated, although not all genes were downregulated. At the functional level, we found that the changes in gene expression were accompanied by parallel changes in ligand-evoked calcium signaling. These findings are consistent with and extend previous reports regarding the extensive repertoire of GPCRs expressed by astrocytes (Duffy and MacVicar, 1995; Verkhratsky et al., 1998; Calì and Bezzi, 2010). Importantly, our findings demonstrate that inflammatory stimuli can significantly alter astrocyte calcium signaling elicited by multiple GPCRs. Although the downstream functional implications of these changes in astrocyte calcium signaling are not yet clear, alterations in GPCR-evoked astrocyte [Ca2+]i transients may represent mechanisms whereby CNS inflammation influences various functions in reactive astrocytes. Recent findings suggest that reactive astrogliosis in response to viral infection can significantly alter neuronal function (Ortinski et al., 2010). Our findings extend these observations by demonstrating that inflammatory stimuli significantly modulate astrocyte calcium signaling in multiple ways. The large number of different GPCRs that trigger distinct forms of astrocyte [Ca2+]i transients that are modulated by inflammatory stimuli points toward the potential for selective pharmacological manipulation and novel therapeutic strategies that may be relevant in diverse CNS disorders.
Footnotes
This work was supported by grants from National Institutes of Health NS057624 (M.V.S.), NS060677, NS071292, and NS063186 (B.S.K.); T32-MH19925 through the Cousins Center for Psychoneuroimmunology at University of California Los Angeles (UCLA) (M.E.H.); The Dr. Miriam and Sheldon G. Adelson Medical Foundation (M.V.S., G.C., and D.H.G.); and National Institute of Neurological Disorders and Stroke P30NS062691 through the UCLA Informatics Center for Neurogenetics and Neurogenomics. Thanks to Dr. Eiji Shigetomi for guidance on calcium imaging and to Fuying Gao for help with data analysis.
The authors declare no competing financial interests.
References
- Abbracchio MP, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Miras-Portugal MT, King BF, Gachet C, Jacobson KA, Weisman GA, Burnstock G. Characterization of the UDP-glucose receptor (re-named here the P2Y14 receptor) adds diversity to the P2Y receptor family. Trends Pharmacol Sci. 2003;24:52–55. doi: 10.1016/S0165-6147(02)00038-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ata AK, Funa K, Olsson Y. Expression of various TGF-beta isoforms and type I receptor in necrotizing human brain lesions. Acta Neuropathol. 1997;93:326–333. doi: 10.1007/s004010050623. [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calì C, Bezzi P. CXCR4-mediated glutamate exocytosis from astrocytes. J Neuroimmunol. 2010;224:13–21. doi: 10.1016/j.jneuroim.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Chihara N, Aranami T, Sato W, Miyazaki Y, Miyake S, Okamoto T, Ogawa M, Toda T, Yamamura T. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci U S A. 2011;108:3701–3706. doi: 10.1073/pnas.1017385108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Coppola G. Designing, performing, and interpreting a microarray-based gene expression study. Methods Mol Biol. 2011;793:417–439. doi: 10.1007/978-1-61779-328-8_28. [DOI] [PubMed] [Google Scholar]
- Coppola G, Marmolino D, Lu D, Wang Q, Cnop M, Rai M, Acquaviva F, Cocozza S, Pandolfo M, Geschwind DH. Functional genomic analysis of frataxin deficiency reveals tissue-specific alterations and identifies the PPARgamma pathway as a therapeutic target in Friedreich's ataxia. Hum Mol Genet. 2009;18:2452–2461. doi: 10.1093/hmg/ddp183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daginakatte GC, Gadzinski A, Emnett RJ, Stark JL, Gonzales ER, Yan P, Lee JM, Cross AH, Gutmann DH. Expression profiling identifies a molecular signature of reactive astrocytes stimulated by cyclic AMP or proinflammatory cytokines. Exp Neurol. 2008;210:261–267. doi: 10.1016/j.expneurol.2007.10.016. [DOI] [PubMed] [Google Scholar]
- Delbro D, Westerlund A, Björklund U, Hansson E. In inflammatory reactive astrocytes co-cultured with brain endothelial cells nicotine-evoked Ca(2+) transients are attenuated due to interleukin-1beta release and rearrangement of actin filaments. Neuroscience. 2009;159:770–779. doi: 10.1016/j.neuroscience.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Duffy S, MacVicar BA. Adrenergic calcium signaling in astrocyte networks within the hippocampal slice. J Neurosci. 1995;15:5535–5550. doi: 10.1523/JNEUROSCI.15-08-05535.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S, MacVicar BA. In vitro ischemia promotes calcium influx and intracellular calcium release in hippocampal astrocytes. J Neurosci. 1996;16:71–81. doi: 10.1523/JNEUROSCI.16-01-00071.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddleston M, Mucke L. Molecular profile of reactive astrocytes—implications for their role in neurological disease. Neuroscience. 