Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer (original) (raw)
. Author manuscript; available in PMC: 2013 Jan 14.
Published in final edited form as: N Engl J Med. 2012 Jun 2;366(26):2443–2454. doi: 10.1056/NEJMoa1200690
Abstract
Background
Blockade of programmed death 1 (PD-1), an inhibitory receptor expressed by T cells, can overcome immune resistance. We assessed the antitumor activity and safety of BMS-936558, an antibody that specifically blocks PD-1.
Methods
We enrolled patients with advanced melanoma, non–small-cell lung cancer, castration-resistant prostate cancer, or renal-cell or colorectal cancer to receive anti–PD-1 antibody at a dose of 0.1 to 10.0 mg per kilogram of body weight every 2 weeks. Response was assessed after each 8-week treatment cycle. Patients received up to 12 cycles until disease progression or a complete response occurred.
Results
A total of 296 patients received treatment through February 24, 2012. Grade 3 or 4 drug-related adverse events occurred in 14% of patients; there were three deaths from pulmonary toxicity. No maximum tolerated dose was defined. Adverse events consistent with immune-related causes were observed. Among 236 patients in whom response could be evaluated, objective responses (complete or partial responses) were observed in those with non–small-cell lung cancer, melanoma, or renal-cell cancer. Cumulative response rates (all doses) were 18% among patients with non–small-cell lung cancer (14 of 76 patients), 28% among patients with melanoma (26 of 94 patients), and 27% among patients with renal-cell cancer (9 of 33 patients). Responses were durable; 20 of 31 responses lasted 1 year or more in patients with 1 year or more of follow-up. To assess the role of intratumoral PD-1 ligand (PD-L1) expression in the modulation of the PD-1–PD-L1 pathway, immunohistochemical analysis was performed on pretreatment tumor specimens obtained from 42 patients. Of 17 patients with PD-L1–negative tumors, none had an objective response; 9 of 25 patients (36%) with PD-L1–positive tumors had an objective response (P = 0.006).
Conclusions
Anti–PD-1 antibody produced objective responses in approximately one in four to one in five patients with non–small-cell lung cancer, melanoma, or renal-cell cancer; the adverse-event profile does not appear to preclude its use. Preliminary data suggest a relationship between PD-L1 expression on tumor cells and objective response. (Funded by Bristol-Myers Squibb and others; ClinicalTrials.gov number, NCT00730639.)
Human cancers harbor numerous genetic and epigenetic alterations, generating neoantigens that are potentially recognizable by the immune system.1 Although an endogenous immune response to cancer is observed in preclinical models and patients, this response is ineffective, because tumors develop multiple resistance mechanisms, including local immune suppression, induction of tolerance, and systemic dysfunction in T-cell signaling.2-5 Moreover, tumors may exploit several distinct pathways to actively evade immune destruction, including endogenous “immune checkpoints” that normally terminate immune responses after antigen activation. These observations have resulted in intensive efforts to develop immunotherapeutic approaches for cancer, including immune-checkpoint-pathway inhibitors such as anti–CTLA-4 antibody (ipilimumab) for the treatment of patients with advanced melanoma.6-8
Programmed death 1 (PD-1) is a key immune-checkpoint receptor expressed by activated T cells, and it mediates immunosuppression. PD-1 functions primarily in peripheral tissues, where T cells may encounter the immunosuppressive PD-1 ligands PD-L1 (B7-H1) and PD-L2 (B7-DC), which are expressed by tumor cells, stromal cells, or both.9-12 Inhibition of the interaction between PD-1 and PD-L1 can enhance T-cell responses in vitro and mediate preclinical antitumor activity.11,13 In a dose-escalation study, the anti–PD-1 monoclonal antibody BMS-936558 (also known as MDX-1106 and ONO-4538) was administered as a single dose in 39 patients with advanced solid tumors.14 A favorable safety profile and preliminary evidence of clinical activity were shown in this pilot study, establishing the basis for the current multiple-dose trial involving patients with diverse cancers. We report clinical results for 296 patients in this trial.
Methods
Study Design
This study was sponsored by Bristol-Myers Squibb, which provided the study drug and worked jointly with the senior academic authors to design, collect, analyze, and interpret the study results. All the authors signed a confidentiality agreement with the sponsor. The protocol, including a detailed statistical analysis plan, is available with the full text of this article at NEJM.org. All drafts of the manuscript were prepared by the authors with editorial assistance from a professional medical writer paid by the sponsor. All the authors vouch for the accuracy and completeness of the reported data and for the fidelity of this report to the trial protocol, and all the authors made the decision to submit the manuscript for publication.
This phase 1 study assessed the safety, anti-tumor activity, and pharmacokinetics of BMS-936558, a fully human IgG4-blocking monoclonal antibody directed against PD-1, in patients with selected advanced solid tumors. All patients (or their legal representatives) gave written informed consent before enrollment. The antibody was administered as an intravenous infusion every 2 weeks of each 8-week treatment cycle. Response was assessed after each treatment cycle. Patients received treatment for up to 2 years (12 cycles), unless they had a complete response, unacceptable adverse effects, or progressive disease or they withdrew consent. In clinically stable patients, study treatment could be continued beyond apparent initial disease progression until progression was confirmed, as outlined by proposed immune-response criteria.15 Patients with stable disease or an ongoing objective response (complete or partial response) at the end of treatment were followed for up to 1 year and were offered retreatment for 1 additional year in the event of disease progression.
Safety evaluations (clinical examination and laboratory assessments) were conducted for all treated patients at baseline and regular intervals. The severity of adverse events was graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0.16
Dose Escalation
Patients with advanced melanoma, non–small-cell lung cancer, renal-cell cancer, castration-resistant prostate cancer, or colorectal cancer were enrolled. Cohorts of three to six patients per dose level were enrolled sequentially at doses of 1.0, 3.0, or 10.0 mg per kilogram of body weight. Dose escalation proceeded when a minimum of three patients had completed the safety-evaluation period (56 days) at a given dose level, with dose-limiting toxicity in less than one third of patients. Intrapatient dose escalation was not permitted.
