Functional consequences of prolactin signalling in endothelial cells: a potential link with angiogenesis in pathophysiology? (original) (raw)
Abstract
Prolactin is best known as the polypeptide anterior pituitary hormone, which regulates the development of the mammary gland. However, it became clear over the last decade that prolactin contributes to a broad range of pathologies, including breast cancer. Prolactin is also involved in angiogenesis via the release of pro-angiogenic factors by leukocytes and epithelial cells. However, whether prolactin also influences endothelial cells, and whether there are functional consequences of prolactin-induced signalling in the perspective of angiogenesis, remains so far elusive. In the present study, we show that prolactin induces phosphorylation of ERK1/2 and STAT5 and induces tube formation of endothelial cells on Matrigel. These effects are blocked by a specific prolactin receptor antagonist, del1-9-G129R-hPRL. Moreover, in an in vivo model of the chorioallantoic membrane of the chicken embryo, prolactin enhances vessel density and the tortuosity of the vasculature and pillar formation, which are hallmarks of intussusceptive angiogenesis. Interestingly, while prolactin has only little effect on endothelial cell proliferation, it markedly stimulates endothelial cell migration. Again, migration was reverted by del1-9-G129R-hPRL, indicating a direct effect of prolactin on its receptor. Immunohistochemistry and spectral imaging revealed that the prolactin receptor is present in the microvasculature of human breast carcinoma tissue. Altogether, these results suggest that prolactin may directly stimulate angiogenesis, which could be one of the mechanisms by which prolactin contributes to breast cancer progression, thereby providing a potential tool for intervention.
Keywords: prolactin, prolactin receptor, endothelial cells, angiogenesis, pathophysiology
Introduction
Prolactin is best known as the polypeptide anterior pituitary hormone, which regulates the development of the mammary gland throughout female reproductive life. During pregnancy, prolactin stimulates the expansion and differentiation of the lobuloalveolar system. After delivery, prolactin induces the final induction of milk protein gene expression and lactation [1]. Together with growth hormone and placental lactogen, prolactin forms a family of hormones that probably resulted from the duplication of one ancestral gene [2]. Besides this very well-known role in lactation, prolactin is also produced in many other healthy as well as diseased tissues, such as the mammary gland, prostate, skin, decidua, adipocytes and some immune cells, which all express the prolactin receptor [3]. Based on the almost ubiquitous distribution of prolactin receptors, and an ever-growing list of extrapituitary prolactin-expressing tissues, a wide array of functions beyond lactation has been documented or claimed for prolactin. In illustration, prolactin has been shown to regulate the immune responses [2, 4, 5], indicated, for instance, by marked proliferation of the pre-T lymphoma Nb2 cells in response to prolactin [6].
Recent evidence also suggests that prolactin may be involved in cancer progression. Large epidemiological prospective studies, conducted in patients with breast cancer, showed that high prolactin levels prior to treatment are associated with treatment failure, earlier recurrence and worse overall survival [7]. In addition, recent genetic data imply that polymorphisms in prolactin and the prolactin receptor genes may contribute to the development of breast adenomas and cancer [8–10]. These results are further supported by in vivo studies. Prolactin has the ability to induce tumour growth in an autocrine/paracrine fashion in murine models of prostate and breast cancer, which may help to understand its role in human tumourigenesis [11]. In accordance with these observations, prolactin stimulates the growth and motility of human breast cancer cells _in vitro_[12]. The actions of prolactin are mediated by at least six recognized prolactin receptor isoforms (resulting from mRNA splicing variants) found on human breast epithelium [12]. These different isoforms share an identical extracellular and transmembrane domain [3]. Since the in vivo relevance of the short prolactin receptor isoforms are unclear, the long prolactin receptor isoform is considered the major isoform through which prolactin transmits its signals, although the expression of the isoforms may differ between tissues and may depend on the estrous cycle [2, 3, 13, 14]. The prolactin/prolactin-receptor complex associates with and activates several signalling pathways, such as STAT5 and ERK1/2, that are shared with other members of the cytokine receptor superfamily [12]. Interestingly, over the last decade, human prolactin analogues have been developed that down-regulate the effects of either local prolactin (competitive antagonism) or of the constitutively active receptor variants (inverse agonism) [15]. Prolactin also plays an important role in the dynamic process of angiogenesis. For instance, Erdmann and co-workers elegantly showed that prolactin is involved in the regression of angiogenesis during luteolysis [16], while genetic ablation of the prolactin receptor induces angiogenesis defects in the corpus luteum in mice, during the process of luteal transition [17]. More studies support these findings. Indeed, when rat prolactin cDNA, fused to the cytomegalovirus promoter, was introduced into mouse muscle by direct injection, evidence of marked angiogenesis was found in the testis of these mice [18]. In a late-stage chicken chorioallantoic membrane (CAM) bioassay, prolactin stimulated blood vessel formation [19]. In the above-mentioned studies, the underlying cellular mechanisms of prolactin-induced angiogenesis have not been investigated. Interestingly, prolactin can stimulate the expression of angiogenic factors, such as vascular endothelial growth factor (VEGF), by the epithelium, macrophages and leukocytes [20, 21]. Moreover, the prolactin receptor is expressed in the endothelium of the pulmonary artery, aorta, corpus luteum and umbilical vein from bovine origin [22, 23]. Therefore, it was previously postulated that the angiogenic effects of prolactin may be mediated through a direct or an indirect effect (or both) on endothelial cells [24]. Against this background, we aimed to investigate the functional consequences of prolactin receptor signalling in endothelial cells with regard to angiogenesis, in the setting of breast cancer.
Materials and methods
Cells and culture
The murine endothelial cell line 2H11 was purchased from American Type Culture Collection (ATCC; Manassas, VA, USA), and maintained in Dulbecco's minimal essential media (DMEM; Lonza, Basel, Switzerland) supplemented with glutamate, 1% penicillin/streptomycin and 10% foetal calf serum (FCS). This cell line has been shown to be useful in in vitro angiogenesis assays for evaluating the potential angiogenic properties of novel compounds [25, 26]. Human umbilical vein endothelial cells (HUVEC) were isolated from fresh human umbilical cord veins and maintained in RPMI-1640 (Invitrogen, Carlsbad, CA, USA) supplemented with 10% human serum, 10% FCS, 1% glutamin (Invitrogen), 100 IU/ml penicillin (Sigma-Aldrich, St. Louis, MO, USA) and 100 μg/ml streptomycin (Sigma-Aldrich) as previously described [27].
Human recombinant prolactin and the pure prolactin receptor antagonist del1-9-G129R-hPRL were prepared in the French laboratory as previously described [28]. Both human prolactin and del1-9-G129R are ligands of the human and mouse prolactin receptor [28, 29]. The antagonist was demonstrated to block activity of the long isoforms of mouse and human prolactin receptors, using several in vitro bioassays [28]. It also acts as a pure antagonist in vivo, as was assessed by competitive inhibition of prolactin-triggered signalling cascades in various mouse target tissues known to express the prolactin receptor endogenously, such as liver, mammary gland and prostate [28, 29]. As the extracellular ligand binding of the prolactin receptor is common to all its isoforms, del1-9-G129R is assumed to inhibit actions mediated by short and long isoforms.
Cell stimulation and Western blot
Cells were plated in 24-well plates and maintained in DMEM supplemented with 10% FCS. The next day, cells were washed twice with phosphate-buffered saline, and then serum starved for 8 hrs prior to stimulation. Cells were stimulated for 30 min. with different concentrations of prolactin (10, 100, 500, 1000 and 10,000 μg/l) in serum-free medium at 37°C/5% CO2, with or without the prolactin receptor antagonist del1-9-G129R-hPRL (2000 and 10,000 μg/l). Next, cells were lysed in Laemmli lysis buffer, incubated for 5 min. at 95°C, and whole cell lysates were separated by 10% SDS-PAGE. After electrophoresis, proteins were transferred onto immobilon-P PVDF membranes (Millipore, Billerica, MA, USA). Membranes were incubated overnight at 4°C with primary antibodies against tubulin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), phospho-STAT5 or phospho-ERK1/2 (both Cell Signalling, Beverly, MA, USA) diluted in TBS with 0.1% Tween (TBS-T)/1% BSA. All secondary horseradish peroxydase (HRP)-conjugated antibodies were from DakoCytomation (Glostrup, Denmark). Blots were imaged using Lumilight Plus ECL substrate (Roche, Basel, Switzerland) on a GeneGnome imager (Syngene, Cambridge, UK). Blots were repeated three times.
