Ring1b bookmarks genes in pancreatic embryonic progenitors for repression in adult β cells (original) (raw)
Polycomb complexes are important for multiple differentiation processes. Using stage-specific knockouts, van Arensbergen et al. show that during embryonic pancreatic β-cell differentiation, Ring1b (a subunit of Polycomb-repressive complex 1) acts to mark genes that are later repressed in terminally differentiated β cells. Interestingly, repression is maintained through Ring1b-independent mechanisms. This study reveals separate mechanisms that either establish or maintain gene repression in a cellular lineage.
Keywords: Ring1b, Polycomb, pancreatic β cells, cellular programming, epigenetics
Abstract
Polycomb-mediated gene repression is essential for embryonic development, yet its precise role in lineage-specific programming is poorly understood. Here we inactivated Ring1b, encoding a polycomb-repressive complex 1 subunit, in pancreatic multipotent progenitors (_Ring1b_progKO). This caused transcriptional derepression of a subset of direct Ring1b target genes in differentiated pancreatic islet cells. Unexpectedly, Ring1b inactivation in differentiated islet β cells (_Ring1b_βKO) did not cause derepression, even after multiple rounds of cell division, suggesting a role for Ring1b in the establishment but not the maintenance of repression. Consistent with this notion, derepression in _Ring1b_progKO islets occurred preferentially in genes that were targeted de novo by Ring1b during pancreas development. The results support a model in which Ring1b bookmarks its target genes during embryonic development, and these genes are maintained in a repressed state through Ring1b-independent mechanisms in terminally differentiated cells. This work provides novel insights into how epigenetic mechanisms contribute to shaping the transcriptional identity of differentiated lineages.
Throughout embryonic development, an interplay between positive and negative regulatory mechanisms promotes dynamic changes in chromatin structure that set up lineage-specific transcriptional programs. Although current knowledge supports this general model, the exact manner in which chromatin regulatory complexes contribute to differentiation programs remains to be defined.
One of the key mechanisms to create repressive chromatin states is mediated by Polycomb group (PcG) proteins (Sparmann and van Lohuizen 2006; Schwartz and Pirrotta 2007; Schuettengruber and Cavalli 2009; Vidal 2009; Margueron and Reinberg 2011). PcG proteins form two major families of complexes, named Polycomb-repressive complex 1 (PRC1) and PRC2. PRC2 comprises Eed, Suz12, and two related subunits, Ezh1 and Ezh2, that catalyze the trimethylation of histone H3K27 (H3K27me3). PRC1 complexes include Bmi1, Cbx proteins, Mel18, and the ring finger proteins Ring1a and Ring1b (Vidal 2009). Among these, Ring1b has a primary role in catalyzing another post-translational histone modification, H2A K119 ubiquitination (de Napoles et al. 2004; Wang et al. 2004). Ring1b-mediated repression is linked to H2A ubiquitination, although Ring1b also promotes chromatin compaction independently of this histone-modifying activity (Eskeland et al. 2010). Thus, many of the molecular components and mechanisms underlying PcG-mediated repression have begun to be unraveled.
Multiple lines of evidence indicate that PcG proteins are pivotal for embryonic developmental programs. Notably, several Drosophila and mouse PcG mutants have shown embryonic lethality or homeotic transformations due to derepression of Hox genes (e.g., Lewis 1978; O'Carroll et al. 2001; Voncken et al. 2003). Furthermore, PcG proteins show a strong preference to bind inactive genes that encode for developmental regulators, and PcG-dependent modifications are dynamically placed and removed at key lineage regulatory genes throughout pancreatic and neural differentiation (Boyer et al. 2006; Bracken et al. 2006; Lee et al. 2006; Mohn et al. 2008; van Arensbergen et al. 2010). The precise role of PcG proteins in developmental programs, however, is still poorly understood. In part, this is because many germline PcG mutant models exhibit early lethality (Faust et al. 1998; O'Carroll et al. 2001; Voncken et al. 2003; Pirity et al. 2005). A good example is provided by Ring1b mutations, which (unlike Ring1a deficiency) causes defective gastrulation and embryonic lethality (del Mar Lorente et al. 2000; Voncken et al. 2003). For viable genetic mutations, including _Bmi1_−/− or conditionally inactive Ezh2 mice, studies have so far largely uncovered severe self-renewal defects (e.g., Jacobs et al. 1999; Chen et al. 2009; Dhawan et al. 2009). Recent studies, however, have begun to address how PcG proteins control cellular differentiation from tissue-specific progenitors. For example, during normal differentiation of epidermal progenitors, Ezh2 levels are down-regulated, leading to the activation of genes associated with epidermis differentiation (Ezhkova et al. 2009). Consequently, inactivation of Ezh2 in basal skin progenitors has led to premature epidermal differentiation. In the ventral foregut endoderm, Ezh2 has been shown to restrict the pancreatic fate choice (Xu et al. 2011). Other experiments suggest that Ezh2 and Ring1b restrict neurogenesis to early developmental stages by repressing the proneural genes Neurogenin1 and NeuroD1 during the later astrogenic stage (Hirabayashi et al. 2009; Roman-Trufero et al. 2009). It is thus evident that PcG-dependent repression plays crucial developmental roles, although there is still a limited understanding of the detailed roles that PcG-mediated repression plays throughout different stages of lineage-specific programs.
After the specification and differentiation of cellular lineages, transcriptional states are maintained throughout multiple rounds of cell division. PcG-dependent repressive mechanisms have also been proposed to underlie long-term maintenance of cellular identity (Ringrose and Paro 2007). However, ablation of PcG genes in differentiated cells has led to proliferative defects without obvious loss of cellular identity, although genetic studies reported so far have not directly addressed effects on the transcriptional programs of differentiated lineages (e.g., Chen et al. 2009; Juan et al. 2011). Thus, further studies are required to address whether PcG proteins maintain the cellular identity of differentiated cells.
In this study, we created conditional mutations of the PRC1 subunit gene Ring1b to address the stage-specific functions of PcG-mediated repression during the embryonic differentiation of pancreatic β cells. Our results show that during embryonic differentiation, Ring1b is required to establish the transcriptional repression of target genes in the differentiated β-cell lineage, despite the fact that the maintenance of this repression in terminally differentiated β cells is independent of Ring1b. We created cell lines from mice with stage-specific mutations to demonstrate that the transcriptional phenotypes are mitotically stable and integrated expression and occupancy studies to show that they reflect a direct function of Ring1b. The results therefore reveal separate mechanisms that either establish or maintain the repression of a discrete set of genes in a cellular lineage and provide novel insights into how PcG-mediated repression contributes to shaping the transcriptional identity of pancreatic β cells.