Cohort Expansion
A maximum tolerated dose was not reached. Initially, five expansion cohorts of approximately 16 patients each were enrolled at doses of 10.0 mg per kilogram for melanoma, non–small-cell lung cancer, renal-cell cancer, castration-resistant prostate cancer, and colorectal cancer. On the basis of initial signals of activity, additional expansion cohorts of approximately 16 patients each were enrolled for melanoma (at a dose of 1.0 or 3.0 mg per kilogram, followed by cohorts randomly assigned to 0.1, 0.3, or 1.0 mg per kilogram), lung cancer (patients with the squamous or nonsquamous subtype, randomly assigned to a dose of 1.0, 3.0, or 10.0 mg per kilogram), and renal-cell cancer (at a dose of 1.0 mg per kilogram).
Patients
Eligible patients had documented advanced solid tumors; an age of 18 years or older; a life expectancy of 12 weeks or more; an Eastern Cooperative Oncology Group performance status of 0, 1, or 2 (on a scale from 0 to 5, with 0 indicating that the patient is asymptomatic, 1 that the patient is restricted in strenuous activity, and 2 that the patient is ambulatory but unable to work)17; measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST), version 1.0,18 with modification (see Methods S1 in the Supplementary Appendix, available at NEJM.org; and the protocol); adequate hematologic, hepatic, and renal function; and a history of one to five systemic treatment regimens. Patients with radiographically stable treated brain metastases were enrolled. Patients with a history of chronic autoimmune disease, prior therapy with antibodies that modulate T-cell function (e.g., anti–CTLA-4, anti–PD-1, and anti–PD-L1), conditions requiring immunosuppressive medications, or chronic infection (e.g., human immunodeficiency virus infection and hepatitis B or C) were excluded.
Immunohistochemical Analysis for PD-L1
Immunohistochemical analysis for PD-L1 was performed on archival or newly obtained pretreatment formalin-fixed, paraffin-embedded tumor specimens with the use of the murine antihuman PD-L1 monoclonal antibody 5H1.11,19 The percentage of tumor cells exhibiting cell-surface staining for PD-L1 was scored by two independent pathologists who were unaware of outcomes. PD-L1 positivity was defined per specimen by a 5% expression threshold19,20; patients with multiple specimens were considered PD-L1–positive if any specimen met this criterion.
Pharmacokinetics and Pharmacodynamics
For pharmacokinetic analysis, serum concentrations of anti–PD-1 antibody were quantified with the use of an enzyme-linked immunosorbent assay. For pharmacodynamic analysis, peripheral-blood mononuclear cells (PBMCs) were isolated from patients at baseline and after the first treatment cycle to estimate PD-1–receptor occupancy by the antibody on circulating CD3+ T cells by means of flow cytometry.14
Statistical Analysis
Data on all 296 patients treated as of the date of analysis for this report (February 24, 2012) were used for summaries of baseline characteristics and adverse events. Pharmacokinetic and molecular-marker analyses included treated patients with available data as of February 24, 2012. The efficacy analysis included the 236 patients who could be evaluated for a response and who began treatment by July 1, 2011. Adverse events were coded with the use of the Medical Dictionary for Regulatory Activities (MedDRA), version 14.1. Adverse events of special interest, with a potential immune-related cause, were identified with the use of a predefined list of MedDRA terms. The best responses in individual patients were derived from investigator-reported data per modified RECIST, version 1.0. Objective responses were confirmed by at least one sequential tumor assessment, and objective response rates were calculated as [(complete responses + partial responses) ÷ number of patients] × 100. Fisher's exact test was used to assess the association between PD-L1 expression and objective response.
Results
Baseline Patient Characteristics
A total of 296 patients with advanced solid tumors, including melanoma (104 patients), non– small-cell lung cancer (122), renal-cell cancer (34), castration-resistant prostate cancer (17), and colorectal cancer (19), began treatment with anti– PD-1 antibody between October 2008 and February 24, 2012. The majority of patients were heavily pretreated; 47% had received at least three prior regimens (Table S1-A in the Supplementary Appendix). Notable prior therapies included immunotherapy and BRAF inhibitors in patients with melanoma (64% and 8% of patients, respectively); platinum-based chemotherapy and tyrosine kinase inhibitors in patients with lung cancer (94% and 34%, respectively); and nephrectomy, immunotherapy, and antiangiogenic therapy in patients with renal-cell cancer (94%, 59%, and 74%, respectively) (Tables S1-B, S1-C, and S1-D in the Supplementary Appendix). Baseline characteristics of the total treated population (296 patients) were similar to those of the efficacy population (236 patients).
Safety
A maximum tolerated dose was not defined at the doses tested in this study. A relative dose intensity (the proportion of administered doses relative to planned doses) of 90% or more was achieved in 86% of patients (Table S2-A in the Supplementary Appendix). Fifteen of 296 patients (5%) discontinued treatment owing to treatment-related adverse events (Tables S2-B and S3-A in the Supplementary Appendix). As of the date of analysis, 62 patients (21%) had died; disease progression was the most common cause of death (Table S2-C in the Supplementary Appendix).
The most common adverse events, regardless of causality, were fatigue, decreased appetite, diarrhea, nausea, cough, dyspnea, constipation, vomiting, rash, pyrexia, and headache (Table S3-A in the Supplementary Appendix). Common treatment-related adverse events included fatigue, rash, diarrhea, pruritus, decreased appetite, and nausea (Tables S3-A and S3-B in the Supplementary Appendix). Grade 3 or 4 treatment-related adverse events were observed in 41 of 296 patients (14%). Drug-related serious adverse events (as defined in Table S4 in the Supplementary Appendix) occurred in 32 of 296 patients (11%). The spectrum, frequency, and severity of treatment-related adverse events were generally similar across the dose levels tested. Drug-related adverse events of special interest (e.g., those with potential immune-related causes) included pneumonitis, vitiligo, colitis, hepatitis, hypophysitis, and thyroiditis (Table 1 and Fig. 1C).
Table 1.