Chicken chorioallantoic membrane assay (CAM)
Physiologically developing chicken embryo CAM, at embryo development day (EDD) 7 and 8 as previously described in detail [30], were exposed to human prolactin and/or its antagonist (prolactin:antagonist; 1:10 μg/embryo/day). Prolactin was topically applied (20 μl) at a concentration of 1 or 10 μg/embryo/day in 0.9% NaCl either alone or mixed with its receptor antagonist del1-9-G129R-hPRL (10 μg/embryo/day). The control eggs received (each time 20 μl) 0.9% NaCl at the same days. Each condition was included fourfold. At EDD9, the CAMs were evaluated in vivo by means of FITC-dextran (20 kD, 25 mg/ml, Sigma-Aldrich) epi-fluorescence angiography as described previously [30]. For detecting FITC, light was filtered for excitation at 470 ± 20 nm and a long-pass emission filter was used for detection of the fluorescence (λ > 520 nm, Nikon, Japan). Fluorescence images were acquired with an F-view II 12-bit monochrome Peltier-cooled digital CCD camera driven with analySIS DOCU software from Soft Imaging System GmbH (Münster, Germany). Image-processing quantification was performed using ImageJ-based macro as described previously [31] using the following descriptors: the number of branching points/mm2 and the number of segments per mm2 (number of vessels between two branch points). Tortuosity was assessed by visual determination. Experiments were performed two times.
Proliferation assays (MTT and CellTiter-Glo#x00AE; viability assays)
2H11 endothelial cells seeded at a density of 104/cm2 in 96-well cell culture plates in DMEM supplemented with 1% FCS were treated with the indicated concentrations of prolactin (10, 100, 1000 and 10,000 μg/l) or serum-free medium as negative control, for 18 hrs at 37°C/5% CO2. Cell survival was determined 2 hrs after addition of MTT reagent as described before [32]. Proliferation assays with 2H11 were done six times and each condition was performed in octuplicate.
Additionally, HUVEC (5*103 cells/well) were seeded in gelatin-coated 96-well cell culture plates as described previously [33, 34]. Briefly, 24 hrs after seeding, serum-free medium alone (control) or serum-free medium supplemented with a range of prolactin concentrations (10, 100, 1000 and 10,000 μg/l) was added and cells were grown for a further 72 hrs at 37°C/5% CO2. Cell viability was assessed using the CellTiter-Glo#x00AE; Luminescent Cell Viability Assay (Promega, Madison, WI, USA). Proliferation assays with HUVECs were done two times and each condition was performed in duplicate or quintuplicate.
Migration assays
2H11 endothelial cells were grown to 90% confluence in DMEM supplemented with 3% FCS in T25 cell culture flasks and were subsequently incubated with serum-free medium alone (control) or serum-free medium supplemented with prolactin (1000 μg/l), with the antagonist del1-9-G129R-hPRL, or with prolactin (1000 μg/l) in combination with del1-9-G129R-hPRL (20,000 μg/l) for 18 hrs at 37°C/5% CO2. Next, cells were labelled for 1 hr with 10 μmol/l CellTracker Green 5-chloromethylfluorescein diacetate (CMFDA) (Molecular Probes, Eugene, OR, USA) in serum-free medium. The dye was fixed by 1 hr incubation in medium with 10% FCS. Subsequently, cells were washed and detached with a cell scraper, resuspended in serum-free medium until a concentration of 0.5χ10E6 cells/ml and transferred to 8-μm pore size HTS FluoroBlok cell culture inserts (BD Falcon, Franklin Lakes, NJ, USA). Serum-free medium was added to the bottom compartment and cells were allowed to migrate from the upper compartment to the bottom compartment. Cell migration was assessed as described before [32, 35, 36]. Briefly, fluorescence values representing the number of cells on the bottom side of the insert were read during 71 cycles (each cycle comprising four readings spanning 2 min.) at 37°C. The raw fluorescence data were corrected for background fluorescence and fading of the fluorophore. Migration assays with 2H11 were repeated three times and each condition was included three- to sixfold.
The migration capability of HUVECs was measured using wound scratch assays [37]. In brief, HUVECs were grown to confluence in gelatin-coated wells. Scratch wounds were made in the monolayer by removing cells with a sterile scratch tool (Peira Scientific Instruments, Beerse, Belgium). Cells were washed with PBS and the medium was replaced by serum-free medium alone (control) or serum-free medium supplemented with prolactin (1000 μg/l) for 8 hrs at 37°C/5% CO2. Plates were scanned using an Acumen eX3 laser scanner cytometer (TTP LabTech Ltd, Royston, UK) to acquire images for computational analysis of scratch sizes using UGR Scratch Assay 6.2 software (DCI Labs, Peira Scientific Instruments, Beerse, Belgium). Narrowing of the sizes of the scratches is referred to as ‘wound closure’. In this assay, phase-contrast micrographs of six marked points along the wounded area for each condition were compared at t = 0 hr and t = 18 hrs. More specifically, we rated at each time point the wound recovery in control condition, compared it in cells stimulated with prolactin, and assessed the recovery induced by prolactin as percentage of the control condition at t = 18 hrs. Migration assays with HUVECs were repeated three times and each condition was included at least fivefold.
Endothelial tube formation assay
Endothelial tube formation assays were essentially performed as described before [25]. Briefly, cells (5*104/well) in serum-free medium alone (control) or serum-free medium with prolactin (10, 100, 1000 and 10,000 μg/l) and/or del1-9-G129R-hPRL (20,000 μg/l) were resuspended in 0.3 ml of the indicated medium, and plated on 24-well plates pre-coated with growth factor-reduced Matrigel (0.15 ml). After 18 hrs at 37°C/5% CO2, each well was examined for endothelial cells alignment using a phase contrast tube microscope (Carl Zeiss MicroImaging, Inc. Thornwood, NY, USA). As described in a paper by Aranda and Owen [38], a tube is a capillary-like structure, defined by the remodelling of endothelial cells in Matrigel, resulting from the general process of cell elongation and reorganization. We adapted this definition with more stringent criteria and we therefore quantified in each field the number of branching polygons (enclosed structures in Matrigel with internal holes) formed by connected cells. This method is well established and validated [39–42]. For quantification of endothelial tube formation, the number of tubes was counted in each well (divided in five fields) in a blinded fashion. The tube formation observed after incubation with serum-free medium only was set at 100% and used as reference. Tube formation assays were repeated three times.
Immunohistochemistry and spectral imaging
Breast carcinoma tissue [invasive ductal carcinoma, T2N1, oestrogen receptor negative, progesteron receptor negative, Human Epidermal growth factor Receptor 2 (HER2/neu): negative] was retrospectively obtained from the diagnostic archive of the department of pathology (Academic Medical Center Amsterdam, The Netherlands), in accordance with guidelines set out by the ‘Code for Proper Secondary Use of Human Tissue’ of the Dutch Federation of Biomedical Scientific Societies (Federa). Routinely formalin-fixed and paraffin-embedded tissue blocks were cut (4 μm sections), deparaffinized in xylene and rehydrated in graded series of alcohol washings. Endogenous peroxidase activity was blocked with methanol plus 0.3% peroxide for 20 min. at room temperature [43]. Tissue pretreatment with Tris-EDTA pH 9.0 (Thermo Fisher/Labvision, Fremont, CA, USA) was performed in the PTModule (Thermo Fisher Scientific/Labvision) for 20 min. at 98°C followed by a cool-down to 75°C. Ultra V Block (Thermo Fisher Scientific/Labvision) was applied for 10 min. at room temperature. After blocking, the primary monoclonal antibody recognizing the prolactin receptor (clone 1A2B1, mouse IgG2b; Zymed/Invitrogen, Carlsbad, CA, USA) was diluted (1:500 v/v) in antibody buffer (Scytek Labs, Logan, UT, USA) and incubated overnight at 4°C. This antibody reacts with the long form of the human prolactin receptor and may identify the human prolactin receptor intermediate and γS1 isoforms (Zymed/Invitrogen datasheet). Isotype-matched negative control antibody (mouse IgG2b; Dako, Glostrup, Denmark) was applied in a concentration that matched with the prolactin receptor antibody. Tris-buffered saline was used for washing (3χ2 min.). A three-step HRP polymer immunohistochemistry (IHC) detection technique was applied: post-antibody blocking, 1:1 diluted in Scytek antibody diluent (30 min., room temperature) followed by anti-mouse/rabbit/rat Powervision polymer/HRP (both ImmunoVision, ImmunoLogic, Duiven, The Netherlands), 1:1 diluted in Scytek antibody diluent (30 min., room temperature); HRP activity was visualized with diaminobenzidine (Bright DAB+, ImmunoLogic) after 8 min. Sections were counterstained with haematoxylin (Klinipath, Duiven, The Netherlands) and mounted with VectaMount (Vector Labs, Burlingame, CA, USA). To confirm prolactin receptor expression in endothelial cells, a sequential double staining was performed with the prolactin receptor antibody [44] and an antibody directed against CD34 (clone QBend10, mouse monoclonal, Thermo Fisher Scientific/Labvision). Prolactin receptor antibody binding was detected with a two-step HRP polymer technique as described above. HRP activity was visualized in grey with Vector ImmPACT SG (Vector Labs, Burlingame, CA, USA). Next, CD34 antibody binding was detected with an anti-mouse AP-conjugated polymer (ImmunoLogic) and AP activity was visualized in red using Vector Red (Vector Labs). Potential cross-reactivity between both stainings was tested in control slides leaving out the CD34 antibody. Sections were counterstained with 0.1% methylgreen and mounted with VectaMount. Slides were analysed by light microscopy (Olympus BX51 microscope, Olympus UPlanFl 20χ objective, Olympus DP70 digital camera; Olympus, Zoeterwoude, The Netherlands). In addition, double-stained IHC slides were analysed by the Nuance multispectral imaging system and software (Caliper Life Sciences/Cambridge Research and Instrumentation, Inc., Woburn, MA, USA) [45]. Datasets were acquired from 420 to 720 nm at 20 nm intervals. Using the Nuance software version 3.0 and a spectral library of single Vector ImmPACT SG, single Vector Red and methylgreen multiple staining tissue specimens were unmixed into the individual components. The Nuance software was also used for making an exclusive image of prolactin receptor and CD34 co-localization. All slides were analysed by an independent pathologist.
