Genetic Characterization of Betacoronavirus Lineage C Viruses in Bats Reveals Marked Sequence Divergence in the Spike Protein of Pipistrellus Bat Coronavirus HKU5 in Japanese Pipistrelle: Implications for the Origin of the Novel Middle East Respiratory Syndrome Coronavirus (original) (raw)
Abstract
While the novel Middle East respiratory syndrome coronavirus (MERS-CoV) is closely related to Tylonycteris bat CoV HKU4 (Ty-BatCoV HKU4) and Pipistrellus bat CoV HKU5 (Pi-BatCoV HKU5) in bats from Hong Kong, and other potential lineage C betacoronaviruses in bats from Africa, Europe, and America, its animal origin remains obscure. To better understand the role of bats in its origin, we examined the molecular epidemiology and evolution of lineage C betacoronaviruses among bats. Ty-BatCoV HKU4 and Pi-BatCoV HKU5 were detected in 29% and 25% of alimentary samples from lesser bamboo bat (Tylonycteris pachypus) and Japanese pipistrelle (Pipistrellus abramus), respectively. Sequencing of their RNA polymerase (RdRp), spike (S), and nucleocapsid (N) genes revealed that MERS-CoV is more closely related to Pi-BatCoV HKU5 in RdRp (92.1% to 92.3% amino acid [aa] identity) but is more closely related to Ty-BatCoV HKU4 in S (66.8% to 67.4% aa identity) and N (71.9% to 72.3% aa identity). Although both viruses were under purifying selection, the S of Pi-BatCoV HKU5 displayed marked sequence polymorphisms and more positively selected sites than that of Ty-BatCoV HKU4, suggesting that Pi-BatCoV HKU5 may generate variants to occupy new ecological niches along with its host in diverse habitats. Molecular clock analysis showed that they diverged from a common ancestor with MERS-CoV at least several centuries ago. Although MERS-CoV may have diverged from potential lineage C betacoronaviruses in European bats more recently, these bat viruses were unlikely to be the direct ancestor of MERS-CoV. Intensive surveillance for lineage C betaCoVs in Pipistrellus and related bats with diverse habitats and other animals in the Middle East may fill the evolutionary gap.
INTRODUCTION
Coronaviruses (CoVs) infect humans and a wide variety of animals, causing respiratory, enteric, hepatic, and neurological diseases of various degrees of severity. They have been classified traditionally into groups 1, 2, and 3 based on genotypic and serological characteristics (1, 2). Recently, the nomenclature and taxonomy of CoVs have been revised by the Coronavirus Study Group of the International Committee for Taxonomy of Viruses (ICTV). They are now classified into three genera, Alphacoronavirus, Betacoronavirus, and Gammacoronavirus, replacing the three traditional groups (3). Novel CoVs, which represented a novel genus, Deltacoronavirus, have also been identified (4, 5). While CoVs from all four genera can be found in mammals, bat CoVs are likely the gene source of Alphacoronavirus and Betacoronavirus, and avian CoVs are the gene source of Gammacoronavirus and Deltacoronavirus (5–7).
CoVs are well known for their high frequency of recombination and mutation rates, which may allow them to adapt to new hosts and ecological niches (1, 8–12). This is best exemplified by the severe acute respiratory syndrome (SARS) epidemic, which was caused by SARS CoV (13, 14). The virus has been shown to have originated from animals, with horseshoe bats as the natural reservoir and palm civet as the intermediate host allowing animal-to-human transmission (15–18). Since the SARS epidemic, many other novel CoVs in both humans and animals have been discovered (4, 7, 19–24). In particular, a previously unknown diversity of CoVs has been described in bats from China and other countries, suggesting that bats are important reservoirs of alphaCoVs and betaCoVs (16, 18, 25–32).
In September 2012, two cases of severe community-acquired pneumonia were reported in Saudi Arabia which were subsequently found to have been caused by a novel CoV, Middle East respiratory syndrome coronavirus (MERS-CoV), previously known as human betaCoV 2c EMC/2012 (33, 34, 35). As of May 2013, a total of 49 laboratory-confirmed cases of MERS-CoV infection have been reported with 27 deaths (36), giving a crude fatality rate of 55%. So far, most cases of MERS-CoV infection presented with severe acute respiratory illness (36, 37). A macaque model for MERS-CoV infection has also been established which showed that the virus caused localized-to-widespread pneumonia in all infected animals (38). The viral virulence may be related to the ability of MERS-CoV to evade the innate immunity with an attenuated beta interferon response (39–41). Moreover, the ability to cause human-to-human transmission has raised the possibility of another SARS-like epidemic (36, 37). However, the source of this novel CoV is still obscure, which has hindered public health and infection control strategies for disease prevention. Phylogenetically, MERS-CoV belongs to Betacoronavirus lineage C, being closely related to Tylonycteris bat CoV HKU4 (Ty-BatCoV HKU4) and Pipistrellus bat CoV HKU5 (Pi-BatCoV HKU5), previously discovered in lesser bamboo bat (Tylonycteris pachypus) and Japanese pipistrelle (Pipistrellus abramus) in Hong Kong, China, respectively (31, 32, 42, 43). Moreover, potential viruses with partial gene sequences closely related to MERS-CoV have also been detected in bats from Africa, Europe, and America, although complete genome sequences were not available (44, 45). MERS-CoV is able to infect various mammalian cell lines, including primate, porcine, bat, and rabbit cells, which may be explained by the use of the evolutionarily conserved dipeptidyl peptidase 4 (DPP4) as its functional receptor (46, 47). These results suggested that MERS-CoV may possess broad species tropism and may have emerged from animals. However, the direct ancestor virus and animal reservoir of MERS-CoV are yet to be identified.
To better understand the evolutionary origin of MERS-CoV and the possible role of bats as the reservoir for its ancestral viruses, studies on the genetic diversity and evolution of lineage C betaCoVs in bats would be important. We attempted to study the epidemiology of lineage C betaCoVs, including Ty-BatCoV HKU4 and Pi-BatCoV HKU5, among various bat species in Hong Kong, China. The complete RNA-dependent RNA polymerase (RdRp), spike (S), and nucleocapsid (N) genes of 13 Ty-BatCoV HKU4 and 15 Pi-BatCoV HKU5 strains were sequenced to assess their genetic diversity and evolution. The results revealed that the two viruses were stably evolving in their respective hosts and diverged from their common ancestor a long time ago. However, the S protein of Pi-BatCoV HKU5 exhibited marked sequence divergence and many more positively selected sites than that of Ty-BatCoV HKU4, which may suggest the ability of Pi-BatCoV HKU5 along with its host to occupy new ecological niches. The potential implications on the animal origin of MERS-CoV are also discussed.
MATERIALS AND METHODS
Collection of bat samples.
Various bat species were captured from different locations in Hong Kong, China, over a 7-year period (April 2005 to August 2012). Their respiratory and alimentary specimens were collected using procedures described previously (16, 48). To prevent cross-contamination, specimens were collected using disposable swabs and protective gloves that were changed between samples. All specimens were immediately placed in viral transport medium containing Earle's balanced salt solution (Invitrogen, New York, NY), 20% glucose, 4.4% NaHCO3, 5% bovine albumin, 50,000 μg/ml vancomycin, 50,000 μg/ml amikacin, and 10,000 U/ml nystatin before transportation to the laboratory for RNA extraction.
RNA extraction.
Viral RNA was extracted from the respiratory and alimentary specimens using a QIAamp viral RNA Mini Kit (QIAgen, Hilden, Germany). The RNA was eluted in 50 μl of AVE buffer (QIAgen) and was used as the template for reverse transcription-PCR (RT-PCR).
RT-PCR for CoV and DNA sequencing.
CoV detection was performed by amplifying a 440-bp fragment of the RdRp gene of the CoVs using conserved primers (5′-GGTTGGGACTATCCTAAGTGTGA-3′ and 5′-CCATCATCAGATAGAATCATCATA-3′) designed by multiple alignments of the nucleotide sequences of available RdRp genes of known CoVs as described previously (17, 24). Reverse transcription was performed using a SuperScript III kit (Invitrogen, San Diego, CA). The PCR mixture (25 μl) contained cDNA, PCR buffer (10 mM Tris-HCl [pH 8.3], 50 mM KCl, 3 mM MgCl2, and 0.01% gelatin), 200 μM (each) deoxynucleoside triphosphates (dNTPs), and 1.0 U Taq polymerase (Applied Biosystems, Foster City, CA). The mixtures were amplified in 60 cycles of 94°C for 1 min, 48°C for 1 min, and 72°C for 1 min and a final extension at 72°C for 10 min in an automated thermal cycler (Applied Biosystems, Foster City, CA). Standard precautions were taken to avoid PCR contamination, and no false-positive result was observed in negative controls.
The PCR products were gel purified using a QIAquick gel extraction kit (QIAgen, Hilden, Germany). Both strands of the PCR products were sequenced twice with an ABI Prism 3700 DNA Analyzer (Applied Biosystems, Foster City, CA), using the two PCR primers. The sequences of the PCR products were compared with known sequences of the RdRp genes of CoVs in the GenBank database to identify lineage C betaCoVs.
Sequencing and analysis of the complete RdRp, S, and N genes of Ty-BatCoV HKU4 and Pi-BatCoV HKU5 strains.
To study the genetic diversity and evolution of Ty-BatCoV HKU4 and Pi-BatCoV HKU5 detected in bats, the complete RdRp, S, and N genes of 13 Ty-BatCoV HKU4 strains and 15 Pi-BatCoV HKU5 strains detected at different times and/or places, in addition to those of the nine previous strains with complete genome sequences, were amplified and sequenced using primers designed according to available genome sequences (Table 1) (32). The sequences of the PCR products were assembled manually to produce the complete RdRp, S, and N gene sequences. Multiple sequence alignments were constructed using MUSCLE in MEGA version 5 (49, 50). Phylogenetic trees were constructed using the maximum-likelihood method (51), with bootstrap values calculated from 100 trees. Protein family analysis was performed using PFAM and InterProScan (52, 53). Prediction of transmembrane domains was performed using TMHMM (54). The heptad repeat (HR) regions were predicted by using the coiled-coil prediction program MultiCoil2 (55).
Table 1.
Primers used in this study
| Coronavirus | Primer | |
|---|---|---|
| Forward | Backward | |
| Ty-BatCoV HKU4 | ||
| RdRp | LPW3283 5′-GTAATGTCTGTCAGTATTGGGTT-3′ | LPW3232 5′-AACTAATATGCTCTTTAACACTTCAC-3′ |
| LPW2771 5′-TGYTAYGCTTTAMGNCAYTTYGA-3′ | LPW2773 5′-GTTGGGTAATAACAAAATCACCAA-3′ | |
| LPW2626 5′-GTTTTAACACTYGATAAYGARGA-3′ | LPW2630 5′-AGTATATTGAARTTNGCACARTG-3′ | |
| LPW2738 5′-CCACCCTAATTGTGTTAATTGTA-3′ | LPW2775 5′-TAACTGAAGACCCTTCCTTGAAA-3′ | |
| LPW3233 5′-GGCAATTTTAATAAAGATTTTTATGA-3′ | LPW3234 5′-GCCAAAATCAATGACGCTAAAAT-3′ | |
| LPW1507 5′-GGTTGGGACTATCCTAAGTGTGA-3′ | LPW1508 5′-CCATCATCAGATAGAATCATCATA-3′ | |
| LPW1037 5′-WTATKTKAARCCWGGTGG-3′ | LPW1040 5′-KYDBWRTTRTARCAMACAAC-3′ | |
| LPW3235 5′-CTTAATAAACACTTTTCTATGATGAT-3′ | LPW2678 5′-TACTCACCGAGCTGTACTTTACTA-3′ | |
| S | LPW3797 5′-AGATTTATATAAAATTATGGGAA-3′ | LPW4102 5′-TACGTGGTTTTAATATGCAATAAAA-3′ |
| LPW3899 5′-TCTCTTACTAATACATCGGCT-3′ | LPW3900 5′-AAGACCTGACCATCTTCAGAAA-3′ | |
| LPW4103 5′-TGGTGCAAACCAAGATGTTGAAA-3′ | LPW3712 5′-CTAGCGCTATAACTTCTAAAAGTA-3′ | |
| LPW3720 5′-CATTAGTAGTTAGTGATTGTAAA-3′ | LPW2821 5′-GTCATAAAGTGGTGGTAAAACTT-3′ | |
| LPW2319 5′-ATTAATGCTAGAGAYCTHMTTTG-3′ | LPW2320 5′-TTTGGGTAACTCCAATNCCRTT-3′ | |
| LPW2824 5′-TTTGCCGCTATACCTTTTGCACAA-3′ | LPW4106 5′-TGAGTTATAGGTTCAGGTTTATAA-3′ | |
| LPW4105 5′-TATTAGTGACATCCTTGCTAGGCTT-3′ | LPW2317 5′-GAGCCAAACATACCANGGCCAYTT-3′ | |
| LPW4107 5′-ATGGTCCTAACTTTGCAGAGATA-3′ | LPW21565 5′-TGCCAGACATGCCACCACAA-3′ | |
| N | LPW21407 5′-AACGAATCTTAATAACTCATTGTT-3′ | LPW21408 5′-CTCTTGTTACTCTTCATTGGCAT-3′ |
| Pi-BatCoV HKU5 | ||
| RdRp | LPW3350 5′-TTTGTCAATTTTGGATAGGACAT-3′ | LPW3352 5′-TGATGCATCACAGCARCCATA-3′ |
| LPW3351 5′-ATCAGAATAACTGTGAAGTGCTT-3′ | LPW3275 5′-GACAATTGGACCAAAAGACGTT-3′ | |
| LPW3382 5′-CAAATTGTGTGAACTGTACTGAT-3′ | LPW3387 5′-ATATATCTCGAAGTAACGATCAA-3′ | |
| LPW3172 5′-GTCCTGGCAACTTTAATAAAGATT-3′ | LPW3130 5′-CTAATATGAGAGATGCAAAGA-3′ | |
| LPW1507 5′-GGTTGGGACTATCCTAAGTGTGA-3′ | LPW1508 5′-CCATCATCAGATAGAATCATCATA-3′ | |
| LPW3384 5′-CTAAATTTGTGGACAGGTATTAT-3′ | LPW3399 5′-CTTCGTATACACGTACCACAA-3′ | |
| S | LPW21416 5′-CTCTTGTCGCAGGGTAAACTT-3′ | LPW4284 5′-AAAGACTCTACCTGTGCAGAATA-3′ |
| LPW4086 5′-TAACTTATACTGGACTGTACCCAAA-3′ | LPW4193 5′-AAGCCATTTGAAGGTTACCATT-3′ | |
| LPW4192 5′-ACTTTGCTACTTTACCTGTGTAT-3′ | LPW4137 5′-AGTAACACCAAATGTGAAATT-3′ | |
| LPW4285 5′-AATCGCCACTCTAAACTTTACTA-3′ | LPW4286 5′-AAGAGGCTGGGTATTCTGGGTT-3′ | |
| LPW4138 5′-AAGATGAGTCTATTGCTAATCTAT-3′ | LPW4139 5′-AGCTTCCATATAGGGGTCATA-3′ | |
| LPW4287 5′-TGTGCACAATATGTTGCTGGCTA-3′ | LPW4288 5′-AAAGAACTACCAGTATAATACCAA-3′ | |
| LPW4140 5′-AACACTGAGAATCCACCAAA-3′ | LPW21417 5′-CACACGCATCATAAGTTCGTT-3′ | |
| N | LPW21361 5′-GAATCTTATTATCTCATTGTT-3′ | LPW21362 5′-CTATTACGTTCAATTGGCAAT-3′ |
Estimation of synonymous and nonsynonymous substitution rates.
The number of synonymous substitutions per synonymous site, _K_s, and the number of nonsynonymous substitutions per nonsynonymous site, _K_a, for each coding region were calculated using the Nei-Gojobori (Jukes-Cantor) method in MEGA version 5 (50).
Detection of positive selection.
Sites under positive selection in the S gene in Ty-BatCoV-HKU4 and Pi-BatCoV-HKU5 were inferred using the single-likelihood ancestor counting (SLAC), fixed-effects likelihood (FEL), and random-effects likelihood (REL) methods as implemented in DataMonkey server (http://www.datamonkey.org) (56). Positive selection for a site was considered to be statistically significant if the P value was <0.1 for the SLAC and FEL methods or the posterior probability was at the ≥90% level for the REL method. A mixed-effects model of evolution (MEME) was further used to identify positively selected sites under conditions of episodic diversifying selection in particular positions in sublineages within a phylogenetic tree even when positive selection was not evident across the entire tree (57). Positively selected sites with a P value < 0.05 were reported.
Estimation of divergence time.
As RdRp and N genes are relatively conserved across CoVs and therefore most likely reflect viral phylogeny, divergence time was calculated using complete RdRp and N gene sequence data of Ty-BatCoV HKU4, Pi-BatCoV HKU5, and MERS-CoV strains, and 904 bp of partial RdRp sequence data of lineage C betaCoVs from European bats, with the Bayesian Markov chain Monte Carlo (MCMC) approach as implemented in BEAST (Version 1.7.4) and described previously (9, 17, 21, 44, 58, 59). One parametric model (Constant Size) and one nonparametric model (Bayesian Skyline with five groups) tree prior were used for the inference. Analyses were performed using the Hasegawa-Kishino-Yano (HKY) model with coding sequenced partitioned into the first plus second positions versus the third position, and rate variations between sites were described by a four-category discrete gamma distribution using both strict and relaxed (uncorrelated lognormal [Ucld] and uncorrelated exponential [Uced]) molecular clocks. The MCMC run was 2× 108 steps in length, with sampling every 1,000 steps. Convergence was assessed on the basis of the effective sampling size after a 10% burn-in using Tracer software version 1.5 (58). The mean time of the most recent common ancestor (tMRCA) and the highest posterior density regions at 95% (HPD) were calculated, and the best-fitting model was selected by a Bayes factor, using marginal likelihoods implemented in Tracer (60). Bayesian Skyline analysis using a relaxed clock model with Uced was adopted for making inferences, as this model fitted the data better than other models tested by Bayes factor analysis (data not shown) and allowed variations in substitution rates among lineages. All trees were summarized in a target tree by the Tree Annotator program included in the BEAST package by choosing the tree with the maximum sum of posterior probabilities (maximum clade credibility) after a 10% burn-in.
Nucleotide sequence accession numbers.
The nucleotide sequences of the complete RdRp, S, and N genes of Ty-BatCoV HKU4 and Pi-BatCoV HKU5 have been lodged within the GenBank sequence database under accession no. KC522036 to KC522119.
RESULTS
Detection of Ty-BatCoV HKU4 and Pi-BatCoV HKU5 from bat samples.
A total of 5,426 respiratory and 5,260 alimentary specimens from 5,481 bats of 21 different species were obtained. RT-PCR for a 440-bp fragment in the RdRp genes of CoVs detected the presence of lineage C betaCoVs from two bat species, including Ty-BatCoV HKU4 in 29 (29%) of 99 alimentary samples from lesser bamboo bat (Tylonycteris pachypus) and Pi-BatCoV HKU5 in 55 (25%) of 216 alimentary samples from Japanese pipistrelle (Pipistrellus abramus) (Table 2). None of the respiratory samples were positive for lineage C betaCoVs. Bats positive for Ty-BatCoV HKU4 and Pi-BatCoV HKU5 were from 7 and 13 sampling locations in Hong Kong, respectively. No obvious disease was observed in bats positive for Ty-BatCoV HKU4 and Pi-BatCoV HKU5. Ty-BatCoV HKU4 was found only in adult bats, while Pi-BatCoV HKU5 was found in both adult and juvenile bats.
Table 2.
Detection of Ty-BatCoV HKU4 and Pi-BatCoV HKU5 in bats by RT-PCR
| Bat scientific name | Common name | No. of bats tested | No. (%) of bats positive for CoV in respiratory samples | No. (%) of bats positive for CoV in alimentary samples | ||
|---|---|---|---|---|---|---|
| Ty-BatCoV HKU4 | Pi-BatCoV HKU5 | Ty-BatCoV HKU4 | Pi-BatCoV HKU5 | |||
| Megachiroptera | ||||||
| Pteropodidae | ||||||
| Cynopterus sphinx | Short-nosed fruit bat | 26 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Rousettus leschenaulti | Leschenault's rousette | 73 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Microchiroptera | ||||||
| Hipposideridae | ||||||
| Hipposideros armiger | Himalayan leaf-nosed bat | 198 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Hipposideros pomona | Pomona leaf-nosed bat | 642 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Rhinolophidae | ||||||
| Rhinolophus affinus | Intermediate horseshoe bat | 359 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Rhinolophus pusillus | Least horseshoe bat | 89 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Rhinolophus sinicus | Chinese horseshoe bat | 2012 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Vespertilionidae | ||||||
| Hypsugo pulveratus | Chinese pipistrelle | 1 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Miniopterus magnater | Greater bent-winged bat | 15 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Miniopterus pusillus | Lesser bent-winged bat | 450 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Miniopterus schreibersii | Common bent-winged bat | 758 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Myotis chinensis | Chinese myotis | 122 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Myotis horsfieldii | Horsfield's bat | 7 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Myotis muricola | Whiskered myotis | 4 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Myotis ricketti | Rickett's big-footed bat | 307 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Nyctalus noctula | Brown noctule | 54 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Pipistrellus abramus | Japanese pipistrelle | 219 | 0 (0) | 0 (0) | 0 (0) | 55 (25) |
| Pipistrellus tenuis | Least pipistrelle | 11 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Scotophilus kuhlii | Lesser yellow bat | 18 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Tylonycteris pachypus | Lesser bamboo bat | 115 | 0 (0) | 0 (0) | 29 (29) | 0 (0) |
| Tylonycteris robustula | Greater bamboo bat | 1 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Complete RdRp, S, and N gene analysis of Ty-BatCoV HKU4 and Pi-BatCoV HKU5 strains.
To study the genetic diversity and evolution of lineage C betaCoVs in bats, the complete RdRp, S, and N genes of 13 Ty-BatCoV HKU4 strains and 15 Pi-BatCoV HKU5 strains were sequenced. Comparison of the deduced amino acid (aa) sequences of the RdRp, S, and N genes of Ty-BatCoV HKU4 and Pi-BatCoV HKU5 to those of MERS-CoV showed that MERS-CoV is more closely related to Pi-BatCoV HKU5 than to Ty-BatCoV HKU4 (92.1% to 92.3% versus 89.6% to 90% identity) in the RdRp gene but is more closely related to Ty-BatCoV HKU4 than to Pi-BatCoV HKU5 in the S (66.8% to 67.4% versus 63.4% to 64.5% identity) and N (71.9% to 72.3% versus 69.5% to 70.5% identity) genes (Table 3). Moreover, MERS-CoV is more closely related to Ty-BatCoV HKU4 and Pi-BatCoV HKU5 belonging to Betacoronavirus lineage C than to CoVs belonging to Betacoronavirus lineages A, B, and D (Table 3). Phylogenetic analysis of the complete RdRp, S, and N gene sequences of Ty-BatCoV HKU4 and Pi-BatCoV HKU5 showed that the sequences from the 13 Ty-BatCoV HKU4 strains and 15 Pi-BatCoV HKU5 strains formed two distinct clusters in all three genes, being closely related to each other and to MERS-CoV (Fig. 1). Interestingly, unlike the S genes of the 13 Ty-BatCoV HKU4 strains, which shared highly similar sequences with very short branch lengths, the S genes of Pi-BatCoV HKU5 displayed marked sequence polymorphisms among the 15 strains, with up to 14% nucleotide and 12% amino acid (aa) differences.
Table 3.
Pairwise amino acid identities between the RdRp, S, and N genes of Ty-BatCoV HKU4, Pi-BatCoV HKU5, and MERS-CoV and those of other betaCoVs
| Coronavirus | Pairwise amino acid identity (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ty-BatCoV HKU4_2 | Pi-BatCoV HKU5_31 | MERS-CoV | |||||||
| RdRp | S | N | RdRp | S | N | RdRp | S | N | |
| Betacoronavirus lineage A | |||||||||
| HCoV-OC43 | 68.8 | 33.4 | 33.2 | 68.7 | 31.2 | 34.2 | 68.3 | 32 | 35.3 |
| BCoV | 68.7 | 33.5 | 33.2 | 68.6 | 31.3 | 34.8 | 68.2 | 31.3 | 35.6 |
| PHEV | 68.8 | 33.2 | 32.7 | 68.7 | 31.2 | 33.9 | 68.3 | 32.5 | 35.1 |
| GiCoV | 68.7 | 33.9 | 32.8 | 68.6 | 31.6 | 34.8 | 68.2 | 31.4 | 35.3 |
| RCoV | 68.8 | 32.4 | 33.7 | 68.8 | 31.4 | 34.3 | 68.7 | 32 | 34.8 |
| RbCoV HKU14 | 68 | 33.8 | 33.2 | 68 | 30.9 | 34.9 | 68 | 32.2 | 35.3 |
| AntelopeCoV | 68.7 | 33.7 | 32.8 | 68.6 | 31.2 | 34.8 | 68.2 | 31.4 | 35.3 |
| ECoV | 69.1 | 32.4 | 34.9 | 68.7 | 31.5 | 35.6 | 68.3 | 31.6 | 35.7 |
| MHV | 68.7 | 32.7 | 34.1 | 68.8 | 31.9 | 34.7 | 68.6 | 31.5 | 34.3 |
| HCoV-HKU1 | 67.6 | 32.1 | 32.8 | 68.1 | 30.2 | 33.3 | 67.9 | 31.8 | 32.3 |
| Betacoronavirus lineage B | |||||||||
| SARS-CoV | 71.6 | 33.6 | 45.8 | 71.8 | 33.5 | 43.6 | 71.9 | 31.6 | 46.6 |
| SARSr-Rh-BatCoV HKU3 | 71.7 | 33.6 | 45.2 | 71.7 | 32.8 | 43.9 | 71.8 | 30.6 | 46.2 |
| Betacoronavirus lineage C | |||||||||
| Ty-BatCoV HKU4 | 99.5–100 | 97.3–99.6 | 99.5–100 | 92–92.5 | 67.7–68.1 | 73.5–74 | 89.6–90 | 66.8–67.4 | 71.9–72.3 |
| Pi-BatCoV HKU5 | 92.1–92.4 | 67.5–68.4 | 73.7–75.1 | 99.4–99.7 | 88.3–97 | 97.2–98.6 | 92.1–92.3 | 63.4–64.5 | 69.5–70.5 |
| MERS-CoV | 89.9 | 67.3–67.4 | 71.6–72.1 | 92.1 | 64.3 | 68.8–69.5 | |||
| Betacoronavirus lineage D | |||||||||
| Ro-BatCoV HKU9 | 69.3 | 30.8 | 37.3 | 68.7 | 31 | 36.9 | 68.4 | 30.3 | 37.8 |
Fig 1.

Phylogenetic analysis of RdRp, S, and N genes of Ty-BatCoV HKU4 and Pi-BatCoV HKU5 strains and those of other betaCoVs with available complete genome sequences. The trees were constructed by the maximum-likelihood method with bootstrap values calculated from 100 trees. The analysis included 937, 1,535, and 546 aa positions in the RdRp, S, and N genes, respectively. The scale bars indicate the estimated number of substitutions per 5 or 20 aa. HCoV-HKU1, human coronavirus HKU1; HCoV-OC43, human coronavirus OC43; MHV, murine hepatitis virus; BCoV, bovine coronavirus; PHEV, porcine hemagglutinating encephalomyelitis virus; GiCoV, giraffe coronavirus; RCoV, rat coronavirus; ECoV, equine coronavirus; RbCoV HKU14, rabbit coronavirus HKU14; AntelopeCoV, sable antelope coronavirus; SARS-CoV, SARS coronavirus; SARSr-Rh-BatCoV HKU3, SARS-related Rhinolophus bat coronavirus HKU3; SARSr-CiCoV, SAR-related civet coronavirus; SARSr CoV CFB, SARS-related Chinese ferret badger coronavirus; Ty-BatCoV HKU4, Tylonycteris bat coronavirus HKU4; Pi-BatCoV HKU5, Pipistrellus bat coronavirus HKU5; MERS-CoV EMC, Middle East Respiratory Syndrome Coronavirus EMC; MERS-CoV England1, Middle East Respiratory Syndrome Coronavirus England1; Ro-BatCoV HKU9, Rousettus bat coronavirus HKU9.
The S proteins of Ty-BatCoV HKU4 and Pi-BatCoV HKU5 encoded 1,350 to 1,352 and 1352 to 1,359 aa, respectively. A potential cleavage site, though not perfectly conserved, could be present in the S proteins of Ty-BatCoV HKU4 (S[TM]FR) and Pi-BatCoV HKU5 (R[VFL][ALR]R). InterProScan analysis predicted them to be type I membrane glycoproteins, with most of the protein (residues 18/21/22 to 1294/1296/1297 for Ty-BatCoV HKU4 and residues 22 to 1296/1297/1298/1301/1302/1303 for Pi-BatCoV HKU5) exposed outside the virus, a transmembrane domain (residues 1295/1297/1298 to 1317/1319/1320 for Ty-BatCoV HKU4 and residues 1297/1298/1299/1302/1303/1304 to 1319/1320/1321/1324/1325/1326 for Pi-BatCoV HKU5) at the C terminus, followed by a cytoplasmic tail rich in cysteine residues. Two heptad repeats (HR), important for membrane fusion and viral entry (61), were located at residues 978/980 to 1124/1126 (HR1) and 1251/1253 to 1285/1287 (HR2) for Ty-BatCoV HKU4 and residues 978/979/983/984 to 1124/1125/1129/1130 (HR1) and 1253/1254/1258/1259 to 1287/1288/1292/1293 (HR2) for Pi-BatCoV HKU5. All cysteine residues are conserved between the S genes of Ty-BatCoV HKU4, Pi-BatCoV HKU5, and MERS-CoV. While CoVs are known to utilize a variety of host receptors for cell entry, a number of closely related as well as distantly related CoVs may utilize the same receptor. For example, aminopeptidase N (CD13) has been shown to be the receptor for various alphaCoVs, including human Cov (HCoV) 229E, canine CoV (CCoV), feline infectious peritonitis virus (FIPV), porcine epidemic diarrhea coronavirus (PEDV), and transmissible gastroenteritis coronavirus (TGEV) (62, 63). Moreover, human angiotensin-converting enzyme 2 (hACE2) has been found to be the receptor for HCoV NL63, an alphaCoV, as well as for SARS CoV, a betaCoV, although they utilize different receptor binding sites (64, 65). As for lineage A betaCoVs, HCoV OC43 and the closely related bovine CoV utilize N-acetyl-9-O acetyl neuramic acid as a receptor, whereas carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) is the receptor for mouse hepatitis virus (MHV) (66–70). The S proteins of Ty-BatCoV HKU4 and Pi-BatCoV HKU5 as well as MERS-CoV did not exhibit significant sequence homology to the known receptor binding domains (RBDs) of other CoVs, including betaCoVs such as SARS CoV and HCoV OC43 (71–78). Recently, DPP4 has been identified as a functional receptor for MERS-CoV, although the exact RBD is still unknown (47, 79). Based on the X-ray crystal structure of the RBD in the SARS CoV S protein, residues 377 to 662 have been predicted as a possible RBD for MERS-CoV (80). Using the same methodology, residues 387 to 587 in Ty-BatCoV HKU4 S protein and residues 389 to 580 in Pi-BatCoV HKU5 S protein were predicted to be their possible RBDs. However, further studies are required to elucidate the receptors for Ty-BatCoV HKU4 and Pi-BatCoV HKU5 and their RBDs.
Estimation of synonymous and nonsynonymous substitution rates.
In line with the phylogenetic analysis, multiple alignment of the S gene sequences showed that Pi-BatCoV HKU5 possessed more synonymous and nonsynonymous substitutions than Ty-BatCoV HKU4 (Table 4). Compared to Ty-BatCoV HKU4, in which 58 aa positions contained substitutions, 253 aa positions in Pi-BatCoV HKU5 contained substitutions, among which ≥2 aa were encoded at 67 aa positions (Fig. 2 and 3). The _K_a/_K_s ratios for the RdRp, S, and N genes among different strains of Ty-BatCoV HKU4 and Pi-BatCoV HKU5 were determined (Table 4). The _K_a/_K_s ratios were generally low, although the S genes of both viruses showed relatively higher ratios (0.118) than the RdRp and N genes. This suggested that these genes were under purifying selection. Nevertheless, the _K_a and _K_s values for the S genes of Pi-BatCoV HKU5 were relatively high compared to those of Ty-BatCoV HKU4, which reflected the marked sequence polymorphisms among different strains.
Table 4.
Estimation of nonsynonymous and synonymous substitution rates in the RdRp, S, and N genes of Ty-BatCoV HKU4, Pi-BatCoV HKU5, and MERS-CoV
| Gene | Substitution rate (substitutions/site/yr) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ty-BatCoV HKU4 (18 strains) | Pi-BatCoV HKU5 (19 strains) | MERS-CoV (2 strains) | |||||||
| _K_a | _K_s | _K_a/_K_s | _K_a | _K_s | _K_a/_K_s | _K_a | _K_s | _K_a/_K_s | |
| RdRp | 0.001 | 0.033 | 0.03 | 0.001 | 0.128 | 0.0078 | 0 | 0.006 | 0 |
| S | 0.004 | 0.034 | 0.118 | 0.038 | 0.321 | 0.118 | 0.001 | 0.008 | 0.125 |
| N | 0.001 | 0.019 | 0.053 | 0.005 | 0.095 | 0.053 | 0.002 | 0.010 | 0.2 |
Fig 2.
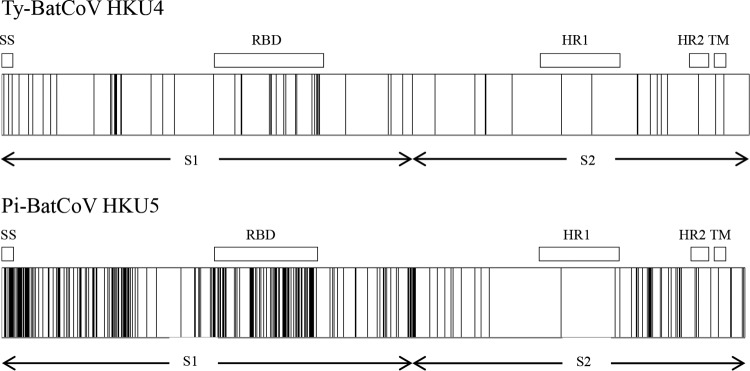
Distribution of amino acid changes in the spike protein of Ty-BatCoV HKU4 (upper panel) and Pi-BatCoV HKU5 (lower panel). The positions of the amino acid changes are depicted by vertical lines. SS, predicted signal peptide; RBD, receptor binding domain; HR1, heptad repeat 1; HR2, heptad repeat 2; TM, transmembrane domain.
Fig 3.

Graphical representation of multiple sequence alignment showing the amino acid changes in the spike protein of Pi-BatCoV HKU5. The height of each symbol indicates the relative frequency of each amino acid at the position. Polar amino acids are indicated in green; neutral amino acids are indicated in purple; basic amino acids are indicated in blue; acidic amino acids are indicated in red; hydrophobic amino acids are indicated in black. The figure was generated using WebLogo (91).
Detection of positive selection in S genes.
The S genes of Pi-BatCoV HKU5 possessed more positively selected sites than the S genes of Ty-BatCoV HKU4 (Fig. 4). Only two and five aa positions in Ty-BatCoV HKU4 were found to be under positive selection using the REL and MEME methods, respectively, whereas no significant positive selection was identified by the SLAC and FEL methods. In contrast, 2, 12, 27, and 43 aa positions in Pi-BatCoV HKU5 were found to be under positive selection using the SLAC, FEL, REL, and MEME methods, respectively. Most of these sites were distributed within the S1 domain, indicating that this domain may have been under functional constraints.
Fig 4.

Distribution of positively selected sites in S proteins identified using REL in Ty-BatCoV HKU4 (upper panel) and Pi-BatCoV HKU5 (lower panel). Positively selected sites with posterior probability greater than 0.5 are shown.
Estimation of divergence time.
To estimate the divergence times of the Ty-BatCoV HKU4, Pi-BatCoV HKU5, and MERS-CoV strains, their complete RdRp and N gene sequences were subjected to molecular clock analysis using the relaxed clock model with Uced. By the use of complete RdRp gene sequences, the tMRCA of MERS-CoV and Pi-BatCoV HKU5 was estimated at 1520.09 (HPDs, 745.73 to 1956.12) (Fig. 5A). Using complete N gene sequences, the tMRCA of MERS-CoV, Ty-BatCoV HKU4, and Pi-BatCoV HKU5 was estimated at 1323.51 (HPDs, 383.58 to 1897.75) (Fig. 5B). Since partial RdRp gene sequences closely related to the corresponding sequence of MERS-CoV have recently been detected in European bats, molecular clock analysis was also performed to estimate their divergence time. Using the 904-bp partial RdRp sequences, the tMRCA of MERS-CoV and three European bat CoV strains (BtCoV 8-691, BtCoV 8-724, and BtCoV UKR-G17) was estimated at 1859.32 (HPDs, 1636.67 to 1987.55) (Fig. 5C). The estimated mean substitution rates of the complete RdRp and N gene and partial RdRp sequence data sets were 5.12 × 10−4, 8.642 × 10−4, and 7.407 × 10−4 substitutions per site per year, comparable to those observed in other CoVs (9, 17, 59, 81, 82).
Fig 5.

Estimation of the tMRCA of Ty-BatCoV HKU4 and Pi-BatCoV HKU5. The time-scaled phylogeny was summarized from all MCMC phylogenies of the complete RdRp (A), complete N (B), and 904-bp RdRp (C) sequence data sets analyzed using the relaxed clock model with an exponential distribution (Uced) in BEAST v 1.7.4. Viruses characterized in this study are bolded.
DISCUSSION
In this study, Ty-BatCoV HKU4 and Pi-BatCoV HKU5 were found to be highly prevalent among lesser bamboo bat and Japanese pipistrelle in Hong Kong, respectively, with detection rates of 25% to 29% in their alimentary samples. In line with previous studies, MERS-CoV is more closely related to Betacoronavirus lineage C than to lineages A, B, and D in the RdRp, S, and N genes (34, 42, 43). Nevertheless, the genetic distance between MERS-CoV and the various strains of Ty-BatCoV HKU4 and Pi-BatCoV HKU5 was still large, with their S proteins having ≤67.4% aa identity. Two recent studies have identified partial gene sequences closely related to MERS-CoV in bats from Africa, Europe, and America, suggesting that lineage C betaCoVs are distributed in bats worldwide (44, 45). In one study, CoVs related to MERS-CoV were detected in 46 (24.9%) Nycteris bats and 40 (14.7%) Pipistrellus bats from Ghana and Europe using RT-PCR targeting a 398-bp fragment of the RdRp gene (44). The extended 904-bp RdRp sequences of three strains from Romania and Ukraine showed that they shared 87.7% to 88.1% nucleotide and 98.3% amino acid identity with those of MERS-CoV compared to 80.3% to 82% and 82.4% to 83.7% nucleotide and 92% to 92.4% and 94 to 94.4% amino acid identity between Ty-BatCoV HKU4/Pi-BatCoV HKU5 and MERS-CoV, respectively, in the corresponding regions. In another study, screening of 606 bats from Mexico also showed the presence of a closely related betaCoV MERS-CoV in a Nyctinomops lacticaudatus bat (45). Although the authors claimed to have used a 329-bp fragment of the RdRp gene for RT-PCR and sequence analysis, the available sequence was in fact within nsp14. Analysis of this partial nsp14 sequence showed that it shared 85.7% nucleotide and 95.5% amino acid identity with that of MERS-CoV (45) compared to 81.9% and 83.4% to 84.2% nucleotide and 88.6% and 92% amino acid identity differences between Ty-BatCoV HUK4/Pi-BatCoV HKU5 and MERS-CoV, respectively, in the corresponding regions. However, complete gene sequences were not available from these bat CoVs to allow more detailed phylogenetic analysis. Molecular clock analysis of the complete RdRp gene dated the tMRCA of MERS-CoV and Pi-BatCoV HKU5 at around 1520, whereas analysis of the N gene dated the tMRCA of MERS-CoV, Ty-BatCoV HKU4, and Pi-BatCoV HKU5 at around 1324. Using the 904-bp RdRp sequences available from the three European strains, the tMRCA of MERS-CoV and European bat CoV strains were dated at around 1859. Our results suggested that Ty-BatCoV HKU4, Pi-BatCoV HKU5, and MERS-CoV diverged at least centuries ago from their common ancestor. Although MERS-CoV and the European bat CoV strains were estimated to have diverged more recently, this is unlike the situation in SARS-related CoVs, which diverged between civet and bat strains only a few years before the SARS epidemic (17). Therefore, these bat lineage C betaCoVs were unlikely to be the direct ancestor of MERS-CoV. However, the present analysis is limited by the lack of more sequences from potential intermediate virus species or strains with widely distributed and well-determined dates, which better reflect the different selective pressures over the long period of time as these viruses evolved. Further studies on bats and other animals are required to fill the gap between these bat lineage C betaCoVs and MERS-CoV during their evolution. Moreover, longer gene or complete genome sequence data from these animal viruses would be important for more accurate taxonomic and evolutionary studies.
The divergent sequences of the S genes of Pi-BatCoV HKU5 may suggest that the virus has a better ability to generate variants to occupy new ecological niches. The S proteins of CoVs are responsible for receptor binding and host adaptation and are therefore among the most variable regions within CoV genomes (16, 18, 28). Studies on SARS CoV have shown that changes in its S protein, both within and outside the receptor binding domain, could govern CoV cross-species transmission and emergence in new host populations (83, 84). We have also previously demonstrated recent interspecies transmission of an alphaCoV, BatCoV HKU10, from Leschenault's rousettes to Pomona leaf-nosed bats, and the virus has been rapidly adapting in the new host by changing its S protein (59). In this study, Ty-BatCoV HKU4 and Pi-BatCoV HKU5 were exclusively detected in lesser bamboo bat (Tylonycteris pachypus) and Japanese pipistrelle (Pipistrellus abramus), respectively. Moreover, the _K_a/_K_s ratios of the RdRp, S, and N genes in both viruses were low, supporting the idea that the two bat species were the respective primary reservoirs for the two CoVs. Nevertheless, in comparison to that of Ty-BatCoV HKU4, the S gene of Pi-BatCoV HKU5 exhibited much higher sequence divergence among different strains due to both synonymous and nonsynonymous substitutions. Moreover, a much higher number of positively selected sites were observed in the S gene of Pi-BatCoV HKU5 than in that of Ty-BatCoV HKU4, with most of the sites under selection being distributed within the S1 region which likely contains the RBD. This suggested that the S1 region of Pi-BatCoV HKU5 may have been under functional constraints in its host species, Japanese pipistrelle, which may have favored adaptation to new hosts or environments.
The marked polymorphisms in the S protein of Pi-BatCoV HKU5 may reflect the biological characteristics of its host species, Japanese pipistrelle, which is a small-size, insectivorous bat with a body weight of 4 to 10 g. It is considered the most common bat species found in urban areas of Hong Kong (85). While it is abundant in wetland areas, its roosts are frequently found in towns and villages as well as in various types of buildings and other man-made structures, such as fans or air conditioners. It is also known to utilize bat houses or boxes as its roosts. Such diverse habitat and adaptability to harsh environments may have favored the mutation of Pi-BatCoV HKU5, especially in its S protein, which is responsible for receptor binding and immunogenicity. Interestingly, this bat species is widely distributed not only in China, Russia, the Korean peninsula, Japan, Vietnam, Burma, and India but also in the Kingdom of Saudi Arabia and neighboring countries (42, 85). Moreover, other Pipistrellus bats, including P. arabicus, P. ariel, P. kuhlii, P. pipistrellus, P. rueppellii, and P. savii, have been recorded in the Arabian Peninsula (http://www.iucn.org/). In fact, partial sequences closely related to those of MERS-CoV detected in bats from Europe also originated from Pipstrellus bats (P. pipistrellus, P. nathusii, and P. pygmaeus) of the family Vespertilionidae, and those from Ghana originated from Nycteris bats (Nycteris cf. gambiensis) of the related family Nycteridae (44). Similarly, the bat betaCoV strain related to MERS-CoV detected in Mexico originated from a N. laticaudatus bat belonging to the Molossidae, a closely related family of Vespertilionidae (45, 86). The difference between this BatCoV and MERS-CoV within the partial nsp14 sequence was also found to be mainly due to substitutions in the third nucleotide positions, suggesting strong purifying selection (45). However, S gene sequences were not available from these bat viruses for further analysis of polymorphisms and selective pressures. Nevertheless, based on our existing data, bats belonging to Vespertilionidae and related families, especially Pipistrellus bats and those with diverse habitats, in the Arabian Peninsula should be intensively sought for potential ancestral viruses of MERS-CoV, which may have evolved through mutations in the S gene, especially in the RBD, allowing efficient transmission to other animals or human. In contrast, lesser bamboo bats, the host species for Ty-BatCoV HKU4 and one of the smallest mammals in the world, with a body weight of 3 to 7 g, have much more restricted habitats. Though this species also belongs to the family Vespertilionidae, it is remarkably adapted to roost inside bamboo stems and is mainly found in rural areas in Hong Kong and various Asian countries (85). This may, in turn, reflect the lower mutation rate observed in the S gene of Ty-BatCoV HKU4.
It remains to be determined if Ty-BatCoV HKU4 and Pi-BatCoV HKU5, as well as other lineage C betaCoVs in bats, utilize the same receptor as MERS-CoV. Recent studies have shown that MERS-CoV utilizes DPP4 as its functional receptor (47, 79). This suggested that these betaCoVs belonging to lineage C may utilize a receptor(s) different from those of other CoVs. Moreover, expression of bat (P. pipistrellus) DPP4 in nonsusceptible cells was found to enable infection by MERS-CoV (47), which is in line with the ability of the virus to replicate in cell lines from Rousettus, Rhinolophus, Pipistrellus, Myotis, and Carollia bats (79). As DPP4 is a evolutionarily conserved protein (47), it may also explain the broad species tropism observed in primate, porcine, and rabbit cell lines and reflect the zoonotic origin of MERS-CoV (46, 79). However, Ty-BatCoV HKU4 and Pi-BatCoV HKU5, as with other bat CoVs, have not been successfully cultured in vitro, which hampers studies on their receptor binding and host adaptation. Further discoveries of lineage C betaCoVs in animals and studies on the receptors of the different animal counterparts in their respective hosts may help further understanding of the mechanism of interspecies transmission and emergence of MERS-CoV.
Bats are increasingly recognized as a reservoir for various zoonotic viruses, including SARS CoV, lyssavirus, and rabies virus and Hendra, Nipah, and Ebola as well as influenza virus (87, 88). While the existence of CoVs in bats was unknown before the SARS epidemic, it is now known that the different bat populations harbor diverse CoVs, which is likely the result of their species diversity, roosting behavior, and migrating ability (16, 18, 29, 31, 32, 89). These warm-blooded flying vertebrates are also ideal hosts to fuel CoV recombination and dissemination (5, 27, 59). It remains to be ascertained if bats could also be the animal origin for the emergence of MERS-CoV either directly or via an intermediate host, the latter as in the case of SARS CoV, where the bat ancestral virus may have jumped to the intermediate host when bats were in contact or mixed with other animals (16). Since the history of contact with animals such as camels and goats has been reported in MERS-CoV-infected cases (90), the virus may have jumped from bats to these animals before infecting humans. Surveillance studies of lineage C betaCoVs from bats and other animals in the Middle East may help identify the origin and chain of transmission of MERS-CoV.
ACKNOWLEDGMENTS
We thank Alan Chi-Kong Wong and Siu-Fai Leung (HKSAR Department of Agriculture, Fisheries, and Conservation [AFCD]) and the Hong Kong Police Force for facilitation and support; Chung-Tong Shek and Joseph W. K. So from AFCD; and King-Shun Lo (Laboratory Animal Unit, The University of Hong Kong) and Cassius Chan for their excellent technical assistance and collection of animal specimens.
We are grateful for the generous support of Carol Yu, Richard Yu, Hui Hoy, and Hui Ming in the genomic sequencing platform. This work was partly supported by the Research Grant Council Grant, University Grant Council; Strategic Research Theme Fund and University Development Fund, The University of Hong Kong; HKSAR Research Fund for the Control of Infectious Diseases of the Food and Health Bureau; Shaw Foundation; Providence Foundation Limited in memory of the late Lui Hac Minh; a donation from Eunice Lam; and the Consultancy Service for Enhancing Laboratory Surveillance of Emerging Infectious Disease for the HKSAR Department of Health.
Footnotes
Published ahead of print 29 May 2013
REFERENCES
- 1.Lai MM, Cavanagh D. 1997. The molecular biology of coronaviruses. Adv. Virus Res. 48:1–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ziebuhr J. 2004. Molecular biology of severe acute respiratory syndrome coronavirus. Curr. Opin. Microbiol. 7:412–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Groot RJ, Baker SC, Baric R, Enjuanes L, Gorbalenya A, Holmes KV, Perlman S, Poon L, Rottier PJ, Talbot PJ, Woo PC, Ziebuhr J. 2011. Coronaviridae, p 806–828_In_King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. (ed), Virus taxonomy, classification and nomenclature of viruses. Ninth report of the International Committee on Taxonomy of Viruses, International Union of Microbiological Societies, Virology Division Elsevier Academic Press, Philadelphia, PA [Google Scholar]
- 4.Woo PC, Lau SK, Lam CS, Lai KK, Huang Y, Lee P, Luk GS, Dyrting KC, Chan KH, Yuen KY. 2009. Comparative analysis of complete genome sequences of three avian coronaviruses reveals a novel group 3c coronavirus. J. Virol. 83:908–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woo PC, Lau SK, Lam CS, Lau CC, Tsang AK, Lau JH, Bai R, Teng JL, Tsang CC, Wang M, Zheng BJ, Chan KH, Yuen KY. 2012. Discovery of seven novel mammalian and avian coronaviruses in the genus Deltacoronavirus supports bat coronaviruses as the gene source of Alphacoronavirus and Betacoronavirus and avian coronaviruses as the gene source of Gammacoronavirus and Deltacoronavirus. J. Virol. 86:3995–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong BQ, Liu W, Fan XH, Vijaykrishna D, Tang XC, Gao F, Li LF, Li GJ, Zhang JX, Yang LQ, Poon LL, Zhang SY, Peiris JS, Smith GJ, Chen H, Guan Y. 2007. Detection of a novel and highly divergent coronavirus from Asian leopard cats and Chinese ferret badgers in Southern China. J. Virol. 81:6920–6926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mihindukulasuriya KA, Wu G, St Leger J, Nordhausen RW, Wang D. 2008. Identification of a novel coronavirus from a beluga whale by using a panviral microarray. J. Virol. 82:5084–5088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herrewegh AA, Smeenk I, Horzinek MC, Rottier PJ, de Groot RJ. 1998. Feline coronavirus type II strains 79-1683 and 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J. Virol. 72:4508–4514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lau SK, Lee P, Tsang AK, Yip CC, Tse H, Lee RA, So LY, Lau YL, Chan KH, Woo PC, Yuen KY. 2011. Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J. Virol. 85:11325–11337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woo PC, Lau SK, Huang Y, Yuen KY. 2009. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. (Maywood) 234:1117–1127 [DOI] [PubMed] [Google Scholar]
- 11.Woo PC, Lau SK, Yip CC, Huang Y, Tsoi HW, Chan KH, Yuen KY. 2006. Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J. Virol. 80:7136–7145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeng Q, Langereis MA, van Vliet AL, Huizinga EG, de Groot RJ. 2008. Structure of coronavirus hemagglutinin-esterase offers insight into corona and influenza virus evolution. Proc. Natl. Acad. Sci. U. S. A. 105:9065–9069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ, SARS Working Group 2003. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348:1953–1966 [DOI] [PubMed] [Google Scholar]
- 14.Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, Nicholls J, Yee WK, Yan WW, Cheung MT, Cheng VC, Chan KH, Tsang DN, Yung RW, Ng TK, Yuen KY. 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Luo SW, Li PH, Zhang LJ, Guan YJ, Butt KM, Wong KL, Chan KW, Lim W, Shortridge KF, Yuen KY, Peiris JS, Poon LL. 2003. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302:276–278 [DOI] [PubMed] [Google Scholar]
- 16.Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, Wong SS, Leung SY, Chan KH, Yuen KY. 2005. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U. S. A. 102:14040–14045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lau SK, Li KS, Huang Y, Shek CT, Tse H, Wang M, Choi GKY, Xu H, Lam CSF, Guo R, Chan KH, Zheng BJ, Woo PC, Yuen KY. 2010. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J. Virol. 84:2808–2819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang LF. 2005. Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676–679 [DOI] [PubMed] [Google Scholar]
- 19.Fouchier RA, Hartwig NG, Bestebroer TM, Niemeyer B, de Jong JC, Simon JH, Osterhaus AD. 2004. A previously undescribed coronavirus associated with respiratory disease in humans. Proc. Natl. Acad. Sci. U. S. A. 101:6212–6216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hasoksuz M, Alekseev K, Vlasova A, Zhang X, Spiro D, Halpin R, Wang S, Ghedin E, Saif LJ. 2007. Biologic, antigenic, and full-length genomic characterization of a bovine-like coronavirus isolated from a giraffe. J. Virol. 81:4981–4990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lau SK, Woo PC, Yip CC, Fan RY, Huang Y, Wang M, Guo R, Lam CS, Tsang AK, Lai KK, Chan KH, Che XY, Zheng BJ, Yuen KY. 2012. Isolation and characterization of a novel Betacoronavirus subgroup A coronavirus, rabbit coronavirus HKU14, from domestic rabbits. J. Virol. 86:5481–5496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJ, Wolthers KC, Wertheim-van Dillen PM, Kaandorp J, Spaargaren J, Berkhout B. 2004. Identification of a new human coronavirus. Nat. Med. 10:368–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vlasova AN, Halpin R, Wang S, Ghedin E, Spiro DJ, Saif LJ. 2011. Molecular characterization of a new species in the genus Alphacoronavirus associated with mink epizootic catarrhal gastroenteritis. J. Gen. Virol. 92:1369–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, Wong BH, Poon RW, Cai JJ, Luk WK, Poon LL, Wong SS, Guan Y, Peiris JS, Yuen KY. 2005. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 79:884–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dominguez SR, O'Shea TJ, Oko LM, Holmes KV. 2007. Detection of group 1 coronaviruses in bats in North America. Emerg. Infect. Dis. 13:1295–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gloza-Rausch F, Ipsen A, Seebens A, Göttsche M, Panning M, Felix Drexler J, Petersen N, Annan A, Grywna K, Müller M, Pfefferle S, Drosten C. 2008. Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg. Infect. Dis. 14:626–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lau SK, Poon RW, Wong BH, Wang M, Huang Y, Xu H, Guo R, Li KS, Gao K, Chan KH, Zheng BJ, Woo PC, Yuen KY. 2010. Coexistence of different genotypes in the same bat and serological characterization of Rousettus bat coronavirus HKU9 belonging to a novel Betacoronavirus subgroup. J. Virol. 84:11385–11394 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Lau SK, Woo PC, Li KS, Huang Y, Wang M, Lam CS, Xu H, Guo R, Chan KH, Zheng BJ, Yuen KY. 2007. Complete genome sequence of bat coronavirus HKU2 from Chinese horseshoe bats revealed a much smaller spike gene with a different evolutionary lineage from the rest of the genome. Virology 367:428–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poon LL, Chu DK, Chan KH, Wong OK, Ellis TM, Leung YH, Lau SK, Woo PC, Suen KY, Yuen KY, Guan Y, Peiris JS. 2005. Identification of a novel coronavirus in bats. J. Virol. 79:2001–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tong S, Conrardy C, Ruone S, Kuzmin IV, Guo X, Tao Y, Niezgoda M, Haynes L, Agwanda B, Breiman RF, Anderson LJ, Rupprecht CE. 2009. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 15:482–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woo PC, Lau SK, Li KS, Poon RW, Wong BH, Tsoi HW, Yip BC, Huang Y, Chan KH, Yuen KY. 2006. Molercular diversity of coronaviruses in bats. Virology 351:180–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woo PC, Wang M, Lau SK, Xu H, Poon RW, Guo R, Wong BH, Gao K, Tsoi HW, Huang Y, Li KS, Lam CS, Chan KH, Zheng BJ, Yuen KY. 2007. Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J. Virol. 81:1574–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bermingham A, Chand MA, Brown CS, Aarons E, Tong C, Langrish C, Hoschler K, Brown K, Galiano M, Myers R, Pebody RG, Green HK, Boddington NL, Gopal R, Price N, Newsholme W, Drosten C, Fouchier RA, Zambon M. 2012. Severe respiratory illness caused by a novel coronavirus, in a patient transferred to the United Kingdom from the Middle East, September 2012. Euro Surveill. 17:20290. [PubMed] [Google Scholar]
- 34.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367:1814–1820 [DOI] [PubMed] [Google Scholar]
- 35.de Groot RJ, Baker SC, Baric RS, Brown CS, Drosten C, Enjuanes L, Fouchier RA, Galiano M, Gorbalenya AE, Memish Z, Perlman S, Poon LL, Snijder EJ, Stephens GM, Woo PC, Zaki AM, Zambon M, Ziebuhr J. 15 May 2013. Middle East respiratory syndrome coronavirus (MERS-CoV); announcement of the coronavirus study group. J. Virol. 10.1128/JVI.01244-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.World Health Organization 2013. Global alert and response: novel coronavirus infection—update. WHO, Geneva, Switzerland: http://www.who.int/csr/don/2013_05_29_ncov/en/index.htmlAccessed 31 May 2013 [Google Scholar]
- 37.Albarrak AM, Stephens GM, Hewson R, Memish ZA. 2012. Recovery from severe novel coronavirus infection. Saudi Med. J. 33:1265–1269 [PubMed] [Google Scholar]
- 38.Munster VJ, de Wit E, Feldmann H. 2013. Pneumonia from human coronavirus in a macaque model. N. Engl. J. Med. 368:1560–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan RW, Chan MC, Agnihothram S, Chan LL, Kuok DI, Fong JH, Guan Y, Poon LL, Baric RS, Nicholls JM, Peiris JS. 3 April 2013. Tropism and innate immune responses of the novel human betacoronavirus lineage C virus in human ex vivo respiratory organ cultures. J. Virol. 10.1128/JVI.00009-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zielecki F, Weber M, Eickmann M, Spiegelberg L, Zaki AM, Matrosovich M, Becker S, Weber F. 2013. Human cell tropism and innate immune system interactions of human respiratory coronavirus EMC compared to those of severe acute respiratory syndrome coronavirus. J. Virol. 87:5300–5304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kindler E, Jónsdóttir HR, Muth D, Hamming OJ, Hartmann R, Rodriguez R, Geffers R, Fouchier RA, Drosten C, Müller MA, Dijkman R, Thiel V. 2013. Efficient replication of the novel human betacoronavirus EMC on primary human epithelium highlights its zoonotic potential. mBio 4:e00611–12. 10.1128/mBio.00611-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, Osterhaus AD, Haagmans BL, Gorbalenya AE, Snijder EJ, Fouchier RA. 2012. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3:e00473–12. 10.1128/mBio.00473-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woo PC, Lau SK, Li KS, Tsang AK, Yuen KY. 2012. Genetic relatedness of the novel human group C betacoronavirus to Tylonycteris bat coronavirus HKU4 and Pipistrellus bat coronavirus HKU5. Emerg. Microbes Infect. 1:e35. 10.1038/emi.2012.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Annan A, Baldwin HJ, Corman VM, Klose SM, Owusu M, Nkrumah EE, Badu EK, Anti P, Agbenyega O, Meyer B, Oppong S, Sarkodie YA, Kalko EKV, Lina PHC, Godlevska EV, Reusken C, Seebens A, Gloza-Rausch F, Vallo P, Tschapka M, Drosten C, Drexler JF. 2013. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 19:456–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anthony S, Ojeda-Flores R, Rico-Chávez O, Navarrete-Macias I, Zambrana-Torrelio C, Rostal MK, Epstein JH, Tipps T, Liang E, Sanchez-Leon M, Sotomayor-Bonilla J, Aguirre AA, Avila R, Medellín RA, Goldstein T, Suzán G, Daszak P, Lipkin WI. 2013. Coronaviruses in bats from Mexico. J. Gen. Virol. 94:1028–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan JF, Chan KH, Choi GK, To KK, Tse H, Cai P, Yeung PM, Cheng VC, Chen H, Che XY, Lau SK, Woo PC, Yuen KY. 2013. Differential susceptibility of different cell lines to the emerging novel human betacoronavirus 2c EMC/2012 (HCoV-EMC): implications on disease pathogenesis and clinical manifestation. J. Infect. Dis. 207:1743–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raj VS, Mou H, Smits SL, Dekkers DH, Müller MA, Dijkman R, Muth D, Demmers JA, Zaki A, Fouchier RA, Thiel V, Drosten C, Rottier PJ, Osterhaus AD, Bosch BJ, Haagmans BL. 2013. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495:251–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yob JM, Field H, Rashdi AM, Morrissy C, van der Heide B, Rota P, bin Adzhar A, White J, Daniels P, Jamaluddin A, Ksiazek T. 2001. Nipah virus infection in bats (order Chiroptera) in peninsular Malaysia. Emerg. Infect. Dis. 7:439–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59:307–321 [DOI] [PubMed] [Google Scholar]
- 52.Apweiler R, Attwood TK, Bairoch A, Bateman A, Birney E, Biswas M, Bucher P, Cerutti L, Corpet F, Croning MD, Durbin R, Falquet L, Fleischmann W, Gouzy J, Hermjakob H, Hulo N, Jonassen I, Kahn D, Kanapin A, Karavidopoulou Y, Lopez R, Marx B, Mulder NJ, Oinn TM, Pagni M, Servant F, Sigrist CJ, Zdobnov EM. 2001. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 29:37–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL. 2002. The Pfam protein families database. Nucleic Acids Res. 30:276–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sonnhammer EL, von Heijne G, Krogh A. 1998. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6:175–182 [PubMed] [Google Scholar]
- 55.Trigg J, Gutwin K, Keating AE, Berger B. 2011. Multicoil2: predicting coiled coils and their oligomerization states from sequence in the twilight zone. PLoS One 6:e23519. 10.1371/journal.pone.0023519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pond SL, Frost SD. 2005. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533 [DOI] [PubMed] [Google Scholar]
- 57.Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL. 2012. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8:e1002764. 10.1371/journal.pgen.1002764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. 10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lau SK, Li KS, Tsang AK, Shek CT, Wang M, Choi GKY, Guo R, Wong BH, Poon RW, Lam CS, Wang SY, Fan RY, Chan KH, Zheng BJ, Woo PC, Yuen KY. 2012. Recent transmission of a novel alphacoronaivurs, bat coronavirus HKU10, from Leschenault's rousettes to Pomona leaf-nosed bats: first evidence of interspecies transmission of coronavirus between bats of different suborders. J. Virol. 86:11906–11918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Suchard MA, Weiss RE, Sinsheimer JS. 2001. Bayesian selection of continuous-time Markov chain evolutionary models. Mol. Biol. Evol. 18:1001–1013 [DOI] [PubMed] [Google Scholar]
- 61.Chan WE, Chuang CK, Yeh SH, Chang MS, Chen SS. 2006. Functional characterization of heptad repeat 1 and 2 mutants of the spike protein of severe acute respiratory syndrome coronavirus. J. Virol. 80:3225–3237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Delmas B, Gelfi J, L'Haridon R, Vogel LK, Sjostrom H, Noren O, Laude H. 1992. Aminopeptidase N is a major receptor for the entero-pathogenic coronavirus TGEV. Nature 357:417–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yeager CL, Ashmun RA, Williams RK, Cardellichio CB, Shapiro LH, Look AT, Holmes KV. 1992. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 357:420–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hofmann H, Pyrc K, van der Hoek L, Geier M, Berkhout B, Pohlmann S. 2005. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. U. S. A. 102:7988–7993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. 2003. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Krempl C, Schultze B, Herrler G. 1995. Analysis of cellular receptors for human coronavirus OC43. Adv. Exp. Med. Biol. 380:371–374 [DOI] [PubMed] [Google Scholar]
- 67.Schultze B, Herrler G. 1992. Bovine coronavirus uses N-acetyl-9-O-acetylneuraminic acid as a receptor determinant to initiate the infection of cultured cells. J. Gen. Virol. 73:901–906 [DOI] [PubMed] [Google Scholar]
- 68.Smits SL, Gerwig GJ, van Vliet AL, Lissenberg A, Briza P, Kamerling JP, Vlasak R, de Groot RJ. 2005. Nidovirus sialate-O-acetylesterases: evolution and substrate specificity of coronaviral and toroviral receptor-destroying enzymes. J. Biol. Chem. 280:6933–6941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vlasak R, Luytjes W, Spaan W, Palese P. 1988. Human and bovine coronaviruses recognize sialic acid-containing receptors similar to those of influenza C viruses. Proc. Natl. Acad. Sci. U. S. A. 85:4526–4529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Williams RK, Jiang GS, Holmes KV. 1991. Receptor for mouse hepatitis virus is a member of the carcinoembryonic antigen family of glycoproteins. Proc. Natl. Acad. Sci. U. S. A. 88:5533–5536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bonavia A, Zelus BD, Wentworth DE, Talbot PJ, Holmes KV. 2003. Identification of a receptor-binding domain of the spike glycoprotein of human coronavirus HCoV-229E. J. Virol. 77:2530–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Godet M, Grosclaude J, Delmas B, Laude H. 1994. Major receptor-binding and neutralization determinants are located within the same domain of the transmissible gastroenteritis virus (coronavirus) spike protein. J. Virol. 68:8008–8016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hofmann H, Simmons G, Rennekamp AJ, Chaipan C, Gramberg T, Heck E, Geier M, Wegele A, Marzi A, Bates P, Pohlmann S. 2006. Highly conserved regions within the spike proteins of human coronaviruses 229E and NL63 determine recognition of their respective cellular receptors. J. Virol. 80:8639–8652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kubo H, Yamada YK, Taguchi F. 1994. Localization of neutralizing epitopes and the receptor-binding site within the amino-terminal 330 amino acids of the murine coronavirus spike protein. J. Virol. 68:5403–5410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peng G, Sun D, Rajashankar KR, Qian Z, Holmes KV, Li F. 2011. Crystal structure of mouse coronavirus receptor-binding domain complexed with its murine receptor. Proc. Natl. Acad. Sci. U. S. A. 108:10696–10701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Prabakaran P, Gan J, Feng Y, Zhu Z, Choudhry V, Xiao X, Ji X, Dimitrov DS. 2006. Structure of severe acute respiratory syndrome coronavirus receptor-binding domain complexed with neutralizing antibody. J. Biol. Chem. 281:15829–15836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu K, Li W, Peng G, Li F. 2009. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. U. S. A. 106:19970–19974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou T, Wang H, Luo D, Rowe T, Wang Z, Hogan RJ, Qiu S, Bunzel RJ, Huang G, Mishra V, Voss TG, Kimberly R, Luo M. 2004. An exposed domain in the severe acute respiratory syndrome coronavirus spike protein induces neutralizing antibodies. J. Virol. 78:7217–7226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Müller MA, Raj VS, Muth D, Meyer B, Kallies S, Smits SL, Wollny R, Bestebroer TM, Specht S, Suliman T, Zimmermann K, Binger T, Eckerle I, Tschapka M, Zaki AM, Osterhaus AD, Fouchier RA, Haagmans BL, Drosten C. 2012. Human coronavirus EMC does not require the SARS-coronavirus receptor and maintains broad replicative capability in mammalian cell lines. mBio 3:e00515–12. 10.1128/mBio.00515-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jiang S, Lu L, Du L, Debnath AK. 2013. A predicted receptor-binding and critical neutralizing domain in S protein of the novel human coronavirus HCoV-EMC. J. Infect. 66:464–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pyrc K, Dijkman R, Deng L, Jebbink MF, Ross HA, Berkhout B, van der Hoek L. 2006. Mosaic structure of human coronavirus NL63, one thousand years of evolution. J. Mol. Biol. 364:964–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vijgen L, Keyaerts E, Moës E, Thoelen I, Wollants E, Lemey P, Vandamme AM, Van Ranst M. 2005. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 79:1595–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Graham RL, Baric RS. 2010. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J. Virol. 84:3134–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 7:439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shek CT. 2006. From Leschenault's Rousette, p 108–112_In_A field guide to the terrestrial mammals of Hong Kong. Friends of Country Park and Cosmos Book Limited, Hong Kong, Hong Kong [Google Scholar]
- 86.Teeling EC, Springer MS, Madsen O, Bates P, O'brien SJ, Murphy WJ. 2005. A molecular phylogeny for bats illuminates biogeography and the fossil record. Science 307:580–584 [DOI] [PubMed] [Google Scholar]
- 87.Leroy EM, Kumulungui B, Pourrut X, Rouquet P, Hassanin A, Yaba P, Délicat A, Paweska JT, Gonzalez JP, Swanepoel R. 2005. Fruit bats as reservoirs of Ebola virus. Nature 438:575–576 [DOI] [PubMed] [Google Scholar]
- 88.Tong S, Li Y, Rivailler P, Conrardy C, Castillo DA, Chen LM, Recuenco S, Ellison JA, Davis CT, York IA, Turmelle AS, Moran D, Rogers S, Shi M, Tao Y, Weil MR, Tang K, Rowe LA, Sammons S, Xu X, Frace M, Lindblade KA, Cox NJ, Anderson LJ, Rupprecht CE, Donis RO. 2012. A distinct lineage of influenza A virus from bats. Proc. Natl. Acad. Sci. U. S. A. 109:4269–4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tang XC, Zhang JX, Zhang SY, Wang P, Fan XH, Li LF, Li G, Dong BQ, Liu W, Cheung CL, Xu KM, Song WJ, Vijaykrishna D, Poon LL, Peiris JS, Smith GJ, Chen H, Guan Y. 2006. Prevalence and genetic diversity of coronaviruses in bats from China. J. Virol. 80:7481–7490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Buchholz U, Müller MA, Nitsche A, Sanewski A, Wevering N, Bauer-Balci T, Bonin F, Drosten C, Schweiger B, Wolff T, Muth D, Meyer B, Buda S, Krause G, Schaade L, Haas W. 2013. Contact investigation of a case of human novel coronavirus infection treated in a German hospital, October-November 2012. Euro Surveill. 18:20406. [PubMed] [Google Scholar]
- 91.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]