1993;54:15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo LC, Allen NJ, Bushong EA, Ventura PB, Chung WS, Zhou L, Cahoy JD, Daneman R, Zong H, Ellisman MH, Barres BA. Development of a method for the purification and culture of rodent astrocytes. Neuron. 2011;71:799–811. doi: 10.1016/j.neuron.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman A, Dingledine R. Molecular cascades that mediate the influence of inflammation on epilepsy. Epilepsia. 2011;52(Suppl 3):33–39. doi: 10.1111/j.1528-1167.2011.03034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GR, Choi HB, Rungta RL, Ellis-Davies GC, MacVicar BA. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008;456:745–749. doi: 10.1038/nature07525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurfein BT, Zhang Y, López CB, Argaw AT, Zameer A, Moran TM, John GR. IL-11 regulates autoimmune demyelination. J Immunol. 2009;183:4229–4240. doi: 10.4049/jimmunol.0900622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol. 2010;72:335–355. doi: 10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamby ME, Sofroniew MV. Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics. 2010;7:494–506. doi: 10.1016/j.nurt.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamby ME, Hewett JA, Hewett SJ. TGF-beta1 potentiates astrocytic nitric oxide production by expanding the population of astrocytes that express NOS-2. Glia. 2006a;54:566–577. doi: 10.1002/glia.20411. [DOI] [PubMed] [Google Scholar]
- Hamby ME, Uliasz TF, Hewett SJ, Hewett JA. Characterization of an improved procedure for the removal of microglia from confluent monolayers of primary astrocytes. J Neurosci Methods. 2006b;150:128–137. doi: 10.1016/j.jneumeth.2005.06.016. [DOI] [PubMed] [Google Scholar]
- Hamby ME, Hewett JA, Hewett SJ. TGF-beta1 reduces the heterogeneity of astrocytic cyclooxygenase-2 and nitric oxide synthase-2 gene expression in a stimulus-independent manner. Prostaglandins Other Lipid Mediat. 2008;85:115–124. doi: 10.1016/j.prostaglandins.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamby ME, Hewett JA, Hewett SJ. Smad3-dependent signaling underlies the TGF-beta1-mediated enhancement in astrocytic iNOS expression. Glia. 2010;58:1282–1891. doi: 10.1002/glia.21005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm TH, Draeby D, Owens T. Microglia are required for astroglial toll-like receptor 4 response and for optimal TLR2 and TLR3 response. Glia. 2012;60:630–638. doi: 10.1002/glia.22296. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- Islam SA, Chang DS, Colvin RA, Byrne MH, McCully ML, Moser B, Lira SA, Charo IF, Luster AD. Mouse CCL8, a CCR8 agonist, promotes atopic dermatitis by recruiting IL-5+ T(H)2 cells. Nat Immunol. 2011;12:167–177. doi: 10.1038/ni.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John GR, Lee SC, Brosnan CF. Cytokines: Powerful regulators of glial cell activation. Neuroscientist. 2003;9:10–22. doi: 10.1177/1073858402239587. [DOI] [PubMed] [Google Scholar]
- John GR, Lee SC, Song X, Rivieccio M, Brosnan CF. IL-1-regulated responses in astrocytes: relevance to injury and recovery. Glia. 2005;49:161–176. doi: 10.1002/glia.20109. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- Krupinski J, Kumar P, Kumar S, Kaluza J. Increased expression of TGF-beta 1 in brain tissue after ischemic stroke in humans. Stroke. 1996;27:852–857. doi: 10.1161/01.str.27.5.852. [DOI] [PubMed] [Google Scholar]
- Lattin J, Zidar DA, Schroder K, Kellie S, Hume DA, Sweet MJ. G-protein-coupled receptor expression, function, and signaling in macrophages. J Leukoc Biol. 2007;82:16–32. doi: 10.1189/jlb.0107051. [DOI] [PubMed] [Google Scholar]
- Li L, Zhang H, Varrin-Doyer M, Zamvil SS, Verkman AS. Proinflammatory role of aquaporin-4 in autoimmune neuroinflammation. FASEB J. 2011;25:1556–1566. doi: 10.1096/fj.10-177279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichter-Konecki U, Mangin JM, Gordish-Dressman H, Hoffman EP, Gallo V. Gene expression profiling of astrocytes from hyperammonemic mice reveals altered pathways for water and potassium homeostasis in vivo. Glia. 2008;56:365–377. doi: 10.1002/glia.20624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm D, Castrén E, Kiefer R, Zafra F, Thoenen H. Transforming growth factor-beta 1 in the rat brain: increase after injury and inhibition of astrocyte proliferation. J Cell Biol. 1992;117:395–400. doi: 10.1083/jcb.117.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovatt D, Sonnewald U, Waagepetersen HS, Schousboe A, He W, Lin JH, Han X, Takano T, Wang S, Sim FJ, Goldman SA, Nedergaard M. The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J Neurosci. 2007;27:12255–12266. doi: 10.1523/JNEUROSCI.3404-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147(Suppl 1):S232–S240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahad DJ, Ransohoff RM. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE) Semin Immunol. 2003;15:23–32. doi: 10.1016/s1044-5323(02)00125-2. [DOI] [PubMed] [Google Scholar]