Statistical analysis
Statistical analyses were conducted using GraphPad Prism version 5.0 software (San Diego, CA, USA). Data were plotted with GraphPad as well and expressed as means ± standard error of the mean. Comparisons between two conditions were analysed using Student's _t_-tests (tube formation and proliferation) when the results were normally distributed, and Mann–Whitney test otherwise. A P value < 0.05 was considered to confer significance.
Results
Activation and blockade of the prolactin receptor
Upon activation of the prolactin receptor, the JAK-STAT signalling pathway is activated, with STAT5 as the primary mediator of prolactin action [2]. Additionally, ERK1/2 phosphorylation is also a major downstream target of prolactin receptor signalling [2]. Hence, we investigated the phosphorylation of STAT5 and ERK1/2 after exposure of 2H11 cells to a dose range of prolactin. As shown in Figure 1, prolactin induced a concentration-dependent phosphorylation of ERK1/2, starting at 10 μg/l and culminating at 1000 μg/l (lane 5–8). Phosphorylation of ERK1/2 was decreased when using 10,000 μg/l prolactin (lane 9). Prolactin also induced the phosphorylation of STAT5, starting at 10 μg/l and peaking at 10,000 μg/l (lane 5–9). Moreover, ERK1/2 and STAT5 phosphorylation induced by prolactin was completely abolished by addition of 20-fold molar excess of the prolactin receptor antagonist del1-9-G129R-hPRL (lane 1 and 2 compared to 6 and 7). The antagonist alone, even at extremely high concentrations (10,000 μg/l), failed to notably induce signalling (lane 3 and 4) (Fig. 1). Overall, these data demonstrate that prolactin has a direct effect on endothelial cells, which is mediated by the prolactin receptor.
Fig 1.
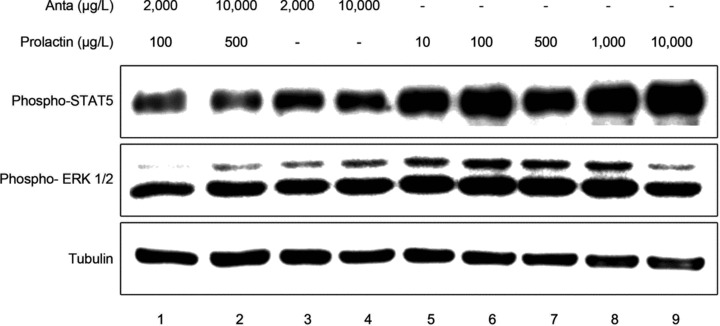
Prolactin exerts a direct effect on endothelial cells. 2H11 endothelial cells, serum starved for 8 hrs, were stimulated for 30 min. with prolactin (10, 100, 500 or 1000 μg/l; lane 5–9) or with prolactin (100 or 500 μg/l) in combination with the prolactin receptor antagonist del1-9-G129R-hPRL (‘Anta’; 2000 or 10,000 μg/l; lane 1 and 2) or with del1-9-G129R-hPRL alone (2000 or 10,000 μg/l; lane 3 and 4). Cell lysates were analysed using Western blot for phosphorylated STAT5 (‘Phospho-STAT5’; top panel), phosphorylated ERK1/2 (‘Phospho-ERK1/2’; middle panel) and tubulin as loading control (bottom panel). A representative picture of an experiment is shown, which was performed three times with similar results.
Prolactin induces angiogenesis in the CAM
The angiogenic activity of prolactin was investigated in vivo using the early chicken embryo CAM assay. In control-treated embryos (0.9% NaCl), the capillary plexus was well developed and a homogeneous vascularization was observed (Fig. 2A). Topical administration of prolactin induced a clear change in the morphology of the vasculature (Fig. 2B). Digital analysis of the images did not reveal a change in the number of branching points: 1987(±58.87)/mm2 in the control condition versus 2124(±190.1)/mm2, _P_= 0.39, after addition of 1 μg prolactin and 2088(±85.71)/mm2, _P_= 0.34, after addition of 10 μg prolactin (Fig. 2D). By contrast, 10 μg prolactin induced a clear increase in vessel density compared to control [3683(±142.4)/mm2_versus_ 3337(±36.5)/mm2, respectively, _P_= 0.0065; Fig. 2E], whilst addition of 1 μg prolactin had no effect on vessel density [3549(±340.2)/mm2, _P_= 0.42]. Combination of 1 μg prolactin with 10 μg of the prolactin receptor antagonist did not significantly affect the number of branching points [1885±(252.3)/mm2, _P_= 0.37; Fig. 2D] in comparison to the control condition. Additionally, prolactin plus antagonist did not influence the number of segments [3314±(398.0)/mm2, _P_= 0.85; Fig. 2E]. Moreover, prolactin enhanced the tortuosity of the vessels (Fig. 2B). The exposure to prolactin induced pillar formation and intussusceptive angiogenesis in the larger blood vessels (arrows in Fig. 2B), phenomena that are known to occur after exposure to the angiogenic growth factor VEGF [46, 47]. These effects of prolactin were markedly diminished by addition of the prolactin receptor antagonist del1-9-G129R-hPRL (Fig. 2C).
Fig 2.
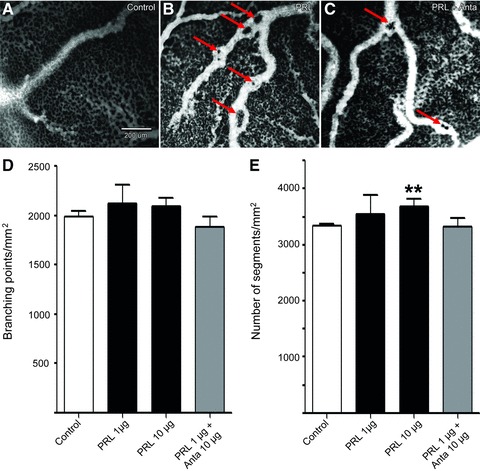
Effect of prolactin on in vivo angiogenesis in the CAM. Images show the CAM (EDD9) with or without treatment with prolactin (PRL) and prolactin with its receptor antagonist (PRL+Anta). The vasculature is visualized by FITC-dextran fluorescence angiography (25 mg/kg, 20 kD, λex= 470 nm). (A) Untreated CAM with small vessels and the capillary network. (B) Angiography of a CAM treated with prolactin (1 μg/embryo/day). (C) Angiography after treatment with prolactin (1 μg/embryo/day) mixed with its receptor antagonist (10 μg/embryo/day). Scale bar in (A) is valid for all three images. Arrows indicate the induced pillar formation and intussusceptive angiogenesis in the larger blood vessels. (D, E) Quantification of digital analysis of the angiography images. (D) Branching points/mm2 for CAM, stimulated with 0.9% NaCl alone (control, white bar) or 0.9% NaCl supplemented with the indicated amounts of prolactin (PRL, black bars) or prolactin with antagonist (PRL + Anta, grey bar). (E) Number of segments/mm2 as a marker of vessel density for CAM, stimulated with 0.9% NaCl alone (control, white bar) or 0.9% NaCl supplemented with the indicated amounts of prolactin (PRL, black bars) or prolactin with antagonist (PRL + Anta, grey bar). Mean values (± standard error of the mean) are shown for two different experiments, in which each condition was included four times (thus eight individual eggs). **P < 0.01.
Effect of prolactin on endothelial cell survival
To investigate whether prolactin has a direct effect on the viability of endothelial cells, cultured cells of the 2H11 cell line were exposed to a dose range of prolactin (from 10 to 10,000 μg/l) for 18 hrs and cell proliferation was assessed using the MTT assay. From 100 μg/l onwards, prolactin induced a subtle, but significant, increase in cell proliferation (105.5 ± 2%, _P_= 0.048; 115.4 ± 3%, P < 0.0001 and 116.9 ± 4%, _P_= 0.0005 for 100, 1000 and 10,000 μg/l prolactin, respectively; Fig. 3A). Similar results were obtained with human endothelial cells of primary origin (HUVEC; Fig. 3B). We observed that 100, 1000 and 10,000 μg/l prolactin induced an even more discrete proliferation of HUVECs (109.2 ± 2%, _P_= 0.003; 105.0 ± 2%, _P_= 0.098 and 105.3 ± 2%, _P_= 0.038, respectively).
