High-molecular weight Aβ oligomers and protofibrils are the predominant Aβ species in the native soluble protein fraction of the AD brain (original) (raw)
Abstract
Alzheimer’s disease (AD) is characterized by the aggregation and deposition of amyloid β protein (Aβ) in the brain. Soluble Aβ oligomers are thought to be toxic. To investigate the predominant species of Aβ protein that may play a role in AD pathogenesis, we performed biochemical analysis of AD and control brains. Sucrose buffer-soluble brain lysates were characterized in native form using blue native (BN)-PAGE and also in denatured form using SDS-PAGE followed by Western blot analysis. BN-PAGE analysis revealed a high-molecular weight smear (>1000 kD) of Aβ42-positive material in the AD brain, whereas low-molecular weight and monomeric Aβ species were not detected. SDS-PAGE analysis, on the other hand, allowed the detection of prominent Aβ monomer and dimer bands in AD cases but not in controls. Immunoelectron microscopy of immunoprecipitated oligomers and protofibrils/fibrils showed spherical and protofibrillar Aβ-positive material, thereby confirming the presence of high-molecular weight Aβ (hiMWAβ) aggregates in the AD brain. In vitro analysis of synthetic Aβ40- and Aβ42 preparations revealed Aβ fibrils, protofibrils, and hiMWAβ oligomers that were detectable at the electron microscopic level and after BN-PAGE. Further, BN-PAGE analysis exhibited a monomer band and less prominent low-molecular weight Aβ (loMWAβ) oligomers. In contrast, SDS-PAGE showed large amounts of loMWAβ but no hiMWAβ40 and strikingly reduced levels of hiMWAβ42. These results indicate that hiMWAβ aggregates, particularly Aβ42 species, are most prevalent in the soluble fraction of the AD brain. Thus, soluble hiMWAβ aggregates may play an important role in the pathogenesis of AD either independently or as a reservoir for release of loMWAβ oligomers.
Keywords: amyloid β protein, protofibrils, fibrils, oligomers, Alzheimer’s disease
Introduction
Alzheimer’s disease (AD) is characterized by the extracellular deposition of amyloid β protein (Aβ) aggregates in the brain [1]. Although high-molecular weight Aβ (hiMWAβ) oligomers, Aβ protofibrils and fibrils, low-molecular weight Aβ (loMWAβ) oligomers, such as dimers, trimers or Aβ*56, have been observed in human AD brain tissue or in mouse models of AD [1–9], it is not entirely clear which Aβ species are the most relevant ones for the development of AD and how these Aβ forms are related to one another in vivo. Some studies have used SDS-PAGE for protein analysis [3–5, 9], which denatures and dissociates proteins into individual polypeptides before determining its molecular weight. By contrast, others have performed only dot blot analysis [6]. Currently, only size exclusion chromatography has been used to study oligomers in non-SDS-treated conditions [3, 9]. However, it is unclear whether interactions with the stationary phase may impact the aggregation state of hiMWAβ species. A detailed analysis of the native Aβ aggregates in the AD brain using blue native-PAGE (BN-PAGE) in comparison with SDS-PAGE analysis that focuses on the identification of the above-mentioned forms of Aβ aggregates is still unavailable.
Antibodies and antibody fragments have been developed to detect specific hiMWAβ oligomers (A11) and protofibrillar/fibrillar conformations (B10AP) [2, 6]. These antibodies and antibody fragments allow isolation of oligomers, protofibrils and fibrils from soluble native protein lysates by immunoprecipitation for further protein analysis. Here, we employed these antibodies and BN-PAGE analysis to clarify whether soluble hiMWAβ oligomers and Aβ protofibrils/fibrils or Aβ dimers and other loMWAβ species represent the predominant Aβ aggregates in the native soluble fraction of the AD brain. SDS-PAGE was used to study the effect of protein denaturation on the spectrum of loMWAβ and hiMWAβ species.
Materials and methods
Neuropathology and human sample characterization
A sample including six AD and four control cases was studied (Table 1). All autopsy brains were collected from individuals who died in the University Hospitals of Bonn or Ulm (Germany). All human tissue was obtained and processed in compliance with German federal laws and with university ethics committee approval.
Table 1.
