De novo mutations in histone modifying genes in congenital heart disease (original) (raw)
. Author manuscript; available in PMC: 2013 Dec 13.
Published in final edited form as: Nature. 2013 May 12;498(7453):220–223. doi: 10.1038/nature12141
Abstract
Congenital heart disease (CHD) is the most frequent birth defect, affecting 0.8% of live births1. Many cases occur sporadically and impair reproductive fitness, suggesting a role for de novo mutations. By analysis of exome sequencing of parent-offspring trios, we compared the incidence of de novo mutations in 362 severe CHD cases and 264 controls. CHD cases showed a significant excess of protein-altering de novo mutations in genes expressed in the developing heart, with an odds ratio of 7.5 for damaging mutations. Similar odds ratios were seen across major classes of severe CHD. We found a marked excess of de novo mutations in genes involved in production, removal or reading of H3K4 methylation (H3K4me), or ubiquitination of H2BK120, which is required for H3K4 methylation2–4. There were also two de novo mutations in SMAD2; SMAD2 signaling in the embryonic left-right organizer induces demethylation of H3K27me5. H3K4me and H3K27me mark `poised' promoters and enhancers that regulate expression of key developmental genes6. These findings implicate de novo point mutations in several hundred genes that collectively contribute to ~10% of severe CHD.
From more than 5000 probands enrolled in the Congenital Heart Disease Genetic Network Study of the National Heart Lung and Blood Institute Pediatric Cardiac Genomics Consortium7, we selected 362 parent-offspring trios comprising a child (proband) with severe CHD and no first-degree relative with identified structural heart disease. Probands with an established genetic diagnosis were excluded. There were 154 probands with conotruncal defects, 132 with left ventricular obstruction, 70 with heterotaxy and six with other diagnoses (Supplementary Table 1).
Genomic DNA samples from trios underwent exome sequencing (see Methods)8. Targeted bases in each sample were sequenced a mean of 107 times by independent reads with 96.0% read eight or more times. In parallel, 264 trios comprising unaffected siblings of autism cases and their unaffected parents (Supplementary Table 1) were sequenced in the same facility using the same protocol and were analyzed as a control group9 (Supplementary Table 2, Supplementary Figure 1). Family relationships were confirmed from sequence data in all trios.
High probability de novo variants in probands were identified using a Bayesian quality score (QS; see Methods). Sanger sequencing of 181 putative de novo mutations across the QS spectrum demonstrated strong correlation of confirmation with QS (R2 = 0.89), with 100% confirmation of 90 calls with QS > 50 (Supplementary Table 3, Supplementary Figure 2). Consequently, de novo mutation calls with QS ≥ 50 were included in the study; this set is estimated to include 90% of mutations with QS > 0, with ~100% specificity; 90% of these have the maximum QS of 100 (Supplementary Figure 3). Sensitivity is further diminished by ~5% due to bases with very low read coverage. We found 0.88 de novo mutations per subject in CHD cases and 0.85 in controls. These mutation rates (1.34 and 1.29 × 10−8 per targeted base) are not significantly different (P = 0.63, binomial test) and are similar to prior estimates10. The set of de novo mutations is shown in Supplementary Table 4.
CHD cases and controls had very similar maternal and paternal ages, which had a small impact on the mutation rate (Supplementary Figure 4). We found no significant effect of geographic ancestry on the mutation rate (Supplementary Figure 5). The number of de novo mutations per subject closely approximated the Poisson distribution, providing no evidence for mutation clustering (Supplementary Figure 6).
Genes contributing to CHD should be expressed in the developing heart/anlagen or tissues that provide developmental cues. We used RNA sequencing of mouse heart at embryonic day (e)14.5 (Supplementary Methods) to partition 16,676 genes with identified human-mouse orthologs into the top quartile of expression (4,169 genes with high heart expression, HHE; threshold >40 reads per million (rpm) mapped reads) and the bottom 75% (12,507 with lower heart expression, LHE). The HHE set included regulatory genes known to be expressed at this stage such as Gata4, Nkx2.5, and Tbx5.
