Low-density Lipoprotein Receptor-related Protein 1 (LRP1)-dependent Cell Signaling Promotes Axonal Regeneration (original) (raw)
Background: LRP1 activation is neuroprotective in vitro. The role of LRP1 in axonal plasticity and regeneration is unknown.
Results: LRP1-dependent cell signaling that includes TrkC activation promotes axonal growth in the CNS.
Conclusion: LRP1 agonists promote regeneration after spinal cord injury.
Significance: A significant role is established for LRP1 in axonal growth and regeneration after CNS injury, identifying a novel class of therapeutic targets for neurological disorders.
Keywords: Axon, Lipoprotein Receptor, MAP Kinases (MAPKs), Neurite Outgrowth, Neurodegeneration
Abstract
Low-density lipoprotein receptors (LRPs) are present extensively on cells outside of the nervous system and classically exert roles in lipoprotein metabolism. It has been reported recently that LRP1 activation could phosphorylate the neurotrophin receptor TrkA in PC12 cells and increase neurite outgrowth from developing cerebellar granule cells. These intriguing findings led us to explore the hypothesis that LRP1 activation would activate canonical neurotrophic factor signaling in adult neurons and promote axonal regeneration after spinal cord injury. We now find that treatment of adult rat dorsal root ganglion neurons in vitro with LRP1 agonists (the receptor binding domain of α-2-macroglobulin or the hemopexin domain of matrix metalloproteinase 9) induces TrkC, Akt, and ERK activation; significantly increases neurite outgrowth (p < 0.01); and overcomes myelin inhibition (p < 0.05). These effects require Src family kinase activation, a classic LRP1-mediated Trk transactivator. Moreover, intrathecal infusions of LRP1 agonists significantly enhance sensory axonal sprouting and regeneration after spinal cord injury in rats compared with control-infused animals (p < 0.05). A significant role is established for lipoprotein receptors in sprouting and regeneration after CNS injury, identifying a novel class of therapeutic targets to explore for traumatic neurological disorders.
Introduction
The LDL receptor-related protein 1 (LRP1) is a large (600-kDa) type I membrane receptor and is a member of the LDL receptor superfamily (1). Although originally known to function as an endocytic receptor for apolipoprotein E (2) and amyloid Aβ (3), LRP1 is currently recognized as an endocytic and cell signaling receptor for diverse ligands, including tissue-type plasminogen activator (tPA), matrix metalloproteinase 9, and activated α2-macroglobulin (4–8). LRP1 agonists activate cell signaling by tyrosine phosphorylation of NP_X_Y motifs present in the LRP1 cytoplasmic domain and by binding of signaling adaptor proteins such as Shc and JNK-interacting protein (JIP) (9, 10). In Schwann cells, LRP1 activates PI3K/Akt-mediated cell survival signaling pathways and counteracts endoplasmic reticulum stress (11, 12). LRP1 also interacts with other receptors, including integrins (13), platelet-derived growth factor receptors (14), and urokinase-type plasminogen activator receptor (15). As a result, LRP1 functions in a diversity of cellular activities, including cell signaling and metabolism, migration, and blood-brain barrier integrity (16, 17).
LRP1 is widely expressed in uninjured central and peripheral neurons (18, 11). Recent reports have suggested a potential role for LRP1 in neural function and growth. LRP1 acts as a prosurvival and migratory receptor in Schwann cells after peripheral nerve injury (11, 19), transduces a signal for phagocytosis of degraded myelin in experimental models of multiple sclerosis (20), and modulates processing of the amyloid precursor protein (20–22, 3). Recently, LRP1 agonists were also reported to induce transactivation of TrkA in a Src family kinase (SFK)3-dependent manner, promoting neurite outgrowth from PC12 cells (23). The latter findings identify a candidate mechanism for potential actions of LRP1 on axonal function and suggest the hypothesis that LRP1 activation could promote axonal sprouting and regeneration in the adult CNS. Synthetic LRP1 ligands have been generated, including several fusion proteins that can be readily infused into the CNS to test potential activities in injury models. We now report potent effects of LRP1-binding ligands on axonal growth in vitro and after spinal cord injury in vivo, identifying a novel LRP1-dependent cell signaling mechanism involved in CNS plasticity and regeneration. These effects result from canonical LRP1 ligand transactivation of Trk.
EXPERIMENTAL PROCEDURES
Experimental Design
In vitro experiments determined whether LRP1 agonists promote neurite outgrowth from cultures of dissociated primary adult dorsal root ganglion (DRG) neurons and on signaling mechanisms activated by LRP1 receptor signaling. A second set of “_ex vivo_” experiments determined whether intrathecal infusion of LRP1 agonists for 3 days would also stimulate growth-related neuronal signaling and neurite outgrowth in DRG neurons that were subsequently removed and cultured in vitro. Finally, in vivo experiments assessed the ability of intrathecal infusions of LRP1 agonists to promote sprouting or regeneration of the central processes of DRG neurons projecting through the dorsal columns after C4 dorsal spinal cord lesions. All experiments examined the same population of large-diameter, neurofilament 200 (NF200)-expressing, NT-3-sensitive DRG neurons.
Reagents
GST fusion proteins that directly bind LRP1 and are LRP1 agonists were prepared as described previously (24, 25). Purified GST and GST fusion proteins were subjected to chromatography on Detoxi-Gel endotoxin-removing columns (Pierce) and endotoxin dialysis decontamination (8). LRP1 agonists include an 18-kDa receptor binding domain of α2-macroglobulin (α2M), henceforth referred to as RBD (25–27), and the 18-kDa LRP1 binding domain of matrix metalloproteinase 9, the hemopexin domain, henceforth referred to as PEX (28, 8). The LRP1 antagonist, receptor-associated protein (RAP), and GST controls were prepared as described previously (27, 29). The resulting protein preparations yielded clearly defined bands with the correct molecular masses when assessed by Coomassie Blue staining of SDS gels or immunoblot analysis with GST-specific antibody. LRP1-dependent cell signaling was confirmed by inhibition of pERK with RAP. The stability of fusion proteins over time at 37 °C (for subsequent in vivo infusions) was assessed by incubating 100 nm RBD or GST in PBS at 37 °C for 0, 24, 48, 72, and 96 h, adding to PC12 cells for 10 min, and examining ERK activation, as described below.
Neurite Outgrowth in Cultures of Adult DRG Neurons
In Vitro Studies
Primary cultures of adult F344 L4-L6 DRG neurons were cultured on poly-L-lysine-coated (16.6 μg/ml) 12-well plates as described previously (30). Cells were treated with 100 nm RBD, PEX, or GST for 18 or 24 h. In some assays, cells were pretreated with GST-RAP (200 nm) 30 min prior to the addition of LRP1 agonists. Some conditions included plating of cells on inhibitory myelin substrates, as described below. Myelin-coated plates were prepared by extracting myelin from rat spinal cord, diluting in water at 10 μg/well, drying overnight at room temperature, and washing with DMEM/F-12 the next day before use.