Fig 3.
Prolactin slightly stimulates proliferation of endothelial cell cultures. (A) MTT assay on a cell culture of the 2H11 endothelial cell line in serum-free medium (control, white bar) or in serum-free medium supplemented with the indicated concentrations of prolactin (PRL, black bars). The mean (± standard error of the mean) of six different experiments performed in octuplicate is shown. (B) CellTiterGlo assay using primary human endothelial cells HUVECs in serum-free medium (control, white bar) or serum-free medium supplemented with the indicated concentrations of prolactin (PRL, black bars). The mean (± standard error of the mean) of two different experiments is shown, in which each condition was included either two or five times. Results shown in (A) and (B) are expressed as relative induction of proliferation (% of control). ***P < 0.001, **P < 0.01, *P < 0.05.
Prolactin stimulates endothelial cell migration
Another hallmark of angiogenesis is the capacity of endothelial cells to migrate. To test for migratory activity, dose ranges of prolactin were tested in a migration assay using 2H11 endothelial cells. While virtually no migration was observed in control conditions, exposure to 1000 μg/l of prolactin significantly enhanced migration (P < 0.0001; Fig. 4A). Again, the antagonist del1-9-G129R-hPRL almost completely reverted the effects of prolactin on cell migration (P < 0.0001; Fig. 4A), demonstrating the involvement of prolactin receptor in prolactin-induced migration.
Fig 4.
Prolactin stimulates endothelial cell migration. (A) Trans-well migration towards serum-free medium of 2H11 cells. Cells were stimulated for 18 hrs either with 1000 μg/l prolactin (PRL), serum-free medium (SF), the prolactin receptor antagonist del1-9-G129R-hPRL (Anta) alone (20,000 μg/l) or prolactin 1000 μg/l in combination with del1-9-G129R-hPRL 20,000 μg/l (PRL+Anta). Fluorescence was measured every 2 min. at the bottom side of the well. The level of fluorescence is shown and is representative for the number of migrated cells in time [relative fluorescence units (RFU)]. A representative example of an experiment performed three times is shown, in which each condition was included three- to sixfold. (B–E) Migration of HUVECs as assessed in the wound scratch assay. After the wound scratch was performed, serum-free medium (control) or serum-free medium supplemented with prolactin (PRL, 1000 μg/l) was added and wound closure was monitored for 8 hrs. (B) and (D) are representative pictures of the wound at t = 0 hr. (C) Representative picture of wound closure after 8-hr incubation in the control condition. (E) Representative picture of wound closure after 8-hr incubation with prolactin (1000 μg/l) over this period. Blue colour represents the digital calculated wound area, used for the analyses. (F) Quantification of wound closure with prolactin (PRL, black bar) expressed as percentage of the control (white bar). Mean values (± standard error of the mean) for three different experiments are shown. Each condition was included at least fivefold. ***P < 0.001, *P < 0.05.
To confirm these results with human endothelial cells of primary origin (HUVEC), a wound scratch assay was performed using the same concentration of prolactin. Wound closure was monitored for 8 hrs. Representative pictures of the experiment showed that prolactin stimulated wound closure as compared to the control (Fig. 4B–E). Quantification of the wound closure confirmed that prolactin significantly enhanced cell migration by 117.0 ± 4%, (_P_= 0.010) (Fig. 4F).
Prolactin stimulates tube formation
We further investigated the effect of prolactin on an in vitro assay of tube formation as defined by others [38–41, 48]. 2H11 cells were cultured on a semi-natural, growth factor reduced, matrix (Matrigel) for 18 hrs. Figure 5A–C are representative pictures of the formation of tube-like structures consisting of anastomosing vasculature-like structures, in response to serum-free medium (A), prolactin 1000 μg/l (B) and prolactin 1000 μg/l in combination with antagonist 20,000 μg/l (C). Addition of prolactin stimulated tube formation in a dose-dependent manner (Fig. 5D), with a trend towards an increase for concentrations as low as 10 and 100 μg/l, and a significant induction from 1000 μg/l (216.6 ± 42%, _P_= 0.036) onwards, with a maximal induction of over threefold at 10,000 μg/l (327.3 ± 78%, _P_= 0.017), as compared to serum-free medium alone. As shown in Figure 5C and E, administration of the prolactin receptor antagonist del1-9-G129R-hPRL at a concentration of 20,000 μg/l dramatically reduced tube formation induced by 1000 μg/l prolactin to the level of the control condition (_P_= 0.017).
Fig 5.
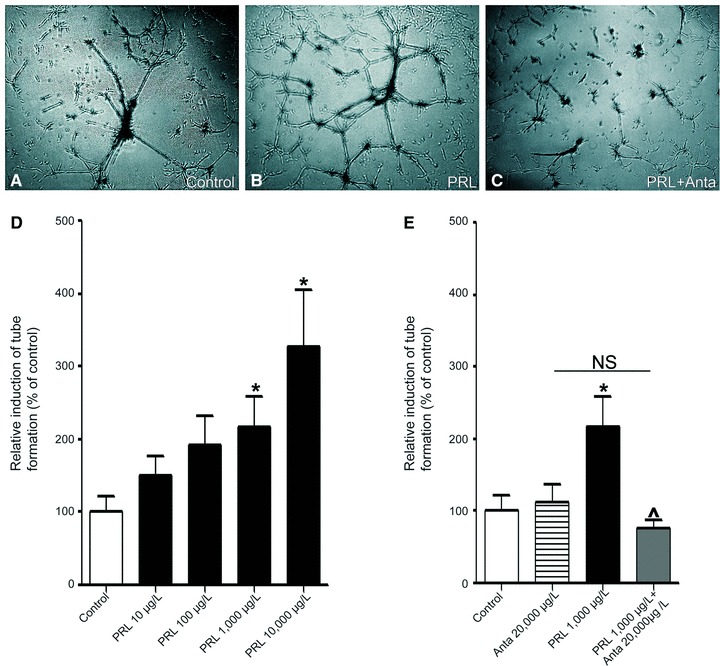
Prolactin promotes endothelial tube formation. The 2H11 endothelial cells (5*104/well) were resuspended in (A) serum-free medium (control), (B) prolactin (PRL, 1000 μg/l) and (C) prolactin (1000 μg/l) in combination with antagonist del1-9-G129R-hPRL (PRL+Anta 20,000 μg/l) and tube formation was assessed after 18 hrs. The representative pictures of an experiment performed three times are shown. (D) Quantification of tube formation in the control condition (white bar) or 10–10,000 μg/l prolactin (PRL, black bars). (E) Quantification of tube formation in the control condition (white bar) or del1-9-G129R-hPRL alone (striped bar) or 1000 μg/l prolactin (PRL, black bar) or prolactin in combination with del1-9-G129R-hPRL (grey bar). (D–E) are shown as mean values (± standard error of the mean) for three different experiments. *P < 0.05 compared to the control, ∧P < 0.05 in comparison to prolactin 1000 μg/l.
Prolactin receptor is expressed on endothelial cells in breast cancer tissue – spectral imaging
Given the recognized stimulating effect of prolactin on the development of breast cancer [7, 10–12, 15], we aimed to investigate the potential clinical relevance of our in vivo and in vitro findings. To this end, we explored whether endothelial cells in breast carcinoma tissue express the prolactin receptor. Hence, we stained breast carcinoma tissue immunohistochemically, using a monoclonal antibody directed against the human prolactin receptor. The antibody was tested on placental tissue and stained positively for decidual cells as previously described [42]. Galsgaard et al. have elegantly evaluated the specificity of the commercially available anti-human PRLR antibodies (B6.2, U5, PRLRi pAb, 1A2B1, 250448 and H-300). The monoclonal antibody 1A2B1 was found to be the most specific for detection of PRLR in immunohistochemistry applications, therefore we choose to use it in our study [44]. The placenta tissue was fully negative with the isotype control [42]. The sequential double staining control did not show any signs of cross-reactivity between both staining sequences (data not shown). As such, this proves that the Vector ImmPACT SG grey reaction product shields the immunoreagents of the first sequence very effectively as is also reported for diaminobenzin reaction product [49].
Figure 6A demonstrates the double staining for prolactin receptor (turquoise) and CD34 (purple) in breast carcinoma tissue. To investigate the expression of the prolactin receptor on CD34-positive endothelial cells, spectral imaging was used to unmix both stains. After spectral unmixing, pseudo-coloured fluorescence-like images, as well as exclusive images of co-localization, were obtained. While Figure 6B shows double staining, this technique is able to show the co-localization of both signals (yellow), indicating endothelial expression of prolactin receptor (Fig. 6C). Figure 6D and E show the single signals for prolactin receptor and endothelial cells, respectively, counterstained with haematoxylin.
Fig 6.
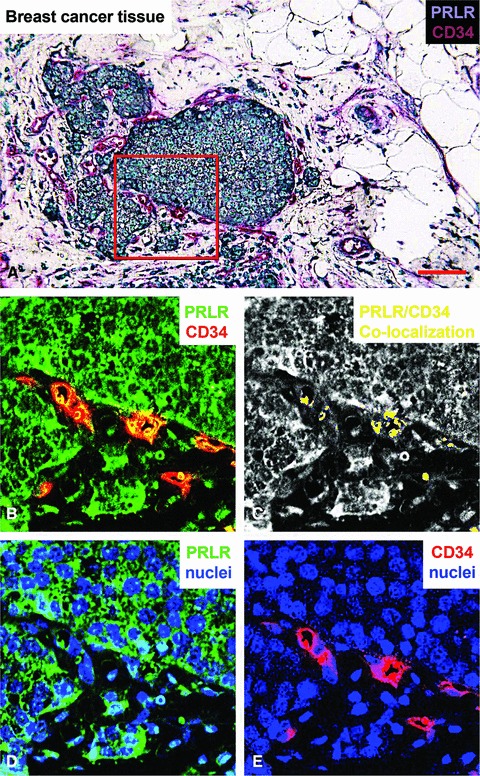
Immunohistochemical staining of the prolactin receptor and CD34 in mammary carcinoma. (A) Staining of the prolactin receptor (turquoise) and CD34 (endothelial cells, purple), counterstaining with haematoxylin (blue). The pictures were digitally processed to unmix the different colours. (B) Fluorescence-like image of both prolactin receptor (green) and CD34 (red) in pseudo-colours. (C) Areas where prolactin receptor and CD34 co-localize, and endothelial expression of prolactin receptor. (D) and (E), respectively, show pseudo-fluorescent staining for the prolactin receptor (green), endothelial cells (red) and nuclei (blue). Scale bar = 0.075 μm in (A–E).
Discussion
Physiologically, angiogenesis plays an important role during embryogenesis, and after birth, it contributes to organ growth. During adulthood, most blood vessels remain quiescent, and physiological angiogenesis occurs only in the cycling ovary, in the placenta during pregnancy and during wound healing [50]. Nonetheless, in many disorders, such as heart failure [51], ischaemic heart disease [52], pre-eclampsia [53], malignancy [54], ocular [55] and inflammatory disorders [56], angiogenesis escapes its self-limiting control thereby leading to the progression of these pathologies.
In this study, we show for the first time that prolactin is able to directly signal to endothelial cells. We observed phosphorylation of STAT5 and ERK1/2, which both have been shown to be involved in the induction of angiogenic events, such as proliferation, migration and tube formation of endothelial cells [57–60]. Additionally, ERK1/2 and Stat 5 activation by other agonists (such as angiopoietin-2, interleukin beta-1, 15(S)-hydroxyeicosatetraenoic acid, etc.) in other cell types (myoblasts, chondrocytes, neuronal cells, etc.) lead to differential regulation of a plethora of genes, related with angiogenesis events [61–64]. For instance, in chondrocytes, IL-1beta induced ERK1/2 phosphorylation but discrete gene expression and ERK1/2 activation by mechanical forces induced VEGF expression [62]. Additionally, observations in human retinal microvascular endothelial cells suggest that 15(S)-HETE-induced angiogenesis requires Jak2-STAT-5B-dependent expression of IL-8 [63]. Interestingly, we repeatedly observed slightly less ERK1/2 phosphorylation in response to 10,000 μg/l prolactin than to 1000 μg/l, whilst such an effect was not detectable for STAT5 phosphorylation. Bell-shaped curves in prolactin receptor mediated dose-response assays have been reported in the past and were referred to as self-antagonism of prolactin at high concentration [65, 66].
We additionally demonstrated that functional consequences of prolactin signalling on endothelial cells relate to angiogenesis. More specifically, we showed that prolactin induces an increase in vessel density and tortuosity in the CAM assay, and rearrangement of endothelial cells into tube structures on Matrigel. We demonstrated that prolactin induces a robust migration of endothelial cells. Importantly, we found that prolactin-induced signalling and angiogenesis were markedly diminished by the specific prolactin receptor antagonist del1-9-G129R-hPRL, thereby suggesting that the observed effects in response to prolactin are prolactin-receptor specific. Finally, we observed that the prolactin receptor is expressed in endothelial cells of vessels in breast carcinoma tissue. Overall, our data provide new insights on how prolactin may contribute to the progression of angiogenesis-related pathologies, such as, among other, breast cancer progression, where the role of prolactin has been particularly well studied. Indeed, it is now known that prolactin is synthesized by mammary epithelial cells [67, 68]. Breast cancer mortality is predominantly due to metastasis, which is partly dependent on cell survival and motility. Previous studies focusing on epithelial breast cancer cells showed that prolactin triggers cytoskeletal reorganization and subsequent cell motility and invasion [69–71]. Moreover, exogenous prolactin is mitogenic and antiapoptotic in breast cancer cells, and overexpression of autocrine prolactin cDNA in breast cancer cell lines has been shown to stimulate their growth and to protect against chemotherapy-induced apoptosis. Autocrine prolactin acts as an inducible survival factor in clonogenic subpopulations of breast cancer cells [72]. In addition, in vitro treatment of human macrophages, the HC11 mouse mammary epithelial cell line and NB2 rat lymphoma cells with prolactin resulted in enhanced release of VEGF, providing an additional mechanism by which prolactin may (indirectly) contribute to tumour growth and metastasis [20, 21]. Our results shed light on a novel potential aspect by which prolactin might influence tumour progression. In addition to targeting epithelial breast cancer cells and inflammatory cells, prolactin exerts a pleiotropic role by affecting endothelial cells and influencing angiogenesis, thereby creating a favourable microenvironment for tumour growth and metastasis. Of note, angiogenesis does not initiate malignancy, but stimulates tumour progression and metastasis [50].
At the cellular level, angiogenesis comprises two different mechanisms: endothelial sprouting and intussusceptive microvascular growth [46]. The sprouting process is based on endothelial cell migration, proliferation and tube formation [73]. Several lines of evidence in our study strongly suggest that the proangiogenic effects of prolactin are rather due to the induction of endothelial cell motility than proliferation. Indeed, its effect on cell proliferation was subtle. In addition, the duration of our migration assays (i.e. 142 min. for Boyden chamber assay and 8 hrs for the wound scratch assay) suggest that the effects of prolactin do not likely rely on cell proliferation. These results are in line with another study demonstrating that high prolactin concentrations alter the actin cytoskeleton of endothelial cells _in vitro_[22]. It is noteworthy that we detected ERK1/2 and Stat5 phosphorylation already with 100 μg/l prolactin. However, it must be kept in mind that a robust and sustained phosphorylation of protein kinases is required to lead to relevant functional consequences. The phosphorylation of ERK1/2 and Stat 5 were analysed at a short time frame (i.e. 30 min.), to decipher whether prolactin could directly induce their activation. Therefore, it is not possible from these particular experiments to draw conclusions on whether 100 μg/l of prolactin is able to induce a signal strong enough to have functional consequences such as cell proliferation, migration or rearrangement. In keeping with this concept, as shown in Figures 3 and 5, 100 μg/l prolactin did not induce significantly tube formation, and led to a very discrete induction of proliferation (105.5 ± 2%, _P_= 0.048). Therefore, whereas the sensitivity of the antibodies used with Western blot allows detecting a signal at this relatively low concentration of prolactin, it is very likely that the induced signalling is not robust enough to induce functional consequences with potential relevance in pathophysiology. We therefore choose to continue working with relatively high concentrations of prolactin in the migration, wound closure and CAM assays.
Growth and remodelling in the angiogenic process, on the other hand, result from intussusceptive angiogenesis alone, which involves transluminal tissue pillar formation and subsequent vascular splitting [46]. Struman and others have already shown that prolactin stimulated new capillary and blood vessel formation in the late-stage CAM assay (day 10–14), whilst they did not observe such an effect of prolactin in the early-stage CAM assay (day 6–8) [19]. These data are in line with ours, since we did not observe an effect of prolactin branching points in the early-stage CAM assay (day 7–9). However, we did find that prolactin increases the number of segments and the tortuosity of vessels in the CAM assay, and induced pillar formation, which is indeed indicative of intussusceptive angiogenesis [46]. Interestingly, the latter is an activity that is also seen in the action spectrum of VEGF [74]. Overall, our data suggest that prolactin affects both sprouting- and intussusceptive angiogenesis. Remarkably, while we did see a stimulatory effect of prolactin on endothelial cell migration, we did not find an effect on branching points in the CAM assay. We favour the view that the angiogenic activity of prolactin is subtle and in the CAM may be masked by the angiogenic cytokine storm of the developmental process.
Several issues should be kept in mind when interpreting the data. First, only a single sample of mamma carcinoma tissue was stained. All together the results of our study support a role for prolactin in angiogenesis, however, future larger studies should assess whether the levels of prolactin receptor expression on endothelial cells in different stages of breast carcinoma tissues. Second, as reviewed by Clapp and co-workers, the roles of prolactin and its receptor on endothelial biology may be tissue-specific [75], which may be explained by a tissue-dependent difference in prolactin receptor heterogeneity and autocrine prolactin expression in vasculature per se. This hypothesis might explain the discrepancies between our and others studies. In line with our data (showing little if any effect of prolactin on endothelial cell proliferation), incubation of 805 μg/l prolactin on bovine brain capillary endothelial cells up to 3 days induced a modest but not significant effect on proliferation [19, 76], and the same results were observed for 48 hrs incubation of rat retinal capillary endothelial cells with 2.3 to 2300 μg/l prolactin [77]. Castilla et al. on the other hand observed a significant effect of proliferation of bovine umbilical vein endothelial cells in response to follicular fluid containing almost 100 μg/l prolactin [78]. Additionally, Malaguarnera et al. showed a dual effect of prolactin on cell proliferation: at low concentration (10 and 25 μg/l), prolactin induced considerable proliferation of human dermal microvessel endothelial cells (approximately 160% and 170%, respectively), while no proliferation was observed in response to 100 μg/l prolactin [79]. Moreover, in contrast with our data, using 10 and 25 μg/l of prolactin, these authors observed only a modest effect on tube formation on Matrigel (approximately 113% and 120%, respectively) [79]. In the present study, we used two different endothelial cell types in our assays. We made use of both the 2H11 endothelial cell line, which are an accepted model of endothelial cells for angiogenesis assays as they express several well-recognized normal tissue endothelial cell markers [25, 26] and HUVECs. The current proliferation and migration data were comparable for HUVECs and 2H11. Third, the concentrations of prolactin we used might seem unusually high. However, one should keep in mind that auto/paracrine prolactin production is increased in breast cancer [80], as epithelial breast cancer cells may be a source of an elevated local prolactin concentration [11]. We therefore chose to use these relatively high doses of prolactin in our experiments. Fourth, other studies showed that modified forms of prolactin (such as phosphorylated prolactin or the 16-kD N-terminal fragment of prolactin) have anti-angiogenic properties [81, 82]. Erdmann and co-workers elegantly showed that the bovine corpus luteum is able to produce prolactin and to process it into antiangiogenic fragments by cathepsin D activity and that prolactin cleavage might mediate angioregression during luteolysis [16]. Nonetheless, it was hypothesized that 16-kD prolactin exerts its effects via a different receptor, possibly belonging to the superfamily of the proliferin receptor [83]. Thus, it might be difficult to compare the results obtained with 16-kD prolactin and the full-length hormone. Phosphorylated prolactin (derived from the pituitary) [82] has been shown to have anti-angiogenic properties. However, autocrine prolactin, secreted by mammary epithelial cells, is unmodified, based on the ability of this autocrine prolactin to promote NB2 cell proliferation [68], thereby explaining the discrepancies in the effects on angiogenesis on the different forms of prolactin.
In conclusion, we propose that prolactin plays a role in the induction of angiogenesis, as a result of direct interaction with endothelial cells. This may provide a potential additional link with the progression of breast cancer. Endothelial cells are considered ideal therapeutic targets that would not become resistant to anti-angiogenic therapy. Indeed, the first anti-angiogenic agents have been approved for the treatment of tumour growth and metastasis by targeting endothelial, mural, stromal, haematopoietic and neoplastic cells, which release an array of chemokines and cytokines and other factors that may promote vessel growth or stabilization [50]. Hence, we suggest that the combination of cytotoxics and del1-9-G129R-hPRL may improve outcomes in breast cancer therapy, not only by targeting the cancer cell population, but also endothelial cells within the tumour could be a target for the prolactin receptor antagonist. In addition, these results can be extrapolated to other angiogenesis-related pathophysiologies [84], where these strategies might be applied as well. Future studies aiming at a detailed in vivo understanding of the interplay between prolactin and angiogenesis, both in pathophysiological and physiological conditions, are warranted to establish the future therapeutic impact of the pathway.
Acknowledgments
Dr. Th. B. Twickler is supported by the Netherlands Organization of Health and Science (NWO-ZonMW). Dr. K. S Borensztajn is supported by an E. Dekker fellowship from the Dutch Heart Society. The authors are grateful for financial support from J. Jacobi Trust.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Horseman ND. Prolactin and mammary gland development. J Mammary Gland Biol Neoplasia. 1999;4:79–88. doi: 10.1023/a:1018708704335. [DOI] [PubMed] [Google Scholar]
- 2.Bole-Feysot C, Goffin V, Edery M, et al. Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr Rev. 1998;19:225–68. doi: 10.1210/edrv.19.3.0334. [DOI] [PubMed] [Google Scholar]
- 3.Ben-Jonathan N, LaPensee CR, LaPensee EW. What can we learn from rodents about prolactin in humans? Endocr Rev. 2008;29:1–41. doi: 10.1210/er.2007-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagano M, Chastre E, Choquet A, et al. Expression of prolactin and growth hormone receptor genes and their isoforms in the gastrointestinal tract. Am J Physiol. 1995;268:G431–42. doi: 10.1152/ajpgi.1995.268.3.G431. [DOI] [PubMed] [Google Scholar]
- 5.Peirce SK, Chen WY, Chen WY. Quantification of prolactin receptor mRNA in multiple human tissues and cancer cell lines by real time RT-PCR. J Endocrinol. 2001;171:R1–4. doi: 10.1677/joe.0.171r001. [DOI] [PubMed] [Google Scholar]
- 6.Gout PW, Beer CT, Noble RL. Prolactin-stimulated growth of cell cultures established from malignant Nb rat lymphomas. Cancer Res. 1980;40:2433–6. [PubMed] [Google Scholar]
- 7.Tworoger SS, Hankinson SE. Prolactin and breast cancer etiology: an epidemiologic perspective. J Mammary Gland Biol Neoplasia. 2008;13:41–53. doi: 10.1007/s10911-008-9063-y. [DOI] [PubMed] [Google Scholar]
- 8.Bogorad RL, Courtillot C, Mestayer C, et al. Identification of a gain-of-function mutation of the prolactin receptor in women with benign breast tumours. Proc Natl Acad Sci USA. 2008;105:14533–8. doi: 10.1073/pnas.0800685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goffin V, Bogorad RL, Touraine P. Identification of gain-of-function variants of the human prolactin receptor. Methods Enzymol. 2010;484:329–55. doi: 10.1016/B978-0-12-381298-8.00017-4. [DOI] [PubMed] [Google Scholar]
- 10.Nyante SJ, Faupel-Badger JM, Sherman ME, et al. Genetic variation in PRL and PRLR, and relationships with serum prolactin levels and breast cancer risk: results from a population-based case-control study in Poland. Breast Cancer Res. 2011;13:R42. doi: 10.1186/bcr2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fernandez I, Touraine P, Goffin V. Prolactin and human tumourogenesis. J Neuroendocrinol. 2010;22:771–7. doi: 10.1111/j.1365-2826.2010.02011.x. [DOI] [PubMed] [Google Scholar]
- 12.Clevenger CV, Furth PA, Hankinson SE, et al. The role of prolactin in mammary carcinoma. Endocr Rev. 2003;24:1–27. doi: 10.1210/er.2001-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corbacho AM, Valacchi G, Kubala L, et al. Tissue-specific gene expression of prolactin receptor in the acute-phase response induced by lipopolysaccharides. Am J Physiol Endocrinol Metab. 2004;287:E750–7. doi: 10.1152/ajpendo.00522.2003. [DOI] [PubMed] [Google Scholar]
- 14.Nagano M, Kelly PA. Tissue distribution and regulation of rat prolactin receptor gene expression. Quantitative analysis by polymerase chain reaction. J Biol Chem. 1994;269:13337–45. [PubMed] [Google Scholar]
- 15.Bernichtein S, Touraine P, Goffin V. New concepts in prolactin biology. J Endocrinol. 2010;206:1–11. doi: 10.1677/JOE-10-0069. [DOI] [PubMed] [Google Scholar]
- 16.Erdmann S, Ricken A, Merkwitz C, et al. The expression of prolactin and its cathepsin D-mediated cleavage in the bovine corpus luteum vary with the estrous cycle. Am J Physiol Endocrinol Metab. 2007;293:E1365–77. doi: 10.1152/ajpendo.00280.2007. [DOI] [PubMed] [Google Scholar]
- 17.Grosdemouge I, Bachelot A, Lucas A, et al. Effects of deletion of the prolactin receptor on ovarian gene expression. Reprod Biol Endocrinol. 2003;1:12. doi: 10.1186/1477-7827-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ko JY, Ahn YL, Cho BN. Angiogenesis and white blood cell proliferation induced in mice by injection of a prolactin-expressing plasmid into muscle. Mol Cells. 2003;15:262–70. [PubMed] [Google Scholar]
- 19.Struman I, Bentzien F, Lee H, et al. Opposing actions of intact and N-terminal fragments of the human prolactin/growth hormone family members on angiogenesis: an efficient mechanism for the regulation of angiogenesis. Proc Natl Acad Sci USA. 1999;96:1246–51. doi: 10.1073/pnas.96.4.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldhar AS, Vonderhaar BK, Trott JF, et al. Prolactin-induced expression of vascular endothelial growth factor via Egr-1. Mol Cell Endocrinol. 2005;232:9–19. doi: 10.1016/j.mce.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 21.Malaguarnera L, Imbesi R, Di RM, et al. Action of prolactin, IFN-gamma, TNF-alpha and LPS on heme oxygenase-1 expression and VEGF release in human monocytes/macrophages. Int Immunopharmacol. 2005;5:1458–69. doi: 10.1016/j.intimp.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 22.Merkle CJ, Schuler LA, Schaeffer RC, Jr, et al. Structural and functional effects of high prolactin levels on injured endothelial cells: evidence for an endothelial prolactin receptor. Endocrine. 2000;13:37–46. doi: 10.1385/ENDO:13:1:37. [DOI] [PubMed] [Google Scholar]
- 23.Ricken AM, Traenkner A, Merkwitz C, et al. The short prolactin receptor predominates in endothelial cells of micro- and macrovascular origin. J Vasc Res. 2007;44:19–30. doi: 10.1159/000097892. [DOI] [PubMed] [Google Scholar]
- 24.Clapp C, Thebault S, de la Escalera MartínezG. Role of prolactin and vasoinhibins in the regulation of vascular function in mammary gland. J Mammary Gland Biol Neoplasia. 2008;13:55–67. doi: 10.1007/s10911-008-9067-7. [DOI] [PubMed] [Google Scholar]
- 25.Borensztajn K, Aberson H, Peppelenbosch MP, et al. FXa-induced intracellular signaling links coagulation to neoangiogenesis: potential implications for fibrosis. Biochim Biophys Acta. 2009;1793:798–805. doi: 10.1016/j.bbamcr.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 26.Walter-Yohrling J, Morgenbesser S, Rouleau C, et al. Murine endothelial cell lines as models of tumour endothelial cells. Clin Cancer Res. 2004;10:2179–89. doi: 10.1158/1078-0432.ccr-03-1013. [DOI] [PubMed] [Google Scholar]
- 27.van Beijnum JR, Dings RP, van der Linden E, et al. Gene expression of tumour angiogenesis dissected: specific targeting of colon cancer angiogenic vasculature. Blood. 2006;108:2339–48. doi: 10.1182/blood-2006-02-004291. [DOI] [PubMed] [Google Scholar]
- 28.Bernichtein S, Kayser C, Dillner K, et al. Development of pure prolactin receptor antagonists. J Biol Chem. 2003;278:35988–99. doi: 10.1074/jbc.M305687200. [DOI] [PubMed] [Google Scholar]
- 29.Rouet V, Bogorad RL, Kayser C, et al. Local prolactin is a target to prevent expansion of basal/stem cells in prostate tumours. Proc Natl Acad Sci USA. 2010;107:15199–204. doi: 10.1073/pnas.0911651107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nowak-Sliwinska P, van Beijnum JR, van Berkel M, et al. Vascular regrowth following photodynamic therapy in the chicken embryo chorioallantoic membrane. Angiogenesis. 2010;13:281–92. doi: 10.1007/s10456-010-9185-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nowak-Sliwinska P, Ballini JP, Wagnieres G, et al. Processing of fluorescence angiograms for the quantification of vascular effects induced by anti-angiogenic agents in the CAM model. Microvasc Res. 2010;79:21–8. doi: 10.1016/j.mvr.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 32.Borensztajn K, Stiekema J, Nijmeijer S, et al. Factor Xa stimulates proinflammatory and profibrotic responses in fibroblasts via protease-activated receptor-2 activation. Am J Pathol. 2008;172:309–20. doi: 10.2353/ajpath.2008.070347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nowak-Sliwinska P, van Beijnum JR, Casini A, et al. Organometallic ruthenium(II) arene compounds with antiangiogenic activity. J Med Chem. 2011;54:3895–902. doi: 10.1021/jm2002074. [DOI] [PubMed] [Google Scholar]
- 34.Brandwijk RJ, Nesmelova I, Dings RP, et al. Cloning an artificial gene encoding angiostatic anginex: from designed peptide to functional recombinant protein. Biochem Biophys Res Commun. 2005;333:1261–8. doi: 10.1016/j.bbrc.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 35.Bijlsma MF, Borensztajn KS, Roelink H, et al. Sonic hedgehog induces transcription-independent cytoskeletal rearrangement and migration regulated by arachidonate metabolites. Cell Signal. 2007;19:2596–604. doi: 10.1016/j.cellsig.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 36.Michail S, Abernathy F. A new model for studying eosinophil migration across cultured intestinal epithelial monolayers. J Pediatr Gastroenterol Nutr. 2004;39:56–63. doi: 10.1097/00005176-200407000-00012. [DOI] [PubMed] [Google Scholar]
- 37.van der Schaft DW, Dings RP, de Lussanet QG, et al. The designer anti-angiogenic peptide anginex targets tumour endothelial cells and inhibits tumour growth in animal models. FASEB J. 2002;16:1991–3. doi: 10.1096/fj.02-0509fje. [DOI] [PubMed] [Google Scholar]
- 38.Aranda E, Owen GI. A semi-quantitative assay to screen for angiogenic compounds and compounds with angiogenic potential using the EA.hy926 endothelial cell line. Biol Res. 2009;42:377–89. [PubMed] [Google Scholar]
- 39.Friedli A, Fischer E, Novak-Hofer I, et al. The soluble form of the cancer-associated Ll cell adhesion molecule is a pro-angiogenic factor. Int J Biochem Cell Biol. 2009;41:1572–80. doi: 10.1016/j.biocel.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 40.Xu H, Czerwinski P, Hortmann M, et al. Protein kinase C alpha promotes angiogenic activity of human endothelial cells via induction of vascular endothelial growth factor. Cardiovasc Res. 2008;78:349–55. doi: 10.1093/cvr/cvm085. [DOI] [PubMed] [Google Scholar]
- 41.Sakai T, Balasubramanian K, Maiti S, et al. Plasmin-cleaved beta-2-glycoprotein 1 is an inhibitor of angiogenesis. Am J Pathol. 2007;171:1659–69. doi: 10.2353/ajpath.2007.070146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Piqueras L, Reynolds AR, Hodivala-Dilke KM, et al. Activation of PPARbeta/delta induces endothelial cell proliferation and angiogenesis. Arterioscler Thromb Vasc Biol. 2007;27:63–9. doi: 10.1161/01.ATV.0000250972.83623.61. [DOI] [PubMed] [Google Scholar]
- 43.Streefkerk JG, de Graaf GA, de Vries RA. Antibody against horse radish peroxidase in lymphoid organs. Acta Morphol Neerl Scand. 1972;10:398–9. [PubMed] [Google Scholar]
- 44.Galsgaard ED, Rasmussen BB, Folkesson CG, et al. Re-evaluation of the prolactin receptor expression in human breast cancer. J Endocrinol. 2009;201:115–28. doi: 10.1677/JOE-08-0479. [DOI] [PubMed] [Google Scholar]
- 45.van der Loos CM. Multiple immunoenzyme staining: methods and visualizations for the observation with spectral imaging. J Histochem Cytochem. 2008;56:313–28. doi: 10.1369/jhc.2007.950170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Makanya AN, Hlushchuk R, Djonov VG. Intussusceptive angiogenesis and its role in vascular morphogenesis, patterning, and remodeling. Angiogenesis. 2009;12:113–23. doi: 10.1007/s10456-009-9129-5. [DOI] [PubMed] [Google Scholar]
- 47.Saaristo A, Veikkola T, Enholm B, et al. Adenoviral VEGF-C overexpression induces blood vessel enlargement, tortuosity, and leakiness but no sprouting angiogenesis in the skin or mucous membranes. FASEB J. 2002;16:1041–9. doi: 10.1096/fj.01-1042com. [DOI] [PubMed] [Google Scholar]
- 48.Reuwer AQ, van Eijk M, Houttuijn-Bloemendaal FM, et al. The prolactin receptor is expressed in macrophages within human carotid atherosclerotic plaques: a role for prolactin in atherogenesis? J Endocrinol. 2011;208:107–17. doi: 10.1677/JOE-10-0076. [DOI] [PubMed] [Google Scholar]
- 49.van der Loos CM, Teeling P. A generally applicable sequential alkaline phosphatase immunohistochemical double staining. J Histotechnol. 2008;31:119–27. [Google Scholar]
- 50.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–6. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 51.Shiojima I, Sato K, Izumiya Y, et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–18. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Molin D, Post MJ. Therapeutic angiogenesis in the heart: protect and serve. Curr Opin Pharmacol. 2007;7:158–63. doi: 10.1016/j.coph.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 53.Levine RJ, Maynard SE, Qian C, et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350:672–83. doi: 10.1056/NEJMoa031884. [DOI] [PubMed] [Google Scholar]
- 54.Liotta LA, Steeg PS, Stetler-Stevenson WG. Cancer metastasis and angiogenesis: an imbalance of positive and negative regulation. Cell. 1991;64:327–36. doi: 10.1016/0092-8674(91)90642-c. [DOI] [PubMed] [Google Scholar]
- 55.Kuiper EJ, Van Nieuwenhoven FA, de Smet MD, et al. The angio-fibrotic switch of VEGF and CTGF in proliferative diabetic retinopathy. PLoS One. 2008;3:e2675. doi: 10.1371/journal.pone.0002675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marrelli A, Cipriani P, Liakouli V, et al. Angiogenesis in rheumatoid arthritis: a disease specific process or a common response to chronic inflammation? Autoimmun Rev. 2011;10:595–8. doi: 10.1016/j.autrev.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 57.Yang X, Qiao D, Meyer K, et al. Signal transducers and activators of transcription mediate fibroblast growth factor-induced vascular endothelial morphogenesis. Cancer Res. 2009;69:1668–77. doi: 10.1158/0008-5472.CAN-07-6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rafiee P, Heidemann J, Ogawa H, et al. Cyclosporin A differentially inhibits multiple steps in VEGF induced angiogenesis in human microvascular endothelial cells through altered intracellular signaling. Cell Commun Signal. 2004;2:3. doi: 10.1186/1478-811X-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang XH, Xu B, Liu JT, et al. Effect of beta-escin sodium on endothelial cells proliferation, migration and apoptosis. Vascul Pharmacol. 2008;49:158–65. doi: 10.1016/j.vph.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 60.Binion DG, Otterson MF, Rafiee P. Curcumin inhibits VEGF-mediated angiogenesis in human intestinal microvascular endothelial cells through COX-2 and MAPK inhibition. Gut. 2008;57:1509–17. doi: 10.1136/gut.2008.152496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mofarrahi M, Hussain SN. Expression and functional roles of angiopoietin-2 in skeletal muscles. PLoS One. 2011;6:e22882. doi: 10.1371/journal.pone.0022882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perera PM, Wypasek E, Madhavan S, et al. Mechanical signals control SOX-9, VEGF, and c-Myc expression and cell proliferation during inflammation via integrin-linked kinase, B-Raf, and ERK1/2-dependent signaling in articular chondrocytes. Arthritis Res Ther. 2010;12:R106. doi: 10.1186/ar3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Farooq F, Molina FA, Hadwen J, et al. Prolactin increases SMN expression and survival in a mouse model of severe spinal muscular atrophy via the STAT5 pathway. J Clin Invest. 2011;121:3042–50. doi: 10.1172/JCI46276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cheranov SY, Wang D, Kundumani-Sridharan V, et al. The 15(S)-hydroxyeicosatetraenoic acid-induced angiogenesis requires Janus kinase 2-signal transducer and activator of transcription-5B-dependent expression of interleukin-8. Blood. 2009;113:6023–33. doi: 10.1182/blood-2008-10-183210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goffin V, Kinet S, Ferrag F, et al. Antagonistic properties of human prolactin analogs that show paradoxical agonistic activity in the Nb2 bioassay. J Biol Chem. 1996;271:16573–9. doi: 10.1074/jbc.271.28.16573. [DOI] [PubMed] [Google Scholar]
- 66.Kinet S, Bernichtein S, Kelly PA, et al. Biological properties of human prolactin analogs depend not only on global hormone affinity, but also on the relative affinities of both receptor binding sites. J Biol Chem. 1999;274:26033–43. doi: 10.1074/jbc.274.37.26033. [DOI] [PubMed] [Google Scholar]
- 67.Kurtz A, Bristol LA, Toth BE, et al. Mammary epithelial cells of lactating rats express prolactin messenger ribonucleic acid. Biol Reprod. 1993;48:1095–103. doi: 10.1095/biolreprod48.5.1095. [DOI] [PubMed] [Google Scholar]
- 68.Mershon J, Sall W, Mitchner N, et al. Prolactin is a local growth factor in rat mammary tumours. Endocrinology. 1995;136:3619–23. doi: 10.1210/endo.136.8.7628401. [DOI] [PubMed] [Google Scholar]
- 69.Doll F, Pfeilschifter J, Huwiler A. Prolactin upregulates sphingosine kinase-1 expression and activity in the human breast cancer cell line MCF7 and triggers enhanced proliferation and migration. Endocr Relat Cancer. 2007;14:325–35. doi: 10.1677/ERC-06-0050. [DOI] [PubMed] [Google Scholar]
- 70.Maus MV, Reilly SC, Clevenger CV. Prolactin as a chemoattractant for human breast carcinoma. Endocrinology. 1999;140:5447–50. doi: 10.1210/endo.140.11.7245. [DOI] [PubMed] [Google Scholar]
- 71.Miller SL, Antico G, Raghunath PN, et al. Nek3 kinase regulates prolactin-mediated cytoskeletal reorganization and motility of breast cancer cells. Oncogene. 2007;26:4668–78. doi: 10.1038/sj.onc.1210264. [DOI] [PubMed] [Google Scholar]
- 72.Howell SJ, Anderson E, Hunter T, et al. Prolactin receptor antagonism reduces the clonogenic capacity of breast cancer cells and potentiates doxorubicin and paclitaxel cytotoxicity. Breast Cancer Res. 2008;10:R68. doi: 10.1186/bcr2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Patan S. Vasculogenesis and angiogenesis. Cancer Treat Res. 2004;117:3–32. doi: 10.1007/978-1-4419-8871-3_1. [DOI] [PubMed] [Google Scholar]
- 74.Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69(Suppl 3):4–10. doi: 10.1159/000088478. [DOI] [PubMed] [Google Scholar]
- 75.Clapp C, Martial JA, Guzman RC, et al. The 16-kilodalton N-terminal fragment of human prolactin is a potent inhibitor of angiogenesis. Endocrinology. 1993;133:1292–9. doi: 10.1210/endo.133.3.7689950. [DOI] [PubMed] [Google Scholar]
- 76.Ferrara N, Clapp C, Weiner R. The 16K fragment of prolactin specifically inhibits basal or fibroblast growth factor stimulated growth of capillary endothelial cells. Endocrinology. 1991;129:896–900. doi: 10.1210/endo-129-2-896. [DOI] [PubMed] [Google Scholar]
- 77.Ochoa A, Montes de Oca P, Rivera JC, et al. Expression of prolactin gene and secretion of prolactin by rat retinal capillary endothelial cells. Invest Ophthalmol Vis Sci. 2001;42:1639–45. [PubMed] [Google Scholar]
- 78.Castilla A, Garcia C, Cruz-Soto M, et al. Prolactin in ovarian follicular fluid stimulates endothelial cell proliferation. J Vasc Res. 2010;47:45–53. doi: 10.1159/000231720. [DOI] [PubMed] [Google Scholar]
- 79.Malaguarnera L, Pilastro MR, Quan S, et al. Significance of heme oxygenase in prolactin-mediated cell proliferation and angiogenesis in human endothelial cells. Int J Mol Med. 2002;10:433–40. [PubMed] [Google Scholar]
- 80.McHale K, Tomaszewski JE, Puthiyaveettil R, et al. Altered expression of prolactin receptor-associated signaling proteins in human breast carcinoma. Mod Pathol. 2008;21:565–71. doi: 10.1038/modpathol.2008.7. [DOI] [PubMed] [Google Scholar]
- 81.Clapp C, Thebault S, Jeziorski MC, et al. Peptide hormone regulation of angiogenesis. Physiol Rev. 2009;89:1177–215. doi: 10.1152/physrev.00024.2009. [DOI] [PubMed] [Google Scholar]
- 82.Huang K, Ueda E, Chen Y, et al. Paradigm-shifters: phosphorylated prolactin and short prolactin receptors. J Mammary Gland Biol Neoplasia. 2008;13:69–79. doi: 10.1007/s10911-008-9072-x. [DOI] [PubMed] [Google Scholar]
- 83.Clapp C, Weiner RI. A specific, high affinity, saturable binding site for the 16-kilodalton fragment of prolactin on capillary endothelial cells. Endocrinology. 1992;130:1380–6. doi: 10.1210/endo.130.3.1311239. [DOI] [PubMed] [Google Scholar]
- 84.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]