We found a significant increase in the rate of protein-altering de novo mutations in HHE genes in patients with CHD compared to controls (P = 0.003, binomial test, OR = 2.53, Table 1). Because it is unlikely that all such de novo mutations alter protein function, we enriched for deleterious de novo mutations, first removing missense mutations at weakly conserved positions among vertebrate orthologs (two or more species with substitutions, median seven), then removing missense mutations at highly conserved positions (zero or one species with substitution; 72% with 0), leaving only damaging mutations (premature termination, splice site and frameshift). This produced successive increases in the OR's to 3.67 and 7.50, with significant differences between cases and controls in each group (Table 1a, Figure 1a). The rise in odds ratio with increasing stringency was significant (P = 0.001, logistic model regression). Other predictors of deleterious mutations, such as Polyphen2, yielded similar results (probably deleterious missense mutations + damaging mutations, P = 0.0007, binomial test). Similar results were found when genes were partitioned across a range of expression thresholds in the developing heart (Supplementary Table 5) and also when analyses used heart RNA expression from e9.5 (Supplementary Table 6). In contrast, there was no significant difference in mutation frequency in CHD cases vs. controls among LHE genes, with OR's near or < 1 in all comparisons (Supplementary Table 7, Figure 1a). Analysis comparing the presence or absence of de novo mutations in each case and control yielded similar results (Supplementary Table 8 and Supplementary Figure 7). Examination of subjects with left ventricular obstruction, conotruncal defects, and heterotaxy demonstrated similarly increased OR's for each group (Supplementary Table 9).
Table 1.
De novo mutations in genes with high expression in developing heart in CHD probands and controls
| Mutations in genes in top quartile of expression at e14.5 | Total # of de novo mutations | De novo mutations/subject | Odds Ratio Cases:Cont (95% Cl)† | P-value†† | ||
|---|---|---|---|---|---|---|
| CHD 362 trios | Controls 264 trios | CHD 362 trios | Controls 264 trios | |||
| Silent | 21 | 21 | 0.06 | 0.08 | N/A | 0.35 |
| Nonconserved Missense | 27 | 17 | 0.07 | 0.06 | 1.59 (0.67–3.74) | 0.76 |
| Silent and Protein Changing | 102 | 53 | 0.28 | 0.20 | N/A | 0.05 |
| All Protein Changing | 81 | 32 | 0.22 | 0.12 | 2.53 (1.22–5.25) | 0.003 |
| Conserved Missense | 39 | 13 | 0.11 | 0.05 | 3.00 (1.25–7.17) | 0.01 |
| Conserved and Damaging Protein Altering | 55 | 15 | 0.15 | 0.06 | 3.67 (1.60–8.42) | 0.0004 |
| Damaging | 15 | 2 | 0.04 | 0.01 | 7.50 (1.52–36.95) | 0.01 |
Figure 1. Enrichment of non-synonymous de novo mutations in heart-expressed genes.

a, Odds ratios (ORs), standard errors and _P_-values (two-tailed binomial exact test) are shown comparing incidence of classes of de novo mutations in CHD cases versus controls for genes in top 25% (red bars) and bottom 75% (blue bars) of expression at e14.5 in developing heart. b, ORs for incidence of mutations in genes in top 25% versus bottom 75% of expression in CHD cases (red bars) and controls (blue bars). `Damaging' denotes premature termination, frameshift or splice site mutations; `Conserved MS' and `Noncons MS' denote mutations at highly or poorly conserved positions, respectively. NS, not significant.
Comparison of de novo mutation frequencies in HHE genes vs. LHE genes in the CHD cohort also revealed a significantly greater rate in HHE genes, again with OR increasing with increasingly stringent filters (Supplementary Table 7, Figure 1b). In contrast, controls showed no significant difference in mutation frequencies in HHE vs. LHE, again with all odds ratios near or < 1 (Supplementary Table 7, Figure 1b).
Strikingly, examination of genes mutated in the CHD set revealed eight involved in production, removal or reading of methylation of histone H3, lysine 4 (H3K4me). Interestingly, three genes in this pathway (MLL2, KDM6A, CHD7) have previously been implicated in severe CHD11,12. In Gene Ontology (GO) analysis (http://david.abcc.ncifcrf.gov/) of the 249 protein-altering de novo mutations in CHD probands, the H3K4me pathway was the only gene set with significant enrichment (P = 4 × 10−7, modified Fisher exact test, P = 4 × 10−4 after Bonferroni correction; see Methods). The number of mutations in this gene set expected by chance was one and controls showed none.
H3K4me is an activating mark found in promoters/enhancers of key developmental genes6. Early in development `poised' promoters/enhancers have both activating H3K4me marks and inactivating H3K27me marks; these promoters/enhancers and their target genes are selectively activated by modification of these marks in different lineages. Mutations in these genes (Table 2, Figure 2) included 27% of the damaging mutations in the HHE gene set. Mutated genes included MLL2 (frameshift mutation) and WDR5 (missense), components of the MLL2 H3K4 _N_-methyl transferase complex2; KDM5A (missense) and KDM5B (splice donor), both H3K4 demethylases3; CHD7 (premature termination), an ATP-dependent helicase that binds H3K4me sites12. There were also de novo mutations in RNF20 (premature termination) and UBE2B (missense), components of a histone H2BK120 ubiquitination complex and in USP44 (missense), encoding an H2B deubiquitinase4. Ubiquitination at H2BK120 is required for H3K4 methylation2.