Treatment-Related Adverse Events of Special Interest That Occurred in at Least 1% of All Treated Patients.
| Event | Anti-PD-1 Antibody, 0.1 mg/kg N = 18) | Anti-PD-1 Antibody, 0.3 mg/kg (N = 19) | Anti-PD-1 Antibody, 1.0mg/ kg(N = 79) | Anti-PD-1 Antibody, 3.0 mg/kg (N = 50) | Anti-PD-1 Antibody, 10.0 mg/kg (N = 130) | Anti-PD-1 Antibody, Total (N = 296) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All Events | Grade 3 or 4 Events | All Events | Grade 3 or 4 Events | All Events | Grade 3 or 4 Events | All Events | Grade 3 or 4 Events | All Events | Grade 3 or 4 Events | All Events | Grade 3 or 4 Events | |
| number of patients (percent) | ||||||||||||
| Any adverse event of special interest* | 8 (44) | 0 | 6 (32) | 0 | 39 (49) | 5 (6) | 19 (38) | 2 (4) | 50 (38) | 11 (8) | 122 (41) | 18 (6) |
| Pulmonary disorders | ||||||||||||
| Pneumonitis | 0 | 0 | 0 | 0 | 3 (4) | 2 (3) | 1 (2) | 0 | 5 (4) | 1 (1) | 9 (3) | 3 (1) |
| Allergic rhinitis | 1 (6) | 0 | 0 | 0 | 2 (3) | 0 | 0 | 0 | 1 (1) | 0 | 4 (1) | 0 |
| Diarrhea | 1 (6) | 0 | 2 (11) | 0 | 15 (19) | 0 | 3 (6) | 0 | 12 (9) | 3 (2) | 33 (11) | 3 (1) |
| Skin events | ||||||||||||
| Rash | 3 (17) | 0 | 2 (11) | 0 | 16 (20) | 0 | 4 (8) | 0 | 11 (8) | 0 | 36 (12) | 0 |
| Pruritus | 0 | 0 | 2 (11) | 0 | 13 (16) | 0 | 4 (8) | 0 | 9 (7) | 1 (1) | 28 (9) | 1 (<1) |
| Vitiligo | 3 (17) | 0 | 0 | 0 | 3 (4) | 0 | 2 (4) | 0 | 0 | 0 | 8 (3) | 0 |
| Pruritic rash | 0 | 0 | 0 | 0 | 1 (1) | 0 | 1 (2) | 0 | 4 (3) | 0 | 6 (2) | 0 |
| Urticaria | 0 | 0 | 0 | 0 | 0 | 0 | 3 (6) | 0 | 2 (2) | 0 | 5 (2) | 0 |
| Macular rash | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2) | 0 | 3 (2) | 1 (1) | 4 (1) | 1 (<1) |
| Alopecia | 0 | 0 | 0 | 0 | 2 (3) | 0 | 0 | 0 | 1 (1) | 0 | 3 (1) | 0 |
| Hypopigmentation | 0 | 0 | 0 | 0 | 2 (3) | 0 | 0 | 0 | 1 (1) | 0 | 3 (1) | 0 |
| Laboratory investigations† | ||||||||||||
| Alanine aminotransferase increased | 0 | 0 | 1 (5) | 0 | 4 (5) | 0 | 2 (4) | 0 | 4 (3) | 2 (2) | 11 (4) | 2 (1) |
| Thyroid-stimulating hormone increased | 2 (11) | 0 | 0 | 0 | 2 (3) | 0 | 2 (4) | 0 | 3 (2) | 1 (1) | 9 (3) | 1 (<1) |
| Aspartate aminotransferase increasec | 0 | 0 | 1 (5) | 0 | 2 (3) | 0 | 2 (4) | 1 (2) | 3 (2) | 1 (1) | 8 (3) | 2 (1) |
| Endocrine disorders | ||||||||||||
| Hypothyroidism | 0 | 0 | 1 (5) | 0 | 2 (3) | 0 | 1 (2) | 0 | 3 (2) | 1 (1) | 7 (2) | 1 (<1) |
| Hyperthyroidism | 1 (6) | 0 | 0 | 0 | 0 | 0 | 1 (2) | 0 | 1 (1) | 1 (1) | 3 (1) | 1 (<1) |
| Infusion-related reaction or hypersensitivty | 0 | 0 | 0 | 0 | 2 (3) | 0 | 3 (6) | 0 | 4 (3) | 1 (1) | 9 (3) | 1 (<1) |
Figure 1. Activity of Anti–Programmed Death 1 (PD-1) Antibody in Patients with Treatment-Refractory Melanoma, Non–Small-Cell Lung Cancer, or Renal-Cell Cancer.
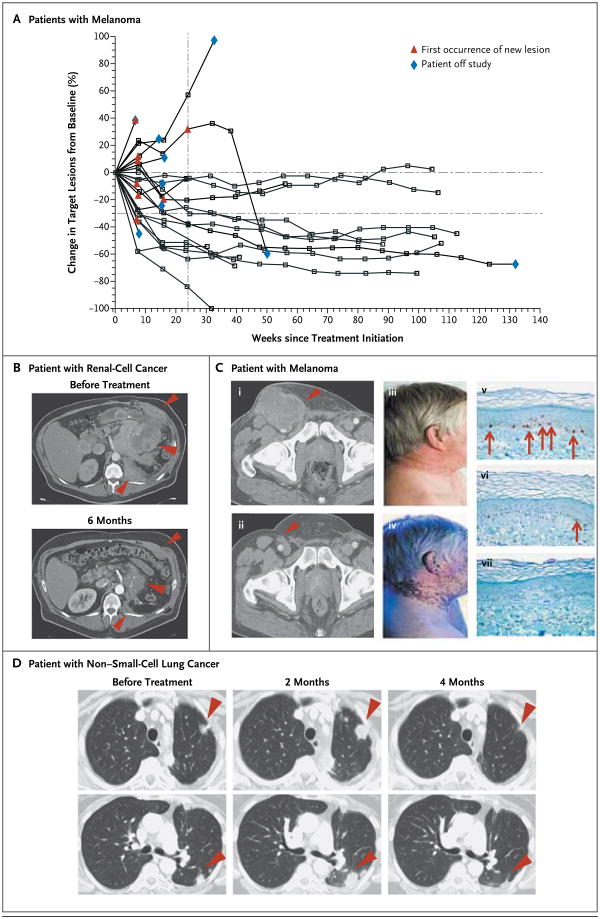
In Panel A, a representative plot shows changes from baseline in the tumor burden, measured as the sum of the longest diameters of target lesions, in 27 patients with melanoma who received anti–PD-1 antibody at a dose of 1.0 mg per kilogram of body weight every 2 weeks. In the majority of patients who had an objective response, responses were durable and evident by the end of cycle 2 (16 weeks) of treatment. The vertical dashed line marks the 24-week time point at which the progression-free survival rate was calculated, and the horizontal dashed line marks the threshold for objective response (partial tumor regression) according to modified Response Evaluation Criteria in Solid Tumors. Tumor regression followed conventional as well as immune-related patterns of response, such as prolonged reduction in tumor burden in the presence of new lesions.15,21 Panel B shows partial regression of metastatic renal-cell cancer in a 57-year-old patient who received anti– PD-1 antibody at a dose of 1.0 mg per kilogram. This patient had previously undergone radical surgery, and progressive disease had developed after treatment with sunitinib, temsirolimus, sorafenib, and pazopanib. The arrowheads show regression of recurrent tumor in the operative field. Panel C shows a complete response in a 62-year-old patient with metastatic melanoma who received anti–PD-1 antibody at a dose of 3.0 mg per kilogram. Pretreatment computed tomographic scanning (i) revealed inguinal-lymph-node metastasis (arrowhead), which regressed completely after 13 months of treatment (ii). Numerous metastases in the subcutaneous tissue and retroperitoneum also regressed completely (not shown). Vitiligo, which developed after 6 months of treatment, is evident in photographs taken at 9 months under visible light (iii) and ultraviolet light (iv). Skin-biopsy specimens with immunohistochemical staining for micro-ophthalmia–associated transcription factor show that melanocytes (arrows) are abundant at the epidermal–dermal junction in normal skin (v), scarce in skin partially affected by vitiligo (vi), and absent in skin fully affected by vitiligo (vii). Panel D shows a partial response in a patient with metastatic non–small-cell lung cancer (nonsquamous histologic type) who received anti–PD-1 antibody at a dose of 10.0 mg per kilogram. The arrowheads show initial progression in pulmonary lesions, followed by regression (an immune-related pattern of response)