- McCandless EE, Zhang B, Diamond MS, Klein RS. CXCR4 antagonism increases T cell trafficking in the central nervous system and improves survival from West Nile virus encephalitis. Proc Natl Acad Sci U S A. 2008;105:11270–11275. doi: 10.1073/pnas.0800898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll NM, Cossoy MB, Fisher E, Staugaitis SM, Tucky BH, Rietsch AM, Chang A, Fox RJ, Trapp BD, Ransohoff RM. Imaging correlates of leukocyte accumulation and CXCR4/CXCL12 in multiple sclerosis. Arch Neurol. 2009;66:44–53. doi: 10.1001/archneurol.2008.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761–2772. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- Neary JT, Kang Y, Willoughby KA, Ellis EF. Activation of extracellular signal-regulated kinase by stretch-induced injury in astrocytes involves extracellular ATP and P2 purinergic receptors. J Neurosci. 2003;23:2348–2356. doi: 10.1523/JNEUROSCI.23-06-02348.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Rodríguez JJ, Verkhratsky A. Glial calcium and diseases of the nervous system. Cell Calcium. 2010;47:140–149. doi: 10.1016/j.ceca.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, Haydon PG, Coulter DA. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci. 2010;13:584–591. doi: 10.1038/nn.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Y, Cai Z, Rhodes PG. Analysis of genes differentially expressed in astrocytes stimulated with lipopolysaccharide using cDNA arrays. Brain Res. 2001;914:15–22. doi: 10.1016/s0006-8993(01)02766-4. [DOI] [PubMed] [Google Scholar]
- Patel JR, McCandless EE, Dorsey D, Klein RS. CXCR4 promotes differentiation of oligodendrocyte progenitors and remyelination. Proc Natl Acad Sci U S A. 2010;107:11062–11067. doi: 10.1073/pnas.1006301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafian TA, Montes C, Imura T, Qi J, Coppola G, Geschwind DH, Sofroniew MV. Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro. PLoS One. 2010;5:e9532. doi: 10.1371/journal.pone.0009532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayeed MM. Signaling mechanisms of altered cellular responses in trauma, burn, and sepsis: role of Ca2+ Arch Surg. 2000;135:1432–1442. doi: 10.1001/archsurg.135.12.1432. [DOI] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- Sharman JL, Mpamhanga CP, Spedding M, Germain P, Staels B, Dacquet C, Laudet V, Harmar AJ. IUPHAR-DB: new receptors and tools for easy searching and visualization of pharmacological data. Nucleic Acids Res. 2011;39:D534–D538. doi: 10.1093/nar/gkq1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Khakh BS. Measuring near plasma membrane and global intracellular calcium dynamics in astrocytes. J Vis Exp. 2009;26:pii:1142. doi: 10.3791/1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Tong X, Kwan KY, Corey DP, Khakh BS. TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat Neurosci. 2012;15:70–80. doi: 10.1038/nn.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitati E, McCandless EE, Klein RS, Diamond MS. CD40-CD40 ligand interactions promote trafficking of CD8+ T cells into the brain and protection against West Nile virus encephalitis. J Virol. 2007;81:9801–9811. doi: 10.1128/JVI.00941-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. Statistical applications in genetics and molecular biology. Berkeley, CA: Berkeley Electronic Press; 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. E-book available at http://www.bepress.com/sagmb/vol3/iss1/art3. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel JC, Waldmann TA, Morris JC. Interleukin-15 biology and its therapeutic implications in cancer. Trends Pharmacol Sci. 2012;33:35–41. doi: 10.1016/j.tips.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swardfager W, Lanct∧ot K, Rothenburg L, Wong A, Cappell J, Herrmann N. A meta-analysis of cytokines in Alzheimer's disease. Biol Psychiatry. 2010;68:930–941. doi: 10.1016/j.biopsych.2010.06.012. [DOI] [PubMed] [Google Scholar]
- Thrane AS, Rappold PM, Fujita T, Torres A, Bekar LK, Takano T, Peng W, Wang F, Thrane VR, Enger R, Haj-Yasein NN, Skare Ø, Holen T, Klungland A, Ottersen OP, Nedergaard M, Nagelhus EA. Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc Natl Acad Sci U S A. 2011;108:846–851. doi: 10.1073/pnas.1015217108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Orkand RK, Kettenmann H. Glial calcium: homeostasis and signaling function. Physiol Rev. 1998;78:99–141. doi: 10.1152/physrev.1998.78.1.99. [DOI] [PubMed] [Google Scholar]
- Voskuhl RR, Peterson RS, Song B, Ao Y, Morales LB, Tiwari-Woodruff S, Sofroniew MV. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci. 2009;29:11511–11522. doi: 10.1523/JNEUROSCI.1514-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]