Table 2.
Genes of interest with de novo mutations in probands
| ID | Gene | Mutation | Dx | Other Structural/Neuro/Ht-Wt |
|---|---|---|---|---|
| 1-00596 | MLL2 † | p.Ser1722Argfs*9 | LVO | Y/Y/N |
| 1-00853 | WDR5 † | p.Lys7Gln | CTD | N/Y/N |
| 1-00534 | CHD7 † | p.Gln1599* | CTD | Y/Y/Y |
| 1-00230 | KDM5A † | p.Arg1508Trp | LVO | N/N/Y |
| 1-01965 | KDM5B † | p.IVS12+1 G>A | LVO | N/N/Y |
| 1-01907 | UBE2B † | p.Arg8Thr | CTD | N/N/N |
| 1-00075 | RNF20 † | p.Gln83* | HTX | Y/Y/Y |
| 1-01260 | USP44 † | p.Glu71Asp | LVO | N/N/N |
| 1-02020 | SMAD2 †† | p.IVS6+1 G>A | HTX | Y/N/N |
| 1-02621 | SMAD2 †† | p.Trp244Cys | HTX | Y/na/N |
| 1-01451 | MED20 | p.IVS2+2 T>C | HTX | N/Y/Y |
| 1-01151 | SUV420H1 | p.Arg143Cys | CTD | N/Y/N |
| 1-00750 | HUWE1 | p.Arg3219Cys | LVO | N/Y/N |
| 1-00577 | CUL3 | p.Iso144Phefs*23 | LVO | Y/Y/N |
| 1-00116 | NUB1 | p.Asp310His | CTD | Y/Y/Y |
| 1-01828 | DAPK3 | p.Pro193Leu | CTD | N/N/na |
| 1-03151 | SUPT5H | p.Glu451Asp | LVO | N/na/N |
| 1-00455 | NAA15 | p.Lys335Lysfs*6 | HTX | Y/Y/N |
| 1-00141 | NAA15 | p.Ser761* | CTD | N/na/Y |
| 1-01138 | USP34 | p.Leu432Pro | LVO | N/na/N |
| 1-00448 | NF1 | p.IVS6+4 delA | CTD | N/na/N |
| 1-00802 | PTCH1 | p.Arg831Gln | LVO | N/na/N |
| 1-02458 | SOS1 | p.Thr266Lys | Other | Y/Y/Y |
| 1-02952 | PITX2 | p.Ala47Val | LVO | N/na/N |
| 1-01913 | RAB10 | p.Asn112Ser | Other | N/na/N |
| 1-00638 | FBN2 | p.Asp2191Asn | CTD | N/na/N |
| 1-00197 | BCL9 | p.Met1395Lys | LVO | N/na/N |
| 1-02598 | LRP2 | p.Gly4372Lys | HTX | N/na/N |
Figure 2. De novo mutations in the H3K4 and H3K27 methylation pathways.
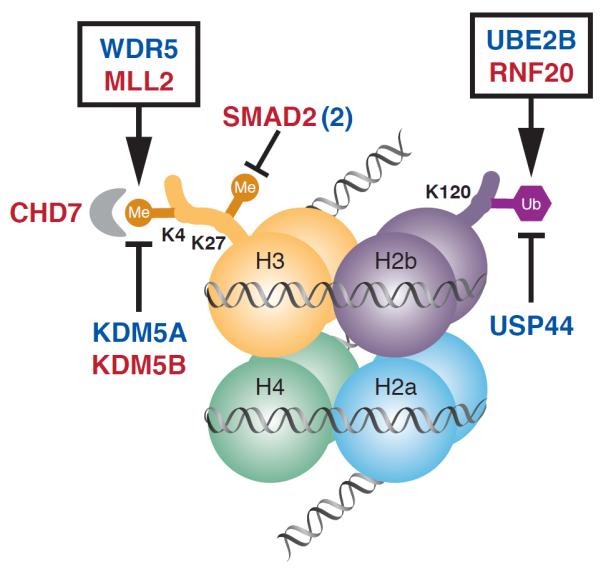
Nucleosome with histone octamer and DNA, with H3K4 methylation bound by CHD7, H3K27 methylation and H2bK120 ubiquitination is shown. Genes mutated in CHD that affect the production, removal and reading of these histone modifications are shown; genes with damaging mutations are shown in red, those with missense mutations are shown in blue. SMAD2 (2) indicates there are two patients with mutation in this gene. Genes whose products are found together in a complex are enclosed in a box.