Hepatic or gastrointestinal adverse events were managed with treatment interruption and, as necessary, with the administration of glucocorticoids. These events (e.g., diarrhea in 33 patients, including three grade 3 or 4 events and elevated alanine aminotransferase levels in 11 patients, including two grade 3 or 4 events) were reversible in all cases. Endocrine disorders were managed with replacement therapy. At the discretion of the treating physician, treatment with anti–PD-1 antibody was reinitiated once the adverse event had been successfully managed. Drug-related pneumonitis occurred in 9 of the 296 patients (3%). Grade 3 or 4 pneumonitis developed in 3 patients (1%). No clear relationship between the occurrence of pneumonitis and tumor type, dose level, or the number of doses received was noted. Early-grade pneumonitis in 6 patients was reversible with treatment discontinuation, glucocorticoid administration, or both. In 3 patients with pneumonitis, infliximab, mycophenolate, or both were used for additional immunosuppression; however, given the small number of patients and variable outcomes, the effectiveness of such treatment was unclear. There were three drug-related deaths (1%) due to pneumonitis (two in patients with non–small-cell lung cancer and one in a patient with colorectal cancer).
Clinical Activity
Antitumor activity was observed at all doses tested. Objective responses were observed in a substantial proportion of patients with non–small-cell lung cancer, melanoma, or renal-cell cancer (Table 2 and Fig. 1) and in various sites of metastasis, including the liver, lung, lymph nodes, and bone. At the time of data analysis, two patients with lung cancer who received 10 mg per kilogram had unconfirmed responses, and eight additional patients (with melanoma, lung cancer, or renal-cell cancer) had a persistent reduction in baseline target lesions in the presence of new lesions (a finding consistent with an immune-related response pattern15). None of these patients were categorized as having had a response for the purpose of calculating objective-response rates. Objective responses, prolonged disease stabilization, or both were observed in patients who had received a variety of prior therapies. No objective responses were observed in patients with colorectal or prostate cancer.
Table 2.
Clinical Activity of Anti–PD-1 Antibody in the Efficacy Population.*
| Dose of Anti–PD-1 Antibody | Objective Response† | Objective-Response Rate‡ | Duration of Response§ | Stable Disease ≥24 wk | Progression-free Survival Rate at 24 wk¶ | |
|---|---|---|---|---|---|---|
| no. of patients/total no. of patients | % (95% CI) | mo | no. of patients/total no. of patients | % (95% CI) | % (95% CI) | |
| Melanoma | ||||||
| 0.1 mg/kg | 4/14 | 29 (8–58) | 7.5+, 5.6+, 5.6, 5.6 | 1/14 | 7 (0.2–34) | 40 (13–66) |
| 0.3 mg/kg | 3/16 | 19 (4–46) | 3.8+, 2.1+, 1.9+ | 1/16 | 6 (0.2–30) | 31 (9–54) |
| 1.0 mg/kg | 8/27 | 30 (14–50) | 24.9+, 22.9, 20.3+, 19.3+, 18.4+, 7.6+, 5.6+, 5.3+ | 3/27 | 11 (2–29) | 45 (26–65) |
| 3.0 mg/kg | 7/17 | 41 (18-67) ║ | 22.4+, 18.3+, 15.2+, 12.9, 11.1, 9.3, 9.2+ | 1/17 | 6 (0.1–29) | 55 (30–80) |
| 10.0 mg/kg | 4/20 | 20 (6–44) | 24.6+, 23.9+, 18.0+, 17.0 | 0/20 | 0 | 30 (9–51) |
| All doses | 26/94 | 28 (19–38) | 6/94 | 6 (2–13) | 41 (30–51) | |
| Non–small-cell lung cancer | ||||||
| Squamous | ||||||
| 1.0 mg/kg | 0/5 | 0 | 0/5 | 0 | 0 | |
| 3.0 mg/kg | 3/6 | 50 (12–88) | ND | 0/6 | 0 | 50 (10–90) |
| 10.0 mg/kg | 3/7 | 43 (10–82) | ND | 0/7 | 0 | 43 (6–80) |
| All doses | 6/18 | 33 (13–59) | ND | 0/18 | 0 | 33 (12–55) |
| Nonsquamous | ||||||
| 1.0 mg/kg | 0/12 | 0 | 1/12 | 8 (0.2–39) | 14 (0–37) | |
| 3.0 mg/kg | 3/13 | 23 (5–54) | ND | 2/13 | 15 (2–45) | 37 (10–64) |
| 10.0 mg/kg | 4/31 | 13 (4–30) | ND | 2/31 | 6 (0.8–21) | 21 (6–36) |
| All doses | 7/56 | 12 (5–24) | ND | 5/56 | 9 (3–20) | 22 (11–34) |
| Unknown type | ||||||
| 1.0 mg/kg | 1/1 | NA | ND | 0/1 | 0 | NA |
| 10.0 mg/kg | 0/1 | 0 | 0/1 | 0 | 0 | |
| All types | ||||||
| 1.0 mg/kg | 1/18 | 6 (0.1–27) | 9.2+ | 1/18 | 6 (0.1–27) | 16 (0–34) |
| 3.0 mg/kg | 6/19 | 32 (13–57) | 30.8+, 7.6+, 5.5+, 3.7+, 1.9+, NA** | 2/19 | 11 (1–33) | 41 (18–64) |
| 10.0 mg/kg | 7/39 | 18 (8–34) | 14.8+, 7.6+, 7.3+, 6.7, 4.2, 3.7+, 3.7 | 2/39 | 5 (0.6–17) | 24 (11–38) |
| All doses | 14/76 | 18 (11–29) | 5/76 | 7 (2–15) | 26 (16–36) | |
| Renal-cell cancer | ||||||
| 1.0 mg/kg | 4/17 | 24 (7–50) | 17.5+, 9.2+, 9.2, 5.6+ | 4/17 | 24 (7–50) | 47 (23–71) |
| 10.0 mg/kg | 5/16 | 31 (11-59) ║ | 22.3+, 21.7+, 12.9, 12.0, 8.4 | 5/16 | 31 (11–59) | 67 (43–91) |
| All doses | 9/33 | 27 (13–46) | 9/33 | 27 (13–46) | 56 (39–73) |
