Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model (original) (raw)
. Author manuscript; available in PMC: 2013 Sep 17.
Published in final edited form as: Nat Genet. 2013 Jul 28;45(9):1021–1028. doi: 10.1038/ng.2713
Abstract
Despite continual progress in the cataloging of vertebrate regulatory elements, little is known about their organization and regulatory architecture. Here we describe a massively parallel experiment to systematically test the impact of copy number, spacing, combination and order of transcription factor binding sites on gene expression. A complex library of ~5,000 synthetic regulatory elements containing patterns from 1 2 liver-specific transcription factor binding sites was assayed in mice and in HepG2 cells. We find that certain transcription factors act as direct drivers of gene expression in homotypic clusters of binding sites, independent of spacing between sites, whereas others function only synergistically. Heterotypic enhancers are stronger than their homotypic analogs and favor specific transcription factor binding site combinations, mimicking putative native enhancers. Exhaustive testing of binding site permutations suggests that there is flexibility in binding site order. Our findings provide quantitative support for a flexible model of regulatory element activity and suggest a framework for the design of synthetic tissue-specific enhancers.
Transcription factors regulate diverse patterns of gene expression by binding cooperatively in clusters at gene promoters, enhancers and other _cis_-regulatory modules1–3. Genetic variations at transcription factor binding sites have been associated with a wide range of human phenotypes4–7. The genome-wide occupancy patterns of transcription factors are readily measured by methods such as chromatin immuno-precipitation followed by sequencing (ChIP-seq)8 that identify regions of open chromatin and transcription factor binding9–12. However, these techniques are limited by the quality of antibodies and, moreover, tend to have poor resolution, preventing a detailed analysis of binding site occupancy, particularly when the binding sites for multiple transcription factors are present in the same _cis_-regulatory module. As a result, very little is known about how the binding of multiple transcription factors in proximity influences the activity of a _cis_-regulatory module on gene expression. For example, a fundamental question concerning gene regulation is whether heterotypic transcription factor binding site clusters constitute a flexible mechanism for fine-tuning robust gene expression, which has been referred to as a `billboard model' (refs. 13–15), or specific patterns of spacing, combination and order are necessary for enhancer function16,17.
The clustering of degenerate transcription factor binding motifs is readily observable in the primary sequence of genomic DNA, a fact that has been exploited to predict distal gene enhancers using probabilistic18 and machine learning19,20 approaches. Although such methods solve the problem of low resolution, they cannot distinguish between functional and neutral sites or assess the combinatorial rules of _cis_-regulatory modules. Nevertheless, a common pattern observed in these clusters is the homotypic grouping of multiple copies of the same motif21, suggesting that multiple copies of the same signal can serve to fine-tune gene expression. Consistent with this hypothesis, several studies found that the synthetic concatenation of key regulatory signals amplified gene expression in reporter assays17,22–24. Such studies demonstrate the value of synthetic approaches in identifying the basic rules underlying regulatory module organization. However, the high cost and low-throughput nature of promoter and enhancer assays have thus far prevented any systematic dissection of mammalian regulatory element architecture in vivo.
We report here the findings of a massively parallel reporter assay in which the functional activity of 4,970 synthetic regulatory element sequences (SRESs), each 168 bp in length, was tested simultaneously in mice and in human hepatocellular carcinoma HepG2 cells. Methodologically, our approach builds on recent experiments that exhaustively tested the effects of mutating every possible base in five mammalian enhancers25,26. Our goal here was to systematically test the rules of regulatory element organization using synthetic elements. We designed a diverse library of SRESs consisting of transcription factor binding sites from 12 known liver-specific transcription factors patterned onto 2 neutral templates. The design comprises three classes of elements that test distinct hypotheses regarding the nature of homotypic clustering, synergy between transcription factors in heterotypic enhancers and the impact of binding site spacing and order on expression (Fig. 1a). Programmable microarrays were used to synthesize the pool of SRESs, which were cloned en masse into a tagged reporter vector library and assayed in vivo using the mouse hydrodynamic tail vein assay27,28 and in vitro by transfection into HepG2 cells. Transcribed tags were identified in liver mRNA or HepG2 cells 24 h after injection or transfection, respectively, by RNA sequencing (RNA-seq) (Fig. 1b). The relative abundance of each of the SRESs was determined via a new analysis pipeline that achieves very high correlation between biological replicates (Online Methods).
Figure 1.

Synthetic enhancer sequence design and controls. (a) SRESs consist of patterns of 12 consensus binding sequences arranged homotypically (class I) or heterotypically (class II and class III) on 1 of 2 neutral, 168-bp templates. (b) Schematic of massively parallel reporter assay methodology. SRESs were cloned upstream of a minimal promoter in a tagged luciferase library and then assayed in vivo using hydrodynamic tail vein injection. Livers were dissected 24 h after injection, mRNA was generated, and tags were reverse transcribed and sequenced. (c) Bimodal distribution of expression values for 4,966 SRESs. Expression values were calculated using the equation shown. (d) Template-template correlation. Expression values for 2,217 pairs of SRESs (not all SRESs had data for both templates owing to quality control measures) containing the exact same patterns of consensus binding sequences on 2 separate templates are plotted. The red line is a linear regression trace, whereas the dashed line is the diagonal. Template 1, hg19 chr. 9: 83,712,599–83,712,766; template 2, hg19 chr. 2: 211,153,238–211,153,405. (e) Expression values from the three mice in which the SRES library was tested, exhibiting a very high level of correlation.
RESULTS
Three classes of synthetic regulatory elements
To maximize our ability to make rigorous conclusions about enhancer organization, we designed three increasingly complex classes of SRESs. Class I SRESs (n = 533) were homotypic, containing 1, 2, 4 or 8 copies of the same transcription factor binding site with different spacing. Class II SRESs (n = 1,797) were heterotypic but still relatively simple, with 2 different types of transcription factor binding sites arranged as 2, 4 or 8 sites that were separated uniformly. Class III SRESs (n = 2,636) were completely heterotypic, with 3–8 types of transcription factor binding sites separated by a fixed distance with only 1 site per transcription factor (Fig. 1a). For all classes, we used consensus binding sequences for 12 transcription factors (AHR/ARNT, CEBPA, FOXA1, GATA4, HNF1A, HNF4A, NR2F2, ONECUT1, PPARA, RXRA, TFAP2C and XBP1) important for liver development and function (Supplementary Table 1). All of these sequences are enriched in putative liver-specific enhancers9, and 10 of 12 matched those used in other transcription factor binding site data sets (Supplementary Table 2). The 12 binding sites were patterned onto 2 different inactive 168-bp genomic DNA templates (template 1: hg19 chr. 9: 83,712,599–83,712,766; template 2: hg19 chr. 2: 211,153,238–211,153,405) (Supplementary Fig. 1a). Template 1 constitutes a portion of a randomly selected element from the VISTA enhancer browser29 with no enhancer activity, and template 2 constitutes a portion of a known muscle enhancer that is not active in liver cells30.