Interestingly, SMAD2 is mutated twice (splice site, conserved missense) a finding unlikely to occur by chance (P = 0.015, Monte Carlo simulation) (Table 2). SMAD2 is asymmetrically phosphorylated downstream of NODAL signaling in the embryonic left-right organizer, resulting in SMAD2 binding to chromatin, recruitment of JMJD3, and demethylation of H3K27me, enabling transcriptional activation at poised sites5. Additional genes of note (Table 2) include SUV420H1 (missense), encoding a histone H4 methylase; MED20 (splice site), a component of the mediator complex; HUWE1 (missense), a ubiquitin ligase targeting histones and TP53; CUL3 (frameshift), a scaffold for assembly of many RING ubiquitin ligases8; NUB1 (missense), which inhibits NEDD8, a cofactor for cullin-based ubiquitin ligases. Lastly, NAA15, an _N_-acetyltransferase13, had two damaging mutations, unlikely a chance event (P = 0.01, Monte Carlo simulation). Among the 17 above genes, ten have no damaging variants and seven have 1–5 among >9500 exomes in NHLBI ESP, 1000 genomes and Yale exome databases.
Phenotypes of the eight patients with de novo mutations in the H3K4me pathway revealed diverse cardiac phenotypes (Table 2, Supplementary Table 10). Other structural, neurodevelopmental and growth abnormalities were common. Additionally, consistent with a role in left-right axis determination5, both patients with SMAD2 mutations had dextrocardia with unbalanced complete AV canal and pulmonary stenosis. For other genes mutated more than once (e.g., NAA15), probands had dissimilar cardiac phenotypes (Supplementary Table 11).
Before initiating exome sequencing, we defined a set of 277 candidate CHD genes (Supplementary Table 12) from human and model system studies. There were 13 CHD probands with de novo mutations in these genes (Table 2, Supplementary Table 13), more than expected by chance (P = 7 × 10−4, Monte Carlo simulation) or in controls (n= 1, P = 0.006, binomial test). This set included several genes known to cause Mendelian CHD, however affected subjects lacked cardinal disease manifestations or had atypical cardiac features. For example, the patient with CHD7 mutation had none of the major criteria (coloboma, choanal atresia or hypoplastic semi-circular canals) for CHARGE syndrome12. Similarly, the patient with MLL2 mutation was not prospectively diagnosed with Kabuki syndrome, however re-evaluation at age 2 after sequencing identified characteristic facial features. Additionally, a patient with an NF1 mutation had a complex conotruncal defect, an unusual finding in neurofibromatosis. These findings support variable expressivity and a broader phenotypic spectrum resulting from mutations at known disease loci. Other genes of interest in this set included RAB10 and BCL9, identified as candidates by rare de novo CNVs14.
Our results implicate de novo point/indel mutations that by chance occur in genes required for normal heart development in the pathogenesis of diverse CHDs. Consistent with this inference, genes with damaging and conserved missense mutations in CHD probands showed higher expression in e14.5 mouse heart compared to controls (Supplementary Figure 8; median 45 vs. 16 rpm, P = 5 × 10−4, Wilcoxon signed-rank test), while expression of genes with silent mutations show no significant difference (median 21 vs. 19 rpm, P = 0.7, Wilcoxon signed-rank test). Expression at e9.5 shows similar results (Supplementary Figure 8). The increased mutation burden of HHE genes in cases is not due to a higher intrinsic mutation rate of these genes because the rate is significantly higher than in controls; moreover, there is no significant difference in mutation rate between HHE and LHE genes in controls. Further, partitioning genes into analogous high and low expression groups for four control adult tissues (brain, heart, liver and lung) showed no significant differences in mutation burden between cases and controls or between high and low expression groups (Supplementary Figure 9).
From the increased fraction of patients with protein-altering mutations in HHE genes in CHD patients (0.22) vs. controls (0.12), we estimate that such mutations play a role in about 10% of these patients (95% confidence interval 5%–15%). This could be somewhat underestimated, since mutation detection is incomplete, analysis is limited to genes with identified mouse orthologs, and the HHE set may not include all trait loci. Similarly, the observed ORs may be somewhat underestimated since not all mutations in cases are likely to confer risk.