Cells were fixed and labeled for NF200, a specific marker of neurites emerging from the NT-3-responsive subpopulation of DRG neurons (31, 32) The in vitro portion of our studies focused on the NT-3-responsive population of DRG neurons because they are the subject of the subsequent spinal cord injury model in vivo. Images were acquired manually at ×100 magnification, and longest neurite length per cell was measured in a minimum of 80–100 neurons/well using ImageJ. Quantification was performed in a blinded manner. Experiments were replicated twice with internal triplicates. Immunolabeling was performed using anti-NF200 (1:4000, Millipore). Dual labeling of NF200 and LRP1 was performed in frozen DRG sections using a polyclonal LRP1 antibody (1:1000, Sigma) (11).
Ex Vivo Studies
Adult F344 female rats weighing 150–200 g (n = 28 rats) were anesthetized with a mixture (2 ml/kg) of ketamine (25 mg/ml), rompun (1.8 mg/ml), and acepromazine (0.25 mg/ml). National Institutes of Health and Institutional Animal Use and Safety Committee guidelines for laboratory animal care and safety were strictly followed for all animal use and post-operation care. Alzet osmotic minipumps (model 1003) were implanted in the spinal cord at the L4–5 level for intrathecal delivery of GST or RBD (5 μm, 1 μl/h) for 3 days. After infusions, DRGs were rapidly isolated, lysed in radioimmune precipitation assay buffer for subsequent immunoblot analysis, or cultured for 18 h, fixed with 4% PFA, stained for NF200, imaged, and quantitated as described above.
Cell Signaling Studies in Primary Adult DRG Neurons
Immunoblot Analyses
L4-L6 rat DRGs were harvested and plated on poly-L-lysine-coated 6-well plates. The cells were treated with 200 nm RBD, PEX, GST, or NT-3 (positive control, Abcam, 20 nm) or vehicle (0.01% Brij35 in PBS) for 30 min in neural basal medium. In some cases, cells were pretreated with the Trk activation inhibitor k252a (10 nm) or with an SFK inhibitor, PP2 (1 μm), 30 min prior to the addition of LRP1 agonists. Cell extracts were prepared using a total protein extraction kit (Millipore). An equivalent amount of cellular protein (20–50 μg/lane) was subjected to SDS-PAGE, immunoblotting, and densitometry, as described previously (11, 12). Primary antibodies used included pTrkC (1:1000, Syd Laboratories), tTrkC (Abcam), pERK or total ERK (1:1000, Cell Signaling Technology). Experiments were replicated five to 11 times. Blots were scanned (Cannoscan), and densitometry was performed using Image J, as previously described (8, 11).
Immunofluorescence
Dissociated DRG neurons were plated on poly-L-lysine-coated coverslips and treated with 100 nm PEX, GST, NT-3 (positive control, 20 nm), or vehicle (0.01% Brij35 in PBS) for 30 min in neural basal medium. In some cases, cells were pretreated with an SFK inhibitor, PP2 (1 μm), 30 min prior to the addition of the LRP1 agonist and then fixed with 4% paraformaldehyde. Dual-label immunofluorescence was performed as described (11). Briefly, cells were incubated with primary antibodies to pTrk (1:500 Tyr-490, Santa Cruz Biotechnology) or pERK (1:1000, Cell Signaling Technology) and with the appropriate fluorescent antibodies (Alexa Fluor 488- or 594-conjugated antibodies). A second primary antibody, NF200 (1:4000; Millipore) and secondary antibody were added. Preparations were mounted on slides using Pro-Long Gold with DAPI for nuclear labeling (Invitrogen). Images were captured on an Olympus Fluoview1000 confocal microscope and quantified using Veloctiy three-dimensional image analysis software (PerkinElmer Life Sciences). A total of 80–110 neurons were counted from four independent experiments.
Spinal Cord Injury and Tissue Processing
17 adult female Fisher 344 rats weighing 150–200 g were deeply anesthetized as described above under animal protocols approved by the Institutional Animal Welfare Committee. Alzet osmotic minipumps were implanted in the spinal cord at the L4–5 level for intrathecal delivery of either 5 μm GST (n = 8 animals) or RBD (n = 9 animals). Three days later, C4 dorsal column spinal cord lesions were placed using a Scouten tungsten wire knife (33). The lesion site was filled with syngeneic bone marrow stromal cells to provide a matrix in the lesion site to which injured axons could attach and grow (34). Rat marrow stromal cells were prepared as described previously (35, 36) and transplanted into the lesion site using glass pipettes and a picospritzer. Pumps were replaced at the same time with fresh RBD. Dorsal column sensory axons were labeled transganglionically by cholera toxin B subunit (CTB) injection into the sciatic nerve (2 μl of 1% solution/sciatic nerve) 3 days before perfusion (36–38). Four weeks after spinal cord lesions, animals were transcardially perfused with 4% paraformaldehyde, post-fixed overnight, and cryoprotected in 30% sucrose at 4 °C. Spinal cords were sectioned sagittally on a cryostat set to 30-μm intervals. All sections were processed free-floating. CTB-labeled sensory axons were visualized as described previously (36). GFAP was detected subsequently in the same sections by fluorescence labeling. We quantified the mean number of dorsal column sensory axons penetrating the lesion site, the total length of axons penetrating the lesion site, and the mean number of axons reaching the midportion of the lesion site using serial 30-μm-thick sections (a series of one-in-seven) labeled for CTB and GFAP. The number of CTB-labeled axons encountered at a virtual line drawn in the midportion of the graft, identified by GFAP labeling (Fig. 4A), was counted using a ×10 ocular with a calibrated grid and a ×40 objective. The total length of CTB-labeled axons within the lesion site was quantified by acquiring images of entire grafts at ×200 magnification, capturing the images in the Neuron plugin of ImageJ and tracing every individual axon within the graft. The total length of all axons in each CTB-labeled section from every animal was summed, and mean values across groups were compared. In addition, total axons in the graft that could be resolved individually were quantified. Multiple axons that could not be resolved as separate axons were quantified as a single axon count. To account for potential differences between CTB axonal labeling efficiency in different animals, the total number of CTB-labeled axons in the dorsal columns approaching the lesion site (500 μm caudal to the lesion) was also quantified in each subject. The total numbers of axons in the graft and reaching the midportion of the graft was then divided by this value. Mean values for axon number and midline-crossing axons were averaged per subject and expressed as mean ± S.E.
FIGURE 4.

Intrathecal infusion of RBD promotes TrkC-mediated cell signaling ERK activation in adult DRGs. A, immunoblot analysis of pERK showing the duration of bioactivity of RBD (100 nm) or GST (100 nm) after incubation at 37 °C for 0, 24, 48, 72, and 96 h, followed by addition to PC12 cells (see “Experimental Procedures”). Bioactivity persisted for 96 h. Total ERK was used as a loading control. Images represent two independent studies. B, schematic illustrating intrathecal implantation of the catheter in the lumbar subarachnoid space for three-day infusions of RBD. The catheter is connected to an osmotic minipump. C, immunoblot analysis of pTrkC and pERK in isolated DRGs after completion of intrathecal infusions. Each lane represents individual rat L4 and L5 DRGs. D, quantification of pTrkC and pERK ratios by densitometry (*, p < 0.05 compared with naïve). Data are mean ± S.E.; n = 3–4 rats/group.