We took several steps to ensure the confidence of our expression measures (Fig. 1c). First, we included in the library two 168-bp negative controls (hg19 chr. 3: 197,439,137–197,439,304 and hg19 chr. 5: 172,177,154–172,177,321) independently validated as such by the tail vein assay (Supplementary Fig. 1a). Two validated 168-bp positive controls were also included in the library: a core region of the Ltv1 enhancer26 (mm9 chr. 7: 29,161,577–29,161,744), as well as a strong liver-specific enhancer (hg19 chr. 19: 35,531,985–35,532,152) in the first intron of HPN (encoding hepsin; Supplementary Fig. 1a). Second, each SRES was paired with an average of 90 tags (median of 67 tags), each 20 bp in length, to facilitate accurate quantification and to minimize tag sequence–specific biases. Finally, we injected the SRES library into three mice to assess reproducibility and verified for each SRES that the aggregate luciferase activity for the library was much stronger than for empty vector control (Supplementary Fig. 1b). Our original design included 5,838 sequences arranged across the 2 templates, and nearly all of these were represented by at least 1 tag in each replicate liver sample. Using a stringent informatics pipeline (Online Methods), we obtained high-quality expression data for 4,966 SRES, as well as for the 4 controls, corresponding to an average of 103,835 individual tags recovered per replicate (Supplementary Table 3). Our final expression measure for each SRES, which varied between 0 and 1, reflects the ratio between the number of transcribed tags and the total number of tags for that SRES in the library. A complete listing of SRESs along with their composition and expression data is provided in Supplementary Table 4.
The four control sequences in the SRES library exhibited the same expression trends as observed in tail vein assays performed with individual plasmids (Supplementary Fig. 1c). We observed high correlation between expression measures from the two templates used for patterning (Spearman's ρ = 0.75, P = 0; Fig. 1d). We identified 123 transcription factor binding site patterns (6%) that resulted in discordant expression in the 2 templates (Supplementary Fig. 2a,b). Discordant patterns had more binding sites (an average of 6.3 binding sites/pattern versus 5.5 binding sites/pattern for concordant patterns), were predominantly class III sites (75% versus 50% for concordant patterns) and were enriched for HNF1A (false discovery rate (FDR)-adjusted P = 1 × 10−6, Fisher's exact test) and NR2F2 (P = 0.002) binding sites (Supplementary Fig. 2c), which were both strong determinants of expression. Together, these data are consistent with the idea that the discordance we observed is predominantly due to noise at higher expression levels, rather than to some intrinsic difference between the templates. As a result, we considered data from both templates for all further analyses.
We also observed strong correlation between the expression measures from the three mouse samples used in the assay (Spearman's ρ = 0.88–0.89, P = 0; Fig. 1e), demonstrating the reproducibility of our results. To ascertain the robustness of SRES architecture in different liver cell types, we also transfected the library into HepG2 cells (a human hepatocellular carcinoma line) and processed tag sequencing data using the same pipeline. As with mouse liver, we observed strong correlation between replicates (Spearman's ρ = 0.79–0.84), good template correlation (Spearman's ρ = 0.69) and trends in the complexity of transcription factor binding sites (Supplementary Fig. 3a–c). Moreover, we observed strong agreement between SRES-driven expression in the mouse liver and HepG2 cells (Spearman's ρ = 0.81) across the entire data set (Supplementary Fig. 3d).
Homotypic amplification is compatible with a subset of sites
Several studies have reported that the concatenation of functional sequences containing transcription factor binding sites can lead to stronger expression of a reporter gene17,22,23. Using class I SRESs, we addressed the universality of this principle for each of the 12 transcription factor binding sites. For five binding sites (CEBPA, FOXA1, HNF1A, ONECUT1 and XBP1 transcription factors), we observed a significant correlation (Spearman's ρ = 0.32, P = 1 × 10−18) between expression and binding site copy number (Fig. 2a). Of these binding sites, the one for HNF1A produced the strongest effect on expression (Spearman's ρ = 0.68), which seemed to be saturated beyond four copies of the binding site. For example, clusters of 4 HNF1A binding sites resulted on average in 1.9-fold higher expression than clusters with 2 binding sites, whereas SRESs containing 8 binding sites resulted in only a 1.2-fold increase in expression relative to SRESs containing 4 binding sites. This finding suggests that some sites were rendered non-functional by crowding or that a biochemical saturation mechanism might exist. For the remaining seven transcription factors, no homotypic clustering effects were observed. Several of these transcription factors (for example, PPARA, RXRA and TFAP2C) are known to function in heterodimeric complexes31,32 and probably require additional cofactors or sequences to drive expression, as we later observed for heterotypic SRESs.
Figure 2.

Homotypic amplification of expression is compatible with a subset of transcription factor binding sites, independent of their spacing. (a) We observed significant correlation between expression and the size of the homotypic cluster for 5 of the 12 transcription factor binding sites (CEBPA, FOXA1, HNF1A, ONECUT1 and XBP1). The PPARA binding site is included as an example of a site that could not be homotypically amplified. Included on the right are box plots for the background expression of all SRESs with a single binding site (B), as well as for the positive (+) and negative (−) controls. Red boxes denote groups of SRESs with significantly higher expression compared to background (Wilcoxon rank-sum test P ≤ 0.05), which is a slightly more stringent test than comparison against negative controls. P values refer to Spearman's correlation coefficients (corrected for multiple testing using FDR). In the box plots, the central rectangle spans the first and third quartiles, the line inside the rectangle is the median, and the lines beyond the box indicate the locations of the minimum and maximum values. (b) In the vast majority of cases, the strength of expression was not dependent on the distance between binding sites, as observed for class I elements. Shown are examples of SRESs, each with two copies of one of the three strongest transcription factor binding sites, including sites for CEBPA, HNF1A and XBP1. P values refer to Spearman's correlation coefficients, and the dashed gray lines are the regression traces.