In patients with lung cancer, 14 objective responses were observed at doses of 1.0, 3.0, or 10.0 mg per kilogram, with response rates of 6%, 32%, and 18%, respectively. Objective responses were observed across non–small-cell histologic types: in 6 of 18 patients (33%) with squamous tumors, 7 of 56 (12%) with nonsquamous tumors, and 1 of 2 with tumors of unknown type. All 14 patients with objective responses started treatment 24 weeks or more before data analysis, and of these, 8 had a response that lasted 24 weeks or more (Table 2). Five of 14 patients with objective responses started treatment 1 year or more before data analysis, and of these, 2 had a response that lasted 1 year or more. Stable disease lasting 24 weeks or more was observed in 5 patients (7%) with lung cancer, all of whom had nonsquamous tumors.
In patients with melanoma, 26 objective responses were observed at doses ranging from 0.1 to 10.0 mg per kilogram, with response rates ranging from 19 to 41% per dose level. At a dose of 3.0 mg per kilogram, objective responses were noted in 7 of 17 patients (41%). Of 26 patients with melanoma who had an objective response, 18 started treatment 1 year or more before February 24, 2012, and of these, 13 had a response that lasted 1 year or more. The remaining 8 patients with objective responses received study medication for less than 1 year, and 6 had responses ranging from 1.9 to 5.6 months. Stable disease lasting 24 weeks or more was observed in 6 patients (6%).
Among patients with renal-cell cancer, objective responses occurred in 4 of 17 patients (24%) treated with a dose of 1.0 mg per kilogram and 5 of 16 (31%) treated with 10.0 mg per kilogram. Of 8 patients with objective responses who started treatment 1 year or more before data analysis, 5 had a response that lasted 1 year or more. Stable disease lasting 24 weeks or more was observed in an additional 9 patients (27%).
Pharmacokinetics and Pharmacodynamics
The median time to the peak concentration of anti PD-1 antibody was 1 to 4 hours after the start of infusion. The pharmacokinetics of the antibody were linear, with a dose-proportional increase in the peak concentration and area under the curve calculated from day 1 to day 14 in the dose range of 0.1 to 10.0 mg per kilogram (35 patients). The pharmacodynamics of anti–PD-1 antibody were assessed according to PD-1–receptor occupancy on circulating CD3+ T cells. In PBMCs from 65 patients with melanoma who were treated with one cycle of anti–PD-1 antibody at a dose of 0.1 to 10.0 mg per kilogram every 2 weeks, the median PD-1–receptor occupancy by anti–PD-1 antibody was 64 to 70% according to dose level (Fig. 2A).
Figure 2. Pharmacodynamic and Molecular-Marker Assessments.
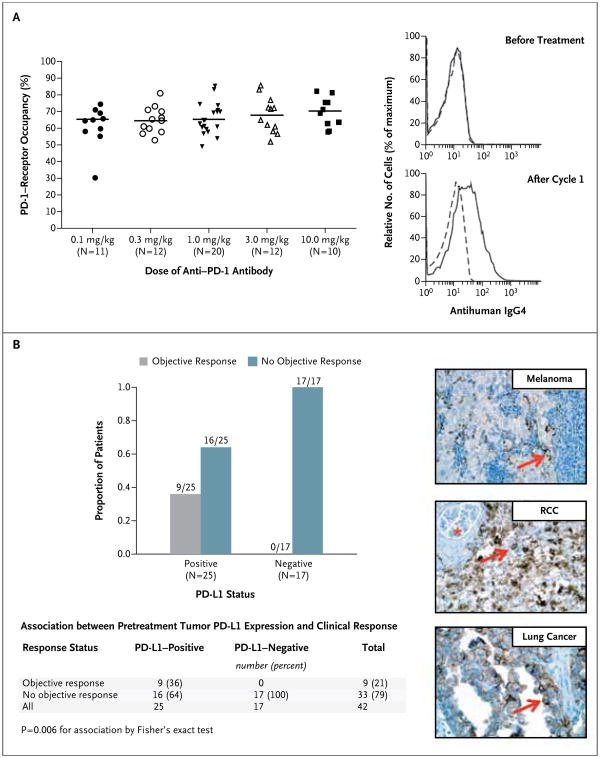
Panel A shows PD-1–receptor occupancy by anti–PD-1 antibody. The graph at the left shows PD-1–receptor occupancy on circulating T cells in 65 patients with melanoma after one cycle (8 weeks) of treatment at a dose of 0.1 to 10.0 mg per kilogram every 2 weeks. Bars indicate median values. The graphs at the right show PD-1–receptor occupancy on CD3-gated peripheral-blood mononuclear cells from a patient with melanoma who received 0.1 mg per kilogram, before treatment (top) and after one treatment cycle (bottom). Cells were stained with biotinylated antihuman IgG4 to detect infused anti–PD-1 antibody bound to PD-1 molecules on the cell surface. Detection was accomplished with the use of streptavidin–phycoerythrin, followed by flow-cytometric analysis. Dashed lines indicate isotype staining controls, and solid lines antihuman IgG4. Panel B shows the correlation of pretreatment tumor cell-surface expression of PD-1 ligand (PD-L1), as determined with immunohistochemical analysis of formalin-fixed, paraffin-embedded specimens, with an objective response to PD-1 blockade in 42 patients with advanced cancers: 18 with melanoma, 10 with non–small-cell lung cancer, 7 with colorectal cancer, 5 with renal-cell cancer, and 2 with castration-resistant prostate cancer. Tumor cell-surface expression of PD-L1 was significantly correlated with an objective clinical response (graph at the left). No patients with PD-L1–negative tumors had an objective response. Of the 25 patients with PD-L1–positive tumors, 2 who were categorized as not having had a response at the time of data analysis are still under evaluation. Shown at the right are immunohistochemical analysis with the anti–PD-L1 monoclonal antibody 5H1 in a specimen of a lymph-node metastasis from a patient with melanoma (top), a nephrectomy specimen from a patient with renal-cell cancer (RCC) (middle), and a specimen of a brain metastasis from a patient with lung adenocarcinoma (bottom). The arrow in each specimen indicates one of many tumor cells with surface-membrane staining for PD-L1. The asterisk indicates a normal glomerulus in the nephrectomy specimen, which was negative for PD-L1 staining.