Statistical Analysis
Differences between multiple treatment groups were assessed using one-way analysis of variance. Individual group differences were assessed using post hoc Fisher's or Newman-Keuls multiple comparisons tests. Comparison of two groups were made using two-tailed Student's t test with a significance criterion of p < 0.05. All procedures were conducted in a blinded fashion. All animals entered into this study were reported and used in the subsequent analyses of results (39).
RESULTS
LRP1 Is Expressed in Adult DRG Neurons, and LRP1 Activation Increases Neurite Outgrowth
Double labeling for LRP1 and NF200, a marker for large-diameter neurons (30), demonstrated that LRP1 is expressed by intact adult, large-diameter DRG neurons and some smaller DRG neurons (Fig. 1A). We then determined whether LRP1 activation promotes growth of cultured adult DRG neurons. Dissociated DRG cultures from naïve adult rats were treated with the LRP1 agonists RBD and PEX, the control protein GST, or were left untreated. RBD and PEX significantly increased neurite outgrowth of NF200-labeled neurites greater than 2-fold compared with the GST control (Fig. 1, B and C, p < 0.005 comparing PEX or RBD to GST) after 18 h. To test whether LRP1 is responsible for the effects of RBD and PEX on neurite outgrowth, cells were pretreated with GST-RAP, a well established LRP1 antagonist that binds to LRP1 and precludes binding of other ligands (29) or with GST as a control. When added alone, GST-RAP did not activate neurite outgrowth (p = 0.9). However, GST-RAP significantly reduced the growth-promoting effects of RBD and PEX, indicating specific activity through LRP1 (Fig. 1, B and C, p < 0.05 comparing PEX to PEX + RAP, and p < 0.05 comparing RBD to RBD + RAP). In a separate set of experiments, effects of LRP1 signaling in permissive (poly-L-lysine) or inhibitory (myelin) environments were examined after 24 h. The addition of RBD significantly increased neurite outgrowth (4-fold) on poly-L-lysine (Fig. 1D, p < 0.01). Moreover, RBD significantly overcame myelin-mediated inhibition, increasing neurite outgrowth 3-fold on the myelin substrate (Fig. 1D, p < 0.05). Thus, LRP1 activation significantly promotes neurite outgrowth from adult DRG sensory neurons under both permissive and non-permissive conditions.
FIGURE 1.

LRP1 agonists promote neurite outgrowth from adult DRG neurons. A, double immunolabeling for LRP1 (green) and NF200 (red) in adult naïve DRGs. LRP1 colocalizes with NF200 neurons and other neuronal populations. B, images of cultured primary adult DRG neurons immunolabeled with NF200. DRG cultures are treated with RBD (100 nm), PEX (100 nm), or GST (100 nm) or untreated with or without RAP (200 nm) pretreatment. 18 h after explantation, RBD- and PEX-treated neurons elaborate a greater number and length of neurites. Scale bar = 5 μm. C, maximum neurite length after 18 h in vitro and addition of the control peptide GST or the LRP1 agonists PEX or RBD. Both PEX and RBD significantly increased maximum neurite length (**, p < 0.005 compared with GST). The LRP1 antagonist RAP significantly reduced maximum neurite length after treatment with either PEX or RBD (*, p < 0.05). The subtotal effect of RAP in reducing neurite outgrowth may be due to the use of relatively low RAP concentrations in the setting of abundant LRP1 expression on DRG neurons (A). ns, not significant. D, in separate experiments, the LRP1 agonist RBD also significantly overcomes myelin inhibition after 24 h. PLL, poly-L-lysine substrate; Myelin, myelin substrate. *, p < 0.05; **, p < 0.005. Data are mean ± S.E.
LRP1 Ligands Elicit TrkC Signaling in DRG Neurons by Transactivation of the LRP1 Receptor
Previous reports indicate that LRP1-dependent cell signaling facilitates neurite outgrowth by transactivating TrkA in PC12 cells (23). To understand the cell signaling mechanisms underlying neurite outgrowth in adult DRG neurons, we tested whether LRP1 agonists activate TrkC. We focused on TrkC-expressing populations of DRG neurons because their axons are lesioned by the dorsal column spinal cord injury used in our in vivo studies, whereas TrkA- and TrkB-expressing DRG neurons are not. Addition of either NT-3, serving as a positive control, or the LRP1 ligand PEX for 30 min to adult DRG cultures resulted in activation of TrkC compared with GST and vehicle-treated controls (Fig. 2, A and B, p < 0.01). Both 170-kDa and 150-kDa pTrkC bands were identified, as described previously (40). Next, we measured ERK activation after treatment with PEX. Activation was increased compared with controls (Fig. 2C, quantified in D). To test whether Trk activation was required for LRP1-dependent cell signaling, we added the Trk inhibitor k252a in the presence of PEX and measured ERK activation (Fig. 2C). PEX significantly activated ERK (p < 0.01), and this activation was blocked by k252a (Fig. 2D). Taking the results of Fig. 2 together, we conclude that LRP-1-dependent cell signaling requires TrkC activation, at least in part. That is, PEX activates TrkC (Fig. 2, A and B), which canonically activates ERK. Addition of k252a in the presence of PEX reduces ERK levels to near base-line. Although it is possible that LRP1 ligands also activated ERK through TrkA and B receptors in these mixed DRG cultures, the nearly complete reduction of pERK by the addition of k252a indicates that all PEX-induced Trk signaling is blocked by k252a, including the portion generated by TrkC activation.
FIGURE 2.

LRP1 agonists activate TrkC-mediated signaling in adult DRGs. A, immunoblot analysis of pTrkC. Primary adult DRGs were stimulated with vehicle, PEX (200 nm), NT-3 (positive control, 4 nm), or GST (200 nm) for 30 min. B, quantification of pTrkC by densitometry. Total TrkC was used as a loading control (**, p < 0.01; *, p < 0.05 compared with GST). C, immunoblot analysis of pERK. Adult DRGs were pretreated with or without k252A (10 nm) and subsequently stimulated with PEX for 30 min. D, quantification of pERK by densitometry. Total ERK was used as a loading control (**, p < 0.01 compared with K252A). Equal amounts of cellular protein (20–50 μg) were loaded into each lane, subjected to SDS-PAGE, and electrotransferred to nitrocellulose for detection with specific antibodies. Data are mean ± S.E. Blots represent n = 4–5 independent experiments.
To further investigate mechanisms through which LRP1 agonists activate signaling in adult DRG neurons, we treated adult DRG neurons with the SFK inhibitor PP2 in the presence of PEX or RBD. Previously, PP2 has been reported to block LRP1-induced transactivation of TrkA (23). PP2 significantly inhibited both PEX- and RBD-induced ERK activation (p < 0.05, Fig. 3, A–C). In contrast, PP2 did not inhibit ERK phosphorylation induced by NT-3, and when added alone, PP2 did not activate cell signaling. RBD also activated Akt signaling (Fig. 3D), and this canonical Trk-related signaling was inhibited by PP2 (D). We next showed that PP2 specifically blocks LRP1-dependent cell signaling in TrkC-expressing neurons using double labeling for NF200 (expressed only by TrkC-bearing DRG neurons) (31, 32) and pTrk. NT-3 treatment induced pTrk labeling, and this was not affected by addition of PP2 (data not shown; PEX also induced pTrk labeling in the NF200-labeled population of DRG neurons, and this labeling was attenuated by addition of PP2 (Fig. 3E, p < 0.05)). These data are further confirmed by quantification in Fig. 3F. Double labeling for NF200 and pERK revealed that PEX induced pERK in NF200-expressing adult DRG neurons, and this labeling was eliminated by addition of PP2. Collectively, these findings strongly suggest that LRP1 transactivates TrkC in an SFK-dependent manner.