In addition to systematically testing the role of transcription factor binding site copy number, class I SRESs have a wide range of spacing between binding sites. To determine whether the expression driven by homotypic clusters was dependent on the spacing of transcription factor binding sites, we examined class I SRESs containing 2 or 4 copies of each of the 12 liver-specific binding sites. For 11 of 12 binding sites, we observed no significant correlation between binding site spacing and expression (Spearman's correlation P > 0.05) (Fig. 2b and Supplementary Figs. 4 and 5). The binding site for NR2F2 was the only exception, showing slightly stronger expression with increasing distance between copies of the binding site in two- and four-site SRESs.
Enhancer predictions defined by low-resolution methods such as ChIP-seq tend to be quite long (often >1 kb). However, many enhancers have shorter, core elements26 (as short as 44 bp33) that are sufficient to drive tissue-specific expression in vivo. We decided to investigate how many copies of each transcription factor binding site were required to yield reproducible expression. Because each of the SRESs was cloned upstream of a 31-bp minimal promoter element containing a TATA box that could recruit transcriptional complexes, it is conceivable that the impact of a single transcription factor binding site could be detected. SRESs with the same number and type of binding sites were grouped for this comparison to reduce the impact of potential novel motifs created by the positioning of sequences on the templates. We compared the expression driven by all 48 groups of homotypic SRESs (12 transcription factor binding sites with 1, 2, 4 and 8 sites per SRES) against a background group of all SRESs with only 1 site. Unexpectedly, we found that a single copy of the 17-bp HNF1A (FDR-adjusted P = 0.006, Wilcoxon rank-sum test) or XBP1 (P = 0.002) consensus sequence produced significant levels of expression. In contrast, two CEBPA sites (P = 1 × 10−5), four FOXA1 sites (P = 0.002) and eight ONECUT1 sites (P = 0.0002) were necessary to achieve significant levels of expression. Together, these findings demonstrate that consistent expression can be derived from a small number of transcription factor binding sites.
Increased binding site complexity leads to stronger expression
Homotypic clusters of transcription factor binding sites are observed throughout vertebrate genomes21 and are often sufficient to drive robust expression in reporter assays17,22,23. We were interested in the impact of regulatory element heterogeneity on gene expression. In general, we observed the strongest expression from completely heterotypic class III SRESs, lower levels of expression from simple heterotypic class II SRESs (with sites for two transcription factors) and the lowest expression from homotypic class I SRESs (Fig. 3a), even when controlling for the number of patterned sites (Fig. 3b). Compared to negative controls (mean expression = 0.055), 198 (37%) class I SRESs, 1,116 (62%) class II SRESs and 2,229 (85%) class III SRESs resulted in significantly higher levels of expression (P < 0.05, Wilcoxon rank-sum test). The mean expression of the top 10% of class I SRESs was 0.32, whereas mean expression was 0.50 for class II SRESs and 0.59 for class III SRESs. Together, these results suggest that synergy in heterotypic clusters has a role in driving higher levels of expression compared with homotypic clusters. These trends were identical in HepG2 cells (Supplementary Fig. 3b).
Figure 3.
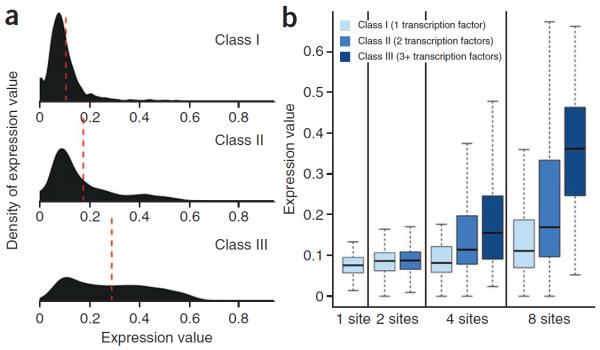
Heterotypic elements drive stronger expression than homotypic ones. (a) Density of expression by SRES class. The density of expression is plotted for each class of SRES. Red dashed lines denote the mean expression value for each class. (b) Box plot of expression by class and number of sites. Note that there are no class II SRESs with 1 site or class III SRESs with <3 sites. In the box plots, the central rectangle spans the first and third quartiles, the line inside the rectangle is the median, and the lines beyond the box indicate the locations of the minimum and maximum values.
Strong reporter expression favors specific motif combinations
Although heterotypic SRESs on average resulted in stronger expression than homotypic ones, there was still considerable variability in expression driven by heterotypic SRESs. This variability suggests that specific configurations of the same transcription factor binding sites that lead to stronger or weaker expression could exist. To identify factors resulting in favorable and unfavorable configurations for expression, we modeled the expression of class I and II SRESs as a function of the number and type of transcription factor binding sites, including a synergy term for all pairs of transcription factors in the sequence (Online Methods), with the model independent of the positioning of binding sites. The model was trained using class I and II data, and terms were exhaustively removed to minimize the Akaike information criterion34 (AIC) and to avoid overfitting. To further address the possibility of overfitting, we evaluated the model using 10-fold cross-validation on the entire set of 2,330 class I and II SRESs (Supplementary Fig. 6a) as well as on a non-redundant set of 234 unique transcription factor binding site combinations. In both cases, the estimate of the standard error of the model on the test data was negligible. In the same setting, we examined the ability of the model to distinguish between active and inactive SRESs (Online Methods) by computing the area under the receiver operating characteristic (ROC) curve (AUC), obtaining values of 0.78 and 0.84, respectively (Supplementary Fig. 6b,c). These values indicate that the model can accurately describe the activity of the SRESs using information on the composition of transcription factor binding sites.
Transcription factor binding sites that drove strong expression in class I SRESs were also significant contributors in our combinatorial model (Supplementary Fig. 7). We also observed a significant role for the RXRA binding site (P = 0.03, Wald χ2 test) and found that increasing copy number of the AHR/ARNT binding site negatively affected expression (P = 0.006).
By examining cooperative terms in the model that made significant contributions to expression, we identified four transcription factor binding site interactions (FOXA1-NR2F2, NR2F2-ONECUT1, NR2F2-XBP1 and RXRA-XBP1) (Fig. 4a). Particularly notable in this set was the binding site for NR2F2 (also known as COUP-TFII), which did not affect expression when additional copies were present (P = 0.64, Wald χ2 test) but cooperated with the FOXA1 (P = 0.006), ONECUT1 (P = 0.02) and XBP1 (P = 0.04) binding sites. We also observed highly significant interference between the HNF1A and XBP1 binding sites (P = 0.0002, Wald χ2 test), suggesting that the transcription factors that recognize these sites may compete for cofactors to drive different modes of transcription.
Figure 4.