PD-L1 Expression in Tumors
Sixty-one pretreatment tumor specimens from 42 patients (18 with melanoma, 10 with non–small-cell lung cancer, 7 with colorectal cancer, 5 with renal-cell cancer, and 2 with prostate cancer) (Table S5 in the Supplementary Appendix) were analyzed for PD-L1 expression on the surface of tumor cells (Fig. 2B). Biopsy specimens from 25 of the 42 patients were positive for PD-L1 expression by immunohistochemical analysis. Of these 25 patients, 9 (36%) had an objective response. None of the 17 patients with PD-L1– negative tumors had an objective response. This analysis is based on optional biopsies in a non-random subset of the population, and testing of a statistical hypothesis was not prespecified. These preliminary results must therefore be interpreted with caution.
Discussion
Our data show that approximately one in four to one in five patients treated with anti–PD-1 antibody had objective responses with durability; these occurred in heavily pretreated patients with diverse tumor types. PD-1 blockade extends the spectrum of clinical activity by immunotherapy beyond immunogenic tumor types, such as melanoma and renal-cell cancer, to treatment-refractory, meta-static non–small-cell lung cancer, a tumor type that is generally not considered to be responsive to immunotherapy. The level of activity seen with anti–PD-1 antibody in patients with lung cancer who had received substantial amounts of prior therapy (55% with at least three lines of previous therapy) (Table S1-B in the Supplementary Appendix) and across histologic types is of interest, particularly in the patients with squamous tumors.22,23 These unexpected findings underscore the possibility that any neoplasm could be immunogenic with proper immune activation; however, the reason why only a minority of patients had a response (i.e., tumor or patient ‘host’ factors or both) is not known. The full therapeutic potential of PD-1 blockade across other tumor types remains to be defined.
The durability of objective responses across multiple cancer types in patients treated with anti–PD-1 antibody was also notable. Although anti–PD-1 antibody was not compared with other therapies in this study, this durability contrasts with the relatively modest durability of objective responses observed in many patients with non–small-cell lung cancer, melanoma, or renal-cell cancer who are treated with conventional chemotherapy, tyrosine kinase inhibitors, or both. The response durability in heavily pretreated patients with lung cancer who received anti–PD-1 antibody is particularly interesting, because standard salvage therapies historically have shown modest benefit in these patients.24 As measured by standard RECIST criteria in this study, objective responses were long-lasting, with response durations of 1 year or more in 20 of 31 patients who had a response with 1 year or more of follow-up. In addition, patterns of tumor regression consistent with immune-related patterns of response15,21,25 were observed. That is, index lesions often responded as previously undetected lesions became detectable, a finding that is possibly related to lymphocyte infiltration of previously unknown small nests of tumor cells. Although the full effect of these unconventional response patterns remains to be defined in randomized trials with survival end points, these observations are reminiscent of findings with ipilimumab in which a significant extension of overall survival was observed in treated patients.7,8
Drug-related grade 3 or 4 toxic effects occurred in 14% of patients who received anti–PD-1 antibody, suggesting that therapy can be delivered in an outpatient setting with minimal supportive care. Among adverse events of special interest, pneumonitis was observed, with findings ranging from isolated radiographic abnormalities to progressive, diffuse infiltrates associated with clinical symptoms in a small number of patients. Although three deaths occurred, mild-to-moderate pneumonitis was managed successfully with either observation or glucocorticoids.
A particular challenge in cancer immunotherapy has been the identification of mechanism-based predictive biomarkers that could be used to identify candidates for such treatment and guide disease-management decisions. Our findings suggest that PD-L1 expression in tumors is a candidate molecular marker that warrants further exploration for use in selecting patients for immunotherapy with anti–PD-1 antibody. Our observation of an objective response in 36% of the patients with PD-L1–positive tumors and in none of those with PD-L1–negative tumors suggests that PD-L1 expression on the surface of tumor cells in pretreatment tumor specimens may be associated with an objective response. Although tumor-cell expression of PD-L1 may be driven by constitutive oncogenic pathways, recent research suggests that it may also reflect adaptive immune resistance in response to an endogenous antitumor immune response, which may remain in check unless it is unleashed through blockade of the PD-1–PD-L1 pathway.19 Although our data on PD-L1 expression are consistent with current knowledge of the role of PD-L1 in tumor immune resistance, additional studies will be needed to define the role of PD-L1 as a potential predictive marker of response to anti–PD-1 antibody therapy.
This study and a companion study of anti–PD-L1 antibody, now reported in the Journal,26 describe clinical activity with these agents that validates the importance of the PD-1–PD-L1 pathway for the treatment of some cancers. The signals of clinical activity in patients with non–small-cell lung cancer, melanoma, and renal-cell cancer treated with anti–PD-1 antibody and the possibility of basing patient selection for such treatment on PD-L1 expression in tumors need to be prospectively assessed. Phase 2 trials involving immunologic and molecular-marker correlates (ClinicalTrials.gov numbers, NCT01354431 and NCT01358721) are under way, and phase 3 studies of anti–PD-1 antibody for the treatment of non–small-cell lung cancer, melanoma, and renal-cell cancer are being planned. An assessment of such treatment for other tumor types is also of interest.