These findings establish that mutations in many genes in the H3K4me-H3K27me pathway disrupt cardiac development and are consistent with prior evidence implicating these chromatin marks in regulating key developmental genes6, including those involved in cardiac development15,16. Targeted sequencing in larger CHD cohorts will enable assessment of the role of each individual gene in this pathway. These findings imply dosage sensitivity for these chromatin marks in CHD, similar to recent findings implicating haploinsufficiency for chromatin modifying/remodeling genes in diverse cancers17,18. Investigation of the consequences of these mutations on specific enhancers/promoters and the genes they regulate will likely provide further insight into the CHD pathogenesis.
The demonstration that point/indel mutations contribute to ~10% of CHD patients and the finding that six genes were mutated twice (Supplementary Table 11) enables an estimate of the size of the gene set that contributes to these CHDs (see Methods). The point-wise estimate is 401 genes (95% confidence interval 197–813), indicating that many more CHD-related genes and pathways remain to be discovered.
Exome sequencing of probands with autism have revealed broadly similar results: de novo mutations in a large set of genes occur in a significant fraction of patients, with relatively high OR's for damaging mutations in genes expressed in the brain9,19–21. Most interestingly, CHD8, which like CHD7 reads H3K4me marks, is frequently mutated in autism22, raising the question of whether the H3K4me pathway may play a role in many congenital diseases. Among 249 protein-altering de novo mutations in CHD (Supplementary Table 4) and 570 such mutations in autism9,19,20,23, there were two genes, CUL3 and NCKAP1, with damaging mutations in both CHD and autism and none in controls (P = 0.001, Monte Carlo simulation), and several others with mutations in both (e.g., SUV40H1 and CHD7). Similarly, rare copy number variants at 22q11.2, 1q21, and 16p11 are found in patients with autism, CHD or both diseases24–26. These observations suggest variable expressivity of mutations in key developmental genes. Identification of the complete set of these developmental genes and the full spectrum of the resulting phenotypes will likely be important for patient care and genetic counseling.
Our findings do not resolve the pathogenesis of most CHD cases. Rare and de novo copy number variants appear to account for a small fraction14,27; rare or common transmitted variants are expected to also make significant contributions. Additionally, considering the role of H3K4me and H3K27me marks in promoter/enhancer regulation, non-coding mutations cannot be dismissed. Lastly, evidence of dosage sensitivity of many chromatin-modifying genes raises the possibility that environmental perturbations of these pathways in critical developmental windows might phenocopy the effects of these mutations.
METHODS
Patient cohorts
Probands with or without parents were recruited from 9 centers in the United States and the United Kingdom into the Congenital Heart Disease Genetic Network Study of the Pediatric Cardiac Genomics Consortium (CHD Genes: NCT01196182)7. The protocol was approved by the Institutional Review Boards of Boston Children's Hospital, Brigham and Women's Hospital, Great Ormond St. Hospital, Children's Hospital of Los Angeles, Children's Hospital of Philadelphia, Columbia University Medical Center, Icahn School of Medicine and Mt. Sinai, Rochester School of Medicine and Dentistry, Steven and Alexandra Cohen Children's Medical Center of New York, and Yale School of Medicine. Written informed consent was obtained from each participating subject or their parent/guardian. Probands were selected for severe congenital heart disease (excluding isolated VSDs, ASDs, PDAs or PSs), availability of both parents, and absence of any CHD in first-degree relatives. Cardiac diagnoses were obtained from review of echocardiogram, catheterization and operative reports; extracardiac findings were extracted from medical records. Controls were from 264 previously studied quartets that included one offspring with autism, an unaffected sibling and unaffected parents, all recruited with written informed consent by the Simons Foundation Autism Research Initiative28. Parents and their unaffected sibling from this cohort were analyzed in the current study.
Exome sequencing
Trios were sequenced at the Yale Center for Genome Analysis following the same protocol. Genomic DNA from venous blood was captured with the NimbleGen v2.0 exome capture reagent (Roche) and sequenced (Illumina HiSeq 2000, 75 base paired-end reads). Reads were mapped to the reference genome using Eland. SNV and indel calls were assigned quality scores (QS) using SAMtools8 and annotated for novelty using dbSNP, build 135, 1000 genomes, May 2011 release and the Yale Exome Database, for impact on encoded proteins, and conservation of variant position.