Combinatorial effects in heterotypic clusters of two different transcription factor binding sites. (a) Significant interactions that were identified from a general model of class I and class II SRES–driven expression using the χ2 goodness-of-fit test or direct comparisons between class I and class II data sets. Dotted gray lines refer to the 19 combinations (of a possible 121) that were sampled in class II SRESs. Red lines indicate significant synergy, and a black line indicates significant interference (P values ≤ 0.05 in all cases). (b) Direct comparisons between the eight pairs of interacting binding sites and predicted expression based on class I data alone (black lines). Five interactions (NR2F2-XBP1, NR2F2-ONECUT1, NR2F2-FOXA1, RXRA-XBP1 and HNF1A-XBP1) were identified by including combinatorial terms in the model (Wald χ2 test P values shown at top right), whereas three (FOXA1-RXRA, FOXA1-PPARA and TFAP2C-RXRA) were identified by directly comparing expression from binding site pairs in combinations with homotypic sequences containing an equal number of the binding sites in isolation (Wilcoxon rank-sum test). A single example is shown for each combination, corresponding to a fixed number of sites (the number of sites in each SRES is given in parentheses, and asterisks to the left of P values indicate those that are significant). In the box plots, the central rectangle spans the first and third quartiles, the line inside the rectangle is the median, and the lines beyond the box indicate the locations of the minimum and maximum values.
Finally, we speculated that synergy and interference between factors could manifest as sequences with specific densities of transcription factor binding sites but not appear as overall trends. Therefore, we also looked at these interactions using direct comparisons between binding site pairs for different densities of binding sites (2, 4 or 8 sites per SRES). We employed a stringent test for synergy that compares the expression driven by a heterotypic combination of two transcription factor binding sites to that resulting from equally sized homotypic clusters of both binding sites. By comparing data from homotypic and heterotypic SRESs from class I and II in such a manner (independent of binding site spacing and order), we identified three additional cooperative interactions of binding sites (FOXA1-PPARA, FOXA1-RXRA and RXRA-TFAP2C; P < 0.05, Wilcoxon rank-sum test) (Fig. 4b). Together with the interactions predicted by the model itself, these two methods provide a map of eight combinatorial interactions (Fig. 4a).
Synthetic elements mimic putative liver enhancers
Because our library design was completely synthetic, we wanted to see whether the regulatory architecture we inferred from it is relevant to native genomic regulatory elements. We examined a collection of 51,850 putative mouse liver enhancers identified by ChIP-seq experiments9 and employed the same 12 position weight matrixes (PWMs) used to derive consensus binding sequences in our SRES library to map potential transcription factor binding sites in them. In general, these PWMs were enriched within a ~600-bp window centered on the peak position of putative liver enhancers but not in cerebellar enhancers9, which served as a control (Supplementary Fig. 8). We categorized 40,617 (78%) of these putative enhancers into 1 of the 3 classes of SRESs on the basis of transcription factor binding site heterogeneity (Fig. 5a), demonstrating that our library segments exhibited similarity to native configurations of binding sites.
Figure 5.
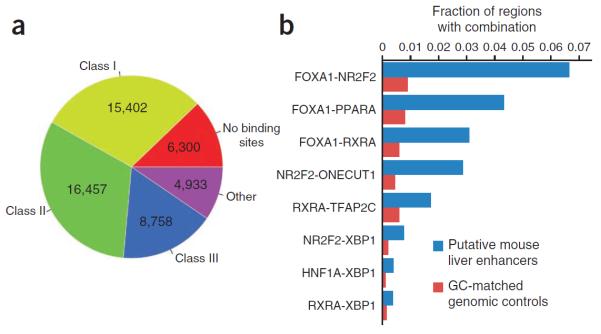
Synthetic enhancers mimic mouse liver enhancers. (a) Categorization of 51,850 putative mouse liver enhancers into the 3 SRES classes on the basis of the distribution of the 12 transcription factor binding sites. (b) Frequencies of transcription factor binding site pairs for each of the 8 interactions identified in Figure 4 in putative mouse liver enhancers versus 103,700 matched random genomic controls.
To determine whether the combinatorial interactions that we identified in the SRES library (Fig. 4a) were also found and enriched in the mouse genome, we analyzed occurrences of each of the eight interaction pairs in putative liver enhancers. All seven cooperative interactions that we identified were significantly enriched in these regions compared to GC- and length-matched random genomic controls (FDR-adjusted P < 1 × 10−17, Fisher's exact test) (Fig. 5b and Supplementary Fig. 9). We also identified a statistically significant enrichment of pairs of binding sites for HNF1A and XBP1 (P = 2 × 10−28), contrary to our expectations based on the model. However, these pairings were extremely rare, occurring in only 205 (0.4%) of putative liver enhancers.
Reporter expression is influenced by binding site order
Class III SRESs had only one copy of each transcription factor binding site separated by a fixed 3-bp spacer. These sequences thus represent the ideal data set to determine whether the order of these binding sites affects the strength of expression. Owing to limitations on library complexity, we restricted the number of transcription factor binding sites to 9 (eliminating those for CEBPA, GATA4 and TFAP2C) and sampled 2,636 of 623,448 possible permutations of 3–8 binding sites. We identified 211 sets of class III SRESs containing at least 2 permutations of exactly the same binding sites in different orders (Supplementary Table 5). Of these, 87 (41%) had a favorable permutation that resulted in a significantly stronger increase in expression than an unfavorable permutation (FDR < 0.05, Wilcoxon signed-rank test). Favorable permutations produced, on average, 2.8-fold higher expression than unfavorable ones, with some pairs varying by as much as 6.8-fold. These results imply that the relative position of a transcription factor binding site in a cluster can frequently influence expression, perhaps by changing DNA secondary structure and/or altering the affinity of binding sites for cofactors. The binding site most sensitive to changes in position was that for NR2F2, appearing in most sets with a favorable configuration (64/87, 74%), despite being the sixth (out of 9) most prevalent binding site in the 211 class III sets overall (Supplementary Fig. 10). For example, we observed that, in SRESs with 3 transcription factor binding sites, permutations with an NR2F2 site in the center position yielded 2.2-fold higher expression than permutations with a promoter-proximal NR2F2 site (Fig. 6a).
Figure 6.

Effects of binding site order in heterotypic enhancers. (a) Depiction of the 10 combinations (out of 39 total combinations) of 3 transcription factor binding sites with a favorable permutation resulting in significantly stronger expression (FDR < 0.05, Wilcoxon rank-sum test). The difference in expression between the best and worst permutation is depicted on the left. (b) Rank-value plot depicting 9 sets of SRESs containing ~49 permutations, each of the same 8 transcription factor binding sites. (c) HepG2 and mouse liver SRES expression strongly agree for the entire set of 441 permutations of 8 transcription factor binding sites. The red line is the regression trace. A specific example of the best and worst permutation for one of these sets (shown in purple in the plots in b,c) appears at the bottom.
Our class III design also contained 441 SRESs with 8 transcription factor binding sites arranged in different orders, with all other variables kept constant. The 441 SRESs were divided into 9 sets on the basis of the 8 binding sites they contained, and, on average, they contained 49 distinct permutations. For each of these sets, the strongest permutation resulted in significantly higher expression than the weakest permutation (P = 0.002–0.01, Wilcoxon rank-sum test), with an average of 5.3-fold difference in expression. Although the best permutations ranked among some of the strongest sequences in the entire library (average expression = 0.58), the weakest configurations (average expression = 0.11) were on par with SRESs with one transcription factor binding site (average expression = 0.08). These results clearly indicate that the correct ordering of binding sites is important for proper expression. However, a rank-value plot of the entire set of configurations suggests that this relationship is more nuanced (Fig. 6b), consistent with a previous analysis of native transcription factor binding35. In each of the nine cases, there was a gradual response to changes in order: for example, the tenth best permutation was, on average, 78% as robust in driving expression as the strongest one. These trends were also consistent in HepG2 cells, further demonstrating that successful configurations can be detected by conserved transcriptional complexes (Fig. 6c). We interpret this observation to suggest that order is important but still highly accommodating of different permutations, largely consistent with the billboard model of regulatory element organization13,14, and is permissive of evolutionary reshuffling of transcription factor binding sites.
DISCUSSION
Using a collection of 4,970 tagged reporters patterned with different transcription factor binding site arrangements for 12 liver-specific transcription factors, we demonstrate several principles describing the activity of higher vertebrate regulatory elements. First, we show that homotypic clustering of some binding sites (CEBPA, FOXA1, HNF1A, ONECUT1 and XBP1) can be used to amplify enhancer strength. However, this principle is not universal, as homotypic clustering of several binding sites (AHR/ARNT, GATA4, HNF4A, NR2F2, PPARA, RXRA and TFAP2C) did not amplify expression. Not unexpectedly, several of the factors that did not drive expression in homotypic clusters are known to function in heterodimeric complexes31,32. We further show that two different 17-bp consensus binding motifs are sufficient to drive consistent expression in adult liver when paired with a minimal promoter. To our knowledge, these constitute the shortest functional elements characterized in vivo. We also observed that the homotypic amplification effect is prone to saturation (for example, with HNF1A binding sites) and, for almost all of the elements tested here, does not seem to be dependent on the spacing of binding sites. We additionally show that heterotypic elements are in general stronger than homotypic ones, probably owing to the presence of specific combinations and orders of binding sites that are important determinants of robust transcription. This finding was particularly evident for NR2F2 and FOXA1 binding sites, both of which interact with multiple other transcription factor binding sites. This observation is consistent with the reported role for FOXA1 as a pioneer transcription factor, recruiting other factors instead of driving transcription by itself36. Finally, we demonstrate that the synergistic and interfering interactions we identified are, respectively, enriched and depleted from putative mouse liver enhancers.
We were not able to exhaustively test transcription factor binding site permutations on the scale seen for native regulatory elements owing to limitations in library complexity, motivating our selection of a single consensus binding sequence per transcription factor. This approach proved problematic for HNF4A, a known master regulator of liver-specific gene expression37. The consensus sequence selected for this transcription factor did not drive transcription in homotypic elements and did not contribute to our model of heterotypic expression, suggesting that a different representative motif might have had stronger activity in adult mouse liver. Indeed, in vitro binding data suggest that HNF4A binding specificity segregates into two distinct groups of sequences38; however, the biological consequence of this observation is unknown. Future massively parallel reporter assay studies with increased binding site complexity will allow the systematic testing of binding site degeneracy. We also observed negligible activity from the consensus binding site used for GATA4, although this weak activity is likely due to a more developmental role for this factor. GATA4 is essential for the early development of the liver from the ventral foregut endoderm39 but is expressed at low levels in the adult liver and is limited to epithelial cells around the biliary ducts40. Another caveat of our approach is that our SRESs were only 168 bp in length, which is on the scale of a core promoter element or p300 ChIP-seq peak but much shorter than most functionally validated elements (which are 1.5–2 kb in length29). As a result, our analysis is unable to assay regulatory structures that might be present on a sparser scale. Methodological improvements such as long-module synthesis on DNA microarrays, polymerase cycling assembly (PCA)26,41 or in vitro recombination41,42 could be used to test larger elements. A final limitation is that the plasmids containing SRESs do not integrate into the host genome and are not chromatinized. These results should therefore be interpreted in the context of other plasmid-based reporter assays. The development of viral, transposon or recombination-based massively parallel reporter assay methods that permit reporter integration will no doubt help tease apart additional features of regulatory organization.
A large subset of the SRES library is devoted to determining the impact of transcription factor binding site order on expression by heterotypic elements. Of these sets, 41% had a favorable permutation that resulted in a significantly stronger increase in expression than a secondary unfavorable permutation, suggesting a key role for binding site order in driving optimal transcription. This percentage is particularly notable considering that the median number of permutations tested was only two, suggesting that, in many cases, we simply did not test a strong permutation. To look at the impact of binding site order more systematically, we examined the expression patterns of 441 SRESs with 8 transcription factor binding sites arranged in different permutations. These patterns conclusively show that there are multiple arrangements that drive strong expression but several weak ones as well. These data are consistent with the notion that there may be no generalized motif-positioning model3,15. Instead, the data support a flexible regulatory architecture for the organization of _cis_-regulatory modules—a loosely organized billboard13,14. An important caveat to this interpretation is that, by analyzing the role of our SRES library in adult liver cells, we may have missed developmental and/or environment-sensitive aspects of the regulatory architecture of transcription factor binding sites. Future studies using massively parallel reporter assay libraries at different time points and under different conditions might be able to address this question more fully.
URL
All analyses were performed using the R statistical software package, http://www.r-project.org/.
ONLINE METHODS
Consensus sequences
In cases where the motif databases contained multiple PWMs representing the binding site of a single transcription factor, we first determined the number of hits for each PWM on a random 10-Mb human sequence and then selected the PWM resulting in the median number of hits. Hits were determined by running the tool tfSearch43 with default parameters. For each PWM, we calculated the consensus sequence on the basis of the log-odds matrix. Such a log-odds matrix was generated by calculating log2 (p i/f i) at each position of the matrix, where p i is the probability of a particular nucleotide i at that position in the matrix and f i is the average frequency of that nucleotide in the genome. Average frequencies were calculated on a random 10-Mb human sequence. In the event of a tie between two bases, we chose one at random. All of our selected sequences were also tested for enrichment in putative mouse liver enhancers9 and matches in other transcription factor binding data sets (Supplementary Table 2).