Results
Stage-specific inactivation of Ring1b during β-cell differentiation
To study the stage-specific functions of PRC1 during pancreatic β-cell development, we crossed mice with a conditional Ring1b LoxP allele and either Pdx1-Cre or Ins-Cre transgenic lines (Fig. 1A; Herrera 2000; Gu et al. 2002; Cales et al. 2008). Ring1b is expressed in multipotent embryonic pancreatic progenitors, the mesenchyme, and the adult islet β cells (Fig. 1B,E,G,I). The Pdx1-Cre transgene efficiently deleted Ring1b in embryonic pancreatic progenitors and adult β cells (_Ring1b_progKO), while Ins-Cre caused efficient recombination in lineage-committed β cells (_Ring1b_βKO) (Fig. 1C,D,F,H,J).
Figure 1.

Stage-specific inactivation of Ring1b in β-cell development. (A) Schematic of the Cre transgenic models used for Ring1b inactivation. Pdx1-Cre inactivates Ring1b in embryonic multipotent pancreatic progenitors (_Ring1b_progKO) and Ins-Cre in lineage-committed β cells (_Ring1b_βKO). (B) Ring1b is expressed in embryonic day 11.5 (E11.5) pancreatic progenitors (n = 2) and in islets from young (14 wk; n = 4) and 1-yr-old (n = 2) mice. (C,D) Ring1b is efficiently excised in _Ring1b_progKO and _Ring1b_βKO islets (n = 5). Levels were normalized for Actb mRNA and are expressed relative to _Ring1b_f/f littermate controls. Note that β cells represent ∼60% of islet cells. Error bars indicate the SEM. (E,F) Immunofluorescence analysis of Ring1b (green), Pdx1 (red), and Topro (blue) shows enriched Ring1b expression in the multipotent progenitor trunk domain of _Ring1b_f/f E13.5 embryos (E), whereas it is absent in _Ring1b_progKO embryos (F). (G,H) Immunofluorescence analysis of Ring1b (green), insulin (red), and Topro (blue) shows enriched Ring1b expression in the pancreatic islets of 3-mo-old _Ring1b_f/f animals (G), whereas it is absent in _Ring1b_progKO islets (H). (I,J) Same as G and H, but for _Ring1b_βKO islets. The white arrows indicate “escaper” cells that have not lost Ring1b.
Early but not late Ring1b inactivation leads to impaired islet endocrine function
To assess the consequences of stage-specific ablation of Ring1b in pancreatic cells, we first studied glucose tolerance in 4-mo-old mice. _Ring1b_progKO mice displayed increased blood glucose levels during a meal test (Fig. 2A) and after intraperitoneal injection of glucose (Fig. 2B). This impairment was due to decreased blood insulin levels (Fig. 2C). In sharp contrast, _Ring1b_βKO mice showed normal glucose tolerance and insulin secretion (Fig. 2E–G). Thus, early but not late Ring1b inactivation caused decreased insulin output and glucose intolerance.
Figure 2.

Early but not late inactivation of Ring1b leads to glucose intolerance. (A,E) Post-prandial blood glucose is significantly increased in _Ring1b_progKO (n = 7) versus _Ring1b_f/f littermate controls (n = 10; *P = 0.0046), whereas no differences were observed in _Ring1b_βKO (n = 10) versus _Ring1b_f/f littermate controls (n = 10). (B,F) Impaired intraperitoneal glucose tolerance in _Ring1b_progKO (n = 7) versus _Ring1b_f/f littermates controls (n = 8) but not in _Ring1b_βKO (n = 8) versus _Ring1b_f/f littermates controls (n = 7; *P < 0.05). (C,G) Impaired insulin levels in response to an intraperitoneal glucose challenge in _Ring1b_progKO (n = 11) versus _Ring1b_f/f littermate controls (n = 9) but not in _Ring1b_βKO mice (*P = 0.01). (D,H) No significant differences in β-cell mass were observed between _Ring1b_progKO (n = 4) and _Ring1b_f/f littermate controls (n = 4; P = 0.54) or _Ring1b_βKO (n = 3) and _Ring1b_f/f littermate controls (n = 3; P = 0.50). Error bars indicate the SEM.
We next assessed whether glucose intolerance in _Ring1b_progKO mice resulted from an abnormal number of β cells. Previous studies showed that deficiency of other PcG subunits induces transcripts from the Cdkn2a locus, leading to reduced β-cell growth (Chen et al. 2009; Dhawan et al. 2009). _Ring1b_progKO and _Ring1b_βKO islets showed moderately increased Cdkn2a transcripts (Supplemental Fig. 1), yet this was not sufficient to cause significant changes in β-cell mass (Fig. 2D,H; Supplemental Fig. 2A). Thus, the inactivation of Ring1b during early pancreatic development results in a functional impairment of adult β cells, whereas Ring1b inactivation in lineage-committed β cells does not.
Early but not late Ring1b inactivation causes derepression of a subset of Ring1b targets
To investigate whether the impairment of islet endocrine function resulted from a transcriptional perturbation of Ring1b targets, we assessed transcriptional changes in _Ring1b_progKO islets. Consistent with the fact that Ring1b is a component of a transcriptional repressive complex, we observed a marked derepression phenotype in _Ring1b_progKO islets, with significantly more probes showing up-regulation (n = 398) than down-regulation (n = 179; P < 2.2 × 10−16) (Fig 3A; Supplemental Table 1).
Figure 3.
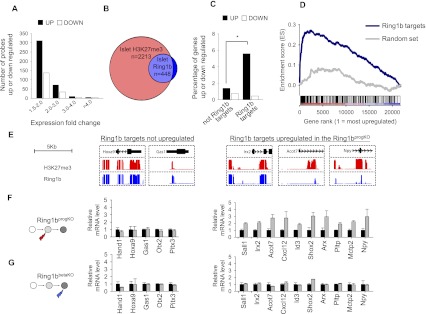
Derepression of Ring1b targets in _Ring1b_progKO islets. (A) _Ring1b_progKO islets exhibit a transcriptional derepression phenotype. GeneChip expression analysis was performed with islets from adult _Ring1b_progKO mice (n = 3) and control littermates (n = 3). Probes showing at least 1.5-fold up-regulation or down-regulation at P < 0.05 in _Ring1b_progKO islets were binned in four groups according to their fold changes. Up-regulated probes were significantly more common than down-regulated probes (P < 2.2 × 10−16). (B) Venn diagram showing the overlap of H3K27me3 targets and Ring1b targets in islets (the overlap is 407 out of 448). (C) Ring1b targets were preferentially up-regulated in _Ring1b_progKO islets. Transcript up-regulation in Ring1b_progKO islets was approximately fourfold more frequent in Ring1b-bound than nonbound genes ([*_] P = 1.2 × 10−12), whereas transcript down-regulation did not differ between the two groups. (D) GSEA showed that genes bound by Ring1b in islets were enriched among genes that showed a high expression fold change in _Ring1b_progKO islets (P < 0.001). An analogous enrichment analysis is shown for a random gene set of the same size. (E) H3K27me3 and Ring1b-binding patterns in mouse islets for selected Ring1b target genes that were either up-regulated or unperturbed in _Ring1b_progKO islets. (F,G) qRT–PCR assessment of direct Ring1b target genes in _Ring1b_progKO (F, gray bars; n = 3) and Ring1bβKO (G, gray bars; n = 3) islets. Levels were normalized for Actb mRNA and are expressed relative to _Ring1b_f/f littermate controls (black bars; n = 3). This confirmed that many direct Ring1b target genes were unaffected in both Ring1b-deficient models (left panels), whereas a subset of targets were selectively up-regulated in _Ring1b_progKO but not _Ring1b_βKO islets (right panels). Error bars indicate the SEM.