Identification and confirmation of de novo mutations
Heterozygous SNVs and indels in the proband that showed QS ≥ 60 and 600, respectively, and rare non-reference calls in both parents were selected. Read plots of all putative indels were visually inspected in trio members to eliminate false calls. A Bayesian algorithm was used to assist de novo mutation calls. Elements included probability of the proband being heterozygous at the test position; probability that parents are homozygous for the reference allele, given frequency of reference and non-reference reads and probability of heterozygosity in offspring; probability that a variant is de novo given its population frequency. Resulting QSs scaled from 0 to 100. Their correlation with bona fide de novo mutations was determined by Sanger sequencing of PCR amplicons harboring 181 putative mutations distributed across the QS spectrum. Additionally, all six de novo indels with QS > 50 in the HHE gene set were tested and confirmed by Sanger sequencing.
RNA sequencing and analysis
Hearts from e14.5 mouse embryos (strain 129SvEv) were isolated, rinsed, and immersed in RNALater. Left and right atria, left ventricle (with interventricular septum, aortic and mitral valves), and right ventricle (with pulmonary and tricuspid valves) were dissected. Chamber-specific RNAs were extracted and pooled from 5 embryos, selected with oligo-dT, copied into double stranded DNA, and ligated to adaptors. 150–250 bp fragments were isolated after acrylamide gel electrophoresis, amplified and sequenced (Illumina HiSeq2000), with > 40 million paired-end 50 base reads per library as previously described29. Reads were aligned to the mouse genome (mm9)30, and reads per gene per million mapped reads (rpm) was determined. The average of rpm of each gene from each chamber was used as the measure of heart expression. RNA from atria, ventricle and truncus/outflow tract at e9.5 was prepared, sequenced and analyzed by an analogous approach. RNA sequencing of control human adult tissues- lung, liver, heart and brain- from the Illumina Human Body Map (http://www.ebi.ac.uk/arrayexpress/browse.html?keywords=E-MTAB-513) was similarly performed and analyzed as reads per gene per million reads per kb of transcript.
Principal component analysis
The EIGENSTRAT program was used to compare SNP genotypes of probands and individuals of known ancestry in HapMap3 (http://hapmap.ncbi.nlm.nih.gov/). SNPs with MAF >5% without significant linkage disequilibrium with other SNPs were analyzed. The results of analysis correctly distinguished ancestry groups in HapMap3 samples; ancestries of CHD subjects were assigned accordingly.
Statistical analyses
The significance of mutation frequency differences between groups was tested with two-tailed binomial exact tests; two-tailed Fisher exact tests assessed differences in numbers of patients with one or more de novo mutations; tests among 3 groups was by Chi-square analysis. Gene expression at e14.5 of genes mutated in cases and controls was compared by Wilcoxon signed-rank test. Correlation of mutation rate and parental age was tested by Pearson's correlation. The expected number of genes with more than one de novo mutation was determined by Monte Carlo simulation (108 iterations) specifying the total number of protein-altering mutations and 21,000 genes of observed coding length. Analogous approaches were used to determine probabilities of any gene having ≥ 2 damaging mutations, ≥ 1 damaging and ≥ 1 mutation at a conserved position, and ≥ 13 genes mutated in both CHD and autism. The fit to the Poisson distribution of the observed numbers of de novo mutations per subject was assessed by Chi-square test.
Overrepresentation of de novo mutations in the H3K4me pathway and the presence of significant enrichment of other gene pathways was tested via Gene Ontology (GO) analysis, using a modified Fisher's exact test with Bonferroni correction as implemented in DAVID (http://david.abcc.ncifcrf.gov/). Input was all genes with protein-altering de novo mutations in CHD or control subjects, and all genes sequenced. The H3K4me gene set was: CHD8, MLL3, SETD7, WHSC1L1, CDC73, WHSC1, SETD1A, MLL2, KDM5A, MLL4, MLL5, UBE2B, ASH1L, SETD1B, MLL, LEO1, PAF1, KDM5C, CTR9, PRDM9, MEN1, CHD7, RNF20, KDM1A, RNF40, SMYD3, KDM6A, KDM5B, USP44, WDR5. The expected number of mutations in the H3K4me set was calculated from the fraction of the exome coding region attributable to this gene set and the total number of de novo mutations.