Library synthesis and cloning
Two neutral 168-bp DNA templates were selected on the basis of a low score in a liver classification model (data not shown) and were validated using the tail vein injection assay (Supplementary Fig. 1). SRESs were ordered as 200-nt oligonucleotides (Custom Array), including a 168-nt variable sequence flanked on either side by 16 nt of constant sequence to enable amplification of the oligonucleotide library by PCR. Sequences were prescreened for XbaI or HindIII restriction enzyme sites. The oligonucleotide library was amplified using primers (OLIGO_AMP_F and OLIGO_AMP_R) that targeted the constant flanking sequences and also introduced 15 bp of sequence homology with the vector to make the amplified product competent for downstream cloning. The amplified library was run on a PAGE gel, and the 240-bp band was excised and transferred to a siliconized 0.5-ml microcentrifuge tube (Ambion) with a hole in the bottom introduced through puncture by a 20-gauge needle. This tube was placed in a 1.5-ml siliconized microcentrifuge tube (Ambion) and centrifuged in a tabletop microcentrifuge at 16,110_g_ for 5 min to create a gel slurry that was then resuspended in 200 μl of 1× Tris-EDTA and incubated at 65 °C for 2 h with periodic vortexing. The aqueous phase was separated from gel fragments by centrifugation through 0.2-μm NanoSep columns (Pall Life Sciences). DNA was recovered by standard QIAquick column purification and was subjected to an additional round of amplification using short outer primers (SS_F and SS_R). The SRES library was cloned into the EcoRV site of a tagged pGL4.23 library described previously26, using the standard InFusion (Clontech) protocol and Stellar competent cells (Clontech). Seven transformations were performed, and bacteria were grown overnight in two 50-ml liquid cultures (3.5 transformations per culture) at 37 °C in a shaking incubator. DNA was extracted using the Invitrogen ChargeSwitch Midi Prep kit. A complete listing of all primer sequences used is provided in Supplementary Table 6.
Tail vein injections
Templates and control sequences were validated individually using previously described methods27. The SRES library was injected using essentially the same methods, with the exception that RNA was collected from dissected livers. Briefly, 10 μg of plasmid or SRES library diluted in TransIT EE Hydrodynamic Gene Delivery System (Mirus Bio) was injected into three male CD1 mice weighing between 20 and 24 g and 13–18 weeks of age (Charles River) following the manufacturer's protocol. To measure the aggregate injection efficiency of the SRES library, an additional three mice were injected with the library along with 2 μg of pGL4.74 (hRluc/TK) (Promega) to correct for differences in injection efficiency. After 24 h, mice were sacrificed, and livers were dissected. Total RNA was purified using the RNeasy Maxi kit (Qiagen) with on-column DNase digestion, and 500 μg was used as input for the Oligotex mRNA Midi kit (Qiagen), yielding ~3% mRNA. For the mice injected with the library and pGL4.74, firefly luciferase and Renilla luciferase activities in the supernatant (diluted 1:20) were measured on a Synergy 2 microplate reader (BioTek) in technical replicates of four using the Dual-Luciferase Reporter Assay System (Promega). All animal work was approved by the UCSF Institutional Animal Care and Use Committee. No statistical method was used to predetermine sample size.
Cell culture
HepG2 cells (ATCC) were maintained in DMEM supplemented with 10% FCS, glutamine (2 mM), penicillin (100 U/ml) and streptomycin (50 μg/ml). HepG2 cells (5 × 106) were plated in 15-cm plates and incubated for 24 h. Cells were transfected with 15 μg of DNA using X-tremeGENE HP (Roche) according to the manufacturer's protocol. The X-tremeGENE:DNA ratio was 2:1. Three independent replicate cultures were transfected with the SRES library and sequenced.
Sequencing of RNA-derived tags
We identified 20-bp tags in liver and HepG2 cell mRNA using previously described methods26. For livers, four RT-PCR runs were performed for each of the three biological replicates and were then multiplexed and sequenced together on a single lane of an Illumina Genome Analyzer IIx using a custom sequencing primer (TAG_SEQ_F). For HepG2 cells, two RT-PCR runs were performed for each replicate and sequenced. Each run was 36 cycles, with an additional 6 cycles to read the indexing tag using the index sequencing primer (TAG_SEQ_INDEX). For each aliquot, reads were filtered on the basis of the quality scores for the first 20 bases, which correspond to the degenerate tag. The number of occurrences of each tag were counted, and tags whose occurrence was supported by at least two reads were classified as being present in that aliquot.
Associating SRESs with tags
SRESs were associated with tags ostensibly as previously described26. Briefly, ~1,000-bp segments separating SRESs and tags on the pGL4.23 plasmid were excised by digesting with HindIII, which digests both 3′ of the SRES and 5′ of the tag. The digested plasmid was purified and recircularized using intramolecular ligation, resulting in the tag being adjacent to the 3′ end of the SRES. The region spanning the SRES and tag was amplified from recircularized plasmids by PCR with the forward primer targeting the region immediately 5′ of the SRES (SRES_PE_F) and the reverse primer targeting the region immediately 3′ of the tag (TAG_PE_R). PCR products were purified using QIAquick columns and sequenced on a HiSeq 2000 (Illumina). Forward and reverse reads (sequenced using custom sequencing primers SRES_SEQ_F and SRES_SEQ_R, respectively) covered 101 bp of each side of the SRES, and the index read covered the 20-bp tag sequence (index read sequencing primer TAG_SEQ_F). Read pairs where all bases had a Phred score of >25 were aligned to the SRES library with Burrows-Wheeler Aligner (BWA)44 (version 0.6.2; using default options). Each sequence in the SRES library was aligned with an average of 2,213 reads. Each tag was associated with an average of nine reads. We uniquely mapped each tag to the sequence aligned with the highest number of reads associated with that tag. We discarded the tag in the event of a tie or if it was mapped to a sequence aligned with less than two reads. Finally, we discarded sequences associated with fewer than ten tags. Further detail is provided in the Supplementary Note.