To assess whether this transcriptional response reflected a derepression of direct Ring1b targets, we used tiling arrays to profile Ring1b binding in islet cells (Supplemental Table 1). We identified 448 genes bound by Ring1b in islets and, consistent with previous studies in embryonic stem (ES) cells, found that they form a subset of the genes that are enriched in H3K27me3 in islets (Fig. 3B; Ku et al. 2008).
Of all of the Ring1b target genes, 5.6% were significantly up-regulated in _Ring1b_progKO islets, in contrast to only 1.4% of non-Ring1b target genes (P = 1.2 × 10−12) (Fig. 3C). Gene set enrichment analysis (GSEA) consistently showed that Ring1b-bound genes were enriched among up-regulated genes (P < 0.001) (Fig. 3D). Likewise, 28% of genes that were up-regulated _Ring1b_progKO islets showed H3K27me3 enrichment in wild-type islets, indicating that they were PcG targets, whereas H3K27me3 enrichment was observed in 15% of all genes (P = 6.9 × 10−11). Thus, genes bound by Ring1b—and, more generally, PcG targets—were preferentially derepressed in _Ring1b_progKO islets, although only a subset of Ring1b targets was perturbed.
We next selected 15 genes that were bound by Ring1b in islets (Fig. 3E), of which 10 showed up-regulation and five showed no change in _Ring1b_progKO islets in the gene chip analysis, and confirmed these findings in all cases by quantitative RT–PCR (qRT–PCR) (Fig. 3F). All 15 Ring1b target genes were then examined in islets from adult _Ring1b_βKO mice. In keeping with their normal glucose tolerance, the expression of Ring1b target genes was unperturbed (Fig. 3G). Thus, Ring1b deletion in pancreatic progenitors led to derepression of a subset of Ring1b target genes in differentiated islet cells, whereas no changes were encountered when Ring1b was deleted in lineage-committed β cells.
Early Ring1b inactivation causes islet misexpression of neural and disallowed genes
To further understand the role of Ring1b-dependent repression in pancreatic endocrine programming, we focused on the subset of genes that were derepressed in _Ring1b_progKO islets. This gene set was enriched in neural developmental regulators (P = 5.4 × 10−7) (Fig. 4A) and in genes that showed a tissue-specific gene expression pattern in the brain and cerebellum (P < 0.05) (Fig. 4B; Supplemental Fig. 3A). Thus, neural-enriched genes that are normally silent in islets, such as Rab3c, Mkx, and Kcnj3, were up-regulated in _Ring1b_progKO islets. Of note, Npy is normally detected in scarce control β cells but was expressed in a sixfold higher number of _Ring1b_progKO β cells (Supplemental Fig. 3B–D). Thus, Ring1b deletion in pancreatic progenitors causes abnormal islet expression of neural-enriched and neuroendocrine genes.
Figure 4.

Misexpression of neural and disallowed genes in _Ring1b_progKO islets. (A) The top categories of gene ontology analysis of up-regulated genes in _Ring1b_progKO islets. P < 0.05 after Benjamini correction for all uncorrected _P_-values shown in the table. (B) Genes that were up-regulated in _Ring1b_progKO islets were frequently neural-specific ([*] P < 0.05 for all comparisons of cortex or cerebellum with all other tissues). Tissue-specific genes were defined as those expressed in that tissue and no more than two other tissues. (C) GSEA showed that genes that have been classified as disallowed in β cells were enriched among genes up-regulated in _Ring1b_progKO islets (P < 0.001). An analogous enrichment analysis is shown for a random gene set of the same size (P = 0.661).
We recently showed that “β-cell disallowed genes”—a term that refers to genes that are selectively inactive in β cells and have been proposed to be deleterious for β-cell function (Pullen et al. 2010; Thorrez et al. 2011)—are selectively targeted by PcG-mediated repression during late differentiation (van Arensbergen et al. 2010). The current analysis showed that this set of genes was also enriched among genes that are up-regulated in _Ring1b_progKO islets (P < 0.001) (Fig. 4C). Thus, during embryonic development, Ring1b establishes repressed states that refine the neuroendocrine phenotype of islet cells and prevent inappropriate gene activity in differentiated cells.
Early Ring1b inactivation prevents repression of de novo Ring1b targets
We next examined why only a subset of Ring1b targets was derepressed in _Ring1b_progKO islets. We reasoned that if Ring1b is solely required to establish repressed chromatin rather than maintain it, genes that were already targeted by Ring1b in pluripotent cells prior to inactivation of Ring1b in pancreatic progenitors should not be affected in _Ring1b_progKO islets. In contrast, genes that are targeted by Ring1b de novo during pancreas development should be perturbed if Ring1b is inactivated at this early stage.
To identify Ring1b targets that are established de novo during embryonic development, we profiled Ring1b in ES cells. We found that of all 448 islet Ring1b targets, 90 (20.1%) were not bound in ES cells (Supplemental Table 1). These were classified as de novo Ring1b targets (Fig. 5A). In keeping with our predictions, de novo islet Ring1b targets were more frequently up-regulated in _Ring1b_progKO islets than targets that were already bound by Ring1b in ES cells (12.2 vs. 3.9%, respectively; P = 0.005) (Fig. 5B). Likewise, GSEA showed that de novo Ring1b targets were preferentially enriched among up-regulated genes (Fig. 5C). Thus, loss of Ring1b at the pancreatic progenitor stage preferentially affects genes that are targeted de novo by PcG proteins during embryogenesis, rather than those that were already targeted during earlier stages. Consistent with this idea, islet Ring1b targets that showed up-regulation in _Ring1b_progKO islets acquired H3K27me3 de novo throughout normal pancreas development, whereas islet Ring1b targets that were unaffected in _Ring1b_progKO mice already displayed H3K27me3 enrichment at the pancreatic progenitor stage (Fig. 5D,E; Supplemental Fig. 4). Thus, Ring1b inactivation in pancreatic progenitors preferentially alters the expression of genes that are targeted de novo by Ring1b during late pancreatic development while leaving genes that are targeted by Ring1b prior to Ring1b deletion in pancreatic progenitors largely unperturbed. These findings further support a role for Ring1b in establishing rather than maintaining β-cell-repressive programs.