Supplementary Material
Supplement1
Acknowledgments
Supported by Bristol-Myers Squibb, Ono Pharmaceutical, and grants from the National Institutes of Health (5R01 CA142779, to Drs. Topalian and Pardoll) and the Melanoma Research Alliance (to Drs. Topalian and Pardoll).
Dr. Topalian reports receiving grant support and reimbursement for travel expenses from Bristol-Myers Squibb. Dr. Hodi reports receiving reimbursement for travel expenses from Bristol-Myers Squibb and funding for clinical trials from Bristol-Myers Squibb, Genentech, Pfizer, Novartis, and Synta Pharmaceuticals on behalf of his institution. Dr. Brahmer reports receiving grant support from MedImmune, Regeneron, Merck, and Synta Pharmaceuticals, consulting fees and reimbursement for travel expenses from Genentech, consulting fees from Eli Lilly, reimbursement for travel expenses from Bristol-Myers Squibb, and payment for the development of educational presentations from Quintiles and the Network for Continuing Medical Education. Dr. Smith reports receiving grant support from Bristol-Myers Squibb. Dr. McDermott reports receiving grant support and consulting fees from Prometheus Laboratories and consulting fees from Bristol-Myers Squibb and Roche/Genentech. Dr. Powderly reports receiving grant support (including for the development of educational presentations) and lecture fees from Bristol-Myers Squibb; receiving grant support, consulting fees, and lecture fees from Genentech; receiving grant support and consulting fees from Celldex; receiving grant support and consulting fees from Amplimmune; receiving grant support from ImClone; receiving consulting fees and lecture fees from Genoptix; receiving consulting fees from Veridex; receiving lecture fees from Dendreon; and being the founder of and holding stock in BioCytics, which has received contract work from Genentech and Millennium. Dr. Carvajal reports receiving consulting fees from Morphotek and Novartis and lecture fees from the Florida Society of Dermatology and Dermatologic Surgery and Imedex. Dr. Atkins reports receiving consulting fees from Bristol-Myers Squibb, Genentech, CureTech, Merck, Prometheus Laboratories, Nektar, and AstraZeneca. Dr. Spigel reports receiving consulting fees from Bristol-Myers Squibb on behalf of his institution. Dr. Drake reports receiving consulting fees and payment for the development of educational presentations from Bristol-Myers Squibb, receiving consulting fees from Dendreon and Pfizer, holding stock or stock options in Amplimmune, holding patents regarding the blockade of lymphocyte checkpoint molecules, and receiving patent royalties from Bristol-Myers Squibb and Amplimmune on behalf of his institution. Dr. Pardoll reports receiving consulting fees from Aduro Biotech, Immune Design, NexImmune, ImmuneXcite, Amplimmune, GlaxoSmithKline, Abbott, Jennerex, Pfizer, and Anza Therapeutics; receiving reimbursement for travel expenses from Bristol-Myers Squibb; holding patents regarding immunotherapy with the B7-DC gene and its products, combinatorial therapy of cancer with vaccines plus anti–B7-H1 antibodies, vaccine strategies, and the blockade of lymphocyte checkpoint molecules; and receiving patent royalties from Bristol-Myers Squibb, Amplimmune, GlaxoSmithKline, Aduro Biotech, and BioSante on behalf of his institution. Dr.Chen reports serving as a board observer of, receiving consulting fees from, and holding stock options in Amplimmune; receiving patent licensing payment and research funding from Amplimmune on behalf of Johns Hopkins University and Yale University; receiving patent licensing payment regarding B7-H1– related technologies from Bristol-Myers Squibb on behalf of the Mayo Clinic; receiving funding for research on B7-H1 immunohistochemistry from Bristol-Myers Squibb; and receiving patent licensing payment from ImmuNext on behalf of the Mayo Clinic. Dr. Sharfman reports receiving consulting fees from Genentech and Merck and lecture fees from Prometheus Laboratories. Dr. Anders reports receiving grant support and reimbursement for travel expenses from Bristol-Myers Squibb. Dr. Taube reports receiving grant support from Bristol-Myers Squibb on behalf of her institution. Ms. McMiller reports receiving compensation for sponsored laboratory research from Bristol-Myers Squibb on behalf of her institution. Dr. Korman reports being an employee of, holding stock or stock options in, and receiving reimbursement for travel expenses from Bristol-Myers Squibb. Drs. Jure-Kunkel, Agrawal, Kollia, Gupta, and Wigginton report being employees of and holding stock or stock options in Bristol-Myers Squibb. Mr. McDonald reports being an employee of, holding stock or stock options in, and receiving reimbursement for travel expenses from Bristol-Myers Squibb. Dr. Sznol reports receiving consulting fees from and owning stock options in Genesis Biopharma; receiving consulting fees from Bristol-Myers Squibb, Anaeropharma Science, Prometheus Laboratories, Nektar, and Abbott; and receiving lecture fees from the Center for Biomedical Continuing Education and Institute for Medical Education and Research, both of which are sponsored in part by Bristol-Myers Squibb.
We thank the patients who participated in this study; clinical faculty and personnel, including Ashley Bagheri, Cherylann Carr, Tianna Dauses, Robert Gray, Hans Hammers, Marina Laiko, Dung Le, Evan Lipson, Christian Meyer, Alice Pons, Pritish John, Joanne Riemer, and Theresa M. Salay, of the Johns Hopkins University School of Medicine and the Sidney Kimmel Comprehensive Cancer Center; Donald Lawrence of Massachusetts General Hospital; Toni Choueiri, Leena Gandhi, and David Jackman of Dana–Farber Cancer Institute; Daniel Cho, Natasha Coleman, Rose Marujo, and James Mier of Beth Israel Deaconess Medical Center; Miriam Akita, Marianne Davies, Emily Duffield, Christina Lakomski, Daniel Morgensztern, Isabel Oliva, Von Potter, Elin Rowen, Rebecca Sipples, and Danielle Wanik of Yale Cancer Center; Suzanne Burke, Kim Feldhaus, Elaine Granch, Nabeela Iqbal, and Prathima Koppolu of the University of Michigan; Jahleen Byers, Bryan Greene, Jill Hinson, Eric Keller, Lori Lipocky, and Vanica Pharoah of Carolina BioOncology Institute; and David Carbone, Ken Hande, William Pao, Igor Puzanov, and Serena Rucker of Vanderbilt University Medical Center; Qiankun Sun, statistician, and Mubing Li, lead statistical programmer, of Bristol-Myers Squibb; and Susan Leinbach, medical writer, of Clinical Solutions Group.