Expression measure
Read counts associated with each tag and sequence in the SRES library were quantile normalized across the 12 RT-PCR pools (all 3 mouse replicates) and then normalized again within each replicate. For HepG2 cells, read counts were quantile normalized between the two RT-PCR pools for a single replicate. A tag was considered expressed if it was represented by at least two (normalized) reads in a single pool. For each sequence in the SRES library, the expression value was given by the fraction of the tags in the library that were expressed in that sample. P values from Spearman's correlations and Wilcoxon rank-sum tests were corrected using the Benjamini-Hochberg FDR method45 in cases where there were multiple comparisons.
Template correlation
Discordance between expression data for templates was measured using Cook's distance (D)46. This value measures the influence of each SRES pattern in the regression model describing the relationship between expression on template 1 and expression on template 2. We considered 123 SRESs to be discordant between the templates because they had
where n is the number of SRES template pairs (2,217) and k is the number of independent variables (1).
Quantitative SRES expression modeling
We modeled the expression of all class I and II SRESs (in triplicate, constituting 6,990 data points) as a function of the transcription factor binding site they comprise using generalized linear models (GLMs). A GLM consists of three components: (i) the random component, which consists of the response variable Y and its probability distribution; (ii) the systematic component, which represents the predictor variables in the model _X_1, _X_2, …, _X_12; and (iii) the link function, which links the expected value of Y and the predictor variables. In our case, the response variable was the observed expression value of a sequence, and the predictor variables indicated the number of occurrences in the sequence of each of the 12 transcription factor binding sites considered. Given that the expression values are distributed between 0 and 1, we used a GLM with binomial family and a logit link
and
i^g(i^)=a^0+a^1×X1+a^2×X2+…+a~1×X1×X1+a~2×X2×X2+a¨1×X1×X2+…+a¨66×X11×X12
where g(i^) is the link function and a^0,a^1,…,a~1,a~2,…,a¨1,a¨2,… are the parameters to be estimated. This approach is similar to a standard logistic regression18 but differs in that GLMs can accept observed values of 0% and 100% and take into consideration the sample size when estimating the coefficients and their errors. In each case, we started from a complete model with all variables included with linear and quadratic powers, as well as all possible interactions between linear terms. We then applied a stepwise procedure, optimizing the models by taking into consideration the AIC. The AIC is based on the goodness of fit, but it is penalized by the number of estimated parameters. Predictor variables were successively removed from the starting model according to the deviance explained by the predictor variable when fitted individually, with the least significant predictor variable being removed first.
For cross-validation, we averaged the expression data for all class I and II SRESs, resulting in a total of 2,330 data points. Each of these was designated positive or negative on the basis of whether it exceeded the threshold equal to the average expression of all one-site SRESs (this value is higher than that for the negative controls). Because this data set was highly redundant (the same combination of transcription factor binding sites was present several times and was associated with different expression values), we performed cross-validation in two different ways. First, we performed a standard tenfold cross-validation, training the model on nine-tenths of the data and testing it on the remaining one-tenth. Second, we reduced the data set to 234 unique combinations of transcription factor binding sites, where each combination was associated with the average expression value of the corresponding SRESs. On this data set, we performed a standard tenfold cross-validation, training the model on nine-tenths of the data and testing it on the remaining one-tenth. To ascertain the stability of the model, we determined the numerical values of the model coefficients for each of the cross-validation folds. On the basis of coefficient deviation, we concluded that the model produced stable coefficients for all transcription factor binding sites. All analyses were carried out using the R statistical software package (see URL).
Transcription factor binding site analysis
Putative transcription factor binding sites were identified by searching the sequences with MAST47 for motifs listed in Supplementary Table 1. MAST was run independently on each individual sequence with default parameters, using either putative liver or cerebellum enhancers from the same source9. Enrichment in putative liver enhancers was evaluated in 600-bp windows (±300 bp from the peak center) on the basis of the distribution in Supplementary Figure 9. Enrichment was tested in putative liver enhancers against a background of 103,700 GC- and length-matched genomic control regions and was evaluated by Fisher's exact test.
Supplementary Material
Supplementary table 4
Supplementary table 5
Supplementary text & figures
ACKNOWLEDGMENTS
This work was supported by National Human Genome Research Institute (NHGRI) grant 1R01HG006768 (N.A. and J.S.) and the Pilot/Feasibility grant from the University of California, San Francisco (UCSF) Liver Center (P30 DK026743). N.A. is also supported by National Institute of Child and Human Development grant R01HD059862, NHGRI grant R01HG005058, National Institute of General Medical Sciences grant GM61390, National Institute of Neurological Disorders and Stroke grant 1R01NS079231, National Institute of Diabetes and Digestive and Kidney Diseases grant 1R01DK090382 and the Simons Foundation (SFARI 256769). R.P.S. was supported in part by a Canadian Institutes of Health Research (CIHR) fellowship in hepatology. M.J.K. was supported in part by US National Institutes of Health training grant T32 GM007175, the UCSF Quantitative Biosciences Consortium fellowship for Interdisciplinary Research and the Amgen Research Excellence in Bioengineering and Therapeutic Sciences fellowship. This work was funded in part by the Intramural Research Program of the US National Institutes of Health, National Library of Medicine (I.O.).
Footnotes
Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS R.P.S., L.T., R.P.P., J.S., I.O. and N.A. conceived key aspects of the project and planned experiments. R.P.S., R.P.P., M.J.K. and F.I. performed experiments. L.T., R.P.S. and R.P.P. analyzed data. R.P.S., L.T., R.P.P., I.O., J.S. and N.A. wrote the manuscript. All authors commented on and revised the manuscript.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
Accession code. All raw data, including SRES-tag association and tag abundance data, have been deposited in the Sequence Read Archive (SRA) under accession SRP018414.