Figure 5.
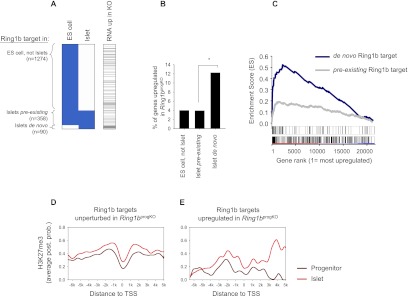
Early loss of Ring1b leads to misexpression of de novo Ring1b targets. (A) Cluster representation of Ring1b-bound genes (blue) based on binding in ES cells and islets. The right column shows genes up-regulated in _Ring1b_progKO islets in black. (B) Percentage of up-regulated genes among the three groups of Ring1b targets. De novo Ring1b targets are more frequently up-regulated than pre-existing Ring1b targets ([*] P = 0.005). (C) GSEA of de novo and pre-existing islet Ring1b targets among genes ranked according to their fold change in expression in _Ring1b_progKO islets. Islet de novo Ring1b targets displayed strong enrichment among up-regulated genes (P < 0.001, maximum enrichment score = 0.52), while weaker enrichment was observed for pre-existing Ring1b targets (P = 0.0011, maximum enrichment score = 0.19). The two bars below the graph depict the rank position of de novo (top) and pre-existing (bottom) Ring1b targets. (D,E) H3K27me3 enrichment in wild-type pancreatic progenitors (dark red) and wild-type islets (light red) for Ring1b targets that were unperturbed (n = 418) (D) or up-regulated (n = 28) (E) in _Ring1b_progKO islets. Enrichment is quantified as average posterior probability (post. prob.). Plots that show H3K27me3 as a fold enrichment value show the same pattern (Supplemental Fig. 4).
Silencing of Ring1b targets is mitotically inherited in _Ring1b_βKO β cells
The observation that Ring1b targets are derepressed after gene ablation in progenitors but not after gene ablation in differentiated islets could theoretically reflect a defect in mitotic transmission of repressed states, given that more rounds of cell divisions occur from gene deletion to analysis in the early ablation model. To address this possibility, we generated immortalized β-cell lines from _Ring1b_progKO and _Ring1b_βKO mice (Fig. 6A) using RIP-TAg transgenic mice expressing the SV40 large T-antigen in β cells (Hanahan 1985). β-Cell lines were maintained in culture for >20 estimated cell divisions. We analyzed five cell lines from separate animals for each genotype and tested six genes that were up-regulated in the _Ring1b_progKO islets. In keeping with our observations in native islets, five out of six of these genes were consistently up-regulated in the _Ring1b_progKO β-cell lines (Fig. 6B–E; Supplemental Fig. 5). Remarkably, all of these genes remained repressed in _Ring1b_βKO β-cell lines, despite multiple rounds of cell divisions. Thus, _Ring1b_-deficient β cells are capable of mitotically propagating repressed states that were established in a Ring1b-dependent manner during pancreatic development. This finding further supports that Ring1b is not essential to maintain transcriptional repression of its target genes in differentiated β cells.
Figure 6.
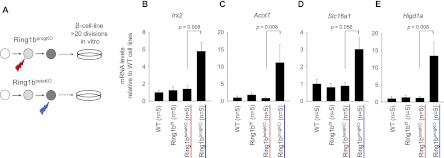
Mitotic inheritance of repression patterns. (A) Schematic representing the generation of β-cell lines. We crossed _Ring1b_progKO and _Ring1b_βKO mice with RIP-TAg transgenics to generate insulinomas. _Ring1b_-deficient β-cell lines were established from individual adult animals per genotype. (B–E) qRT–PCR analysis of Irx2, Acot7, Slc16a1, and Higd1a in cell lines established from wild-type (n = 5), _Ring1b_f/f littermate control (n = 5), _Ring1b_βKO (n = 5), and _Ring1b_progKO (n = 5) mice. _Ring1b_progKO cell lines displayed derepression of Ring1b targets, whereas repression was maintained in the _Ring1b_βKO cell lines despite >20 rounds of cell division. Error bars indicate the SEM.
Ring1b is required in pancreatic progenitors for target gene DNA methylation in differentiated islet cells
We next assessed molecular mechanisms that could underlie the maintenance of repression in the _Ring1b_βKO β-cell lines in genes that showed up-regulation in the _Ring1b_progKO β-cell lines. We first confirmed that Ring1b was bound to these genes in the cell lines (Fig. 7A). Interestingly, the related protein Ring1a was not strongly enriched at these same genes in control β cells, and, importantly, neither Ring1a nor Bmi1 (another PRC1 subunit) showed increased binding in mutant islets (Fig. 7B; Supplemental Fig. 6A). This result argued that gene silencing in _Ring1b_βKO β cells was unlikely to be maintained by PcG-mediated repressive mechanisms.
Figure 7.

Ring1b is required in pancreatic progenitors for target gene DNA methylation in differentiated islet cells. (A,B) Chromatin immunoprecipitation (ChIP) analysis of Ring1b and Ring1a occupancy at direct Ring1b targets in cell lines established from _Ring1b_f/f littermate controls (black bar; n = 3) and _Ring1b_βKO (light-gray bar; n = 3) and _Ring1b_progKO (dark-gray bar; n = 3) mice. The dotted line indicates enrichment = 1. (C) As in A and B, but here, MeDIP was performed (n = 4). Enrichments are shown relative to controls after normalization for Tbp. Error bars indicate the SEM.
We next hypothesized that DNA methylation was responsible for maintaining repression in control and _Ring1b_βKO β cells. Accordingly, methylated DNA immunoprecipitation (MeDIP) showed that Ring1b targets had a decreased level of DNA methylation in _Ring1b_progKO cells compared with control and _Ring1b_βKO β cells (Fig. 7C). Furthermore, H3K9me3, a histone mark in certain cases associated with DNA methylation (Hashimoto et al. 2010), displayed a similar trend in _Ring1b_progKO β cells (Supplemental Fig. 6B). These findings thus suggest that Ring1b-independent repressive mechanisms—namely, DNA methylation and H3K9me3—are associated with the maintenance of a repressed state that is initiated in a Ring1b-dependent manner in pancreatic progenitors.