Footnotes
No other potential conflict of interest relevant to this article was reported.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
Contributor Information
Suzanne L. Topalian, Johns Hopkins University School of Medicine and the Sidney Kimmel Comprehensive Cancer Center, Baltimore
F. Stephen Hodi, Dana–Farber Cancer Institute, Boston
Julie R. Brahmer, Johns Hopkins University School of Medicine and the Sidney Kimmel Comprehensive Cancer Center, Baltimore
Scott N. Gettinger, Yale University School of Medicine and Yale Cancer Center, New Haven, CT
David C. Smith, University of Michigan, Ann Arbor
David F. McDermott, Beth Israel Deaconess Medical Center, Boston
John D. Powderly, Carolina BioOncology Institute, Huntersville, NC
Richard D. Carvajal, Memorial Sloan-Kettering Cancer Center, New York
Jeffrey A. Sosman, Vanderbilt University Medical Center, Nashville
Michael B. Atkins, Beth Israel Deaconess Medical Center, Boston
Philip D. Leming, Cincinnati Hematology-Oncology, Cincinnati
David R. Spigel, Sarah Cannon Research Institute/Tennessee Oncology, Nashville
Scott J. Antonia, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL
Leora Horn, Vanderbilt University Medical Center, Nashville
Charles G. Drake, Johns Hopkins University School of Medicine and the Sidney Kimmel Comprehensive Cancer Center, Baltimore
Drew M. Pardoll, Johns Hopkins University School of Medicine and the Sidney Kimmel Comprehensive Cancer Center, Baltimore
Lieping Chen, Yale University School of Medicine and Yale Cancer Center, New Haven, CT
William H. Sharfman, Johns Hopkins University School of Medicine and the Sidney Kimmel Comprehensive Cancer Center, Baltimore
Robert A. Anders, Johns Hopkins University School of Medicine and the Sidney Kimmel Comprehensive Cancer Center, Baltimore
Janis M. Taube, Johns Hopkins University School of Medicine and the Sidney Kimmel Comprehensive Cancer Center, Baltimore
Tracee L. McMiller, Johns Hopkins University School of Medicine and the Sidney Kimmel Comprehensive Cancer Center, Baltimore
Haiying Xu, Johns Hopkins University School of Medicine and the Sidney Kimmel Comprehensive Cancer Center, Baltimore
Alan J. Korman, Bristol-Myers Squibb, Milpitas, CA
Maria Jure-Kunkel, Bristol-Myers Squibb, Princeton, NJ
Shruti Agrawal, Bristol-Myers Squibb, Princeton, NJ
Daniel McDonald, Bristol-Myers Squibb, Princeton, NJ
Georgia D. Kollia, Bristol-Myers Squibb, Princeton, NJ
Ashok Gupta, Bristol-Myers Squibb, Princeton, NJ
Jon M. Wigginton, Bristol-Myers Squibb, Princeton, NJ
Mario Sznol, Yale University School of Medicine and Yale Cancer Center, New Haven, CT
References
- 1.Sjöblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 2.Topalian SL, Weiner GJ, Pardoll DM. Cancer immunotherapy comes of age. J Clin Oncol. 2011;29:4828–36. doi: 10.1200/JCO.2011.38.0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–9. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. 2006;90:51–81. doi: 10.1016/S0065-2776(06)90002-9. [DOI] [PubMed] [Google Scholar]
- 5.Mizoguchi H, O'Shea JJ, Longo DL, Loeffler CM, McVicar DW, Ochoa AC. Alterations in signal transduction molecules in T lymphocytes from tumor-bearing mice. Science. 1992;258:1795–8. doi: 10.1126/science.1465616. [DOI] [PubMed] [Google Scholar]
- 6.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–6. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 7.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 9.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleu-kin-10 secretion. Nat Med. 1999;5:1365–9. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 10.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. Erratum, Nat Med, 2002,8:1039. [DOI] [PubMed] [Google Scholar]
- 12.Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24:207–12. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99:12293–7. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–75. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolchok JD, Hoos A, O'Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412–20. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 16.Cancer Therapy Evaluation Program (CTEP) Common Terminology Criteria for Adverse Events (CTCAE), Version 3.0. Bethesda, MD: National Cancer Institute; Apr 2003, http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. [Google Scholar]
- 17.Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5:649–55. [PubMed] [Google Scholar]
- 18.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors: European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 19.Taube JM, Anders RA, Young GD, et al. Colocalization of inflammatory response with B7-H1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Science Transl Med. 2012;4:127ra37. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson RH, Kuntz SM, Leibovich BC, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow-up. Cancer Res. 2006;66:3381–5. doi: 10.1158/0008-5472.CAN-05-4303. [DOI] [PubMed] [Google Scholar]
- 21.Ribas A, Chmielowski B, Glaspy JA. Do we need a different set of response assessment criteria for tumor immunotherapy? Clin Cancer Res. 2009;15:7116–8. doi: 10.1158/1078-0432.CCR-09-2376. [DOI] [PubMed] [Google Scholar]
- 22.Gridelli C, Ardizzoni A, Ciardiello F, et al. Second-line treatment of advanced non-small cell lung cancer. J Thorac Oncol. 2008;3:430–40. doi: 10.1097/JTO.0b013e318168c815. [DOI] [PubMed] [Google Scholar]
- 23.Miller VA. Optimizing therapy in previously treated non-small cell lung cancer. Semin Oncol. 2006;33(1):S25–S31. doi: 10.1053/j.seminoncol.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 24.Scagliotti G, Brodowicz T, Shepherd FA, et al. Treatment-by-histology interaction analyses in three phase III trials show superiority of pemetrexed in nonsquamous non-small cell lung cancer. J Thorac Oncol. 2011;6:64–70. doi: 10.1097/JTO.0b013e3181f7c6d4. [DOI] [PubMed] [Google Scholar]
- 25.Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer. 2011;11:805–12. doi: 10.1038/nrc3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brahmer JR, Tykodi SS, Chow LQM, et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement1