De novo DNA demethylation and noncoding transcription define active intergenic regulatory elements (original) (raw)
Abstract
Deep sequencing of mammalian DNA methylomes has uncovered a previously unpredicted number of discrete hypomethylated regions in intergenic space (iHMRs). Here, we combined whole-genome bisulfite sequencing data with extensive gene expression and chromatin-state data to define functional classes of iHMRs, and to reconstruct the dynamics of their establishment in a developmental setting. Comparing HMR profiles in embryonic stem and primary blood cells, we show that iHMRs mark an exclusive subset of active DNase hypersensitive sites (DHS), and that both developmentally constitutive and cell-type-specific iHMRs display chromatin states typical of distinct regulatory elements. We also observe that iHMR changes are more predictive of nearby gene activity than the promoter HMR itself, and that expression of noncoding RNAs within the iHMR accompanies full activation and complete demethylation of mature B cell enhancers. Conserved sequence features corresponding to iHMR transcript start sites, including a discernible TATA motif, suggest a conserved, functional role for transcription in these regions. Similarly, we explored both primate-specific and human population variation at iHMRs, finding that while enhancer iHMRs are more variable in sequence and methylation status than any other functional class, conservation of the TATA box is highly predictive of iHMR maintenance, reflecting the impact of sequence plasticity and transcriptional signals on iHMR establishment. Overall, our analysis allowed us to construct a three-step timeline in which (1) intergenic DHS are pre-established in the stem cell, (2) partial demethylation of blood-specific intergenic DHSs occurs in blood progenitors, and (3) complete iHMR formation and transcription coincide with enhancer activation in lymphoid-specified cells.
Until recently, our knowledge of genome function has been focused on protein-coding genes. Yet, analyses of evolutionary constraint across vertebrates and eutherian mammals reveal millions of bases in the human genome that have undergone purifying selection, of which only a fraction are within known protein-coding sequences (The Encode Project Consortium 2007). Many (∼40%) are bundled into conserved units as small as tens of nucleotides that are scattered across vast intergenic space (Lindblad-Toh et al. 2011). Interestingly, most of these do not coincide with sequence features characteristic of protein-coding or structural elements, but instead, suggest a regulatory function based, for example, on an enrichment of transcription factor binding motifs (TFBS).
Combined profiles of modified histones and DNase accessibility have charted chromatin states across diverse cell types from fly (Kharchenko et al. 2011), human (Ernst et al. 2011), and mouse (Shen et al. 2012). These have exposed numerous putative _cis_-regulatory elements, most notably enhancers and insulators. The activity of such elements is frequently specific to cell type and context, and therefore a synthesis of developmentally diverse, tissue-specific genomic data sets is required to detect and interpret regulatory function.
Enhancers and other distal _cis_-regulatory elements are transcription-factor (TF) docking sites for long-distance gene regulation. They help to establish alternate gene expression programs that in turn guide cell fate decisions and control cellular phenotypes (Bulger and Groudine 2011). For many TFs, stable occupancy at target sites relies on a DNA sequence free of cytosine methylation and nucleosome interference. Further, recent studies addressing single receptor models propose significant interplay between DNA methylation, transcription factor binding, and the activity of enhancers (Stadler et al. 2011; Wiench et al. 2011).
DNA methylation itself is thought to be a critical component of the mechanisms that define and stabilize cell-type identity and developmental state. In a typical mammalian genome, methylation is the default state, with 70%–80% of all CpG sites modified. Unmethylated CpGs frequently occur in areas of high CpG density, so-called CpG Islands (CGIs), which often overlap gene promoters. Recently, we described an unbiased, empirical model for detecting hypomethylated regions (HMRs) based on clustering of largely unmethylated CpG sites in the genome (Molaro et al. 2011), independent of predefined CGIs. Using this approach, we identified a new class of intergenic and intronic HMRs (iHMRs) that differ in several ways from those observed near promoters, including size and sequence composition. With few specific exceptions, promoter HMRs (pHMRs), whether coinciding with CGIs or not, are shared (constitutive) across diverse cell types. For this reason, their presence does not specifically flag genes that are transcriptionally active (Hodges et al. 2011). In contrast, many iHMRs tend to be either stem cell specific or “de novo” demethylated in specific differentiated cells. Thus, we sought to understand the role of hypomethylation at these sites in relation to different regulatory activities. In particular, we asked whether iHMRs designate select classes of regulatory elements, and whether their component features are prognostic of gene activity.
Here, we show that differential comparisons of HMR profiles across multiple cell types can provide detailed information about the presence and status of regulatory elements in individual cell types. Using HMR profiles obtained from bisulfite sequencing (BS-seq) of five different human and three chimpanzee cell types, we defined sets of constitutive and cell-type-specific iHMRs. The cells represented both embryonic (H1 ESC) (Lister et al. 2009) and adult somatic stem cell stages (hematopoietic stem and progenitor cells, HSPCs) in addition to differentiated states from two divergent hematopoietic lineages (B lymphocytes, the GM12878 lymphoblastoid cell line, and neutrophils) (Hodges et al. 2011). We superimposed the HMR profiles on available ChIP-seq and DNase-seq data sets from The ENCODE Project Consortium (2012), finding a remarkably precise, highly cell-type-specific overlap between HMR calls and modified histone peaks. This enabled us to clearly distinguish enhancer-, insulator-, and promoter-like iHMRs, and their respective methylation dynamics, during differentiation from stem to mature cells. Furthermore, using CAGE and RNA-seq data sets from ENCODE, we find a strong connection between the methylation status of iHMRs and transcription of noncoding RNAs at the intergenic site. We show that transcription at iHMRs originates from defined sequence elements, and gives rise to distinct classes of RNAs that reflect the iHMR's regulatory activity. Next, we compared the human methylation profiles with data corresponding to orthologous blood cell types from chimpanzee. Methylation states at HMRs are generally conserved between human and chimp, with variation depending on the HMR type and divergence of the underlying sequence elements. Overall, enhancer iHMRs are the most variable between species, individuals, and developmental states. Our data indicate that progressive demethylation and transcription together define the most active enhancer elements in mature cells. Thus, HMRs reveal accurate cues to the activity of regulatory elements and RNA polymerase II genome wide, providing a strong framework to analyze methylation changes during development. Our integrated analysis of HMRs, and in particular iHMRs, can therefore determine cell-type-specific regulatory centers, including signatures of differentiation.
Results
Recently, using genome-wide DNA methylation data sets we have shown that intergenic domains of hypomethylation (HMRs) are more widespread and more variable during cellular differentiation than had been previously appreciated (Hodges et al. 2011). In fact, HMRs occur throughout the genome, differing in their sequence context (e.g., CpG density) and their dynamic behavior during development. Because of this, HMRs, unlike CGIs, cannot be predicted by sequence characteristics alone and must instead be identified empirically using cell-type-specific methylation profiles. Here, using whole-genome BS-seq data, we compared sets of HMRs in H1 ESCs and primary human B cells. Differential HMR analysis revealed three broad categories of sites with different features: (1) shared, (2) specific, or (3) shared and significantly expanded in length in one cell type (Fig. 1A; Supplemental Fig. S1A–D). These broad classifications alone permit some initial inferences regarding the potential behavior of the HMR. For example, HMRs overlapping gene promoters (“pHMRs”) have high CpG density and are predominantly shared between the two cell types (shared or both shared and expanding in one cell type relative to the other). Most intergenic and intronic HMRs (iHMRs), on the other hand, have lower CpG density than canonical CGIs (Supplemental Fig. S1A–D) and are often cell-type specific. The exceptions are CTCF-bound iHMRs, which are predominantly shared between the cell types. Examples of a shared, expanding pHMR and an intergenic iHMR for two B cell-associated loci are shown in Supplemental Figure S2.
Figure 1.

Distribution of hypomethylated regions in stem and differentiated cells. (A) Genomic distribution of hypomethylated regions (HMR) in H1 ESCs and B cells. Colors in each bar indicate whether an HMR is shared between the two cell types, specific to one, or shared but expanding, i.e., significantly larger in one cell type than in the other. (B) Overlap between iHMRs and DHS in the different cell-types. (C) Most iHMRs contain regions of DNase hypersensitivity. The heatmap shows the enrichment of DNase-seq signal at H1 ESC iHMRs. iHMRs are aligned between the black lines, white points indicate genomic locations not mappable with short DNase-seq reads. Rows are sorted by hierarchical clustering. (D) A subset of high CpG density DNase HS is hypomethylated. Distribution of average methylation levels for DHS in H1 ESC split by CpG density (observed/expected; O/E). (E) Hypomethylated DHS are marked by histone modifications. Log fold enrichment over genomic background for H3K4me2 at intergenic H1 ESC DHS with high (>0.4 O/E) CpG density is shown. (F) Hypomethylated DHS have higher sequence conservation. The fraction of positions with phastCons scores over 0.9 in intergenic DHS depending on methylation state is shown.
Overall, the number of cell-type-specific iHMRs is fourfold higher in the mature cells (5106 in H1 ESC vs. 20,556 in B cells), indicating that lineage-specific loss of methylation at intergenic sites occurs during differentiation. We have previously found an enrichment for binding motifs of lymphoid-specific TFs at B cell iHMRs (Hodges et al. 2011). In addition, recent work has shown that binding of some transcription factors is directly linked to intergenic hypomethylation (Stadler et al. 2011). Therefore, we hypothesized that these iHMRs designate a lymphoid lineage-specific class of distal regulatory elements.
iHMRs discriminate a class of highly active, conserved DNase hypersensitive sites
DNase I hypersensitivity sequencing (DNase-seq) is the established method for determining chromatin accessibility genome wide, and hence for identifying putative regulatory elements. Since a correlation between DNase sensitivity and methylation levels at some CpG sites has been reported (Thurman et al. 2012), we investigated the relationship between iHMRs and intergenic DNase hypersensitivity sites (DHS) in H1 ESCs and the GM12878 lymphoblastoid cell line (LCL, using data from LCLs as a proxy for primary B cells; see Methods and Supplemental Fig. S8). A majority of iHMRs overlaps significant DHS peaks in mature and stem cells (57% and 84%, respectively) (Fig. 1B), and at least weak DHS signal enrichment can be detected in almost all iHMRs (Fig. 1C). In contrast, DHSs far out-number iHMRs, and only some (11%–33%) DHSs were hypomethylated (Fig. 1B; Supplemental Fig. S6A). CpG density is a strong predictor for the methylation state of a DHS, with low CpG density correlating with high methylation levels (Fig. 1D) (Spearmen r = −0.17; P = 1.6 × 10−133). On the other hand, DHSs with higher CpG density show bimodal methylation levels. Among these high CpG sites, hypomethylation occurs specifically at those DHS positive for CTCF binding (68% vs. 31% without CTCF; P = 3.6 × 10−185, Fisher's exact test) or histone modifications, such as H3K4me2, associated with active regulatory states (Fig. 1E). Hypomethylated DHSs also show on average higher sequence conservation than those that do not overlap HMRs (Fig. 1F) (P = 2.5 × 10−229; Wilcoxon test).
Using MNase-seq data from LCLs, we observed nucleosome-depleted regions matching the length of the iHMRs (Supplemental Fig. S1E). This depletion is dynamic and cell-type specific, as H1 ESC-specific iHMR sites are not depleted for nucleosomes in LCLs. Together, these data suggest that a specific subset of important DHSs are de-novo demethylated during lymphoid lineage specification, and that loss of methylation is associated with local changes to nucleosomes.
Composite features of HMRs reflect a diversity of regulatory states
To investigate what classes of genomic elements give rise to iHMRs and their potentially diverse regulatory functions, we relied on ChIP-seq against modified histones, DNase-seq, and related techniques, to classify iHMRs based on their chromatin signature (Heintzman et al. 2007). We gathered an extensive catalog of chromatin state data sets from the ENCODE project (The ENCODE Project Consortium 2012) in H1 ESC and LCLs. Globally, we defined four major classes of iHMRs in H1 ESCs, based on different combinations of CTCF occupancy, DNase hypersensitivity, RNA polymerase II (RNA Pol II) binding, and histone modifications (Fig. 2A). Their respective chromatin states resembled the typical signatures of insulators (“_CTCF_”), enhancers (“_Enhancer-like_”), active promoters (“_Promoter-like_”), and bivalent elements (“_Bivalent_”) (Ernst et al. 2011). This classification was further supported by a specific enrichment of ENCODE enhancer and promoter predictions (Yip et al. 2012) in their respective iHMR groups (Supplemental Fig. S1D, P < 1.6 × 10−110; Fisher's exact test). iHMRs overlapping sequence-defined CpG islands (outside of annotated gene-promoter regions) are mostly shared between cell types (97% compared with 68% for non-CGI iHMRs; P < 2.2 × 10−16; Fisher's exact test). In H1 ESCs, CGI-overlapping iHMRs were found to be either Promoter-like, with levels of H3K4me3 and RNA Pol II binding reminiscent of pHMRs at annotated genes, or in the bivalent state, with high levels of H3K4me2 and H3K27me3. Enhancer-like iHMRs, on the other hand, are mostly cell-type specific and lie outside of annotated CGIs (Fig. 2A). This classification strategy could also be applied to other cell types. In LCLs, a less prominent class of bivalent iHMRs was present and, instead, we observed a large group of “silent” iHMRs marked by H3K27me3, with little or no H3K4-methylation and lower DNase hypersensitivity (Supplemental Fig. S1F). A second class of “promoter-like” iHMRs in a putatively “inactive” state (no H3K27ac and RNA Pol II) was also observed. As predicted, B cell–specific iHMRs (especially in the “enhancer” class) show highly cell-type-specific chromatin states, while shared iHMRs (mostly in the “CTCF” and “promoter” classes) also share many histone marks between cell types (Supplemental Fig. S1F).
Figure 2.

Hypomethylated regions mark different classes of active genomic regulatory elements. (A) Diverse chromatin states at H1 iHMRs. Heatmap showing the chromatin state within iHMRs. Each column represents one iHMR, sorted by hierarchical clustering, grouped into four main clusters. The top lines indicate overlap of the HMR with functional element predictions from ENCODE, CpG Islands, the iHMR location (intergenic or intronic), and whether it is shared between H1 ESCs and B cells. (B) Average chromatin mark profile in H1 ESC iHMRs of the four different clusters defined in A. HMRs are aligned between the black lines, and the fold-enrichment signals are averaged across all iHMRs at each relative position. Bold lines highlight the histone marks that distinguish each cluster. (C) Barplots show, for each of the defined classes, the fraction of iHMRs occupied by factors associated with different types of elements and chromatin states.
Meta-profiles of chromatin states confirmed distinct patterns of histone modifications specifically within the region identified by hypomethylation at all iHMR types (Fig. 2B). In all cases, H3K4me2 covers the entire hypomethylated region, along with a central narrower peak of DNase hypersensitivity. H3K4me1 enrichment corresponds with the boundaries of enhancer and promoter-like iHMRs, while promoter-like iHMRs also contain a central, sharp peak of H3K4me3. In contrast, the bivalent iHMRs lie within broader domains of H3K27me3, where H3K4 methylation specifically marks the core hypomethylated region. The functional significance of these iHMR classifications is supported by enrichments for pluripotency factors at stem cell enhancers (POU5F1 and NANOG) and polycomb proteins (SUZ12) at bivalent sites (Fig. 2C). Histone acetyltransferase EP300, like RNA polymerase II, is abundant at both enhancer and promoter-like iHMRs, but not present at transcriptionally silent CTCF and bivalent sites. Overall, different types of iHMRs identify regions displaying different chromatin states, suggesting a diversity of regulatory activities associated with hypomethylation in specific developmental contexts.
Coordinated changes at iHMRs and nearby pHMRs can impact the activities of associated genes
Because CpG dense gene promoter HMRs are typically pre-established in embryonic stem cells and shared between different cell types, tissue-specific activation of genes is often difficult to predict from the methylation states of their promoters. Previously, we showed that pHMRs expand upon lineage-specific gene activation during adult blood cell maturation. Illustrating this pattern, differential H1 ESC and B cell methylation shows asymmetric spreading of hypomethylation at pHMRs outside of the constitutive CpG-rich core region (Fig. 3A; Supplemental Fig. S3A,B). The direction and extent of differential hypomethylation differs greatly between genes, but in each case tracks very closely with cell-type-specific spreading of H3K4me2 at these promoters (Fig. 3B; Supplemental Fig. S3C).
Figure 3.

Coordinated changes in pHMRs, iHMRs, and histone marks occur at cell-type specifically regulated genes. (A) Differential hypomethylation at expanding promoter HMRs. B cell pHMRs are aligned between the black lines and color denotes the change in methylation level between H1 ESCs and B cells at each position. (B) As above, differential H3K4me2 ChIP-seq signal in H1 ESCs is displayed for the same sites. Color denotes the fold change in H3K4me2 enrichment between H1 ESCs and LCLs. (C) Enhancer HMRs are enriched near expanding promoter HMRs. Cumulative density plot showing the distances between expanded or constant pHMRs and enhancer or CTCF iHMRs. (D) Genes with both expanding pHMRs and nearby cell-type-specific iHMRs are up-regulated. Median fold expression change (CAGE signal) between LCLs and H1 ESCs for genes grouped by the distance to the closest B cell–specific iHMR and by significant expansion of their promoter HMR. (E) A subset of expanded promoter HMRs are silenced and marked with H3K27me3 in stem cells. Genes with an H1 ESC-specific expanded promoter HMR are split by their expression levels (at 1 CAGE RPM), and fold enrichment for H3K27me3 at the promoter in H1 ESCs is shown. (F) Bivalent promoters have nearby bivalently marked iHMRs. Scatterplot with H3K27me3 signal at pairs of pHMRs and nearby (<25 kb) iHMRs.
Given that most differential methylation occurs distal to gene promoters, and that many of these regions show features of regulatory elements, we asked whether iHMRs are informative about the regulation of nearby genes. First, we observed that expanding pHMRs are more proximal to cell-type-specific iHMRs (P = 2.2 × 10−17; Wilcoxon test), but not to CTCF iHMRs (P = 0.77; Wilcoxon test) (Fig. 3C). This suggested that demethylation of enhancers might be involved in the activation of these genes. Indeed, both enhancer iHMR hypomethylation and promoter pHMR expansion together are more predictive of higher gene expression changes than either on its own. A total of 44% of genes with an expanded promoter HMR and a nearby (<25 kb) iHMR show elevated expression (fold change >2), compared with 23% with an expanded pHMR, but no nearby (>100 kb) iHMR and 12% without a significant expansion of the pHMR, but an iHMR closer than 25 kb. This effect is diluted with iHMR distance from the gene (Spearman r = −0.21; P = 2.93 × 10−125) (Fig. 3D).
Breaking this overall pattern, in H1 ESCs a subset of expanded pHMRs occurs at silent genes (Fig. 3E; Supplemental Fig. S3) (P = 2.6 × 10−26; Wilcoxon test). In those cases, high levels of H3K27me3 cover the pHMR, spreading along the expanded pHMR region, while H3K4me2 remains confined to only the constitutively hypomethylated core region (Supplemental Fig. S3E). Consistently, many of these bivalent pHMRs co-occur with nearby bivalent iHMRs (Fig. 3F), as defined in Figure 1. In B cells, the majority of bivalent iHMRs observed in H1 ESCs remains hypomethylated and are generally resolved into either an active (high H3K4 methylation and RNA Pol II binding) or a silent (some H3K27me3 enrichment and low H3K4 methylation) chromatin state (Fig. 4A; for examples, see Supplemental Fig. S4). While silencing mostly occurs with H3K27 methylation, a smaller subset of H1 ESC bivalent iHMRs are silenced by DNA methylation in B cells. These sites are completely devoid of DNase hypersensitivity and all of the studied histone modifications. These described changes all tend to be coordinated between the iHMR and an associated pHMR. Illustrating these patterns, Figure 4B depicts examples of multiple shared and specific iHMRs near the annexin A2 receptor gene ANXA2R, including a cluster of bivalent H1 ESC iHMRs, that become active in LCLs, as indicated by exchange of trimethylated H3K27 for acetylated H3K27.
Figure 4.
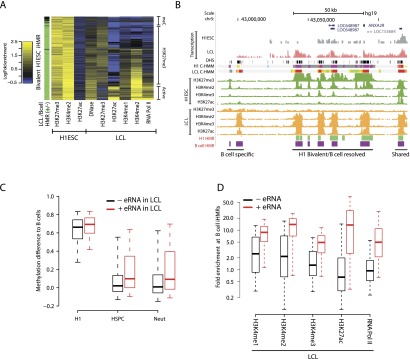
Resolution of bivalent iHMRs during differentiation and stepwise, de novo hypomethylation at transcribed enhancer-like iHMRs. (A) Bivalent iHMRs are resolved to active or silenced chromatin states during differentiation. Heatmap showing the LCL chromatin profile at iHMRs with a bivalent chromatin signature in H1 ESCs. Sidebar colors indicate whether the HMR remains hypomethylated in B cells (green) or becomes fully methylated (black). (B) Example locus surrounding ANXA2R, a gene expressed in lymphocytes and bone marrow, illustrating the coordinated resolution of the H1 ESC bivalent chromatin state in B cells/LCL. ENCODE regulation and transcription tracks are shown along with chromatin states modeled by ChromHMM (Ernst et al. 2011) in H1ES and GM12878 cells. Transcription tracks are presented in log scale. (C) Transcribed, active enhancer-like iHMRs in B cells are fully methylated in H1 ESCs and show intermediate states in other blood cell types. Differences in methylation levels between other cell types and B cell iHMRs are shown. (D) Only B cell iHMRs with eRNA (>0.1 RPKM) show strong enrichment for chromatin marks, suggesting an active regulatory state.
Transcribed, cell-type-specific iHMRs mark likely active enhancers
Many fully methylated regions in H1 ESCs lost DNA methylation in the mature B cell, and we referred to these as de novo demethylated iHMRs. We sought to understand the timing of these changes during cell-fate specification, so we compared the methylation levels in B cells at these iHMRs with other human primary hematopoietic cell types, including hematopoietic stem and progenitor cells (HSPCs) and a nonlymphoid blood cell (neutrophils) (Supplemental Fig. S5A). iHMRs shared between B cells and H1 ESCs also show equally low methylation levels in the other blood cell types. iHMRs not present in H1 ESCs, however, show a large population of lineage-shared sites, which have equally low methylation levels in all three blood cell types, as well as a smaller fraction of B cell iHMRs only partially hypomethylated in the other blood cells. As expected, these iHMRs are enriched for lymphoid-specific TFs such as NFKB, ELF1, EGR1, EBF1, and PAX5 (data not shown) and are proximal to genes involved in lymphocyte development (using GREAT) (McLean et al. 2010).
In accordance with previous observations in HSPCs (Hodges et al. 2011), this suggested that B cell–specific regulatory elements become partially hypomethylated early during hematopoietic differentiation and maintain this intermediate methylation state in blood sister lineages, while undergoing additional hypomethylation specifically in B cells. Consistent with this, the “silent” class of demethylated B cell iHMRs (Supplemental Fig. S1E) is potentially active in other blood cells. These lymphoid-specific iHMRs often are already DNase hypersensitive in stem cells, but not yet hypomethylated, i.e., they are shared DHSs with differential HMRs between H1 ESC and B cells (Supplemental Fig. S6B). This potentially primed state in the stem cell population is characterized by intermediate DNase hypersensitivity, but an almost complete absence of the studied histone marks, except for a slight H3K4me1 enrichment (Supplemental Fig. S6C). In the blood lineage, these sites become partially hypomethylated, while in B cells, a subset acquires the signature of potentially active regulatory elements and is fully hypomethylated. The rest remain “silent” in B cells, showing H3K27me3 enrichment.
To better understand these progressive methylation changes between cell-types, we searched for associated regulatory events. We observed that some de novo iHMRs showed evidence of being transcribed (see example in Fig. 4B). Transcription at some enhancer sites has recently been described as a marker of an active enhancer state (so-called eRNA) (Kim et al. 2010), which prompted us to investigate whether intergenic transcription was linked more generally to cell-type-specific iHMR hypomethylation. We indeed found that transcriptional activity is not an exclusive property of pHMRs at gene promoters or “promoter-like” iHMRs. In H1 ESCs, over 44% of intergenic enhancer-like iHMRs possess CAGE tags, which represent the transcription start site (TSS) of capped transcripts. In LCLs, transcription occurs at those iHMRs with the most B cell–specific hypomethylation (Fig. 4C) (P = 7.4 × 10−35; Wilcoxon test). Compared with the other lineage-shared iHMRs, B cell–specific transcribed intergenic HMRs also display higher levels of several histone modifications, particularly H3K27ac (P = 1.8 × 10−293; Wilcoxon test), suggesting a strong enrichment for active lymphoid enhancers (Fig. 4D; Creyghton et al. 2010). Similarly, among iHMRs, which were bivalent in H1 ESCs, transcription in LCLs marks those regions that lose H3K27me3 (Supplemental Fig. S5B). Importantly, this iHMR transcriptional activation is correlated with the activation of nearby genes (Supplemental Fig. S5C,D). Together these data suggest that iHMR transcription is linked to highly cell-type-specific demethylation of previously primed loci, which may function as cell-type-specific active enhancer elements (see Fig. 7 below and Discussion).
Figure 7.

Model of iHMR behavior at a B cell specifically expressed gene. Shared DHS sites are pre-established in the embryonic stem cell. Hypomethylation at the CpG Island gene promoter (right) and at a CTCF iHMR (left) is constant during development. The enhancer-like iHMR (middle) is fully methylated in H1 ESC. In blood-specified progenitors (HSPCs), it becomes partially demethylated but remains inactive, i.e., lacks H3K4 methylation and RNA transcription. In the B cell state, where the gene is expressed, the promoter HMR expands beyond the core CGI region, and the iHMR becomes fully hypomethylated. The enhancer-like iHMR displays an active enhancer chromatin state (H3K4me1, H3K27ac). It is bound by TBP and RNA Pol II at specific sequence elements (including the TATA box), which initiate eRNA transcription within the iHMR.
Features of transcription at iHMRs reveal positional cues in the primary sequence
We analyzed RNA-seq data sets (Djebali et al. 2012) to further explore the nature of transcription at iHMRs. Consistent with our functional classification scheme, different classes of iHMRs generate distinct types of RNA transcripts (with the exception of the mostly silent CTCF iHMRs) (Fig. 5A–C). All transcripts are less abundant overall than annotated long noncoding RNAs and mRNAs. Capped transcripts from bivalent iHMRs and enhancer iHMRs are even less frequent in the steady-state RNA population than transcripts derived from promoter-like iHMRs (Fig. 5A) (P = 2.6 × 10−10; Wilcoxon test). Bivalent iHMRs are enriched for polyadenylated transcripts at levels comparable to annotated transcripts, while RNAs from enhancer-like iHMRs are mostly, but not exclusively, nuclear and non-poly(A)+ compared with RNAs from promoter-like iHMRs (P = 7.2 × 10−6; Wilcoxon test) (Fig. 5B,C), consistent with previous descriptions of eRNA (Kim et al. 2010). The distinction between enhancer-like and promoter-like transcribed iHMRs is also reflected in their primary genomic sequence. ARTS, a sequence-based promoter prediction tool (Sonnenburg et al. 2006), which was trained on annotated gene-promoters, assigns significantly lower promoter scores to enhancer-like iHMRs than promoter-like iHMRs (Fig. 5D) (P = 1.3 × 10−262; Wilcoxon test).
Figure 5.

iHMRs produce different classes of transcripts from specific transcription start sites. (A_–_D) Different types of RNAs arise from iHMRs classes. Boxplots represent the distribution of values (fifth to 95th percentile) for each iHMR class compared with annotated lincRNAs and mRNAs. Enhancer iHMR transcripts are less expressed and less polyadenylated, while bivalent HMRs make low abundance poly(A) RNAs. Promoter-like, but not enhancer-like iHMRs contain strong promoter sequence signals. (A) Expression level, (B) nuclear localization, (C) polyadenylation levels, (D) genomic sequence-based promoter prediction scores (ARTS). (E–H) eRNA TSS positions match specific sequence and chromatin features. (E) Fraction of positions covered with RNA-seq (transcript body) and CAGE tags (TSS). (F) Specific positional arrangement of histone modifications and DNase hypersensitivity around the eRNA TSS. (G) CpG density is symmetric around the TSS, but GC-skew (strand bias of “G” vs. “C”) occurs specifically in the direction of transcription. (H) ARTS genomic sequence-based TSS prediction scores peak at the experimentally defined eRNA TSS in the sense direction. (I) Transcription is linked with hypomethylation at intergenic DHSs. Methylation levels at DHS with or without transcription are shown. (J) Presence of the TATA motif at DHS is linked with hypomethylation. Methylation levels at DHS with and without an exact TATA motif match are shown. (K) The TATA motif predicts transcription of enhancer-like iHMRs. The barplot depicts the fraction of expressed (CAGE RPM >0.1) and silent iHMRs in different clusters that contain an exact TATAAA match.
To study the relationship between transcription and iHMR features in more detail, we used peaks of CAGE tags to define transcription start sites of capped RNAs within enhancer-like intergenic HMRs. Meta-profiles of RNA and histone data depicted well-defined start sites for these transcripts (Fig. 5E) with CAGE tags, marking the 5′ end of the RNAs, preferentially situated at the center of the iHMR, and RNA-seq coverage, marking the transcript bodies, following the direction of transcription.
This raised the question of whether specific DNA elements signal the action of RNA polymerases inside iHMRs. To answer this, we searched for features that coincided with the observed TSS positions. Histone modifications reveal a clear positional signal, with peaks of H3K4me1, H3K27ac, and DNase hypersensitivity centered slightly upstream of the TSS (Fig. 5F). Similarly, TATA box binding protein (TBP) and TBP associated factors (TAF) are enriched at these loci, peaking immediately upstream of the TSS (Supplemental Fig. S7A) together with RNA Pol II. Given the well-defined start sites, we looked for the TATA box sequence motif within enhancer-like iHMRs. The TATA box is an ancient motif that provides a precise positional cue for initiation by RNA Pol II at a fixed distance from the TSS (Lenhard et al. 2012). We found enrichment for TATA matches in transcribed enhancer-like iHMRs (Fig. 5K; Supplemental Fig. S7B). These matches occur around 20 bp upstream of the eRNA TSS and show evidence of increased evolutionary conservation at these positions (Supplemental Fig. S7C,D).
Interestingly, the distribution of guanines and cytosines is asymmetric around these TSSs (Fig. 5G). This GC skew begins slightly upstream of the CAGE peak and is codirectional with transcription. GC skew allows the formation of thermodynamically stable R-loop structures, and may protect the DNA sequence from methylation (Ginno et al. 2012). This was an unexpected property of enhancer iHMRs, which has previously only been associated with constitutively hypomethylated promoter CGIs rather than with iHMRs that are comparatively CpG poor and dynamically demethylated during differentiation.
Despite overall DNA sequence-based promoter prediction scores being low at enhancer-like iHMRs (Fig. 5D), a distinct peak at the TSS position is seen, which further supports the notion that well-defined sequence features control iHMR transcription (Fig. 5H). The specific TSSs, corresponding to chromatin and conserved sequence features, suggests a link between transcription and the regulatory function of at least some iHMRs, rather than iHMR transcription being a purely random by-product of proximity to high concentrations of RNA Pol II at the promoters of regulated genes.
A close link between intergenic transcription and iHMR establishment is also supported by the observation that DHSs with CAGE tags are significantly more likely to be hypomethylated (Fig. 5I) (P = 9.8 × 10−144 Fisher's exact test). Furthermore, the presence of a TATA box motif in the DNA sequence of a DHS is predictive of hypomethylation (Fig. 5J) (P = 5.2 × 10−20; Fisher's exact test). Interestingly, among the classes of iHMRs defined in Figure 1, the TATA box distinguishes the cell-type-specific subclass of enhancer iHMRs that are transcribed (Fig. 5K). This suggests that recruitment of TBP (Supplemental Fig. S6D), RNA Pol II, and transcription are not purely a consequence of hypomethylation, but instead are directed by sequence features that possibly play a causal role in the establishment of hypomethylation at these sites.
Cross-species and within-species comparison of iHMRs reveals evolving set of putative enhancers
Comparative HMR and chromatin-state analysis between cell types revealed distinct classes of intergenic elements. To test for functional conservation of these iHMRs, we investigated the concordance of methylation states between corresponding blood cell types in chimpanzee and human. In adult blood cells, 77% of human-shared iHMRs also overlap iHMRs in chimpanzee (Fig. 6A). A subset of B cell–specific human iHMRs is also specifically hypomethylated in chimp B cells, but not other blood cell-types (P = 7.2 × 10−287; χ2 test). Human-specific iHMRs show lower sequence conservation than those shared with chimp (P = 2.3 × 10−248; Wilcoxon test) (Fig. 6B), suggesting that species-differential iHMRs may be explained in part by divergence of functional regulatory sequences. Next, for each class of human B cell iHMRs (defined in Supplemental Fig. S1E), we measured the methylation state in chimp. Notably, the methylation state of enhancer-like iHMRs is significantly less conserved in chimp compared with other classes (Fig. 6C) (P < 1.3 × 10−150, Fisher's exact test). At human transcribed iHMRs containing a TATA box, conservation of the TATA motif predicts conservation of hypomethylation in chimp (Fig. 6D) (P = 0.004, Fisher's exact test), consistent with a role for transcriptional signals in establishment and maintenance of iHMRs. Consistently, this effect is not seen at CTCF iHMRs, which are for the most part transcriptionally inert.
Figure 6.

iHMRs are conserved in methylation state and sequence, and are enriched for human population variation in methylation levels. (A) Cell-type-specific hypomethylation is conserved between human and chimp. Overlap between human B cell–specific or shared HMRs with HMRs in different chimpanzee cell types. (B) Intergenic HMRs shared between human and chimp are also more conserved at the sequence level. (C) Enhancer-like iHMRs are more variable between human and chimp. Barplots show the percentage of B cell iHMRs of different classes that overlap a chimp B cell iHMR. (D) Conservation of the TATA motif at enhancer-like iHMRs predicts conservation of hypomethylation. Barplots show the percentage of human iHMRs (containing the TATA motif) shared with chimp for (left) transcribed, enhancer-like iHMRs and (right) CTCF iHMRs, depending on whether the TATA motif is conserved in chimp. (E) Methylation is more variable at enhancer-like iHMRs in the human population. Barplots show the fraction of probed loci in different HMR classes at which methylation levels vary significantly between whole-blood samples from individuals (see Methods). (F) iHMRs that are conserved with chimp are also less variable in methylation level between human individuals.
Since a substantial fraction of cell-type-specific iHMRs showed variability between species, we investigated whether iHMR methylation levels might also be variable in human populations. Using targeted BS-seq data from the whole blood of 44 human individuals (N Plongthongkum, KR van Eijk, S de Jong, T Wang, JH Sul, MPM Boks, RS Kahn, HL Fung, RA Ophoff, and K Zhang, in prep.), methylation levels at individual CpG sites within HMRs were assessed. Variable CpGs, (i.e., sites with variation in methylation levels between individuals, not caused by a SNP disrupting the CpG site itself) are enriched specifically within B cell–specific enhancer-like iHMRs compared with pHMRs or other classes of iHMRs (Fig. 6E) (P < 1.5 × 10−9; Wilcoxon test). Notably, iHMRs conserved with chimp also show reduced methylation variation among human individuals (Fig. 6F) (P = 1.1 × 10−7; Wilcoxon test), suggesting more robust hypomethylation and possibly stronger selective constraint on the methylation state. Together, these data show that intergenic loci, specifically putative enhancers, with methylation states that dynamically change during differentiation are also the most likely to vary both between individuals and between species.