FIGURE 3.

LRP1 transactivates TrkC via a SFK-mediated mechanism in adult DRG neurons. A and B, immunoblot analysis of pERK after stimulation with LRP1 agonists, PEX (200 nm) or RBD (200 nm), NT-3 (positive control, 4 nm), or vehicle with or without pretreatment with PP2 (1 μm) for 30 min. C, quantification of pERK by densitometry. Total ERK was used as a loading control (*, p < 0.05 compared with vehicle; n = 4–8 independent experiments). D, immunoblot analysis of pAkt after stimulation with RBD (200 nm) with or without PP2 (1 μm) pretreatment for 30 min. E, immunofluorescence microscopy for pTrk and pERK in primary DRG neurons after treatment with PEX with or without pretreatment with PP2 (1 μm) for 30 min. NF200 (green) was used to identify neurons as the TrkC-expressing subpopulation. F, quantification of pTrk in NF200-positive neurons (*, p < 0.05 compared with GST). Data are mean ± S.E.; n = 80–100 neurons counted.
Intrathecal Infusions of the LRP1 Ligand RBD Promote Neurite Outgrowth through ERK Activation
To determine whether intrathecal infusions of RBD effectively target and elicit signaling from neurons with central axonal projections, we chronically infused either RBD or GST (control) into the lumbar intrathecal compartment. First, we tested whether LRP1 agonists remain bioactive at body temperature over time. PC12 cells were stimulated with RBD or GST that had been incubated for 0–96 h at 37 °C. RBD activated ERK, confirming bioactivity of RBD even after incubation for 96 h at 37 °C (Fig. 4A). GST alone had no effect, as anticipated. Next, osmotic minipumps were implanted in the back, with the tip of a delivery catheter positioned in the subdural space between L4 and L5 (Fig. 4B). Three days after initiating intrathecal infusions of RBD or GST, L4 DRGs were isolated and lysed in radioimmune precipitation assay buffer for immunoblot analysis. RBD-treated DRGs showed significant increases in phosphorylation of TrkC and ERK compared with GST-treated rats (Fig. 4C). These findings were further confirmed by densitometry (Fig. 4D). We repeated intrathecal infusions of RBD and GST for 3 days and then isolated and cultured DRGs to measure neurite outgrowth after 24 h. Findings were compared with DRGs removed from naïve controls. RBD-treated DRGs extended significantly longer NF200-labeled neurites than neurons from GST or naïve animals (Fig. 5A, p < 0.001 comparing RBD to both GST and naïve groups). GST-infused controls showed a non-significant increase in neurite length compared with naïve controls, a potential consequence of intrathecal catheter placement causing a mild conditioning effect on DRG neurons. RBD markedly and consistently increased ERK phosphorylation compared with both naïve and GST-infused controls (Fig. 5, B and C, p < 0.01). These findings indicate that intrathecal infusions of LRP1 agonists are an effective means of activating downstream canonical Trk receptor signaling and enhancing neurite growth.
FIGURE 5.

Intrathecal infusion of RBD promotes neurite outgrowth and sustains ERK activation in adult DRGs. A, explantation of L4 and L5 DRG neurons after completion of intrathecal infusions following RBD treatment showed a significant increase in maximum neurite length (**, p < 0.005 compared with GST). Neurite length was measured after 18 h in cell culture. GST-infused controls also exhibited an increase in neurite length compared with naïve controls, possibly because of a mild conditioning effect of the catheter implants, but this difference from naïve animals was not statistically significant (ns) (n = 8–9 rats/group). B, intrathecal infusions also markedly increased pERK after 18 h in cell culture compared with both naïve (N) and GST-infused controls. C, quantification of pERK ratios by densitometry. Total ERK was used as a loading control. **, p < 0.01 compared with GST. Data are mean ± S.E. Blots are representative of n = 3 independent experiments.
Intrathecal Infusions of RBD Promote Axonal Sprouting and Regeneration after Spinal Cord Injury
Given the significant effects of LRP1-dependent cell signaling on regeneration-related neurite outgrowth in vitro, we next determined whether targeting of LRP1 receptors was an effective mechanism for influencing axonal growth after spinal cord injury. Rats underwent C4 dorsal column lesions that completely transected all dorsal column sensory axons projecting to the nucleus gracilis (Fig. 6, A and B). This is a standard model for assessing the potential of novel compounds to enhance CNS axonal plasticity and regeneration (37, 41–43). The lesion site was filled with syngeneic bone marrow stromal cells to provide a matrix in the lesion site into which injured axons could attach (34, 36). Without this matrix, even stimulated axons will not regenerate into the lesion. Notably, animals infused with RBD exhibited significant increases in axonal regeneration into the lesion site compared with GST-infused animals (Figs. 6, C–F). Animals treated with RBD showed a 71% increase in the total proportion of all labeled dorsal column axons regenerating into the lesion site compared with control-infused subjects (p = 0.03). The number of CTB-labeled axons in the lesion site constituted 28.7% of all CTB-labeled axons present in the dorsal column projection, compared with 16.8% in controls (Fig. 6G), indicating recruitment of regeneration in more than one-quarter of all labeled axons. The efficiency of axonal labeling did not differ significantly between the RBD- and GST-infused groups (193 ± 3.5 labeled axons/subject in RBD-treated animals versus 204 ± 4.7 in GST-treated animals, p = 0.44). Moreover, the total length of all axons regenerating into the lesion site was 3.6-fold greater in RBD-treated animals compared with controls (p < 0.01, Fig. 6H). In addition, the distance over which axons regenerated also significantly increased in RBD-treated animals, reflected by the number of axons reaching the middle of the lesion site (120% increase, p < 0.001) compared with controls (Fig. 6I).
FIGURE 6.

Intrathecal infusions of LRP1 agonists significantly increase axonal regeneration after spinal cord injury. A, illustration of spinal cord injury model. A C4 dorsal column lesion was surgically placed, transecting all axons of the dorsal column sensory projection (confirmed in Fig. 8). A bone marrow stromal cell graft was placed in the lesion cavity (green) to provide a permissive matrix for axonal growth. The LRP1 agonist RBD was infused intrathecally to stimulate Trk activation in DRG somata. B, GFAP labeling identified the lesion cavity (dashed lines) in this sagittal section encompassing the lesion site. B–F, rostral is left, caudal is right. C, sparse dorsal column sensory axonal penetration of the C4 dorsal column lesion site was observed in control subjects that received intrathecal infusions of GST. Sensory axons were labeled with CTB and approached the host/lesion interface (dashed line) from the caudal aspect of the lesion. The boxed region is shown at a higher magnification in E. D, in contrast, there was extensive axonal regeneration into the lesion site in animals that received RBD, shown at a higher magnification in F. An occasional axon extended beyond the lesion (arrow). Scale bars = 300 μm (A), 200 μm (C and D), and 50 μm (E and F). G, RBD infusion significantly increases the total number and proportion of axons regenerating into the lesion site (*, p = 0.03 compared with GST-treated controls). The proportion of axons in the graft compared with all labeled axons in the dorsal columns approaching the lesion is shown. H, the total length of all axons penetrating the lesion site was increased 3.6-fold in RBD-infused subjects (*, p < 0.01), and RBD infusion also increased the distance over which axons regenerated into the lesion site (I), reflected by the proportion of all labeled axons per subject that reached the lesion midline (**, p < 0.001 compared with GST-controls). Data are mean ± S.E.
Furthermore, analysis of histological sections demonstrated numerous CTB-labeled axons sprouting into the host gray matter as axons approached the lesion site. Quantification revealed a significant 80% increase in the number of these sprouting sensory axons in the host gray matter (p < 0.05, Fig. 7). Growth of transected axons remote from the lesion site has been referred to as “regenerative sprouting” (44). Sectioning of the medulla in all animals confirmed that lesions were complete and no axons were spared because the CTB tracer was not present in the nucleus gracilis (Fig. 8).
FIGURE 7.
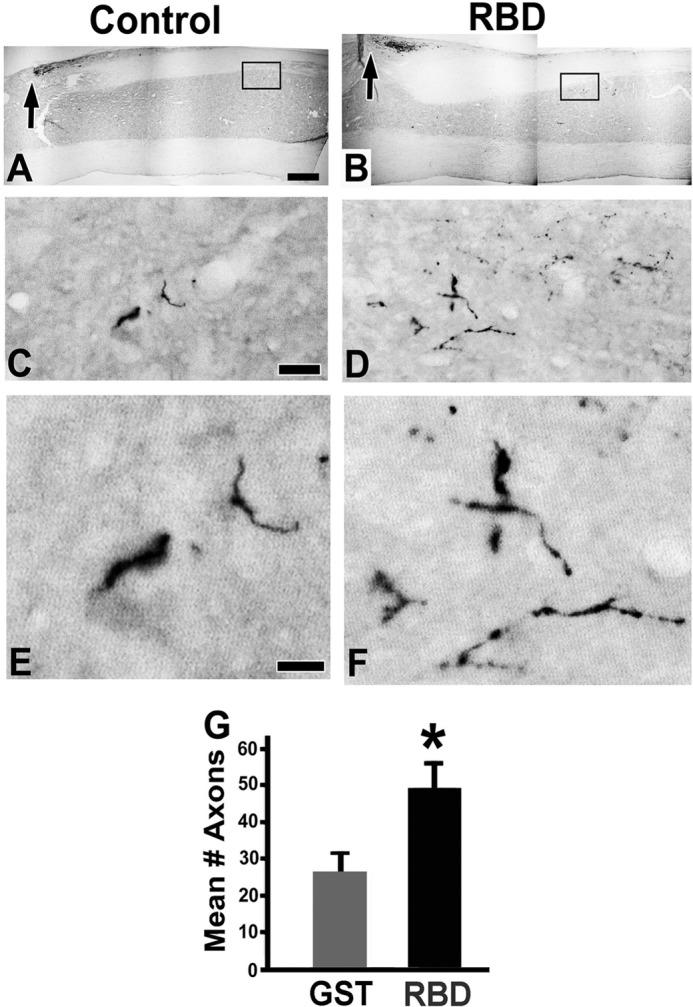
Intrathecal infusions of LRP1 agonists significantly increased axonal sprouting after spinal cord injury. Shown is CTB labeling of ascending dorsal column sensory axons in sagittal spinal cord sections. Low-magnification views show the site of dorsal column lesions (arrows) in control animals (A) and RBD-treated animals (B). Boxed regions are shown at a higher magnification in C–F. C and E, in control-lesioned subjects, very few CTB-labeled dorsal column sensory axons were observed in cervical gray matter below the lesion site (C5-C8 segments). Such axon terminals were not typically detected in intact animals. D and F, in RBD-infused animals, there were greater numbers of CTB-labeled axons sprouting into host gray matter below the lesion. F is an enlargement of the axons in panel D. G, RBD-treated rats exhibited a significant increase in the total number of axons counted in a series of one-in-six, 35-μm-thick sagittal sections. *, p < 0.05 compared with GST. Data are mean ± S.E. Scale bar = 450 μm (A and B), 10 μm (C and D), and 5 μm (E and F).
FIGURE 8.
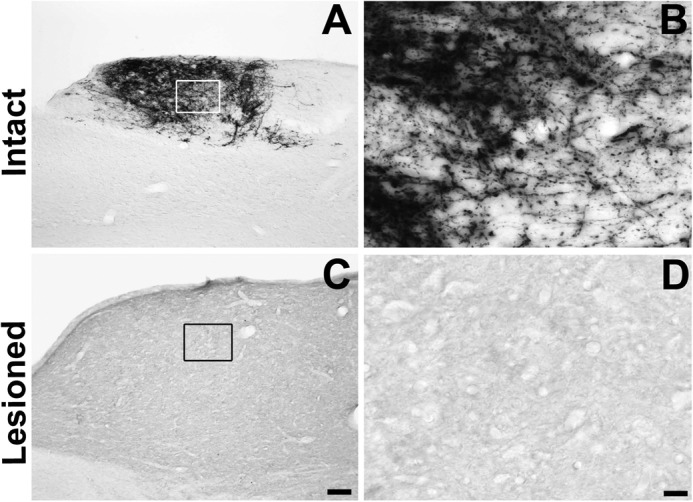
Lesion completeness. Sectioning of the medulla in all animals was performed to determine whether lesions were complete. Incomplete lesions resulting in spared axons would be apparent by CTB labeling in the medullary nucleus gracilis. A and B, in intact animals, numerous CTB labeled axons were present in the nucleus gracilis following intrasciatic injections (examined 72 h later). C and D, in lesioned animals, a complete absence of CTB labeling in the nucleus gracilis was observed, indicating that lesions were complete. Scale bar = 200 μm (A and C) and 20 μm (B and D).
DISCUSSION
The findings of this study identify an entirely novel mechanism for promoting axonal sprouting and regeneration in the CNS in vivo: the targeting of LRP1 receptors. Agonists of this LDL gene family receptor result in activation of canonical Trk signaling in a SFK-dependent manner, resulting in ERK and Akt activation. As a result, there is increased neurite outgrowth in vitro together with amelioration of myelin-mediated inhibition. Intrathecal infusions of LRP1 agonists result in significant increases in axonal sprouting and regeneration after spinal cord injury. Collectively, these findings indicate that targeting of lipoprotein receptors can activate cell signaling relevant to axonal growth in vitro and in vivo.
Although the LDL family of receptors has classically been associated with the modulation of lipoprotein metabolism, modern studies indicate a diversity of roles for this receptor system in various aspects of cellular activities, including cell signaling and function. However, the role of LRP1-dependent cell signaling in CNS injury in vivo has not been extensively examined to date. Mice with conditional loss of LRP1 in post-mitotic neurons exhibit alterations in brain lipid metabolism, synapse loss, and neurodegeneration (45). LRP1 also modulates amyloid precursor protein processing through interaction with apolipoprotein E (46, 3). This may account for the selective vulnerability of specific apolipoprotein E genotypes to the development of Alzheimer's disease. LRP1 receptors are also present on glia. For example, LRP1 is expressed by Schwann cells after peripheral nerve injury (11) and is thought to contribute to the ability of these cells to survive injury and contribute to peripheral nerve regeneration. Our data substantially expand the known roles of LRP1 in the nervous system by demonstrating significant effects on axonal growth and regeneration in in vivo models of central nervous system injury.
We did not study functional outcomes in this study because the ascending dorsal column sensory projection was transected several levels below its normal target of projection, the nucleus gracilis, and axons did not regenerate over the extended distance that would be required to support functional recovery. Rather, we used this model because it is of established value in determining whether candidate therapies enhance the plasticity and regeneration of CNS axons that otherwise fail to grow after spinal cord injury (34, 36, 41–43) Indeed, our findings reveal significant effects of LRP1 agonists on axonal growth after injury in this valuable model system. Future studies will explore outcomes in clinically relevant contusive spinal cord injury lesion models that result in motor and sensory deficits. A hypothetical advantage of LRP1 agonists in these models is the broad expression of LRP1 receptors on numerous CNS neuronal populations (18), suggesting that drugs targeting these receptors could broadly activate the intrinsic CNS neuronal growth response to injury (30, 47).
Mechanisms underlying the modulation of axonal growth include LRP1-initiated cell signaling and transactivation of TrkC. A previous report suggested that α2-macroglobulin may directly bind Trks independently of LRP1 (48). However, our results and others (23) indicate that structurally diverse LRP1 ligands, including matrix metalloproteinase 9-PEX and RBD, all activate Trks, A,and ERK1/2 and that these actions are blocked by RAP. RAP precludes binding of LRP1 ligands and, thus, blocks LRP1-initiated cell signaling in multiple cell culture models (5, 8, 49). Thus, the effects of LRP1 agonists in our models appear to depend on direct LRP1 binding, rather than direct binding to Trk receptors.
A major point of significance in this work lies in the observation that LRP1 activates canonical signaling associated with neurotrophin receptors. Neurotrophins, in turn, exhibit potent effects in models of CNS injury and neurodegeneration (34, 50–52) and potentially provide a new means of stimulating neurotrophin receptors through the use of LRP1 agonists, which have now been shown to stimulate both TrkA- and TrkC-mediated signaling (23 and present findings). The activity of LRP1 agonists on a diversity of Trk receptors could result in therapeutic benefits extending beyond the large-diameter dorsal root ganglion neuronal population targeted in this study. For example, LRP1 transactivation of TrkA (23) could alter aberrant patterns of nociceptive axon sprouting that are thought to contribute to chronic pain after spinal cord injury (53) or in other conditions. On the other hand, as with any therapy that targets axonal sprouting and regeneration, non-targeted sprouting induced by LRP1 agonists could also worsen functional outcomes. These possibilities must be addressed in future studies.
Moreover, intrathecal infusions of LRP1 agonists in this study resulted in enhancement of central axonal regeneration that is similar in nature to the effects of “conditioning lesions.” However, conditioning lesions are clinically impractical, whereas intrathecal infusions of LRP1 agonists are clinically relevant. Future studies will define the therapeutic window for infusion of LRP1 agonists in central and peripheral nerve injury and the effects of LRP1 receptor targeting on other axonal populations, including motor axonal systems of the spinal cord. Future studies will also address whether the combination of LRP1 infusions with NT-3 chemotropic gradients beyond a spinal cord lesion will enhance the number of sensory axons regenerating that fully bridge the lesion site, a combinatorial approach taken in previous studies using conditioning lesions (34, 36–38).
We are attempting to generate small molecules and bioactive peptides that bind and initiate LRP1-dependent cell signaling to further enhance clinical relevance. For example, the GST fusion protein used in these studies, the RBD of α2-macroglobulin, also contains a distinct binding domain for β amyloid (Aβ). This could potentially result in the formation of RBD-Aβ complexes (24) that accelerate β-amyloid-mediated neuronal degeneration (54). Yet other reports indicate that LRP1 agonists might reduce aberrant amyloid precursor protein processing (55) and that LRP1-dependent cell signaling in neurons and glia appears to consistently result in prosurvival effects (56). Moreover, the LRP1 ligand derived from the hemopexin domain of matrix metalloproteinase 9, PEX, does not bind Aβ and would be unlikely to influence Alzheimer's disease-related pathology. We are currently designing novel LRP1-binding molecules that activate cell signaling that is tailored to specific CNS disorders, including molecules that, like PEX, bind LRP1 but not Aβ. Regardless of positive or negative effects of LRP1 agonists on amyloid-mediated neurodegeneration, these effects would be unlikely to generate adverse events over the transient periods that RBD would be administered for neurotrauma. Collectively, findings of this study reveal a potentially new and practical drug class for the treatment of nervous system injury and degeneration.
Acknowledgments
We thank Lori Graham and Drs. Paul Lu and Elisabetta Mantuano for technical assistance.
*
This work was supported, in whole or in part, by National Institutes of Health Grants NINDS-R01NS-057456 and P30NS047101. This work was also supported by the Veterans Administration, by the Neurosciences Imaging Center, and by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation.
3
The abbreviations used are:
SFK
Src family kinase
DRG
dorsal root ganglion
NF200
neurofilament 200
RBD
receptor binding domain
PEX
hemopexin domain
RAP
receptor-associated protein
pERK
phospho-ERK
pTrkC
phospho-TrkC
CTB
cholera toxin subunit B
NT-3
neurotrophin-3
PP2
4-amino-5-(4-chlorophenyl)-7-(dimethylethyl)pyrazolo[3,4-d] pyrimidine
GFAP
glial fibrillary acidic protein.
REFERENCES
- 1.Strickland D. K., Gonias S. L., Argraves W. S. (2002) Diverse roles for the LDL receptor family. Trends Endocrinol. Metab. 13, 66–74 [DOI] [PubMed] [Google Scholar]
- 2.Kowal R. C., Herz J., Goldstein J. L., Esser V., Brown M. S. (1989) Low density lipoprotein receptor-related protein mediates uptake of cholesteryl esters derived from apoprotein E-enriched lipoproteins. Proc. Natl. Acad. Sci. U.S.A. 86, 5810–5814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holtzman D. M., Herz J., Bu G. (2012) Apolipoprotein E and apolipoprotein E receptors. Normal biology and roles in Alzheimer disease. Cold Spring Harbor Perspect. Med. 2, a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herz J., Strickland D. K. (2001) LRP. A multifunctional scavenger and signaling receptor. J. Clin. Invest. 108, 779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu K., Yang J., Tanaka S., Gonias S. L., Mars W. M., Liu Y. (2006) Tissue-type plasminogen activator acts as a cytokine that triggers intracellular signal transduction and induces matrix metalloproteinase-9 gene expression. J. Biol. Chem. 281, 2120–2127 [DOI] [PubMed] [Google Scholar]
- 6.Hayashi H., Campenot R. B., Vance D. E., Vance J. E. (2007) Apolipoprotein E-containing lipoproteins protect neurons from apoptosis via a signaling pathway involving low-density lipoprotein receptor-related protein-1. J. Neurosci. 27, 1933–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Padmasekar M., Nandigama R., Wartenberg M., Schlüter K. D., Sauer H. (2007) The acute phase protein α2-macroglobulin induces rat ventricular cardiomyocyte hypertrophy via ERK1,2 and PI3-kinase/Akt pathways. Cardiovasc. Res. 75, 118–128 [DOI] [PubMed] [Google Scholar]
- 8.Mantuano E., Inoue G., Li X., Takahashi K., Gaultier A., Gonias S. L. (2008) The hemopexin domain of matrix metalloproteinase-9 activates cell signaling and promotes migration of Schwann cells by binding to low-density lipoprotein receptor-related protein. J. Neurosci. 28, 11571–11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kinoshita A., Whelan C. M., Smith C. J., Mikhailenko I., Rebeck G. W., Strickland D. K., Hyman B. T. (2001) Demonstration by fluorescence resonance energy transfer of two sites of interaction between the low-density lipoprotein receptor-related protein and the amyloid precursor protein. Role of the intracellular adapter protein Fe65. J. Neurosci. 21, 8354–8361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su H. P., Nakada-Tsukui K., Tosello-Trampont A. C., Li Y., Bu G., Henson P. M., Ravichandran K. S. (2002) Interaction of CED-6/GULP, an adapter protein involved in engulfment of apoptotic cells with CED-1 and CD91/low density lipoprotein receptor-related protein (LRP). J. Biol. Chem. 277, 11772–11779 [DOI] [PubMed] [Google Scholar]
- 11.Campana W. M., Li X., Dragojlovic N., Janes J., Gaultier A., Gonias S. L. (2006) The low-density lipoprotein receptor-related protein is a pro-survival receptor in Schwann cells. Possible implications in peripheral nerve injury. J. Neurosci. 26, 11197–11207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mantuano E., Henry K., Yamauchi T., Hiramatsu N., Yamauchi K., Orita S. (2011) The unfolded protein response is a major mechanism by which LRP1 regulates Schwann cell survival after injury. J. Neurosci. 31, 13376–13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salicioni A. M., Gaultier A., Brownlee C., Cheezum M. K., Gonias S. L. (2004) Low density lipoprotein receptor-related protein-1 promotes β1 integrin maturation and transport to the cell surface. J. Biol. Chem. 279, 10005–10012 [DOI] [PubMed] [Google Scholar]
- 14.Muratoglu S. C., Mikhailenko I., Newton C., Migliorini M., Strickland D. K. (2010) Low density lipoprotein receptor-related protein 1 (LRP1) forms a signaling complex with platelet-derived growth factor receptor-β in endosomes and regulates activation of the MAPK pathway. J. Biol. Chem. 285, 14308–14317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonias S. L., Gaultier A., Jo M. (2011) Regulation of the urokinase receptor (uPAR) by LDL receptor-related protein-1 (LRP1). Curr. Pharm. Des. 17, 1962–1969 [DOI] [PubMed] [Google Scholar]
- 16.May P., Woldt E., Matz R. L., Boucher P. (2007) The LDL receptor-related protein (LRP) family. An old family of proteins with new physiological functions. Ann. Med. 39, 219–228 [DOI] [PubMed] [Google Scholar]
- 17.Lillis A. P., Van Duyn L. B., Murphy-Ullrich J. E., Strickland D. K. (2008) LDL receptor-related protein 1. Unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 88, 887–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolf B. B., Lopes M. B., VandenBerg S. R., Gonias S. L. (1992) Characterization and immunohistochemical localization of α-2-macroglobulin receptor (low density lipoprotein receptor related protein) in human brain. Am. J. Pathol. 141, 37–42 [PMC free article] [PubMed] [Google Scholar]
- 19.Mantuano E., Jo M., Gonias S. L., Campana W. M. (2010) Low density lipoprotein receptor-related protein (LRP1) regulates Rac1 and RhoA reciprocally to control Schwann cell adhesion and migration. J. Biol. Chem. 285, 14259–14266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaultier A., Wu X., Le Moan N., Takimoto S., Mukandala G., Akassoglou K., Campana W. M., Gonias S. L. (2009) Low-density lipoprotein receptor-related protein 1 is an essential receptor for myelin phagocytosis. J. Cell Sci. 122, 1155–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goto J. J., Tanzi R. E. (2002) The role of the low-density lipoprotein receptor-related protein (LRP1) in Alzheimer's A β generation: development of a cell-based model system. J. Molec. Neurosci. 19, 37–41 [DOI] [PubMed] [Google Scholar]
- 22.Liu Q., Zerbinatti C. V., Zhang J., Hoe H. S., Wang B., Cole S. L., Herz J., Muglia L., Bu G. (2007) Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 56, 66–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi Y., Mantuano E., Inoue G., Campana W. M., Gonias S. L. (2009) Ligand binding to LRP1 transactivates Trk receptors by a Src family kinase-dependent pathway. Sci. Signal. 2, ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mettenburg J. M., Webb D. J., Gonias S. L. (2002) Distinct binding sites in the structure of α 2-macroglobulin mediate the interaction with β-amyloid peptide and growth factors. J. Biol. Chem. 277, 13338–13345 [DOI] [PubMed] [Google Scholar]
- 25.Mantuano E., Mukandala G., Li X., Campana W. M., Gonias S. L. (2008b) Molecular dissection of the human α2-macroglobulin subunit reveals domains with antagonistic activities in cell signaling. J. Biol. Chem. 283, 19904–19911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Howard G. C., Yamaguchi Y., Misra U. K., Gawdi G., Nelsen A., DeCamp D. L., Pizzo S. V. (1996) Selective mutations in cloned and expressed α-macroglobulin receptor binding fragment alter binding to either the α2-macroglobulin signaling receptor or the low density lipoprotein receptor-related protein/α2-macroglobulin receptor. J. Biol. Chem. 271, 14105–14111 [DOI] [PubMed] [Google Scholar]
- 27.Nielsen K. L., Holtet T. L., Etzerodt M., Moestrup S. K., Gliemann J., Sottrup-Jensen L., Thogersen H. C. (1996) Identification of residues in alpha-macroglobulins important for binding to the α2-macroglobulin receptor/low density lipoprotein receptor-related protein. J. Biol. Chem. 271, 12909–12912 [DOI] [PubMed] [Google Scholar]