Discussion
Several distinct mechanisms are known to promote repressed chromatin states. The specific role that each mechanism plays in controlling cell fate decisions, differentiation, and long-term phenotypic maintenance, however, remains elusive. In the present study, we showed that Ring1b acts in pancreatic embryonic progenitors to specify the transcriptional program of differentiated β cells. We demonstrated that Ring1b function in pancreatic progenitors is essential to establish transcriptional repression and DNA methylation of selected target genes in differentiated β cells, whereas after cells have differentiated, Ring1b is no longer required to maintain the repression of these same target genes. The data therefore indicate that during lineage-specific differentiation, Ring1b bookmarks genes that are later locked in a stably repressed state through Ring1b-independent mechanisms.
Ring1b is used to refine the transcriptional program of differentiated β cells
Earlier studies showed that the differentiation of β cells entails the selective removal of PcG-mediated repression from a core set of neural regulatory genes (van Arensbergen et al. 2010). This in turn enables the activation of a transcriptional program in β cells that is remarkably similar to that of ectodermally derived neural cell types (van Arensbergen et al. 2010). The current findings suggest that during the differentiation of β cells, PcG-mediated repression is also employed to suppress the activation of selected neural genes. Plausibly, this fulfills a need to repress the indiscriminate activation of neural genes in cells that express many neural transcriptional activators. Existing data thus suggest that during pancreas development, PcG-dependent repression is selectively removed from neuroendocrine regulatory genes that promote islet cell differentiation, although, concomitantly, PcG-mediated repression is used to repress neural-enriched genes that do not form part of the pancreatic endocrine program.
Epigenomic profiling in pancreatic islet cells has also shown that PcG-mediated repression targets disallowed genes in β cells, defined as genes that are selectively absent and potentially deleterious for β cells (Thiel and Schuit 2008; Pullen et al. 2010; van Arensbergen et al. 2010). The present study shows that Ring1b not only binds to such genes, but is also required in pancreatic progenitors to establish their repression in β cells.
The transcriptional changes observed in _Ring1b_progKO islet cells led to abnormal glucose intolerance due to defective β-cell insulin secretory function. One of the disallowed genes that we identified as up-regulated in Ring1b-deficient islets was Acot7, encoding a type II acyl-CoA thiosterase. Acot7 displays a preference for medium-chain-length acyl-CoA esters (Kirkby et al. 2010), which are implicated in the gating of ATP-sensitive K+ channels and the control of insulin granule exocytosis (Corkey et al. 2000). The absence of Acot7 from normal β cells may thus allow acyl-CoA to reach high levels, modulating both glycolytic flux and late events in insulin granule release. Consistent with this prediction, forced overexpression of Acot7 in β-cell lines leads to impaired glucose and KCl-stimulated insulin secretion (GA Rutter and TJ Pullen, unpubl.). This enzyme is thus a plausible candidate contributor to the defective secretion in _Ring1b_progKO mice. Another gene that is up-regulated in _Ring1b_-deficient islets is Npy, which is known to suppress cAMP levels and inhibit insulin secretion and can therefore also contribute to the in vivo phenotype of _Ring1b_progKO mice (Wang et al. 1994; Myrsen-Axcrona et al. 1997). In addition to Acot7 and Npy, we do not exclude roles of other up-regulated genes that could contribute to abnormal β-cell function due to a collective transcriptional defect of differentiated β cells.
A developmental bookmarking function of Ring1b
PcG complexes control numerous developmental functions, including anteroposterior patterning, gastrulation, X inactivation, stem cell differentiation, and genomic imprinting (Sparmann and van Lohuizen 2006; Margueron and Reinberg 2011). Furthermore, PcG complexes target developmental regulatory genes, consistent with a pivotal role in development (Boyer et al. 2006; Bracken et al. 2006; Lee et al. 2006). However, the precise spatiotemporal roles of PcG proteins in lineage-specific differentiation programs are only beginning to be understood.
One limitation to understanding PcG-dependent programming has been that its deficiency often leads to premature senescence or cell death, resulting in the abrogation of the lineage of interest. Recent studies have nevertheless revealed how the PcG subunit Ezh2 controls the differentiation of lineage-specific precursors. Thus, in skin progenitors, Ezh2 deficiency leads to the activation of genes that promote differentiation, whereas the inactivation of Ezh2 in skeletal muscle or heart progenitors causes altered lineage differentiation (Ezhkova et al. 2009; Juan et al. 2011; Delgado-Olguin et al. 2012). Furthermore, Ezh2 deficiency causes altered cell fate decisions of foregut endoderm multipotent progenitors (Xu et al. 2011). These results have highlighted a crucial role for Ezh2 in establishing the transcriptional fidelity of organ-specific progenitors and their offspring.
In the present study, we exploited the fact that Ring1b deficiency does not disrupt pancreatic organogenesis or cell growth to understand stage-specific functions of PcG-mediated repression during lineage programming. Our results uncouple the mechanisms that establish and maintain the repression of a discrete set of genes in a cellular lineage. Ring1b was essential to set up repression when it first associated with specific loci, whereas repression of those loci thereafter became Ring1b-independent despite maintained Ring1b occupancy. This transient function of Ring1b in pancreatic progenitors was linked to the acquisition of DNA methylation at target loci in terminally differentiated cells. Based on these findings, we propose a model in which Ring1b marks genes for repression as lineages are formed during pancreas organogenesis, and once the terminally differentiated β-cell lineage is established, its transcriptional state is maintained through alternate mechanisms.
Ring1b is not required to maintain the differentiated β-cell phenotype
Ring1b was dispensable to maintain the transcriptional program of differentiated β cells. Interestingly, Ezh2 deficiency in differentiated β cells only causes a cell growth phenotype that appears completely rescued by the inactivation of the cell cycle regulator Cdkn2a (Chen et al. 2009), consistent with other studies showing a critical role of PcG-dependent repression in controlling the growth of differentiated cells (e.g., Jacobs et al. 1999; Bracken et al. 2007). On the other hand, existing studies have shown diverse PcG-dependent functions during development to either control cell fate choices or the timing of differentiation or, as shown in the present study, refine the transcriptional program of differentiated cellular offspring. To our knowledge, however, no study has demonstrated that the inactivation of a PcG subunit in mature cells results in a global modification of its transcriptional program. This contrasts with observations that removal of DNA methylation in differentiated β cells causes transcriptional perturbations that cause cellular transdifferentiation events, suggesting that DNA methylation is a dominant mechanism for maintenance of cell identity in differentiated β cells (Dhawan et al. 2011). Taken together, existing data support a prominent role for PcG-mediated repression during developmental programming of cellular identities, whereas direct evidence for PcG-dependent maintenance of adult terminally differentiated phenotypes is still lacking.