Discussion
Patterns of DNA methylation, and specifically localized hypomethylation, distinguish developmental lineages and the different cell types within them (Hodges et al. 2011; Bock et al. 2012). However, we and others have shown that the degree of differential methylation between cell types is modest among CpG dense promoters (Stadler et al. 2011; Bock et al. 2012). Instead, the most cell-type discriminatory patterns are found in intergenic space, especially outside of CpG Islands. These cell-type-specific iHMRs cannot be predicted by simple sequence characteristics and must instead be identified empirically using cell-type-specific methylation profiles. The importance of this unbiased approach to identify functional regulatory elements has recently been confirmed by comparative methylation analysis across vertebrates (Long et al. 2013).
The extensive sampling of regions with lower CpG density is necessary to capture the diversity of putative, cell-type-specific regulatory elements that might be impacted by DNA methylation. Here, the integration of numerous genome-scale data sets revealed that HMRs fall into distinct functional groups with differing methylation dynamics and regulatory behaviors during development. From this analysis, four dominant classes of intergenic hypomethylated regions emerged: enhancer-like, promoter-like, bivalent, and insulator, related to chromatin patterns found genome wide (Ernst et al. 2011). However, it bears mention that substructure is apparent within each category. This is because each group displays a range of enrichment for distinctive histone marks and other features, resulting in somewhat diffuse cut-offs between clusters and revealing the complexity within groups to be even higher than depicted. This complexity of regulatory elements in the noncoding genome has only recently become visible, and the link between DNA methylation and the diverse element types and chromatin states has not previously been deeply addressed on a genome-wide scale.
Our analyses investigated a number of long-held beliefs about the relationship between DNA methylation and regulatory domains and also revealed new and unexpected insights. For example, DNase hypersensitivity is believed to be a universal signature of active _cis_-regulatory elements and has been shown to be strongly negatively associated with DNA methylation (Thurman et al. 2012). Yet, we observe a class of shared DHS sites that are fully methylated in stem cells and lose methylation in differentiated cells. This suggests that while permissive DHS sites are already set up in embryonic stages, they can still be methylated, until specific regulatory events, including histone modifications and ncRNA transcription, occur during differentiation.
As summarized in Figure 7, we found that one of the primary distinctions between different types of iHMRs is the tendency to be either constitutive throughout development or de novo demethylated in mature cells. This characteristic separates enhancer-like iHMRs from other classes. Our data suggest that these iHMRs are the most cell-type specific and the most dynamically regulated during development. Based on our comparison of the different cell types in this study, we may suggest a timeline of enhancer iHMR formation in which three major steps are observed. First, DHS are pre-established in the embryonic stem cell, but remain methylated. Hematopoietic regulatory elements are partially demethylated during blood-cell commitment, a process that may be initiated at even earlier stages of differentiation than the HSPC stage assessed here. Lastly, lymphoid enhancers become transcribed and completely demethylated specifically in B cells, reaching their cell-type-specific fully active state (Fig. 7).
As most specific iHMRs in adult somatic lymphoid cells are not hypomethylated in the stem cell, this would imply that demethylating mechanisms, whether passive or active, are at work in differentiating cells. This hypothesis is supported by a recent observation that hydroxymethylcytosine marks enhancers in differentiating neural progenitors in mouse (Serandour et al. 2012), though it remains to be seen whether this association extends to other cell types. Alternatively, molecular interactions that protect these regions from maintenance methylation may also play a role. Demethylation associated with TF binding could be one such mechanism, since it was recently shown that CTCF binding is both necessary and sufficient to create distal regions of low methylation resembling iHMRs (Stadler et al. 2011). Our data support this model, since we observe nucleosome displacement and increased frequency of TF occupancy at sites that become hypomethylated in lymphoid cells.
The state of DNA methylation at distal regulatory sites also allowed us to infer the activity of nearby genes and perhaps even more accurately detect links between iHMR methylation and the methylation state at the gene promoter itself. Previously, “active” enhancers were identified by histone marks, i.e., H3K4me1 and H3K27ac, which distinguish distal enhancers from proximal promoters. Here, we show that iHMRs specifically mark active elements that are linked to both pHMR expansion and differential gene expression. Most of these highly active iHMRs also harbor TSS for noncoding transcripts. In fact, our data indicate that transcription is a hallmark of active iHMRs, and recent evidence suggests a putative functional basis for this phenomenon, since disruption of some enhancers, as well as siRNA knockdown of eRNAs, interferes with the expression of eRNAs and enhancer-regulated genes (Ling et al. 2004; Wang et al. 2011; Melo et al. 2013). iHMR transcripts originate from well-defined, position-specific, but generally weak promoter sequence elements in the iHMR, in patterns that separate iHMR types and CGIs. Close examination of iHMR TSSs reveals conservation of transcriptional sequence signals embedded within the iHMR. We observed strand asymmetry (CG skew) in iHMR transcripts, reminiscent of features linking transcription to protection from methylation at promoter CGIs (Ginno et al. 2012). These characteristics suggest that, in addition to simple TF binding, transcription may be involved in establishing or maintaining hypomethylation at dynamic iHMRs. Indeed, we observed that even low CpG density intergenic regulatory regions with transcriptional activity are strongly hypomethylated. Accordingly, while many lymphocyte-specific iHMRs already begin to lose methylation in the early stages of the blood lineage, the complete loss of methylation at iHMRs correlates with increased noncoding transcriptional output and enhancer activation. Whether or not disruption of these RNAs or their transcription alters enhancer methylation states remains to be shown.
iHMRs that show bivalent histone marks in stems cells are typically constitutively hypomethylated. Like enhancer iHMRs, their activity appears tightly coordinated with expanded pHMRs of nearby genes. We found an unexpected degree of coincidence between pairs of bivalent, expanded pHMRs and iHMRs. Previously, “permissive” H3K4me1-marked enhancers have been paired with polycomb-repressed promoters (Rada-Iglesias et al. 2011, Taberlay et al. 2011), but the extent of bivalently marked iHMRs and their widespread co-occurrence with pHMR counterparts had not yet been seen genome wide.
Patterns of methylation at many iHMRs are conserved in chimpanzee in a manner reflected by increased sequence conservation and reduced variation at the epigenetic level within the human population. In contrast, a subset of human-specific enhancer-like iHMRs show increased methylation variation within the human population. This epigenetic variation may be relevant to inter-individual differences in gene regulation and to disease susceptibility (Akhtar-Zaidi et al. 2012; Toperoff et al. 2012). Loss or gain of TF-binding events resulting from sequence divergence may account for cross-species differential methylation at constrained elements (MacArthur and Brookfield 2004; Prabhakar et al. 2008; Wilson and Odom 2009; Schmidt et al. 2010). Indeed, only 40% of human B cell–specific iHMRs overlap iHMRs of the orthologous cell type in chimpanzee compared with constitutive, shared iHMRs of other cell types. This may signify that cell-type-specific iHMRs depend on sequence features with a higher turnover rate than constitutively hypomethylated regions, including CGIs. In this context, we found that loss of a TATA box at enhancer iHMRs predicts loss of hypomethylation in chimp, suggesting a role for TBP and the transcriptional machinery, in addition to the function of other specific TFs, in the establishment of these iHMRs. These comparisons may thus serve to distinguish those enhancer iHMRs that fit an “enhanceosome” model, whereby exclusion of a single binding event can wipe out enhancer function in one species (possibly by loss of hypomethylation), or a “billboard” model in which binding-site rearrangements within the enhancer are tolerated as long as the sum of TF interaction is constant (Arnosti and Kulkarni 2005; Lusk and Eisen 2010).