- 28.Van den Steen P. E., Van Aelst I., Hvidberg V., Piccard H., Fiten P., Jacobsen C., Moestrup S. K., Fry S., Royle L., Wormald M. R., Wallis R., Rudd P. M., Dwek R. A., Opdenakker G. (2006) The hemopexin and _O-_glycosylated domains tune gelatinase B/MMP-9 bioavailability via inhibition and binding to cargo receptors. J. Biol. Chem. 281, 18626–18637 [DOI] [PubMed] [Google Scholar]
- 29.Herz J., Goldstein J. L., Strickland D. K., Ho Y. K., Brown M. S. (1991) 39-kDa protein modulates binding of ligands to low density lipoprotein receptor-related protein/α 2-macroglobulin receptor. J. Biol. Chem. 266, 21232–21238 [PubMed] [Google Scholar]
- 30.Blesch A., Lu P., Tsukada S., Alto L. T., Roet K., Coppola G., Geschwind D., Tuszynski M. H. (2012) Conditioning lesions before or after spinal cord injury recruit broad genetic mechanisms that sustain axonal regeneration: superiority to camp-mediated effects. Exp. Neurol. 235, 162–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lawson S. N., Waddell P. J. (1991) Soma neurofilament immunoreactivity is related to cell size and fibre conduction velocity in rat primary sensory neurons. J. Physiol. 435, 41–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMahon S. B., Armanini M. P., Ling L. H., Phillips H. S. (1994) Expression and coexpression of Trk receptors in subpopulations of adult primary sensory neurons projecting to identified peripheral targets. Neuron 12, 1161–1171 [DOI] [PubMed] [Google Scholar]
- 33.Weidner N., Ner A., Salimi N., Tuszynski M. H. (2001) Spontaneous corticospinal axonal plasticity and functional recovery after adult central nervous system injury. Proc. Natl. Acad. Sci. U.S.A. 98, 3513–3518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alto L. T., Havton L. A., Conner J. M., Hollis E. R., 2nd, Blesch A., Tuszynski M. H. (2009) Chemotropic guidance facilitates axonal regeneration and synapse formation after spinal cord injury. Nat. Neurosci. 12, 1106–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hofstetter C. P., Schwarz E. J., Hess D., Widenfalk J., El Manira A., Prockop D. J., Olson L. (2002) Marrow stromal cells form guiding strands in the injured spinal cord and promote recovery. Proc. Natl. Acad. Sci. U.S.A. 99, 2199–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kadoya K., Tsukada S., Lu P., Coppola G., Geschwind D., Filbin M. T., Blesch A., Tuszynski M. H. (2009) Combined intrinsic and extrinsic neuronal mechanisms facilitate bridging axonal regeneration one year after spinal cord injury. Neuron 64, 165–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu P., Yang H., Jones L. L., Filbin M. T., Tuszynski M. H. (2004) Combinatorial therapy with neurotrophins and cAMP promotes axonal regeneration beyond sites of spinal cord injury. J. Neurosci. 24, 6402–6409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor L., Jones L., Tuszynski M. H., Blesch A. (2006) Neurotrophin-3 gradients established by lentiviral gene delivery promote short-distance axonal bridging beyond cellular grafts in the injured spinal cord. J. Neurosci. 26, 9713–9721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kilkenny C., Browne W. J., Cuthill I. C., Emerson M., Altman D. G. (2010) Improving bioscience research reporting. The ARRIVE guidelines for reporting animal research. PLoS Biol. 8, e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsoulfas P., Stephens R. M., Kaplan D. R., Parada L. F. (1996) TrkC isoforms with inserts in the kinase domain show impaired signaling responses. J. Biol. Chem. 271, 5691–5697 [DOI] [PubMed] [Google Scholar]
- 41.Ramer M. S., Priestley J. V., McMahon S. B. (2000) Functional regeneration of sensory axons into the adult spinal cord. Nature 403, 312–316 [DOI] [PubMed] [Google Scholar]
- 42.Bradbury E. J., Moon L. D., Popat R. J., King V. R., Bennett G. S., Patel P. N., Fawcett J. W., McMahon S. B. (2002) Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 416, 636–640 [DOI] [PubMed] [Google Scholar]
- 43.Neumann S., Bradke F., Tessier-Lavigne M., Basbaum A. I. (2002) Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 34, 885–893 [DOI] [PubMed] [Google Scholar]
- 44.Tuszynski M. H., Steward O. (2012) Concepts and methods for the study of axonal regeneration in the CNS. Neuron 74, 777–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Q., Trotter J., Zhang J., Peters M. M., Cheng H., Bao J., Han X., Weeber E. J., Bu G. (2010) Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive age-dependent synapse loss and neurodegeneration. J. Neurosci. 30, 17068–17078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andersen O. M., Willnow T. E,. (2006) Lipoprotein receptors in Alzheimer's disease. Trends Neurosci. 29, 687–694 [DOI] [PubMed] [Google Scholar]
- 47.Qiu J., Cai D., Dai H., McAtee M., Hoffman P. N., Bregman B. S., Filbin M. T. (2002) Spinal axon regeneration induced by elevation of cyclic AMP. Neuron. 34, 895–903 [DOI] [PubMed] [Google Scholar]
- 48.Chiabrando G. A., Sánchez M. C., Skornicka E. L., Koo P. H. (2002) Low-density lipoprotein receptor-related protein mediates in PC12 cell cultures the inhibition of nerve growth factor-promoted neurite outgrowth by pregnancy zone protein and α2-macroglobulin. J. Neurosci. Res. 70, 57–64 [DOI] [PubMed] [Google Scholar]
- 49.Bacskai B. J., Xia M. Q., Strickland D. K., Rebeck G. W., Hyman B. T. (2000) The endocytic receptor protein LRP also mediates neuronal calcium signaling via _N_-methyl-d-aspartate receptors. Proc. Natl. Acad. Sci. U.S.A. 97, 11551–11556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fischer W., Wictorin K., Björklund A., Williams L. R., Varon S., Gage F. H. (1987) Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature 329, 65–68 [DOI] [PubMed] [Google Scholar]
- 51.Gravel C., Götz R., Lorrain A., Sendtner M. (1997) Adenoviral gene transfer of ciliary neurotrophic factor and brain-derived neurotrophic factor leads to long-term survival of axotomized motor neurons. Nat. Med. 3, 765–770 [DOI] [PubMed] [Google Scholar]
- 52.Nagahara A. H., Merrill D. A., Coppola G., Tsukada S., Schroeder B.E., Shaked G. M., Wang L., Blesch A., Kim A., Conner J. M., Rockenstein E., Chao M. V., Koo E. H., Geschwind D., Masliah E., Chiba A. A., Tuszynski M. H. (2009) Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat. Med. 15, 331–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Christensen M. D., Hulsebosch C. E. (1997) Spinal cord injury and anti-NGF treatment results in changes in CGRP density and distribution in the dorsal hown in the rat. Exp. Neurol. 147, 463–475 [DOI] [PubMed] [Google Scholar]
- 54.Wilhelmus M. M., Otte-Höller I., van Triel J. J., Veerhuis R., Maat-Schieman M. L., Bu G., de Waal R. M., Verbeek M. M. (2007) Lipoprotein receptor-related protein-1 mediates amyloid b mediated cell death of cerebrovascular cells. Am. J. Path. 171, 1989–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ulery P. G., Strickland D. K. (2000) LRP in Alzheimer's disease. Friend or foe? J. Clin. Invest. 106, 1077–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fuentealba R. A., Liu Q., Kanekiyo T., Zhang J., Bu G. (2009) Low-density lipoprotein receptor related protein 1 (LRP1) promotes anti-apoptotic signaling in neurons by activating Akt survival pathway. J. Biol. Chem. 284, 34045–34053 [DOI] [PMC free article] [PubMed] [Google Scholar]