List of autopsy cases studied
| Case no. | Diagnosis | Age | Gender | AD type | Braak-NFT stage | Aβ phase | CERAD plaque score |
|---|---|---|---|---|---|---|---|
| 1 | Control | 60 | m | 0 | 0 | 0 | 0 |
| 2 | Control | 66 | m | 0 | I | 0 | 0 |
| 3 | Control | 69 | f | 0 | I | 0 | 0 |
| 4 | Control | 71 | f | 0 | I | 0 | 0 |
| 5 | AD | 79 | f | 1 | IV | 3 | 2 |
| 6 | AD | 78 | m | 1 | IV | 4 | 1 |
| 7 | AD | 62 | f | 1 | VI | 4 | 3 |
| 8 | AD | 91 | f | 2 | IV | 3 | 1 |
| 9 | AD | 84 | m | 2 | VI | 4 | 3 |
| 10 | AD | 64 | f | 2 | VI | 4 | 3 |
Demented as well as non-demented patients were examined 1–4 weeks prior to death using standardized protocols for routine clinical examination, including neurological status, upon admission to hospital. These data were used to determine whether individuals clinically fulfilled the DSM-IV criteria for dementia [11]. AD was diagnosed when dementia was observed and when the degree of AD-related neuropathology indicated at least a moderate likelihood for AD according to internationally acknowledged criteria [12].
After assessment of unfixed tissue from one hemisphere for biochemical studies, the brains were fixed in a 4% aqueous formaldehyde solution for at least 3 weeks before undergoing neuropathological screening. Presence or absence of gross infarction, haemorrhage, tumour and other findings were recorded. Tissue blocks from the medial temporal lobe (MTL) were excised at the levels of the (i) anterior limit of the dentate gyrus and (ii) lateral geniculate body [13]. These blocks together with tissue blocks from the occipital cortex (Brodmann areas 17–19) were embedded in paraffin. All sections were cut at 10 μm.
Neurofibrillary changes were detected by immunostaining with an antibody directed against abnormal phosphorylated τ protein (AT-8, Pierce, Rockford, IL, USA, 1/1000) [14]. Neuritic plaques were also diagnosed in sections immunostained with this same antibody. The presence of Aβ deposition was assessed using immunohistochemistry with an antibody raised against Aβ17–24 (4G8 [15], Covance, Emeryville, CA, USA, 1/5000, formic acid pre-treatment).
Diagnosis of the stages in the development of neurofibrillary changes (Braak NFT stage) and the semi-quantitative assessment of neuritic plaques (CERAD score) were performed in accordance with published and recommended criteria [12, 14, 16, 17]. For staging of Aβ pathology, we used a previously published protocol for four phases of β-amyloidosis in the MTL [18]. This hierarchically based procedure facilitates study of the topographic distribution pattern of Aβ deposition in additional brain regions [18, 19]: phase 1 represents Aβ deposition that is restricted to the temporal neocortex. Phase 2 is characterized by the presence of additional Aβ plaques in the entorhinal cortex and/or in the hippocampal subiculum-CA1 region. The third phase is marked by the presence of Aβ plaques in the outer zone of the molecular layer of the fascia dentata, subpial band-like amyloid and/or presubicular ‘lake-like’ amyloid. The existence of further Aβ plaques in the hippocampal sector CA4 and/or the pre-α layer of the entorhinal cortex characterize the fourth and final phase of Aβ deposition in the MTL. Reference pathology for all cases was performed by one and the same neuropathologist (D.R.T.).
Biochemical analysis of human AD and control brains
Fresh frozen human brain tissue from the six AD and four control cases was used to assess the presence and types of native Aβ aggregates in AD and control brains (Table 1). Protein extraction from 30 mg of fresh frozen human occipital (Brodmann areas 17–19) and temporal cortex (Brodmann areas 35 and 36) was carried out in 2 ml of 0.32 M sucrose dissolved in 1 M Tris-buffer (pH 7.4) with a protease and phosphatase inhibitor cocktail (Complete and PhosSTOP, Roche, Mannheim, Germany). The tissue was homogenized as previously described [20]. The homogenate was placed on ice for 30 min., and the supernatant was clarified by centrifuging for 30 min. at 14,000 ×g at 4°C. To avoid the segregation of high-molecular weight proteins from the soluble into the insoluble fraction, a centrifuging speed in excess of 14,000 ×g was not used. The resultant supernatant, i.e. the sucrose-soluble fraction, was aliquoted into appropriate volumes and stored at –80°C until use. Protein amounts were determined using BCA Protein Assay (Bio-Rad, Hercules, CA, USA).
For immunoprecipitation, 200 μl of brain lysate was incubated with 1 μl anti-Aβ1–17 (6E10, 1 mg/ml; Covance, Dedham, MA, USA), with 20 μl B10AP antibody fragments coupled to alkaline phosphatase ([2], 0.55 mg/ml) or with 1 μl A11 ([6], 1 mg/ml; Millipore, Temecula, CA, USA) antibodies at 4°C for 4 hrs with gentle agitation. A total of 50 μl of protein G Microbeads (Miltenyi Biotec, Bergisch-Gladbach, Germany) were added to the mixture and incubated overnight at 4°C on a shaking table with gentle agitation. The mixture was then passed through the μColumns which separate the microbeads by retaining them into the column, while the rest of the lysate flows through. After several mild rinsing steps with 1× tris-buffered saline (TBS) buffer (pH 7.4), the microbead-bound proteins were eluted with 1× Lithium dodecyl sulfate (LDS) sample buffer at 95°C (Invitrogen, Carlsbad, CA, USA).