Methods
GM12878 methylation data
Lymphoblastoid cell line (LCL) methylomes were generated from genomic DNA purchased from Coriell (cat #NA12878) according to the methods described previously (Molaro et al. 2011). Four flow cell lanes of bisulfite-converted fragment libraries were sequenced on the Illumina Hiseq platform to obtain paired-end 100-bp read lengths and ∼10× coverage of the human genome.
Methylation data
Whole-genome bisulfite sequencing data for human B cells, Neutrophils, and HSPCs were taken from Hodges et al. (2011) (GEO accession number GSE31971), and data for H1 ESCs was obtained from Lister et al. (2009).
Methylation data for chimpanzee-derived hematopoietic cells
Flow cytometry and DNA extraction were performed according to previously described methods (Hodges et al. 2011). Briefly, peripheral blood was collected from healthy female donors and pooled. After isolation by Ficoll gradient, mononuclear cells were fixed and stained with antibodies against the following human cell surface markers (eBioscience): anti-CD34 conjugated to PE-Cy7, anti-CD38 conjugated to APC, anti-CD45 conjugated to PE, anti-CD19 conjugated to PE, and anti-CD235a (Glycophorin) conjugated to PE. For lineage depletion, either a combination of PE-conjugated antibodies against CD45, CD19, and CD235a, or a commercially available human hematopoietic lineage cocktail was used.
Computational analysis
Mapping BS-seq reads was performed with methods described by Smith and colleagues using tools from the RMAP package (Smith et al. 2009). Mapping statistics for BS-seq libraries generated herein are provided in Supplemental Table S1. Hypomethylated regions were called for each cell type using the Hidden Markov Model described in Hodges et al. (2011). For H1 ESCs, two replicates were available and only HMRs called in both replicates were used (Suplemental Fig. S8A). The methylation level of genomic regions (HMRs, DHS) was computed as the mean ratio of converted to unconverted calls at all CpGs in the interval. Expanding promoter HMRs were detected as an uninterrupted run of at least five significantly differentially methylated CpG sites next to a shared HMR (for details on differential methylation calls see Hodges et al. 2011).
We used ChIP-seq or RNA-seq from LCLs (GM12878) as a proxy for B cells. Comparing methylation levels at iHMRs, we confirmed that B cell iHMRs are strongly shared with LCLs, which hence make a good proxy for B cells (Supplemental Fig. S8B,C). For B cell HMR analysis relying on LCL data, only those HMRs with an average methylation level <20% in the GM12878 bisulfite data were used. We did not use HMR calls from the GM12878 methylome itself since, as an immortalized cell line, it contains a number of features differing from primary cells, including very large domains of hypomethylation and “fuzzy” HMR boundaries (Supplemental Figs. S8C, S9).
Human genome version 19 and gencode (version 7) gene annotations were used, defining any HMR within 250 bp of a gene's 5′ end as a TSS HMR, any HMR over 1 kb from any gene as intergenic, and an HMR within a gene that does not overlap any exon as intronic. CpG Island calls were obtained from the UCSC Genome Browser track “CpG Islands” based on Gardiner-Garden and Frommer (1987). HMRs were considered shared between two cell types if HMRs called in each data set independently overlap by at least 1 bp.
Conservation of HMRs between human and chimp: HMRs were called mapping BS-seq data to the chimpanzee genome (version panTro3 from the UCSC Genome Browser) and lifted over to the human genome (hg19) using the UCSC Genome Browser liftOver tool (version 1.28 with the “panTro3ToHg19” and “hg19ToPanTro3” chains) to assess overlap with human HMRs. Only those human HMRs were considered for analysis that had a unique corresponding position in the chimpanzee genome (based on liftOver to the chimpanzee genome and back resulting in only the original position).
All ChIP, MNase, and DNase-seq signal tracks were taken from ENCODE (The ENCODE Project Consortium 2011) (Broad histone ChIP, Duke DNase-seq, Stanford MNase-seq) and are scored as fold enrichment over the average genomic read density. TF-binding calls are based on ENCODE uniform peak calls (SPP) (The ENCODE Project Consortium 2011). DNase HS sites are based on Duke DNase HS calls in ENCODE. Enhancer and (novel) promoter predictions were taken from the ENCODE elements analysis (Yip et al. 2012) and are based on supervised classification of ENCODE histone-modification and TF-binding data. All ENCODE data sets are available at http://encodeproject.org/ENCODE/downloads.html.
Likely evolutionarily conserved positions were determined using the phastCons mammalian constraint score (from the UCSC track “Vertebrate Multiz Alignment & Conservation”) with a cutoff of 0.9 on the posterior probability of conservation.
Gene expression data was taken from the ENCODE transcriptome group for GM12878 and H1 ESCs (UCSC ENCODE RNA-seq Track). RNA-seq (UCSC CSHL Long RNA-seq) and CAGE (UCSC Riken CAGE) reads in each HMR (based on their 5′ end) were counted and normalized (RPKM: reads per kilobase and million mappable reads for long RNA-seq and RPM for CAGE). Replicates were pooled and for each HMR the highest value of any RNA type [Poly(A)+ or Poly(A)−, whole cell, nuclear, or cytoplasmic] was used. Expression changes are computed as log((1 + RPKM1)/(1 + RPKM2)). To avoid confounding effects from host genes, eRNA analysis was limited to intergenic iHMRs (>1 kb from any gene).
CpG density was computed as the ratio of the observed over expected CpG count based on the local CG-density. GC-skew was computed as (G − C)/(G + C). Both were computed in 50-bp sliding windows. The sequence-based promoter prediction scores were generated using the SVM model implemented in ARTS (Sonnenburg et al. 2006) at single-nucleotide resolution on both strands of the human genome. The predictions are based on a combination of features, including position-specific core promoter motifs and local sequence features, e.g., k-mer stats. For details, see Sonnenburg et al. ( 2006). The scores are the raw SVM outputs (−5 to 5), with positive scores indicating a stronger promoter prediction. TATA-box motifs were called as exact matches to 5′-TATAAA-3′ within the HMR on either DNA strand.
Clustering of HMRs into chromatin groups was performed using hierarchical clustering (using the hclust function of R) with a Manhattan distance and UPGMA linkage on the log-transformed ChIP signal. HMRs with any missing data (unmappable regions) or input control signal more than threefold different from the genomic average were filtered out. The trees were cut at a branch height resulting in 20 groups. Only the major groups, containing at least 10% of the intergenic HMRs, were used for further analysis. Clustering of B cell iHMRs was based on chromatin data from GM12878 as well as the chromatin state of these sites in H1 ESCs, as shown in the heatmap (Supplemental Fig. S1E). A comprehensive BED file listing the coordinates of iHMRs and their functional annotations are provided in Supplemental Tables S2 and S3 for H1 ESCs and B cells, respectively.
Histone profiles at “meta-HMRs” were computed by aligning all selected HMRs at their 5′ and 3′ ends, evaluating the signal inbetween in 250 equal-sized steps and taking the (95th percentile) trimmed mean at each position. For TSS centered plots, the CAGE read density within an HMR was smoothed using a 25-bp sliding window, and the point of highest signal was marked as the TSS peak. All boxplots show the fifth, 25th, 50th, 75th, and 95th percentiles.
CpG methylation variation in the human population was assessed using targeted bisulfite sequencing data from N Plongthongkum, KR van Eijk, S de Jong, T Wang, JH Sul, MPM Boks, RS Kahn, HL Fung, RA Ophoff, and K Zhang (in prep.) in whole-blood samples of 44 individuals. Methylation at individual CpGs within HMRs was assessed in 78,605 regions targeted by padlock probes (BSPP) and compared across individuals. For each CpG site targeted by a probe, the standard deviation of methylation levels between individuals was assessed. Sites with an SD >0.1 were considered variable. The variability of each HMR was then scored as the ratio of probes containing variable CpG sites to total probes overlapping the HMR.
Data access
BS-seq data have been deposited in the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) and the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) and are available through the following accession numbers: SRP022182 and SRP021118. BSPP data have been deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE47614.
Acknowledgments
We thank N. Plongthongkum, Kun Zhang, Tina Wang, and Roel A. Ophoff for sharing unpublished BSPP data. We thank members of the Hannon and Gingeras labs, and in particular, Philippe Batut and Antoine Molaro for helpful discussion. We thank members of the ENCODE project for data access and support, especially Carrie Davis, Alex Dobin, Chris Zaleski, Anshul Kundajee, and Kevin Yip. This work was funded in part by National Human Genome Research Institute Grant 5U54HG004557-05.
Footnotes
[Supplemental material is available for this article.]
References
- Akhtar-Zaidi B, Cowper-Sal Lari R, Corradin O, Saiakhova A, Bartels CF, Balasubramanian D, Myeroff L, Lutterbaugh J, Jarrar A, Kalady MF, et al. 2012. Epigenomic enhancer profiling defines a signature of colon cancer. Science 336: 736–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnosti DN, Kulkarni MM 2005. Transcriptional enhancers: Intelligent enhanceosomes or flexible billboards? J Cell Biochem 94: 890–898 [DOI] [PubMed] [Google Scholar]
- Bock C, Beerman I, Lien WH, Smith ZD, Gu H, Boyle P, Gnirke A, Fuchs E, Rossi DJ, Meissner A 2012. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol Cell 47: 633–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulger M, Groudine M 2011. Functional and mechanistic diversity of distal transcription enhancers. Cell 144: 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp P, et al. 2010. Histone h3k27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 107: 21931–21936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2011. A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 9: e1001046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M 1987. Cpg islands in vertebrate genomes. J Mol Biol 196: 261–282 [DOI] [PubMed] [Google Scholar]
- Ginno PA, Lott PL, Christensen HC, Korf I, Chedin F 2012. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell 45: 814–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318 [DOI] [PubMed] [Google Scholar]
- Hodges E, Molaro A, Dos Santos CO, Thekkat P, Song Q, Uren PJ, Park J, Butler J, Rafii S, McCombie WR, et al. 2011. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol Cell 44: 17–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, et al. 2011. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 471: 480–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. 2010. Widespread transcription at neuronal activity-regulated enhancers. Nature 465: 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenhard B, Sandelin A, Carninci P 2012. Metazoan promoters: Emerging characteristics and insights into transcriptional regulation. Nat Rev Genet 13: 233–245 [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, Kheradpour P, Ernst J, Jordan G, Mauceli E, et al. 2011. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478: 476–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J, Ainol L, Zhang L, Yu X, Pi W, Tuan D 2004. HS2 enhancer function is blocked by a transcriptional terminator inserted between the enhancer and the promoter. J Biol Chem 279: 51704–51713 [DOI] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long HK, Sims D, Heger A, Blackledge NP, Kutter C, Wright ML, Grutzner F, Odom DT, Patient R, Ponting CP, et al. 2013. Epigenetic conservation at gene regulatory elements revealed by non-methylated DNA profiling in seven vertebrates. eLife 2: e00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusk RW, Eisen MB 2010. Evolutionary mirages: Selection on binding site composition creates the illusion of conserved grammars in Drosophila enhancers. PLoS Genet 6: e1000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur S, Brookfield JF 2004. Expected rates and modes of evolution of enhancer sequences. Mol Biol Evol 21: 1064–1073 [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G 2010. GREAT improves functional interpretation of _cis_-regulatory regions. Nat Biotechnol 28: 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo CA, Drost J, Wijchers PJ, van de Werken H, de Wit E, Oude Vrielink JA, Elkon R, Melo SA, Leveille N, Kalluri R, et al. 2013. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell 49: 524–535 [DOI] [PubMed] [Google Scholar]
- Molaro A, Hodges E, Fang F, Song Q, McCombie WR, Hannon GJ, Smith AD 2011. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 146: 1029–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar S, Visel A, Akiyama JA, Shoukry M, Lewis KD, Holt A, Plajzer-Frick I, Morrison H, Fitzpatrick DR, Afzal V, et al. 2008. Human-specific gain of function in a developmental enhancer. Science 321: 1346–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J 2011. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470: 279–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, Kutter C, Watt S, Martinez-Jimenez CP, Mackay S, et al. 2010. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328: 1036–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serandour AA, Avner S, Oger F, Bizot M, Percevault F, Lucchetti-Miganeh C, Palierne G, Gheeraert C, Barloy-Hubler F, Peron CL, et al. 2012. Dynamic hydroxymethylation of deoxyribonucleic acid marks differentiation-associated enhancers. Nucleic Acids Res 40: 8255–8265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. 2012. A map of the _cis_-regulatory sequences in the mouse genome. Nature 488: 116–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AD, Chung WY, Hodges E, Kendall J, Hannon G, Hicks J, Xuan Z, Zhang MQ 2009. Updates to the RMAP short-read mapping software. Bioinformatics 25: 2841–2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg S, Zien A, Ratsch G 2006. Arts: Accurate recognition of transcription starts in human. Bioinformatics 22: e472–e480 [DOI] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. 2011. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480: 490–495 [DOI] [PubMed] [Google Scholar]
- Taberlay PC, Kelly TK, Liu CC, You JS, De Carvalho DD, Miranda TB, Zhou XJ, Liang G, Jones PA 2011. Polycomb-repressed genes have permissive enhancers that initiate reprogramming. Cell 147: 1283–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. 2012. The accessible chromatin landscape of the human genome. Nature 489: 75–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toperoff G, Aran D, Kark JD, Rosenberg M, Dubnikov T, Nissan B, Wainstein J, Friedlander Y, Levy-Lahad E, Glaser B, et al. 2012. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood. Hum Mol Genet 21: 371–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, et al. 2011. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 474: 390–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiench M, John S, Baek S, Johnson TA, Sung M, Escobar T, Simmons CA, Pearce KH, Biddie SC, Sabo PJ, et al. 2011. DNA methylation status predicts cell type-specific enhancer activity. EMBO J 30: 3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MD, Odom DT 2009. Evolution of transcriptional control in mammals. Curr Opin Genet Dev 19: 579–585 [DOI] [PubMed] [Google Scholar]
- Yip KY, Cheng C, Bhardwaj N, Brown JB, Leng J, Kundaje A, Rozowsky J, Birney E, Bickel P, Snyder M, et al. 2012. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol 13: R48. [DOI] [PMC free article] [PubMed] [Google Scholar]