More generally, genetic experiments have provided insight into the complexity of epigenetic regulation of development. They illustrate how different regulatory subunits play highly stage-dependent roles in lineage differentiation. A detailed understanding of the specialized functions of epigenetic regulators, together with ongoing efforts to generate a plethora of compounds that target this class of proteins, should provide opportunities to manipulate differentiation programs for disease modeling and therapeutics.
Materials and methods
Mouse models
Mice with Ring1b LoxP alleles as well as RipTAg, Ins-Cre, and Pdx1-Cre transgenic lines have been described (Hager and Hanahan 1999; Herrera 2000; Gu et al. 2002; Cales et al. 2008). All experiments were approved by the Institutional Animal Care Committee of the University of Barcelona.
RNA analysis
Mouse pancreatic islets were isolated from 12- to 14-wk-old male mice as described previously (Luco et al. 2008). Total RNA was extracted from pancreatic islet preparations of three individual male animals for each genotype using Trizol (Invitrogen). RNA integrity was verified with a 2100 Bioanalyzer (Agilent), and labeled cRNA was hybridized to Affymetrix Mouse Genome 430 2.0 arrays. Data normalization was performed as described (van Arensbergen et al. 2010). Expression data on other mouse tissues were described previously (van Arensbergen et al. 2010).
Chromatin immunoprecipitation (ChIP)
ChIPs were performed essentially as described (van Arensbergen et al. 2010). In short, pancreatic islets or mouse ES cells (CGR8) (Skoudy et al. 2004) were fixed in 1% formaldehyde for 10 min, after which nuclei were purified and sonicated using a Bioruptor (Diagenode) to a length of 200–1000 base pairs (bp). Samples were precleared with protein A+G-Sepharose (1:1) and immunoprecipitated with rabbit anti-Ring1b (Garcia et al. 1999), rabbit anti-Ring1a (Schoorlemmer et al. 1997), rabbit anti-H3K9me3 (Abcam, 8898), rabbit anti-H3K27me3 (Upstate Biotechnology, 07-449), and mouse anti-Bmi1 (Millipore, 05-637) overnight at 4°C. Immune complexes were collected by adsorption to protein A+G-Sepharose for 2 h at 4°C. Beads were washed, and immunocomplexes eluted prior to DNA purification with Qiaquick columns (Qiagen).
For tiling array experiments, ChIP and input DNA were amplified as described previously using the Sigma GenomePlex WGA2 kit while adding dUTPs to a final concentration of 0.4 mM during the amplification reaction to enable subsequent fragmentation (van Arensbergen et al. 2010). We fragmented 6–7.5 μg of DNA, labeled it using the Affymetrix GeneChip WT Double-Stranded DNA Terminal Labeling kit, and hybridized it to GeneChip Mouse Promoter 1.0R arrays. Primary processing of data was performed as described (van Arensbergen et al. 2010). H3K27me3 GeneChip data sets from ES cells, purified pancreatic embryonic progenitors, and islets were described previously (van Arensbergen et al. 2010).
MeDIP
Immunoprecipitation of methylated DNA was performed as described previously with minor modifications (Weber et al. 2005). In short, we isolated the DNA from ∼3 million cells using a Proteinase K digest followed by phenol-chloroform extraction. Four micrograms of DNA was sonicated in 500 μL of TE buffer using a Bioruptor (Diagenode). DNA was then denatured in boiling water for 10 min and cooled on ice. The DNA was then diluted in 500 μL of 2× precipitation buffer, and 2 μL of mouse 5-methylcytidine antibody (Eurogentec, BI-MECY-0100) was added. From this step on, samples were treated as in the normal ChIP procedure described above.
Statistical and integrated data analysis
Significant enrichment in GeneChip ChIP experiments relative to input DNA was determined using Cisgenome (Ji et al. 2008). We applied a hidden Markov model as described (Ji and Wong 2005) and used a posterior probability cutoff of 0.5 in at least five continuous probes. Using identical criteria, <0.05% of sites detected with Ring1b antibodies were observed in IgG control ChIP experiments in pancreatic acinar tissue. Enriched intervals were mapped to RefSeq genes when located within 2000 bp of transcriptional start sites.
Differential RNA expression was assessed using the ANOVA statistical analysis of the Partek software package. Genes were considered differentially expressed with a fold difference of >1.5 and a _P_-value <0.05. The presence and absence values for expression in other mouse tissues used for comparisons were determined as described previously (van Arensbergen et al. 2010)
The Database for Annotation, Visualization, and Integrated Discovery (DAVID) functional annotation tool was used to study the overrepresentation of annotation terms using all RefSeq genes as the background (Huang et al. 2009). Nonredundant gene ontology categories from the top “molecular function” and “biological process” terms were selected.
GSEA was performed on preranked gene expression data sets using the default weighted enrichment (Subramanian et al. 2005). Significance of the enrichment was assessed using 1000 permutations.
Differences in glucose, insulin, β-cell mass, and gene-specific expression in isolated islets were examined with Student's two-sided _t_-test. In the β-cell line studies, a Wilcoxon test was used to account for the nonnormal distribution of the data. When a single _P_-value is given for several comparisons, it represents the _P_-value for the least significant of the comparisons.
Differences in frequency distributions were assessed in R using Pearson's χ2 test with Yates' continuity correction, except for results in Figure 4B, where Fisher's exact test was used because several entries in the table were smaller than five.
qPCR analysis
qPCR of reverse-transcribed RNA, ChIP, or MeDIP samples was performed on a 7300 Real-Time PCR system (Applied Biosystems) using the Power SYBR Green reagent (Applied Biosystems). Quantities were determined using the 2−ΔΔCt method. A full list of the primers used is provided in Supplemental Table 2.
Immunofluorescence
Embryos were collected at indicated times and processed for immunofluorescence analysis of paraffin-embedded pancreas as previously described (Maestro et al. 2003). The primary antibodies were guinea pig anti-insulin (1:5000; C. Van Schravendijk), rabbit anti-glucagon (1:200; Dako), rabbit anti-NPY (1:1000; Sigma, N9528), goat anti-PDX1 (1:1000; Abcam, ab47383), and rabbit anti-Ring1b (1:1000) (Garcia et al. 1999).
Physiological tests
For meal tests, male age-matched animals were fasted for 16 h overnight, and blood glucose was measured before and 1 h after refeeding. For glucose tolerance tests, fasted animals were injected intraperitoneally with 2 mg of glucose per kilogram of body weight, and glucose levels were tested from tail blood at 0, 15, 30, 75, and 120 min after injection. Glucose was measured using the Glucocard G+ meter (Menarini Diagnostics). Insulin ELISA was performed using the Insulin ELISA kit of Mercodia (catalog no. 10-1247-01) and were read using a Synergy HT Multimode microplate reader (BioTek). Control littermates were _Ring1b_f/f littermates without the Cre transgene.