Estimating number of genes in which de novo mutations contribute to CHD
We addressed this question using the `unseen species problem'9. We infer that the number of probands with non-synonymous mutations in the HHE set (81) minus the expected number (44; calculated from the number observed in controls), represents the number of subjects in whom de novo mutations confer CHD risk (37; 10.0% of probands). The number of genes with > 1 protein-altering de novo mutation (six) minus the most likely number expected by chance (three) represents risk-associated genes with more than 1 mutation (three). The number of risk-associated genes (C) is estimated as follows:
- C = c/u + g2×d× (1-u)/u
- c = number of observed risk-associated genes (34)
- c1 = number of genes mutated once (31)
- d = total number of risk-associated mutations (37)
- g = variation in effect size of individual de novo mutations (assumed to be 1, which minimizes underestimation of set size)
- u = 1 – c1/d (probability that newly added mutation hits a previously mutated gene)
- C = 401
From 95% confidence intervals of the number of risk-associated events, the 95% confidence interval for number of risk genes is calculated as 197–837.
Supplementary Material
1
Acknowledgments
The authors are enormously grateful to the patients and families who participated in this research. We thank the following team members for outstanding contributions to patient recruitment: Danielle Awad, Katrina Celia, Davina Etwaru, Rosalind Korsin, Alyssa Lanz, Emma Marquez, Jaswinder K. Sond, Abigail Wilpers, Roslyn Yee (Columbia Medical School); Kari Boardman, Judith Geva, Joshua Gorham, Barbara McDonough, Angela Monafo, Jan Stryker (Harvard Medical School); Nancy Cross (Yale School of Medicine); Sharon M. Edman, Jennifer L. Garbarini, Jessica E. Tusi, Stacy H. Woyciechowski, (Children's Hospital of Philadelphia); Jiffy Ellashek and Nhu Tran (Children's Hospital of Los Angeles); Karen Flack (University College London); Dorota Gruber, Nancy Stellato (Steve and Alexandra Cohen Children's Medical Center of New York); Denise Guevara, Ariel Julian, Meghan Mac Neal, Cassie Mintz, (Icahn School of Medicine at Mount Sinai); Eileen Taillie (University of Rochester School of Medicine and Dentistry); We thank Vanessa Spotlow, Patrick Candrea, Kira Pavlik and Maria Sotiropoulos for their expert production of exome sequences. We thank Bradley Bernstein and Rusty Ryan (Massachusetts General Hospital) and Benoit Bruneau (Gladstone Institute and U. California, San Francisco) for helpful discussions. Supported by the NIH National Heart Lung and Blood Institute Pediatric Cardiac Genomics Consortium (U01-HL098188, U01-HL098147, U01-HL098153, U01-HL098163, U01-HL098123, U01-HL098162) and in part by the Simons Foundation for Autism Research and the NIH Centers for Mendelian Genomics (5U54HG006504).
Footnotes
Supplementary Information is linked to online version of the paper at www.nature.com/nature
Author Contributions. Study design: M.B., W.K.C., B.D.G., E.G., H.H, J.R.K., R.P.L., L.E.M., J.G.S, C.E.S., D.W., P.S.W.; Cohort ascertainment, phenotypic characterization, and recruitment: R.E.B., M.B., W.K.C., J.D., B.D.G., E.G., J.K., R.K., T.L., J.W.N, G.P., A.R.A., H.S.S., C.E.S, I.A.W.; Informatics/data management: R.D.B., R.E.B, N.J.C., M.C., S.D., J.G., H.H., M.J.I., J.L., A.L., S.M.M., J.D.O., M.P., A.E.R., J.G.S., W.W., P.S.W, S.Z.; Exome sequencing production: J.D.O., A.L., R.P.L., S.M.M., M.W.S., I.R.T.; De novo mutation validation: W.K.C, L.M.; Exome sequencing analysis: K.K.B., Y.H.C., M.C., S.D., K.A.F., J.G., J.K.K., R.P.L., I.P., R.S., S.J.S., J.G.S., C.E.S., S.S., W.W., S.Z.; RNA sequence production/analysis: J.J., M.P., C.E.S., J.G.S, H.W.; Statistical analysis: M.C., R.P.L., I.P., A.R., C.E.S, J.G.S, S.Z., H.Z.; Writing of manuscript: M.B., M.C., W.K.C., B.D.G., E.G., J.R.K., R.P.L., C.E.S., S.Z.
Author Information mRNA available at NCBI under accession IDs listed in Table S4; mutation data is available at dbSNP under batch accession 1059065. Reprints and permissions information is available at www.nature.com/reprints. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NHLBI. The authors declare no competing financial interests; details accompany the full-text HTML version of the paper at www.nature.com/nature.
