Human Cytosolic Extracts Stabilize the HIV-1 Core (original) (raw)
Abstract
The stability of the HIV-1 core in the cytoplasm is crucial for productive HIV-1 infection. Mutations that stabilize or destabilize the core showed defects on HIV-1 reverse transcription and infection. We developed a novel and simple assay to measure the stability of _in vitro_-assembled HIV-1 CA-NC complexes. The assay allowed us to demonstrate that cytosolic extracts strongly stabilize the HIV-1 core. Interestingly, stabilization of _in vitro_-assembled HIV-1 CA-NC complexes is not due solely to macromolecular crowding, suggesting the presence of specific cellular factors that stabilize the HIV-1 core. By using our novel assay, we measured the abilities of different drugs, such as PF74, CAP-1, IXN-053, cyclosporine, Bi2 (also known as BI-2), and the peptide CAI, to modulate the stability of _in vitro_-assembled HIV-1 CA-NC complexes. Interestingly, we found that PF74 and Bi2 strongly stabilized HIV-1 CA-NC complexes. On the other hand, the peptide CAI destabilized HIV-1 CA-NC complexes. We also found that purified cyclophilin A destabilizes _in vitro_-assembled HIV-1 CA-NC complexes in the presence of cellular extracts in a cyclosporine-sensitive manner. In agreement with previous observations using the fate-of-the-capsid assay, we also demonstrated the ability of recombinant CPSF6 to stabilize HIV-1 CA-NC complexes. Overall, our findings suggested that cellular extracts specifically stabilize the HIV-1 core. We believe that our assay can be a powerful tool to assess HIV-1 core stability in vitro.
INTRODUCTION
Soon after human immunodeficiency virus type 1 (HIV-1) enters the host cell, a poorly understood process known as uncoating takes place. The viral RNA genome in the nucleoprotein core is reverse transcribed to viral DNA (vDNA) in the reverse transcription complex (RTC), comprised of capsid protein, in addition to matrix proteins, Vpr, reverse transcriptase, and integrase (1). Synthesis of full-length vDNA in the RTC produces the preintegration complex (PIC) (1). Subsequently, the PIC is actively transported to the nucleus, where vDNA is integrated into cellular chromatin. In addition to proviral integration, a fraction of the viral DNA is converted into unintegrated circular forms by host cell enzymes (2). These DNA circles do not support productive infection yet can be effective markers for nuclear import (1, 3). In the late phase of retroviral replication, the integrated provirus produces viral RNA and transcripts translated into viral proteins, which assemble into new viral particles that are subsequently released into the extracellular milieu to initiate new rounds of infection.
In its simplest definition, uncoating is the shedding of monomeric capsids from the retroviral core or ribonucleoprotein complex. Because only ∼40% of the total capsid in the virion comprises the retroviral core (4–6), a simple overview is that the monomeric capsid is in dynamic equilibrium with the assembled capsid (viral core). This implies that the core might exist in a metastable state only when the soluble capsid is in high concentration, keeping the equilibrium shifted toward the core formation by mass action. The fact that complexes containing capsid have been detected in the cytoplasm of cells early during infection implies that cellular factors might be involved in stabilization of the core (7, 8).
The capsid protein is required for the successful completion of early steps of HIV-1 replication: (i) successful infection requires that capsid uncoating occur during or after reverse transcription (9–13), (ii) elegant experiments have shown that capsid is the genetic determinant for the ability of lentiviruses to infect nondividing cells (14–17), and (iii) the stability provided by the capsid protein assembled into the viral core is important for the occurrence of reverse transcription and productive infection (18–20).
Over the years, strong assays to biochemically measure core stability have been developed, such as the fate-of-the-capsid assay, which measures core stability during infection of cells over time (13), and an in vitro assay that follows the disassembly of isolated cores from virions (18). Even though both assays are widely used (10, 21–28), they are laborious. A rapid assay to measure core stability remains elusive.
Because the stability of the HIV-1 core is important for infection, we developed an assay to measure capsid stability in vitro using HIV-1 CA-NC complexes as a surrogate for the HIV-1 core. Using this assay, we found that cellular extracts stabilize the HIV-1 core in vitro. Our studies also revealed that the stability of the core could also be modulated by using macromolecular-crowding agents, such as bovine serum albumin (BSA) and polyethylene glycol (PEG). Furthermore, we demonstrated that cyclophilin (Cyp) A destabilizes the HIV-1 core in vitro in a cyclosporine-sensitive manner. We also tested the stability of the HIV-1 core in the presence of agents that target the HIV-1 capsid, such as PF74, cyclosporine, Bi2, I-XW-053, CAP-1, and the CAI peptide. By using this stability assay, we tested the intrinsic stability of specific capsid mutants. Finally, we tested the ability of purified CPSF6 to stabilize HIV-1 CA-NC complexes. In summary, we have developed an assay to measure the stability of the HIV-1 capsid and demonstrated the ability of cellular extracts to stabilize capsid. We believe that this assay could assist us in assessing the stability and in the future isolation of specific cellular factors that interact with capsid.
MATERIALS AND METHODS
Reagents.
A stock solution of BSA (Fraction V; Sigma) was prepared at 20 mg/ml in destabilization buffer (20 mM Tris-HCl, pH 8, 60 mM NaCl, 10 mM KCl, 2 mM MgCl2, 1% glycerol, 0.1% NP-40, 0.5 mM dithiothreitol [DTT]). Similarly, a stock solution of PEG 8000 (Fisher) was prepared at 80 mg/ml in destabilization buffer. The cyclophilin A protein was a gift from Chris Aiken. The stock solution of cyclosporine (Sigma; PHR1092; 500 mg) was prepared at 10 mM in ethanol. The CAI peptide (amino acid sequence, ITFEDLLDYYGP) and the CAIctrl peptide (amino acid sequence, IYDPTLYGLEFD) were synthesized by Genescript (95% purity), and stock solutions were prepared at 10 mM in dimethyl sulfoxide (DMSO). PF74 (PF-3450074) and Bi2 were gifts from Chris Aiken. The stock solution of PF74 was prepared at 100 mM in DMSO. The stock solution of Bi2 was prepared at 20 mM in DMSO. CAP-1 (_N_-(3-chloro-4-methylphenyl)-_N_′-{2-[({5-[(dimethylamino)-methyl]-2-furyl}-methyl)-sulfanyl]ethyl}-urea) was purchased from Maybridge (HTS 02911). The stock solution of CAP-1 was prepared at 20 mM in DMSO. The stock solution of I-XW-053 [4-(4,5-diphenyl-1H-imidazol-2-yl)benzoic acid] was prepared at 10 mM in DMSO.
Mutagenesis.
The pET11a plasmids expressing CA-NC containing the different mutations were created by site-directed mutagenesis and confirmed by sequencing analysis.
HIV-1 CA-NC expression and purification.