β-Cell mass analysis
The pancreases of 6-mo-old mice were dissected, weighed, folded to reduce their length, fixed, and embedded in OCT. Cryosections (6 μm) were obtained at 30-μm intervals throughout the organs. Approximately 60–70 cryosections were obtained from each pancreas. Immunofluorescence for DAPI and insulin was performed at each fifth slice. Images were taken by automated capturing and reconstruction of ∼50 frames (Leica DMI 600B). Insulin-positive area and total tissue area were determined with in-house developed ImageJ macros and are available on request. In short, after manual curation of the merged RGB images, total tissue surface was determined by binarizing a diffused image for DAPI. β-Cell surface was determined by binarizing the insulin signal based on its relative intensities compared with the other two channels. These binarized images were then again manually curated, after which the β-cell mass was obtained by multiplying the ratio of the β-cell surface to total surface with the pancreas weight.
Ring1b-deficient β-cell lines
We performed crosses to generate mice that carried a RIP-TAg transgene alone or were, in addition, homozygous for the Ring1bLoxP allele and carried either Ins-Cre, Pdx1-Cre, or no Cre transgene. We dissected β-cell tumors from 10- to 14-wk-old mice when they showed a basal blood glucose level <25 mg/dL. Tumors were rinsed in PBS and then ruptured to release the inner cells. These cells were transferred to 20 separate 96-well plates, each containing ∼10,000 cells. Cells were monitored for proliferation and emergence of fibroblasts, and based on this, one cell line per individual animal was used for further analysis. Cells were cultured as normal Min6 β-cell lines in DMEM supplemented with 10% FBS, 70 uM 2-mercaptoethanol, 2 mM L-glutamine, and penicillin–streptomycin. We selected five cell lines at passages 5–8, obtained from five different mice from each genotype, and, in all cases, confirmed genotypes.
Data access
Microarray data for RNA expression and ChIP experiments are publicly available through ArrayExpress under accession numbers E-MTAB-1404 and E-MTAB-1402, respectively.
Supplementary Material
Supplemental Material
Acknowledgments
We thank Finn Cilius Nielsen, Rehannah Borup, and Susanne Smed (RH Microarray Center, Rigshospitalet, Copenhagen) for array hybridizations; Anouchka L. Skoudy for ES cells; Vanessa Grau for mouse colony maintenance; Carme Sanahuja for assistance; and Mark Kalisz for insightful comments. Funding is from Ministerio de Ciencia e Innovación (SAF2011-27086 to J.F., and BFU2010-18146 to M.V.), Fundación Ramón Areces (to M.V.), and the Biology of Liver and Pancreatic Development and Disease Marie Curie Training Program (to M.C. and J.F.).
Footnotes
Supplemental material is available for this article.
References
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441: 349–353 [DOI] [PubMed] [Google Scholar]
- Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K 2006. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev 20: 1123–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, Theilgaard-Monch K, Minucci S, Porse BT, Marine JC, et al. 2007. The Polycomb group proteins bind throughout the INK4A–ARF locus and are disassociated in senescent cells. Genes Dev 21: 525–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cales C, Roman-Trufero M, Pavon L, Serrano I, Melgar T, Endoh M, Perez C, Koseki H, Vidal M 2008. Inactivation of the polycomb group protein Ring1B unveils an antiproliferative role in hematopoietic cell expansion and cooperation with tumorigenesis associated with Ink4a deletion. Mol Cell Biol 28: 1018–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Gu X, Su IH, Bottino R, Contreras JL, Tarakhovsky A, Kim SK 2009. Polycomb protein Ezh2 regulates pancreatic β-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev 23: 975–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corkey BE, Deeney JT, Yaney GC, Tornheim K, Prentki M 2000. The role of long-chain fatty acyl-CoA esters in β-cell signal transduction. J Nutr 130: 299S–304S [DOI] [PubMed] [Google Scholar]
- Delgado-Olguin P, Huang Y, Li X, Christodoulou D, Seidman CE, Seidman JG, Tarakhovsky A, Bruneau BG 2012. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat Genet 44: 343–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Mar Lorente M, Marcos-Gutierrez C, Perez C, Schoorlemmer J, Ramirez A, Magin T, Vidal M 2000. Loss- and gain-of-function mutations show a polycomb group function for Ring1A in mice. Development 127: 5093–5100 [DOI] [PubMed] [Google Scholar]
- de Napoles M, Mermoud JE, Wakao R, Tang YA, Endoh M, Appanah R, Nesterova TB, Silva J, Otte AP, Vidal M, et al. 2004. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev Cell 7: 663–676 [DOI] [PubMed] [Google Scholar]
- Dhawan S, Tschen SI, Bhushan A 2009. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic β-cell proliferation. Genes Dev 23: 906–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan S, Georgia S, Tschen SI, Fan G, Bhushan A 2011. Pancreatic β cell identity is maintained by DNA methylation-mediated repression of Arx. Dev Cell 20: 419–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskeland R, Leeb M, Grimes GR, Kress C, Boyle S, Sproul D, Gilbert N, Fan Y, Skoultchi AI, Wutz A, et al. 2010. Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol Cell 38: 452–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su IH, Hannon G, Tarakhovsky A, Fuchs E 2009. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 136: 1122–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust C, Lawson KA, Schork NJ, Thiel B, Magnuson T 1998. The Polycomb-group gene eed is required for normal morphogenetic movements during gastrulation in the mouse embryo. Development 125: 4495–4506 [DOI] [PubMed] [Google Scholar]
- Garcia E, Marcos-Gutierrez C, del Mar LM, Moreno JC, Vidal M 1999. RYBP, a new repressor protein that interacts with components of the mammalian Polycomb complex, and with the transcription factor YY1. EMBO J 18: 3404–3418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G, Dubauskaite J, Melton DA 2002. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 129: 2447–2457 [DOI] [PubMed] [Google Scholar]
- Hager JH, Hanahan D 1999. Tumor cells utilize multiple pathways to down-modulate apoptosis. Lessons from a mouse model of islet cell carcinogenesis. Ann N Y Acad Sci 887: 150–163 [DOI] [PubMed] [Google Scholar]
- Hanahan D 1985. Heritable formation of pancreatic β-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 315: 115–122 [DOI] [PubMed] [Google Scholar]
- Hashimoto H, Vertino PM, Cheng X 2010. Molecular coupling of DNA methylation and histone methylation. Epigenomics 2: 657–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera PL 2000. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 127: 2317–2322 [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Suzki N, Tsuboi M, Endo TA, Toyoda T, Shinga J, Koseki H, Vidal M, Gotoh Y 2009. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 63: 600–613 [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M 1999. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397: 164–168 [DOI] [PubMed] [Google Scholar]
- Ji H, Wong WH 2005. TileMap: Create chromosomal map of tiling array hybridizations. Bioinformatics 21: 3629–3636 [DOI] [PubMed] [Google Scholar]
- Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH 2008. An integrated software system for analyzing ChIP–chip and ChIP-seq data. Nat Biotechnol 26: 1293–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan AH, Derfoul A, Feng X, Ryall JG, Dell'Orso S, Pasut A, Zare H, Simone JM, Rudnicki MA, Sartorelli V 2011. Polycomb EZH2 controls self-renewal and safeguards the transcriptional identity of skeletal muscle stem cells. Genes Dev 25: 789–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby B, Roman N, Kobe B, Kellie S, Forwood JK 2010. Functional and structural properties of mammalian acyl-coenzyme A thioesterases. Prog Lipid Res 49: 366–377 [DOI] [PubMed] [Google Scholar]
- Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, et al. 2008. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 4: e1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, et al. 2006. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125: 301–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis EB 1978. A gene complex controlling segmentation in Drosophila. Nature 276: 565–570 [DOI] [PubMed] [Google Scholar]
- Luco RF, Maestro MA, Sadoni N, Zink D, Ferrer J 2008. Targeted deficiency of the transcriptional activator Hnf1α alters subnuclear positioning of its genomic targets. PLoS Genet 4: e1000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestro MA, Boj SF, Luco RF, Pierreux CE, Cabedo J, Servitja JM, German MS, Rousseau GG, Lemaigre FP, Ferrer J 2003. Hnf6 and Tcf2 (MODY5) are linked in a gene network operating in a precursor cell domain of the embryonic pancreas. Hum Mol Genet 12: 3307–3314 [DOI] [PubMed] [Google Scholar]
- Margueron R, Reinberg D 2011. The Polycomb complex PRC2 and its mark in life. Nature 469: 343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, Bibel M, Schubeler D 2008. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell 30: 755–766 [DOI] [PubMed] [Google Scholar]
- Myrsen-Axcrona U, Karlsson S, Sundler F, Ahren B 1997. Dexamethasone induces neuropeptide Y (NPY) expression and impairs insulin release in the insulin-producing cell line RINm5F. Release of NPY and insulin through different pathways. J Biol Chem 272: 10790–10796 [DOI] [PubMed] [Google Scholar]
- O'Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T 2001. The polycomb-group gene Ezh2 is required for early mouse development. Mol Cell Biol 21: 4330–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirity MK, Locker J, Schreiber-Agus N 2005. Rybp/DEDAF is required for early postimplantation and for central nervous system development. Mol Cell Biol 25: 7193–7202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen TJ, Khan AM, Barton G, Butcher SA, Sun G, Rutter GA 2010. Identification of genes selectively disallowed in the pancreatic islet. Islets 2: 89–95 [DOI] [PubMed] [Google Scholar]
- Ringrose L, Paro R 2007. Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development 134: 223–232 [DOI] [PubMed] [Google Scholar]
- Roman-Trufero M, Mendez-Gomez HR, Perez C, Hijikata A, Fujimura Y, Endo T, Koseki H, Vicario-Abejon C, Vidal M 2009. Maintenance of undifferentiated state and self-renewal of embryonic neural stem cells by Polycomb protein Ring1B. Stem Cells 27: 1559–1570 [DOI] [PubMed] [Google Scholar]
- Schoorlemmer J, Marcos-Gutierrez C, Were F, Martinez R, Garcia E, Satijn DP, Otte AP, Vidal M 1997. Ring1A is a transcriptional repressor that interacts with the Polycomb-M33 protein and is expressed at rhombomere boundaries in the mouse hindbrain. EMBO J 16: 5930–5942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B, Cavalli G 2009. Recruitment of polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development 136: 3531–3542 [DOI] [PubMed] [Google Scholar]
- Schwartz YB, Pirrotta V 2007. Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet 8: 9–22 [DOI] [PubMed] [Google Scholar]
- Skoudy A, Rovira M, Savatier P, Martin F, Leon-Quinto T, Soria B, Real FX 2004. Transforming growth factor (TGF)β, fibroblast growth factor (FGF) and retinoid signalling pathways promote pancreatic exocrine gene expression in mouse embryonic stem cells. Biochem J 379: 749–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparmann A, van Lohuizen M 2006. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer 6: 846–856 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel G, Schuit F 2008. No REST for healthy β cells. Diabetologia 51: 1343–1346 [DOI] [PubMed] [Google Scholar]
- Thorrez L, Laudadio I, Van Deun K, Quintens R, Hendrickx N, Granvik M, Lemaire K, Schraenen A, Van Lommel L, Lehnert S, et al. 2011. Tissue-specific disallowance of housekeeping genes: The other face of cell differentiation. Genome Res 21: 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Arensbergen J, Garcia-Hurtado J, Moran I, Maestro MA, Xu X, Van de Casteele M, Skoudy AL, Palassini M, Heimberg H, Ferrer J 2010. Derepression of Polycomb targets during pancreatic organogenesis allows insulin-producing β-cells to adopt a neural gene activity program. Genome Res 20: 722–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M 2009. Role of polycomb proteins Ring1A and Ring1B in the epigenetic regulation of gene expression. Int J Dev Biol 53: 355–370 [DOI] [PubMed] [Google Scholar]
- Voncken JW, Roelen BA, Roefs M, de Vries S, Verhoeven E, Marino S, Deschamps J, van Lohuizen M 2003. Rnf2 (Ring1b) deficiency causes gastrulation arrest and cell cycle inhibition. Proc Natl Acad Sci 100: 2468–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZL, Bennet WM, Wang RM, Ghatei MA, Bloom SR 1994. Evidence of a paracrine role of neuropeptide-Y in the regulation of insulin release from pancreatic islets of normal and dexamethasone-treated rats. Endocrinology 135: 200–206 [DOI] [PubMed] [Google Scholar]
- Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y 2004. Role of histone H2A ubiquitination in Polycomb silencing. Nature 431: 873–878 [DOI] [PubMed] [Google Scholar]
- Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schubeler D 2005. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 37: 853–862 [DOI] [PubMed] [Google Scholar]
- Xu CR, Cole PA, Meyers DJ, Kormish J, Dent S, Zaret KS 2011. Chromatin ‘prepattern' and histone modifiers in a fate choice for liver and pancreas. Science 332: 963–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material