References
- 1.Halfon MS, et al. Ras pathway specificity is determined by the integration of multiple signal-activated and tissue-restricted transcription factors. Cell. 2000;103:63–74. doi: 10.1016/s0092-8674(00)00105-7. [DOI] [PubMed] [Google Scholar]
- 2.Lettice LA, et al. Opposing functions of the ETS factor family define Shh spatial expression in limb buds and underlie polydactyly. Dev. Cell. 2012;22:459–467. doi: 10.1016/j.devcel.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- 4.Jeong Y, et al. Regulation of a remote Shh forebrain enhancer by the Six3 homeoprotein. Nat. Genet. 2008;40:1348–1353. doi: 10.1038/ng.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benko S, et al. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat. Genet. 2009;41:359–364. doi: 10.1038/ng.329. [DOI] [PubMed] [Google Scholar]
- 6.Sturm RA, et al. A single SNP in an evolutionary conserved region within intron 86 of the HERC2 gene determines human blue-brown eye color. Am. J. Hum. Genet. 2008;82:424–431. doi: 10.1016/j.ajhg.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harismendy O, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-γ signalling response. Nature. 2011;470:264–268. doi: 10.1038/nature09753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 9.Shen Y, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Visel A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blow MJ, et al. ChIP-Seq identification of weakly conserved heart enhancers. Nat. Genet. 2010;42:806–810. doi: 10.1038/ng.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song L, et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011;21:1757–1767. doi: 10.1101/gr.121541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rastegar S, et al. The words of the regulatory code are arranged in a variable manner in highly conserved enhancers. Dev. Biol. 2008;318:366–377. doi: 10.1016/j.ydbio.2008.03.034. [DOI] [PubMed] [Google Scholar]
- 14.Kulkarni MM, Arnosti DN. Information display by transcriptional enhancers. Development. 2003;130:6569–6575. doi: 10.1242/dev.00890. [DOI] [PubMed] [Google Scholar]
- 15.Brown CD, Johnson DS, Sidow A. Functional architecture and evolution of transcriptional elements that drive gene coexpression. Science. 2007;317:1557–1560. doi: 10.1126/science.1145893. [DOI] [PubMed] [Google Scholar]
- 16.Merika M, Thanos D. Enhanceosomes. Curr. Opin. Genet. Dev. 2001;11:205–208. doi: 10.1016/s0959-437x(00)00180-5. [DOI] [PubMed] [Google Scholar]
- 17.Thanos D, Maniatis T. Virus induction of human IFNb gene expression requires the assembly of an enhanceosome. Cell. 1995;83:1091–1100. doi: 10.1016/0092-8674(95)90136-1. [DOI] [PubMed] [Google Scholar]
- 18.Krivan W, Wasserman WW. A predictive model for regulatory sequences directing liver-specific transcription. Genome Res. 2001;11:1559–1566. doi: 10.1101/gr.180601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee D, Karchin R, Beer MA. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res. 2011;21:2167–2180. doi: 10.1101/gr.121905.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narlikar L, et al. Genome-wide discovery of human heart enhancers. Genome Res. 2010;20:381–392. doi: 10.1101/gr.098657.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gotea V, et al. Homotypic clusters of transcription factor binding sites are a key component of human promoters and enhancers. Genome Res. 2010;20:565–577. doi: 10.1101/gr.104471.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharon E, et al. Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat. Biotechnol. 2012;30:521–530. doi: 10.1038/nbt.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grskovic M, Chaivorapol C, Gaspar-Maia A, Li H, Ramalho-Santos M. Systematic identification of cis-regulatory sequences active in mouse and human embryonic stem cells. PLoS Genet. 2007;3:e145. doi: 10.1371/journal.pgen.0030145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gertz J, Siggia ED, Cohen BA. Analysis of combinatorial cis-regulation in synthetic and genomic promoters. Nature. 2009;457:215–218. doi: 10.1038/nature07521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Melnikov A, et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 2012;30:271–277. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patwardhan RP, et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol. 2012;30:265–270. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim MJ, et al. Functional characterization of liver enhancers that regulate drug-associated transporters. Clin. Pharmacol. Ther. 2011;89:571–578. doi: 10.1038/clpt.2010.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang G, Budker V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum. Gene Ther. 1999;10:1735–1737. doi: 10.1089/10430349950017734. [DOI] [PubMed] [Google Scholar]
- 29.Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35:D88–D92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Donoghue M, Ernst H, Wentworth B, Nadal-Ginard B, Rosenthal N. A muscle-specific enhancer is located at the 3′ end of the myosin light-chain 1/3 gene locus. Genes Dev. 1988;2:1779–1790. doi: 10.1101/gad.2.12b.1779. [DOI] [PubMed] [Google Scholar]
- 31.Issemann I, Prince RA, Tugwood JD, Green S. The peroxisome proliferator–activated receptor:retinoid X receptor heterodimer is activated by fatty acids and fibrate hypolipidaemic drugs. J. Mol. Endocrinol. 1993;11:37–47. doi: 10.1677/jme.0.0110037. [DOI] [PubMed] [Google Scholar]
- 32.Williams T, Tjian R. Characterization of a dimerization motif in AP-2 and its function in heterologous DNA-binding proteins. Science. 1991;251:1067–1071. doi: 10.1126/science.1998122. [DOI] [PubMed] [Google Scholar]
- 33.De Val S, et al. Combinatorial regulation of endothelial gene expression by ets and forkhead transcription factors. Cell. 2008;135:1053–1064. doi: 10.1016/j.cell.2008.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakamoto Y, Ishiguro M, Kitagawa G. Akaike Information Criterion Statistics. KTK Scientific Publishers; Tokyo: 1986. [Google Scholar]
- 35.Tomovic A, Oakeley EJ. Position dependencies in transcription factor binding sites. Bioinformatics. 2007;23:933–941. doi: 10.1093/bioinformatics/btm055. [DOI] [PubMed] [Google Scholar]
- 36.Lupien M, et al. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132:958–970. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sladek FM, Zhong WM, Lai E, Darnell JE., Jr. Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev. 1990;4:2353–2365. doi: 10.1101/gad.4.12b.2353. [DOI] [PubMed] [Google Scholar]
- 38.Badis G, et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009;324:1720–1723. doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watt AJ, Zhao R, Li J, Duncan SA. Development of the mammalian liver and ventral pancreas is dependent on GATA4. BMC Dev. Biol. 2007;7:37. doi: 10.1186/1471-213X-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dame C, et al. Hepatic erythropoietin gene regulation by GATA-4. J. Biol. Chem. 2004;279:2955–2961. doi: 10.1074/jbc.M310404200. [DOI] [PubMed] [Google Scholar]
- 41.Schwartz JJ, Lee C, Shendure J. Accurate gene synthesis with tag-directed retrieval of sequence-verified DNA molecules. Nat. Methods. 2012;9:913–915. doi: 10.1038/nmeth.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y, Werling U, Edelmann W. SLiCE: a novel bacterial cell extract–based DNA cloning method. Nucleic Acids Res. 2012;40:e55. doi: 10.1093/nar/gkr1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ovcharenko I, et al. Mulan: multiple-sequence local alignment and visualization for studying function and evolution. Genome Res. 2005;15:184–194. doi: 10.1101/gr.3007205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc., B. 1995;57:289–300. [Google Scholar]
- 46.Cook D. Detection of influential observation in linear regression. Technometrics. 1977;19:15–18. [Google Scholar]
- 47.Bailey TL, Gribskov M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14:48–54. doi: 10.1093/bioinformatics/14.1.48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table 4
Supplementary table 5
Supplementary text & figures