References
- 1.Reller MD, Strickland MJ, Riehle-Colarusso T, Mahle WT, Correa A. Prevalence of congenital heart defects in metropolitan Atlanta, 1998–2005. J Pediatr. 2008;153:807–813. doi: 10.1016/j.jpeds.2008.05.059. doi:10.1016/j.jpeds.2008.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. doi:10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pedersen MT, Helin K. Histone demethylases in development and disease. Trends Cell Biol. 2010;20:662–671. doi: 10.1016/j.tcb.2010.08.011. doi:10.1016/j.tcb.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 4.Fuchs G, et al. RNF20 and USP44 regulate stem cell differentiation by modulating H2B monoubiquitylation. Mol Cell. 2012;46:662–673. doi: 10.1016/j.molcel.2012.05.023. doi:10.1016/j.molcel.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dahle O, Kumar A, Kuehn MR. Nodal signaling recruits the histone demethylase Jmjd3 to counteract polycomb-mediated repression at target genes. Sci Signal. 2010;3:ra48. doi: 10.1126/scisignal.2000841. doi:10.1126/scisignal.2000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. doi:10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 7.Pediatric Cardiac Genomics Consortium The Congenital Heart Disease Network Study (CHD GENES): Rationale, design and early results. Circulation Research. doi: 10.1161/CIRCRESAHA.111.300297. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyden LM, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482:98–102. doi: 10.1038/nature10814. doi:10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanders SJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. doi:10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scally A, Durbin R. Revising the human mutation rate: implications for understanding human evolution. Nat Rev Genet. 2012;13:745–753. doi: 10.1038/nrg3295. doi:10.1038/nrg3295. [DOI] [PubMed] [Google Scholar]
- 11.Lederer D, et al. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. American journal of human genetics. 2012;90:119–124. doi: 10.1016/j.ajhg.2011.11.021. doi:10.1016/j.ajhg.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vissers LE, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–957. doi: 10.1038/ng1407. doi:10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 13.Gendron RL, Adams LC, Paradis H. Tubedown-1, a novel acetyltransferase associated with blood vessel development. Dev Dyn. 2000;218:300–315. doi: 10.1002/(SICI)1097-0177(200006)218:2<300::AID-DVDY5>3.0.CO;2-K. doi:10.1002/(SICI)1097-0177(200006)218:2<300∷AID-DVDY5>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 14.Greenway SC, et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet. 2009;41:931–935. doi: 10.1038/ng.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wamstad JA, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–220. doi: 10.1016/j.cell.2012.07.035. doi:10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paige SL, et al. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell. 2012;151:221–232. doi: 10.1016/j.cell.2012.08.027. doi:10.1016/j.cell.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ceol CJ, et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471:513–517. doi: 10.1038/nature09806. doi:10.1038/nature09806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sausen M, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nature genetics. 2013;45:12–17. doi: 10.1038/ng.2493. doi:10.1038/ng.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Roak BJ, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. doi:10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iossifov I, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. doi:10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neale BM, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. doi:10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Roak BJ, et al. Multiplex Targeted Sequencing Identifies Recurrently Mutated Genes in Autism Spectrum Disorders. Science. 2012 doi: 10.1126/science.1227764. doi:10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong A, et al. Rate of de novo mutations and the importance of father's age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. doi:10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vorstman JA, Breetvelt EJ, Thode KI, Chow EW, Bassett AS. Expression of autism spectrum and schizophrenia in patients with a 22q11.2 deletion. Schizophrenia research. 2012 doi: 10.1016/j.schres.2012.10.010. doi:10.1016/j.schres.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 25.Mefford HC, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. doi:NEJMoa0805384 [pii]10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghebranious N, Giampietro PF, Wesbrook FP, Rezkalla SH. A novel microdeletion at 16p11.2 harbors candidate genes for aortic valve development, seizure disorder, and mild mental retardation. American journal of medical genetics. Part A. 2007;143A:1462–1471. doi: 10.1002/ajmg.a.31837. doi:10.1002/ajmg.a.31837. [DOI] [PubMed] [Google Scholar]
- 27.Soemedi R, et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. American journal of human genetics. 2012;91:489–501. doi: 10.1016/j.ajhg.2012.08.003. doi:10.1016/j.ajhg.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischbach GD, Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010;68:192–195. doi: 10.1016/j.neuron.2010.10.006. doi:10.1016/j.neuron.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 29.Christodoulou DC, Gorham JM, Herman DS, Seidman JG. Construction of normalized RNA-seq libraries for next-generation sequencing using the crab duplex-specific nuclease. Curr Protoc Mol Biol. 2011;Chapter 4(Unit4):12. doi: 10.1002/0471142727.mb0412s94. doi:10.1002/0471142727.mb0412s94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herman DS, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. doi:10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1