The HIV-1 CA-NC protein was expressed, purified, and assembled as previously described (29). The pET11a expression vector (Novagen) expressing the CA-NC protein of HIV-1 was used to transform Escherichia coli BL-21(DE3). CA-NC expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) when the culture reached an optical density of 0.6 at 600 nm. After 4 h of induction, the cells were harvested and resuspended in 20 mM Tris-HCl (pH 7.5), 10 mM 2-mercaptoethanol, and protease inhibitors (Roche). Lysis was performed by sonication, and debris was pelleted for 30 min at 35,000 × g. Nucleic acids were stripped from the solution by using 0.11 volume of 2 M (NH4)2SO4 and the same volume of 10% polyethylenimine. Nucleic acids were pelleted by centrifugation at 29,500 × g for 15 min. The protein was recovered by addition of 0.35 equivalent of saturated (NH4)2SO4. The protein was centrifuged at 10,000 × g for 15 min and resuspended in 20 mM Tris-HCl (pH 7.5), 100 mM NaCl, 10 mM β-mercaptoethanol. The protein was dialyzed against 20 mM Tris-HCl (pH 7.5) and 10 mM β-mercaptoethanol and applied to a 5-ml HiTrap Q HP anion-exchange column (GE Healthcare). The column was eluted using a linear gradient of 0 to 1 M NaCl in the buffer described above using ÄKTA purifier fast protein liquid chromatography (FPLC) (GE). Finally, the CA-NC protein was dialyzed against 20 mM Tris-HCl (pH 7.5), 50 mM NaCl, 1 μM ZnCl2, and 10 mM β-mercaptoethanol and stored at −80°C.
In vitro assembly of CA-NC complexes.
HIV-1 CA-NC particles were assembled in vitro by diluting the CA-NC protein to a concentration of 10 mg/ml in 50 mM Tris-HCl (pH 8.0), 0.5 M NaCl, and 2 mg/ml DNA oligo(TG)25 (50 bp long) (IDT). The pellet was stored at 4°C until needed.
CA-NC stability assay.
To test for stability, 5 μl of CA-NC particles assembled in vitro was incubated in 500 μl of either stabilization buffer (10 mM Tris-HCl, pH 8, 10 mM KCl, 2 mM MgCl2, 0.5 mM DTT) or destabilization buffer (20 mM Tris-HCl, pH 8, 60 mM NaCl, 10 mM KCl, 2 mM MgCl2, 0.5 mM DTT, 1% glycerol, 0.1% NP-40) at room temperature for 1 h. An aliquot of this mixture, henceforth referred to as “input,” was stored. The mixture was spun through a 3.5-ml 70% sucrose cushion (70% sucrose, 1× phosphate-buffered saline [PBS], and 0.5 mM DTT) at 100,000 × g in an SW55 rotor (Beckman) for 1 h at 4°C. After centrifugation, the sucrose cushion was carefully removed and the pellet was resuspended in 1× SDS-PAGE loading buffer. The levels of HIV-1 CA-NC protein in the input and the pellet were assessed by Western blotting using anti-p24 antibodies. The level of cyclophilin A was determined by polyacrylamide electrophoresis, followed by staining with Coomassie blue. The sucrose solution for the cushion was prepared as weight per volume.
Preparation of cytosolic cell extracts.
Cytosolic cell extracts were prepared as follows. 293T cells (106) were washed in PBS, resuspended in stabilization buffer (10 mM Tris-HCl, pH 8, 10 mM KCl, 2 mM MgCl2, 0.5 mM DTT), and incubated for 15 min at 4°C. By using trypan blue staining, which stains lysed cells, we determined that 40% of the cells were lysed. The lysate was centrifuged in a refrigerated Eppendorf microcentrifuge (∼14,000 × g) for 15 min. The cell lysates were then adjusted to a final concentration of 20 mM Tris-HCl, pH 8, 60 mM NaCl, 10 mM KCl, 2 mM MgCl2, 0.5 mM DTT, 1% glycerol, and 0.1% NP-40. Protein levels were measured using the BMA Protein Assay Kit (Pierce). Cell extracts that were treated with 0.1 mg/ml of trypsin (1 h at 37°C) were subsequently incubated with 1 mM phenylmethylsulfonyl fluoride (PMSF) protease inhibitor (30 min at 4°C).
Sucrose gradient fractionation.
Five microliters of CA-NC particles assembled in vitro was incubated in 600 μl of the specified buffer at room temperature for 1 h. A portion of this mixture was stored and referred to as input. The mixture was spun through a 3-ml 40 to 80% sucrose cushion (600 μl each 40%, 50%, 60%, 70%, and 80% sucrose in 1× PBS and 0.5 mM DTT) at 100,000 × g in an SW55 rotor (Beckman) for 45 min at 4°C. All sucrose solutions for the gradient were prepared as weight per volume. After centrifugation, 600-μl fractions were carefully collected. The protein of the fractions was pelleted using methanol-chloroform precipitation as follows. Six hundred microliters of methanol was added to the fractions, followed by 150 μl of chloroform and 450 μl of H2O. The mixture was centrifuged in a refrigerated Eppendorf microcentrifuge (∼14,000 × g) for 15 min. The upper phase was removed, and 650 μl of methanol was added. The mixture was centrifuged in a refrigerated Eppendorf microcentrifuge (∼14,000 × g) for 15 min. The supernatant was removed, and the protein pellet was dried for 15 min. The pellet was resuspended in 1× SDS-PAGE loading buffer, and the levels of HIV-1 CA-NC protein were assessed by Western blotting using anti-p24 antibodies.
Western blot analysis.
Detection of proteins by Western blotting was performed using anti-p24 (Immuno Diagnostics). Secondary antibodies against mouse conjugated to IRDye 680LT or IRDye 800CW were obtained from Li-Cor. Bands were detected by scanning blots using the Li-Cor Odyssey Imaging System in the 700-nm channel.
Construction of recombinant baculovirus expressing OSF-CPSF6(1-321).
Recombinant baculovirus was generated using the Bac-to-Bac system (Invitrogen) following the manufacturer's instructions. For construction of the transfer vector, the CPSF6(1-321) region was amplified by PCR using specific primers and cloned into the EcoRI and XbaI sites of the pFastBac-1 vector containing an OSF (One-Strep and Flag) epitope previously cloned into the BamHI and EcoRI sites of the plasmid. The correctness of the construct was confirmed by sequencing. The pFastBac1-OSF-CPSF6(1-321) vector was recombined into the bacmid by transformation of DH10 Bac cells (Invitogen). The recombinant bacmid was obtained according to the supplier's protocol. Single colonies were grown to stationary phase in 2 ml of LB medium supplemented with the necessary antibiotics (50 mg/ml kanamycin, 7 mg/ml gentamicin, and 10 mg/ml tetracycline with shaking at 300 rpm) for up to 24 h at 37°C. The bacterial pellets were resuspended in 0.3 ml of solution I (15 mM Tris-HCl [pH 8.0], 10 mM EDTA, 100 mg/ml RNase A) and 0.3 ml of solution II (0.2 N NaOH, 1% SDS) and incubated at room temperature for 5 min. Samples were treated with 0.3 ml of 3 M potassium acetate (pH 5.5) on ice for 10 min and centrifuged for 10 min at 14,000 × g. The supernatant was transferred to a tube containing isopropanol and incubated on ice for 10 min. The DNA was pelleted by centrifugation for 15 min at 14,000 × g at room temperature, and the pellets were washed with 0.5 ml of 70% ethanol and air dried at room temperature. DNA samples were resuspended in 40 ml Milli-Q water. The presence of the CPSF6(1-321) gene in the bacmids was confirmed by PCR using the pUC/M13 forward and reverse primers described by Invitrogen. The PCR products were sequenced to confirm the presence of the insert containing the OSF-CPSF6(1-321) construct. The bacmid was then used to generate the corresponding recombinant baculovirus according to the supplier's protocol as described previously (30, 31). Briefly, the bacmid was transfected into Spodoptera frugiperda (SF9) cells using Lipofectamine Plus reagent (Invitrogen). The cells were incubated at 28°C in SF-900 II serum-free insect cell medium (Invitrogen) for 3 days, and recombinant viruses expressing CPSF6(1-321) were harvested (P1 viral stock). For amplification of the P1 viral stock, a monolayer culture (2 × 106 cells/ml) was infected with 0.5 ml of P1 viral stock. The P2 viral stock was harvested 3 days postinfection. Expression of OSF-CPSF6(1-321) was analyzed in SF9 cells infected with P2 viral stocks by Coomassie staining and Western blotting using anti-FLAG antibodies.
CPSF6(1-321) expression in insect cells.
Sf9 insect cells (7.5 × 108) growing in suspension were infected with 5 PFU/cell of the recombinant baculovirus and incubated at 28°C for 72 h. All subsequent steps were carried out at 4°C. Cells were collected by centrifugation at 500 × g for 8 min, resuspended in 60 ml of lysis buffer [250 mM NaCl, 50 mM Tris (pH 8.0), 1.5% Triton X-100, 1 mM Tris-(2-carboxyethyl)phosphine hydrochloride (TCEP), and mammalian protease inhibitor cocktail (Sigma)]. The lysate was clarified by centrifugation (20,000 × g; 40 min), filtered (0.45 μm), and incubated in 1.5 ml StrepTactin Superflow affinity resin (Qiagen). The bound protein was washed three times with 15 ml of buffer (50 mM NaCl, 50 mM Tris [pH 8.0], 1 mM TCEP) and eluted with washing buffer supplemented with 2.5 mM d-desthiobiotin. The eluate was analyzed by SDS-PAGE and Western blotting using anti-FLAG antibodies.
RESULTS
Novel assay to measure the stability of HIV-1 CA-NC complexes in vitro.
To measure HIV-1 core stability, we developed an assay that measures the stability of HIV-1 CA-NC complexes in vitro (Fig. 1). The assay consists of incubating HIV-1 CA-NC complexes with different agents and subsequently measuring the remaining amount of assembled HIV-1 CA-NC complexes using a sucrose cushion. Using this assay, we initially screened for a buffer with the capability to destabilize _in vitro_-assembled HIV-1 CA-NC complexes. We incubated _in vitro_-assembled HIV-1 CA-NC complexes with different buffers and measured the remaining amount of assembled HIV-1 CA-NC complexes using a 70% sucrose cushion (Fig. 1); only assembled HIV-1 CA-NC complexes pelleted in a 70% sucrose cushion. We found a buffer that completely destabilized the _in vitro_-assembled HIV-1 CA-NC complexes and called it destabilization buffer (DB): 20 mM Tris-HCl, pH 8, 60 mM NaCl, 10 mM KCl, 2 mM MgCl2, 0.5 mM DTT, 1% glycerol, 0.1% NP-40. The buffer that preserved the stability of HIV-1 CA-NC complexes was named stabilization buffer (SB): 10 mM Tris-HCl, pH 8, 10 mM KCl, 2 mM MgCl2, 0.5 mM DTT. This assay measures the stability of _in vitro_-assembled HIV-1 CA-NC complexes. Next, we decided to explore the conditions that stabilize or destabilize _in vitro_-assembled HIV-1 CA-NC complexes.
Fig 1.
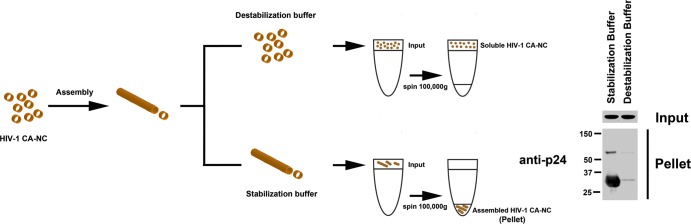
Diagram of the assay to measure the stability of HIV-1 CA-NC complexes. _In vitro_-assembled HIV-1 CA-NC complexes, depicted as tubular structures, are formed by monomeric recombinant HIV-1 CA-NC fusion proteins under highly ionic conditions and recapitulate the surface of the HIV-1 core (29). When HIV-1 CA-NC complexes are incubated in destabilization buffer, they disassemble spontaneously. Disassembled capsids are layered on top of a 70% sucrose cushion; however, the disassembled capsid does not cross the cushion after spinning at 100,000 × g for 1 h. In contrast, incubation of HIV-1 CA-NC in stabilization buffer preserves the assembled structures, which pellet when layered onto a 70% sucrose cushion with spinning at 100,000 × g for 1 h. Input, a fraction of the sample layered onto the sucrose cushion before the centrifugation step. Pellet, a fraction of the capsid pelleted after the sample has been centrifuged at 100,000 × g for 1 h. Input and pellet samples were analyzed by Western blotting using anti-p24 antibodies.
Cellular extracts stabilized _in vitro_-assembled HIV-1 CA-NC complexes.
To test the ability of cellular extracts to stabilize _in vitro_-assembled HIV-1 CA-NC complexes, we incubated HIV-1 CA-NC complexes in the DB buffer supplemented with cellular extracts from 293T cells. As shown in Fig. 2A, DB containing ∼2.5 mg/ml of protein extracts from 293T cells stabilized HIV-1 CA-NC complexes. These results suggested that cellular extracts stabilized HIV-1 CA-NC complexes. Interestingly, heat inactivation of cellular extracts for 30 min at 100°C destroyed their ability to stabilize HIV-1 CA-NC complexes. Heat inactivation of cellular extracts at 50°C or 70°C for 30 min was less effective in eliminating their ability to stabilize HIV-1 CA-NC complexes (data not shown). In agreement with these results, treatment of cellular extracts with 0.1 mg/ml of trypsin decreased their ability to stabilize HIV-1 CA-NC complexes to a minor extent compared to heat inactivation.
Fig 2.

Cellular extracts stabilize _in vitro_-assembled HIV-1 CA-NC complexes. (A) HIV-1 CA-NC complexes were incubated in DB supplemented or not with cellular extracts for 1 h, and the mixture was layered onto a 70% sucrose cushion as described in Materials and Methods. A fraction of the sample was saved as input. After the cushion was spun at 100,000 × g for 1 h, the pellet was resuspended and used for further analysis. Input and pellet samples were analyzed by Western blotting using anti-p24 antibodies. Where indicated, cellular extracts were heat inactivated at 100°C for 30 min or treated with trypsin at a final concentration of 0.1 mg/ml. As a control, HIV-1 CA-NC complexes were incubated in SB. (B) HIV-1 CA-NC complexes were also incubated with cellular extracts pretreated with the nuclease Benzonase. After the mixture was layered onto a 70% sucrose cushion, the pellet was resuspended and used for further analysis. Input and pellet samples were analyzed by Western blotting using anti-p24 antibodies. (C) Similarly, HIV-1 CA-NC complexes were incubated in destabilization buffer supplemented or not with cellular extracts for 1 h at room temperature. Subsequently, samples were layered onto a 40 to 80% sucrose gradient and centrifuged at 100,000 × g for 45 min as described in Materials and Methods. After centrifugation, the different fractions from the gradient were analyzed by Western blotting using anti-p24 antibodies. As a control, a gradient analysis was also performed on HIV-1 CA-NC complexes incubated in stabilization buffer for 1 h at room temperature. (D) Protein concentrations for cell extracts in destabilization buffer under different conditions as described above. Experiments were repeated at least three times, and fluorescence quantification of the percentage of pelleted CA-NC with respect to input is shown with standard deviations.
Next, we explored the possibility that nucleic acids from the extracts stabilize HIV-1 CA-NC complexes. For this purpose, we treated cellular extracts with 100 U/ml of the nuclease Benzonase (Novagen), which digests DNA and RNA. As shown in Fig. 2B, treatment with Benzonase did not affect the ability of cellular extracts to stabilize HIV-1 CA-NC complexes.
To further characterize the stabilization properties of cellular extracts, we separated the HIV-1 CA-NC complexes using a 40 to 80% sucrose gradient (Fig. 2C). Similarly, HIV-1 CA-NC complexes were stabilized in the presence of DB supplemented with cellular extracts. Treatment of cellular extracts by heat or trypsin did not affect the overall protein concentrations (Fig. 2D). Taken together, these results suggested that cellular extracts contain factors that stabilized the HIV-1 core.
Stabilization of HIV-1 CA-NC complexes by cellular extracts could be due to a specific activity in the cellular extracts or to macromolecular crowding (32, 33). To distinguish between these two possibilities, we studied the ability of DB buffer supplemented with 3 mg/ml of BSA to stabilize HIV-1 CA-NC complexes. As shown in Fig. 3A, DB buffer supplemented with 3 mg/ml of BSA showed a minor contribution to the stability of HIV-1 CA-NC complexes compared to cellular extracts. Cellular extracts at 3 mg/ml of total protein showed a more potent stabilization effect than BSA (Fig. 3A and B). These results suggested that the stabilizing effects of cellular extracts are the result of specific factors and macromolecular crowding.
Fig 3.

Effect of macromolecular crowding on the stability of _in vitro_-assembled HIV-1 CA-NC complexes. (A) HIV-1 CA-NC complexes were incubated for 1 h in destabilization buffer supplemented with a final concentration of 3 mg/ml of cellular extracts or BSA. A fraction of each sample was stored as input. Samples were layered onto a 70% sucrose cushion and centrifuged at 100,000 × g for 1 h. The pellets were resuspended and used for further testing. Input and pellet were analyzed by Western blotting using anti-p24 antibodies. As a control, the stability of HIV-1 CA-NC complexes was measured in stabilization buffer. (B) Protein concentrations for destabilization buffer supplemented with cell extracts and BSA. (C) Similarly, the stability of HIV-1 CA-NC complexes was studied in destabilization buffer supplemented with increasing concentrations of BSA. As a control, the stability of HIV-1 CA-NC complexes incubated in stabilization buffer was measured. (D) HIV-1 CA-NC complexes were incubated in DB supplemented with a final concentration of 3 mg/ml cellular extracts or 10 mg/ml BSA for 1 h at room temperature. A fraction of each sample was stored as input. Subsequently, samples were layered onto a 40 to 80% sucrose gradient and centrifuged at 100,000 × g for 45 min as described in Materials and Methods. After centrifugation, the different fractions from the gradient were analyzed by Western blotting using anti-p24 antibodies. As a control, the stability of HIV-1 CA-NC complexes incubated in SB was measured. (E) Stability of HIV-1 CA-NC complexes in destabilization buffer supplemented with increasing concentrations of the crowding agent PEG 8000. As a control, the stability of HIV-1 CA-NC complexes incubated in stabilization buffer was measured. Experiments were repeated at least three times, and fluorescence quantification of the percentage of pelleted CA-NC with respect to input is shown with standard deviations.
To further characterize the macromolecular-crowding effect, we studied the stability of HIV-1 CA-NC complexes on DB supplemented with increasing concentrations of BSA (Fig. 3C). Increasing concentrations of BSA strongly stabilized the HIV-1 CA-NC complexes, suggesting that protein-crowding effects play an important role in the stability of HIV-1 CA-NC. We also explored the effect of BSA using a sucrose gradient (Fig. 3D). These results showed that 10 mg/ml BSA strongly stabilized the HIV-1 CA-NC complexes, comparable to the stability provided by the SB. In addition, the crowding effect was also tested using PEG 8000. As shown in Fig. 3E, DB supplemented with increasing amounts of PEG 8000 stabilized the HIV-1 CA-NC complexes, suggesting that macromolecular crowding increases the stability of the HIV-1 core in the cytosol.
Cyclophilin A decreases the stability of HIV-1 CA-NC complexes in vitro.
To test the ability of cyclophilin A to affect the stability of the _in vitro_-assembled HIV-1 CA-NC complexes, we incubated HIV-1 CA-NC complexes in DB buffer supplemented with cellular extracts using increasing amounts of purified cyclophilin A (Fig. 4A). Interestingly, cyclophilin A destabilized HIV-1 CA-NC complexes, and the effect could be prevented by the use of 10 μM cyclosporine (Fig. 4A). These experiments suggested that the binding of cyclophilin A to capsid destabilizes HIV-1 CA-NC complexes. As a control, we performed the same experiments in DB without cellular extracts and showed the inability of recombinant Cyp A to stabilize HIV-1 CA-NC complexes (Fig. 4B).
Fig 4.

Cyclophilin A decreases the stability of HIV-1 CA-NC complexes. (A and B) The stability of HIV-1 CA-NC complexes in destabilization buffer supplemented (A) or not (B) with cell extracts using increasing amounts of the protein cyclophilin A was measured. Similar experiments were performed in the presence of cyclosporine. As a control, the stability of HIV-1 CA-NC complexes incubated in stabilization buffer was measured. (C and D) Similarly, the stability of HIV-1 CA-NC complexes in destabilization buffer supplemented (C) or not (D) with cell extracts using increasing amounts of cyclosporine was measured. As a control, the stability of HIV-1 CA-NC complexes incubated in stabilization buffer was measured. Experiments were repeated at least three times, and fluorescence quantification of the percentage of pelleted CA-NC with respect to input is shown with standard deviations.
Because cyclophilin A is in cellular extracts, we tested the stability of HIV-1 CA-NC complexes in cellular extracts using increasing concentrations of cyclosporine (Fig. 4C). Our results suggested that cyclosporine does not have an impact on the ability of cell extracts to stabilize HIV-1 CA-NC complexes. As a control, we also tested whether cyclosporine increases the stability of HIV-1 CA-NC complexes in DB alone. These results showed a very small increase in stability when cyclosporine was used at increasing concentrations, which might be due to a crowding effect (Fig. 4D).
The peptide CAI decreases the stability of HIV-1 CA-NC complexes.
Previous work demonstrated that the peptide CAI is an inhibitor of HIV-1 assembly in vitro (34); however, the effect of CAI on the stability of HIV-1 CA-NC complexes has not been studied, and therefore, we explored it. For this purpose, we tested the stability of HIV-1 CA-NC complexes in DB buffer supplemented with cellular extracts using increasing amounts of the CAI peptide. Interestingly, CAI destabilized the HIV-1 CA-NC complexes (Fig. 5A). In contrast, the CAI peptide with a randomized sequence (CAIctrl) did not show an effect on the stability of HIV-1 CA-NC complexes (Fig. 5A). We also tested the ability of CAI to increase the stability of HIV-1 CA-NC complexes in DB buffer alone. In agreement with the ability of CAI to decrease stability, we found that CAI caused minimal or no increase in the stability of HIV-1 CA-NC complexes in DB alone, which might be due to a crowding effect (Fig. 5B).
Fig 5.
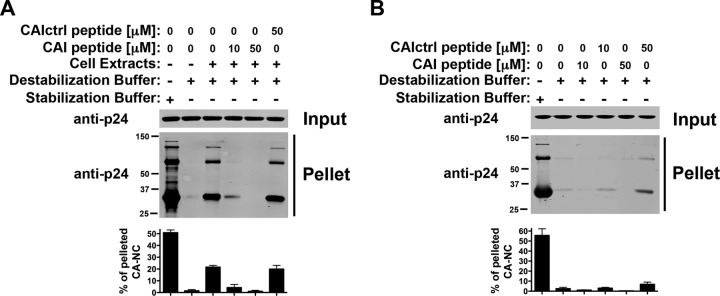
The CAI peptide decreases the stability of HIV-1 CA-NC complexes. The stability of HIV-1 CA-NC complexes in destabilization buffer supplemented (A) or not (B) with cellular extracts using increasing concentrations of the peptide CAI was measured. As a control, the stability of HIV-1 CA-NC complexes incubated in stabilization buffer was measured. Experiments were repeated at least three times, and fluorescence quantification of the percentage of pelleted CA-NC with respect to input is shown with standard deviations.
The small molecule PF74 increases the stability of HIV-1 CA-NC complexes.
The small molecule PF74 decreases the stability of the HIV-1 core (28), which is composed of hexamers and pentamers (35). Here, we tested whether PF74 can modulate the stability of HIV-1 CA-NC complexes, which are formed only of hexamers (29). Interestingly, PF74 increases the stability of HIV-1 CA-NC complexes in a concentration-dependent manner (Fig. 6A). In agreement with this, PF74 further increased the stability of HIV-1 CA-NC complexes already stabilized by cell extracts (Fig. 6B). These results suggested that PF74 stabilizes HIV-1 CA-NC complexes.
Fig 6.
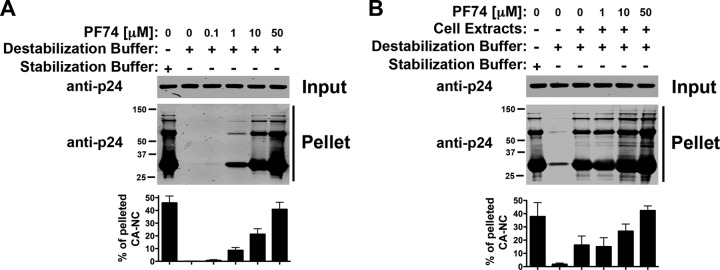
The HIV-1 inhibitor PF74 increases the stability of HIV-1 CA-NC complexes. The stability of HIV-1 CA-NC complexes in destabilization buffer without (A) or with (B) cellular extracts in the presence of increasing concentrations of PF74 was measured. As a control, the stability of HIV-1 CA-NC complexes incubated in stabilization buffer was measured. Experiments were repeated at least three times, and fluorescence quantification of the percentage of pelleted CA-NC with respect to input is shown with standard deviations.
The small molecule Bi2 (also known as BI-2) increases the stability of HIV-1 CA-NC complexes.
We tested the ability of the HIV-1 inhibitor Bi2 (36, 48), which also targets the HIV-1 capsid, to affect the stability of HIV-1 CA-NC complexes. For this purpose, we tested the stability of HIV-1 CA-NC complexes in DB buffer using increasing concentrations of Bi2. As shown in Fig. 7A, Bi2 increases the stability of HIV-1 CA-NC complexes in a concentration-dependent manner in DB alone. Similar results were observed when Bi2 was tested in DB supplemented with cellular extracts (Fig. 7B). These results suggested that Bi2 increases the stability of HIV-1 CA-NC complexes.
Fig 7.
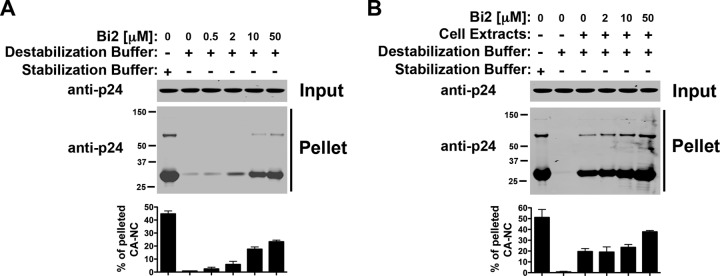
The HIV-1 inhibitor Bi2 (also known as BI-2) increases the stability of HIV-1 CA-NC complexes. Shown is the stability of HIV-1 CA-NC complexes in destabilization buffer without (A) or with (B) cellular extracts using increasing concentrations of Bi2. As a control, the stability of HIV-1 CA-NC complexes incubated in stabilization buffer was measured. Experiments were repeated at least three times, and fluorescence quantification of the percentage of pelleted CA-NC with respect to input is shown with standard deviations.
I-XW-053 and CAP-1 did not show an effect on the stability of HIV-1 CA-NC complexes.
The recently described HIV-1 capsid interactor I-XW-053 blocks HIV-1 infection (37). To understand the mechanism by which I-XW-053 blocks HIV-1 infection, we tested whether I-XW-053 influences the stability of HIV-1 CA-NC complexes in cellular extracts. I-XW-053 showed no effect on the stability of HIV-1 CA-NC complexes in DB supplemented with cellular extracts (data not shown). As a control, we also showed that I-XW-053 has no effect on HIV-1 CA-NC complexes when tested in DB alone (data not shown).
We also explored the ability of the HIV-1 capsid inhibitor CAP-1 to influence the stability of HIV-1 CA-NC complexes in DB supplemented with cellular extracts (references 38 and 39 and data not shown). As a control, we tested the effect of CAP-1 on the stability of HIV-1 CA-NC complexes in DB alone (data not shown). Our results suggested that CAP-1 does not affect the stability of HIV-1 CA-NC complexes in vitro.
Stability of _in vitro_-assembled HIV-1 CA-NC complexes bearing different capsid mutations.
To determine whether the assay can measure intrinsic stability defects in different capsid mutants, we measured the stability of HIV-1 CA-NC complexes bearing different mutants. We initially analyzed the stability of the capsid variant 5Mut (Q67H, K70R, H87P, T107N, and L111I), which has been shown to exhibit greater stability (28). For this purpose, we tested the stability of HIV-1 CA-NC complexes bearing the 5Mut changes (Fig. 8A). Interestingly, the 5Mut HIV-1 CA-NC complexes in DB were as stable as in SB. These results showed the great intrinsic stability of 5Mut, as previously suggested by the fate-of-the-capsid assay (28).
Fig 8.

Stability of _in vitro_-assembled HIV-1 CA-NC complexes bearing different capsid mutations. (A) The stability of HIV-1 CA-NC complexes bearing the 5Mut changes was measured in DB and SB. As a control, the stability of wild-type HIV-1 CA-NC complexes was measured. (B) The stability of HIV-1 CA-NC complexes bearing the 5Mut changes was analyzed in the presence of 10 μM PF74. (C and D) Similarly, the stability of HIV-1 CA-NC complexes bearing the T107N (C) and N74D (D) mutations in DB and SB was measured. As a control, the stability of HIV-1 CA-NC complexes incubated in SB was measured. Experiments were repeated at least three times, and fluorescence quantification of the percentage of pelleted CA-NC with respect to input is shown with standard deviations.
Previous observations had shown that 5Mut changes in capsid results in an HIV-1 strain insensitive to the drug PF74 (28, 40). For this reason, we tested the ability of PF74 to destabilize HIV-1 CA-NC complexes bearing the 5Mut changes. As shown in Fig. 8B, the use of 10 μM PF74 did not further increase or decrease the stability of HIV-1 CA-NC complexes bearing the 5Mut changes when incubated in DB or SB. We also tested the stability of the T107N capsid mutant. We found that the T107N capsid variant is more stable than the wild type (Fig. 8C).
We also tested the stability of the N74D capsid variant. As shown in Fig. 8D, N74D was similar to wild type HIV-1 CA-NC complexes in terms of stability. These results indicated that N74D intrinsic stability is not changed.
Ability of CPSF6 to stabilize HIV-1 CA-NC complexes.
Previous observations have demonstrated that CPSF6 expressed in the cytosol of cells blocks HIV-1 infection and dramatically increases the stability of the HIV-1 core during infection, as measured by the fate-of-the-capsid assay (12, 22, 41). To explore the ability of CPSF6 to stabilize HIV-1 CA-NC complexes, we expressed and purified a fragment of CPSF6(1-321) from insect cells (Fig. 9A). The CPSF6(1-321) fragment was the result of a screen for soluble and well-expressed constructs in insect cells with the ability to bind _in vitro_-assembled HIV-1 CA-NC complexes. Next, we tested the ability of CPSF6(1-321) to stabilized HIV-1 CA-NC complexes. As shown in Fig. 9B, the use of increasing amounts of CPSF6(1-321) increased the stability of HIV-1 CA-NC complexes in DB buffer. These results demonstrated that purified CPSF6 stabilizes HIV-1 CA-NC complexes, in agreement with the results showing that CPSF6 stabilizes the HIV-1 core during infection (22, 41).
Fig 9.
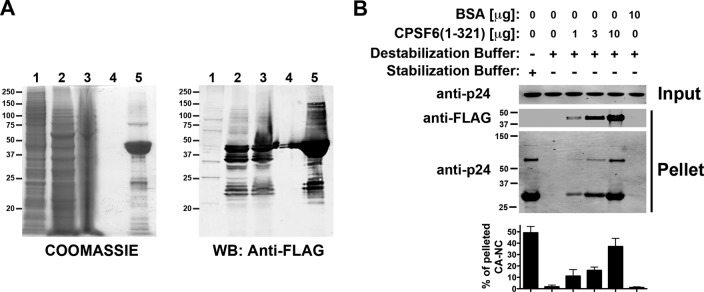
Stabilization of HIV-1 CA-NC complexes by CPSF6(1-321). (A) Coomassie blue-stained SDS-PAGE and Western blot (WB) showing the stepwise purification of recombinant CPSF(1-321) protein from baculovirus-infected Sf9 insect cells. Lanes 1, soluble lysate from control Sf9 cells; lanes 2, soluble lysate from Sf9 cells infected with baculovirus expressing CPSF6(1-321); lanes 3, soluble lysate after filtration (0.45 μm); lanes 4, final wash before elution; lanes 5, elution of CPSF6(1-321) with 2.5 mM d-desthiobiotin. (B) The stability of HIV-1 CA-NC complexes in destabilization buffer in the presence of increasing concentrations of CPSF6(1-321) was measured. As a control, the stability of HIV-1 CA-NC complexes incubated in destabilization buffer was measured in the presence of 10 μg of BSA. Experiments were repeated at least three times, and fluorescence quantification of the percentage of pelleted CA-NC with respect to input is shown with standard deviations.
DISCUSSION
Here, we created a simple and fast method to test HIV-1 CA-NC complex stability in vitro. Using this novel assay, we have demonstrated that cellular extracts contain stabilizing properties. Interestingly, heat inactivation or trypsinization of cellular extracts eliminated the ability of the extracts to stabilize HIV-1 CA-NC complexes. These results suggested that cellular extracts contain a factor(s) with the ability to stabilize HIV-1 CA-NC complexes in vitro. This assay could be instrumental in the identification of cellular factors that stabilize the HIV-1 core or assist the uncoating process of HIV-1.
At the same time, we explored the role of macromolecular crowding in HIV-1 CA-NC stabilization. The cellular cytosol contains a high total concentration of different proteins, so it is known as “crowded” rather than “concentrated.” Because macromolecular crowding has been shown to affect the properties of proteins (32, 33), we tested whether crowding affects the stability of HIV-1 CA-NC complexes in vitro. By using increasing concentrations of BSA and polyethylene glycol, we demonstrated that macromolecular crowding increases the stability of HIV-1 CA-NC complexes. These results suggested that macromolecular crowding plays an important role in stabilizing the HIV-1 core in the cytosol in vivo.
We also explored the role of cyclophilin A in the stability of HIV-1 CA-NC complexes. Cyclophilin A is a cytosolic protein that exhibits isoprolyl isomerase activity and interacts with the HIV-1 capsid protein (42–46). Cyclophilin A is believed to modulate the disassembly process of the HIV-1 core (47). To directly test whether cyclophilin A modulates the stability of HIV-1 CA-NC complexes, we treated HIV-1 CA-NC complexes with increasing amounts of purified recombinant cyclophilin A. The presence of cyclophilin A destabilized HIV-1 CA-NC complexes in vitro in a cyclosporine-dependent manner. These results suggested that the binding of cyclophilin A to capsid and/or its enzymatic activity destabilizes HIV-1 CA-NC complexes. This work showed for the first time the ability of cyclophilin A to destabilize HIV-1 CA-NC complexes in vitro.
The physiological relevance of the in vitro ability of cyclophilin A to destabilize HIV-1 CA-NC complexes is not straightforward, since the use of cyclosporine in cellular extracts did not increase the stability of HIV-1 CA-NC complexes. One possibility is that cyclophilin A might play a role in facilitating uncoating in vivo; however, this effect is difficult to recapitulate in cell extracts, since the cellular structures have been perturbed.
The peptide CAI decreased the stability of HIV-1 CA-NC complexes. These results are in agreement with the notion that CAI is an assembly inhibitor (34). Since CAI binds to residues 169 to 191 of capsid, our results suggested that these residues are important for the stability of _in vitro_-assembled HIV-1 CA-NC complexes.
The capsid-targeting drug PF74, which inhibits infection before reverse transcription (28), increased the stability of HIV-1 CA-NC complexes in vitro. However, the fact that PF74 decreases the stability of the HIV-1 capsid measured by the fate-of-the-capsid assay suggested the existence of a difference (28). Unlike the fate-of-the-capsid assay, which measures the stability of cores, which are composed of capsid hexamers and pentamers (35), our assay measures the stability of HIV-1 CA-NC complexes, which are mainly composed of hexamers. Our results suggested that PF74 stabilizes hexameric capsids. The ability of PF74 to stabilize HIV-1 CA-NC complexes is in agreement with previous findings suggesting that PF74 increases the rate of hexameric capsid assembly (40). Future experiments will attempt to discover the mechanism by which PF74 stabilizes the HIV-1 capsid hexameric array.
We also found that the recently discovered HIV-1 infection inhibitor Bi2 stabilizes _in vitro_-assembled HIV-1 CA-NC complexes, similar to PF74. These results suggested that Bi2 inhibits infection by stabilization of the HIV-1 core.
Furthermore, we tested whether our assay can measure the stability of capsid variants, such as 5Mut, which is known for its increased stability compared to the wild type (28). In agreement with this, 5Mut showed increased stability in our assay. Interestingly, the CA-NC T107N variant also showed increased stability. In contrast, the CA-NC N74D variant showed the same stability as the wild type.
Recent reports have suggested that expression of a cytosolic form of CPSF6 dramatically increases the stability of the HIV-1 core during infection, as measured by the fate-of-the-capsid assay (22, 41). Here, we demonstrated that a purified fragment of CPSF6 stabilizes HIV-1 CA-NC complexes in vitro at concentrations of 20 μg/ml. This unique reagent could facilitate the stabilization of cores isolated from infected cells and help the search for novel factors that bind the HIV-1 core.
Overall, this work represents the development of an assay to measure the stability of HIV-1 CA-NC complexes and allowed us to demonstrate that cellular extracts contain a capsid-stabilizing factor. We believe this novel assay might be instrumental in future experiments attempting to isolate factors that interact with the HIV-1 core.
ACKNOWLEDGMENTS
We thank Chris Aiken and Owen Pornillos for providing reagents. We are grateful to the NIH/AIDS reagents repository program.
This work was supported by an NIH R01 award AI087390 to F.D.-G. X.W. and A.B.S. III were supported by a PO-1 grant (GM-56550).
Footnotes
Published ahead of print 24 July 2013
REFERENCES
- 1.Suzuki Y, Craigie R. 2007. The road to chromatin: nuclear entry of retroviruses. Nat. Rev. Microbiol. 5:187–196 [DOI] [PubMed] [Google Scholar]
- 2.Li L, Olvera JM, Yoder KE, Mitchell RS, Butler SL, Lieber M, Martin SL, Bushman FD. 2001. Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. EMBO J. 20:3272–3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butler SL, Hansen MS, Bushman FD. 2001. A quantitative assay for HIV DNA integration in vivo. Nat. Med. 7:631–634 [DOI] [PubMed] [Google Scholar]
- 4.Briggs JA, Simon MN, Gross I, Krausslich HG, Fuller SD, Vogt VM, Johnson MC. 2004. The stoichiometry of Gag protein in HIV-1. Nat. Struct. Mol. Biol. 11:672–675 [DOI] [PubMed] [Google Scholar]
- 5.Briggs JA, Wilk T, Welker R, Krausslich HG, Fuller SD. 2003. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 22:1707–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lanman J, Lam TT, Emmett MR, Marshall AG, Sakalian M, Prevelige PE., Jr 2004. Key interactions in HIV-1 maturation identified by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 11:676–677 [DOI] [PubMed] [Google Scholar]
- 7.Fassati A, Goff SP. 2001. Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1. J. Virol. 75:3626–3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDonald D, Vodicka MA, Lucero G, Svitkina TM, Borisy GG, Emerman M, Hope TJ. 2002. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 159:441–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arfi V, Lienard J, Nguyen XN, Berger G, Rigal D, Darlix JL, Cimarelli A. 2009. Characterization of the behavior of functional viral genomes during the early steps of human immunodeficiency virus type 1 infection. J. Virol. 83:7524–7535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diaz-Griffero F, Kar A, Lee M, Stremlau M, Poeschla E, Sodroski J. 2007. Comparative requirements for the restriction of retrovirus infection by TRIM5alpha and TRIMCyp. Virology 369:400–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hulme AE, Perez O, Hope TJ. 2011. Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proc. Natl. Acad. Sci. U. S. A. 108:9975–9980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roa A, Hayashi F, Yang Y, Lienlaf M, Zhou J, Shi J, Watanabe S, Kigawa T, Yokoyama S, Aiken C, Diaz-Griffero F. 2012. RING domain mutations uncouple TRIM5alpha restriction of HIV-1 from inhibition of reverse transcription and acceleration of uncoating. J. Virol. 86:1717–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, Sodroski J. 2006. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. U. S. A. 103:5514–5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diaz-Griffero F. 2012. The role of TNPO3 in HIV-1 replication. Mol. Biol. Int. 2012:868597. 10.1155/2012/868597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamashita M, Emerman M. 2004. Capsid is a dominant determinant of retrovirus infectivity in nondividing cells. J. Virol. 78:5670–5678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamashita M, Emerman M. 2006. Retroviral infection of non-dividing cells: old and new perspectives. Virology 344:88–93 [DOI] [PubMed] [Google Scholar]
- 17.Yamashita M, Perez O, Hope TJ, Emerman M. 2007. Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS Pathog. 3:1502–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forshey BM, von Schwedler U, Sundquist WI, Aiken C. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 76:5667–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohagen A, Gabuzda D. 2000. Role of Vif in stability of the human immunodeficiency virus type 1 core. J. Virol. 74:11055–11066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Y, Fricke T, Diaz-Griffero F. 2013. Inhibition of reverse transcriptase activity increases stability of the HIV-1 core. J. Virol. 87:683–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berube J, Bouchard A, Berthoux L. 2007. Both TRIM5alpha and TRIMCyp have only weak antiviral activity in canine D17 cells. Retrovirology 4:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Iaco A, Santoni F, Vannier A, Guipponi M, Antonarakis S, Luban J. 2013. TNPO3 protects HIV-1 replication from CPSF6-mediated capsid stabilization in the host cell cytoplasm. Retrovirology 10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diaz-Griffero F, Kar A, Perron M, Xiang SH, Javanbakht H, Li X, Sodroski J. 2007. Modulation of retroviral restriction and proteasome inhibitor-resistant turnover by changes in the TRIM5alpha B-box 2 domain. J. Virol. 81:10362–10378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diaz-Griffero F, Perron M, McGee-Estrada K, Hanna R, Maillard PV, Trono D, Sodroski J. 2008. A human TRIM5alpha B30.2/SPRY domain mutant gains the ability to restrict and prematurely uncoat B-tropic murine leukemia virus. Virology 378:233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forshey BM, Shi J, Aiken C. 2005. Structural requirements for recognition of the human immunodeficiency virus type 1 core during host restriction in owl monkey cells. J. Virol. 79:869–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohkura S, Goldstone DC, Yap MW, Holden-Dye K, Taylor IA, Stoye JP. 2011. Novel escape mutants suggest an extensive TRIM5alpha binding site spanning the entire outer surface of the murine leukemia virus capsid protein. PLoS Pathog. 7:e1002011. 10.1371/journal.ppat.1002011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perron MJ, Stremlau M, Lee M, Javanbakht H, Song B, Sodroski J. 2007. The human TRIM5alpha restriction factor mediates accelerated uncoating of the N-tropic murine leukemia virus capsid. J. Virol. 81:2138–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi J, Zhou J, Shah VB, Aiken C, Whitby K. 2011. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J. Virol. 85:542–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. 1999. Assembly and analysis of conical models for the HIV-1 core. Science 283:80–83 [DOI] [PubMed] [Google Scholar]
- 30.Brandariz-Nunez A, Menaya-Vargas R, Benavente J, Martinez-Costas J. 2010. A versatile molecular tagging method for targeting proteins to avian reovirus muNS inclusions. Use in protein immobilization and purification. PLoS One 5:e13961. 10.1371/journal.pone.0013961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.White TE, Brandariz-Nunez A, Carlos Valle-Casuso J, Amie S, Nguyen L, Kim B, Brojatsch J, Diaz-Griffero F. 2013. Contribution of SAM and HD domains to retroviral restriction mediated by human SAMHD1. Virology 436:81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ellis RJ. 2001. Macromolecular crowding: an important but neglected aspect of the intracellular environment. Curr. Opin. Struct. Biol. 11:114–119 [DOI] [PubMed] [Google Scholar]
- 33.Ellis RJ. 2001. Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 26:597–604 [DOI] [PubMed] [Google Scholar]
- 34.Sticht J, Humbert M, Findlow S, Bodem J, Muller B, Dietrich U, Werner J, Krausslich HG. 2005. A peptide inhibitor of HIV-1 assembly in vitro. Nat. Struct. Mol. Biol. 12:671–677 [DOI] [PubMed] [Google Scholar]
- 35.Ganser-Pornillos BK, Cheng A, Yeager M. 2007. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 131:70–79 [DOI] [PubMed] [Google Scholar]
- 36.Lepage O, Bhardwaj P, Faucher A, Grand-Maître C, Lacoste J, Lamorte L, Mercier J. 24 November 2011. Inhibitors of HIV replication. US patent WO 2011/143772 A1
- 37.Kortagere S, Madani N, Mankowski MK, Schon A, Zentner I, Swaminathan G, Princiotto A, Anthony K, Oza A, Sierra LJ, Passic SR, Wang X, Jones DM, Stavale E, Krebs FC, Martin-Garcia J, Freire E, Ptak RG, Sodroski J, Cocklin S, Smith AB., III 2012. Inhibiting early-stage events in HIV-1 replication by small-molecule targeting of the HIV-1 capsid. J. Virol. 86:8472–8481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelly BN, Kyere S, Kinde I, Tang C, Howard BR, Robinson H, Sundquist WI, Summers MF, Hill CP. 2007. Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J. Mol. Biol. 373:355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang C, Loeliger E, Kinde I, Kyere S, Mayo K, Barklis E, Sun Y, Huang M, Summers MF. 2003. Antiviral inhibition of the HIV-1 capsid protein. J. Mol. Biol. 327:1013–1020 [DOI] [PubMed] [Google Scholar]
- 40.Blair WS, Pickford C, Irving SL, Brown DG, Anderson M, Bazin R, Cao J, Ciaramella G, Isaacson J, Jackson L, Hunt R, Kjerrstrom A, Nieman JA, Patick AK, Perros M, Scott AD, Whitby K, Wu H, Butler SL. 2010. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 6:e1001220. 10.1371/journal.ppat.1001220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fricke T, Valle-Casuso JC, White TE, Brandariz-Nunez A, Bosche WJ, Reszka N, Gorelick R, Diaz-Griffero F. 2013. The ability of TNPO3-depleted cells to inhibit HIV-1 infection requires CPSF6. Retrovirology 10:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J, Tan Z, Tang S, Hewlett I, Pang R, He M, He S, Tian B, Chen K, Yang M. 2009. Discovery of dual inhibitors targeting both HIV-1 capsid and human cyclophilin A to inhibit the assembly and uncoating of the viral capsid. Bioorg. Med. Chem. 17:3177–3188 [DOI] [PubMed] [Google Scholar]
- 43.Luban J. 2007. Cyclophilin A, TRIM5, and resistance to human immunodeficiency virus type 1 infection. J. Virol. 81:1054–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP. 1993. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73:1067–1078 [DOI] [PubMed] [Google Scholar]
- 45.Rits MA, van Dort KA, Kootstra NA. 2008. Polymorphisms in the regulatory region of the cyclophilin A gene influence the susceptibility for HIV-1 infection. PLoS One 3:e3975. 10.1371/journal.pone.0003975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sokolskaja E, Luban J. 2006. Cyclophilin, TRIM5, and innate immunity to HIV-1. Curr. Opin. Microbiol. 9:404–408 [DOI] [PubMed] [Google Scholar]
- 47.Li Y, Kar AK, Sodroski J. 2009. Target cell type-dependent modulation of human immunodeficiency virus type 1 capsid disassembly by cyclophilin A. J. Virol. 83:10951–10962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lamorte L, Titolo S, Lemke CT, Goudreau N, Mercier J-F, Wardrop E, Shah VB, von Schwedler UK, Langelier C, Banik SS, Aiken C, Sundquist WI, Mason SW. Discovery of novel small-molecule HIV-1 replication inhibitors that stabilize capsid complexes. Antimicrob. Agents Chemother., in press [DOI] [PMC free article] [PubMed] [Google Scholar]