For BN-PAGE of the sucrose fraction, 50 μg of total protein was prepared with 4× NativePAGE sample buffer (Invitrogen) and subjected to native PAGE 4–16% Bis-Tris gel electrophoresis according to the manufacturer’s protocol (Invitrogen). Native-Mark unstained protein standards (Invitrogen) were used as molecular weight markers. The gel was equilibrated in transfer buffer containing 0.2% SDS for 10 min. After protein transfer onto the nitrocellulose membranes (Bio-Rad), the membrane was boiled in phosphate-buffered saline (PBS) buffer in microwave oven for 6 min. Washing buffer and antibody dilution buffer contained 1 M PBS (pH 7.4) with 0.02% Tween (BioRad). A total of 3% non-fat dry milk (Roth, Karlsruhe, Germany) diluted in antibody-dilution buffer was used to block unspecific binding for 1 hr at room temperature.
For SDS-PAGE, sucrose fractions (50 μg total protein) and immunoprecipitation products were electrophoretically resolved in a precast NuPAGE 4–12% Bis-Tris gel system (Invitrogen). The protein load was controlled either by Ponceau S staining or β-actin (C4, 1/1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) immunoblotting. The proteins were transferred to nitrocellulose membranes and the membranes were boiled with PBS for 6 min. followed by blocking with 5% non-fat dry milk (Roth; diluted in antibody-dilution buffer) for 1 hr at room temperature.
For immunodetection of the blotted proteins, the membranes were incubated for 24 hrs at 4°C with the primary antibodies: anti-Aβ1−17 (6E10, 1/1000), anti-Aβ42 (MBC-42, [21] 1/500), anti-Aβ40 (MBC-40, [21] 1/1000) and anti-amyloid precursor protein (APP) (22C11, 1/500; Millipore). The 22C11 anti-APP antibody is directed against an N-terminal part of the APP molecule outside the Aβ region [22]. After washing steps, the corresponding secondary antibodies (EIA grade affinity purified goat antimouse/rabbit IgG-HRP, 1/20000; Bio-Rad) were applied for 2 hrs at room temperature. Blots were developed with an enhanced chemiluminescence (ECL) detection system (Supersignal Pico Western system, ThermoScientific-Pierce, Waltham, MA, USA) and illuminated in ECL Hyperfilm (GE Healthcare, Buckinghamshire, UK). Aβ42- and Aβ40 preparations were used as positive and/or negative controls. All BN-PAGE blots were developed with standard chemiluminescence exposure time of 2–5 min. up to maximum exposure times of 2–3 hrs to detect even minimal amounts of Aβ aggregates. For SDS-PAGE blots, exposure time of 2–5 min. was used except when otherwise indicated.
Electron microscopy of immunoprecipitated oligomeric and fibrillar/protofibrillar proteins from human AD and control brains
For electron microscopy, 5 μl of immunoprecipitated and redissolved A11-positive oligomers or B10AP-positive protofibrils/fibrils were placed on formvar-coated grids. After 1 min. incubation, the excess liquid was wiped off and the grid dried. The grid then was treated with Na-Borhydrite (0.1% in water for 1 min.) followed by blocking with 5% bovine serum albumin, 5% normal goat serum and 0.1% cold-washed fish gelatin in 1 M PBS. The grids were incubated with anti-Aβ1–17 (6E10, 1/50) for 30 min. After washing, the primary antibody was visualized by 15 nm gold-labelled secondary antibodies (1/30, diluted in 1 M PBS; Aurion Immuno Gold Reagents & Accessories, Wageningen, The Netherlands). Then, the grid was post-fixed in 2% glutaraldehyde and block-stained with a 2% aqueous solution of uranyl acetate (Merck, Darmstadt, Germany) for 1 min. followed by five rinsing steps in H2O2. The sections were viewed with a Philips EM400T 120KV (Eindhoven, The Netherlands) and with a Zeiss EM10 (Oberkochen, Germany).
Analysis of synthetic Aβ42 and Aβ40 aggregates in native state and after SDS denaturation
To determine whether synthetic Aβ aggregates primarily form loMWAβ- and hiMWAβ aggregates, we dissolved 15 μmol synthetic Aβ40-peptide (Peptides International, Louisville, KY, USA) in 1 ml cell culture medium (Quantum 263; PAA Laboratories, Pasching, Austria) for 30 min. at 4°C [23]. Aβ42-peptide (Bachem, Bubendorf, Switzerland) was also dissolved in cell culture medium (RPMI1640; GIBCO, Invitrogen) [23]. Aggregation was permitted to occur for 4 hrs at 22°C. To identify oligomers, fibrils and protofibrils structurally we used electron microscopy. For this purpose, 5 μl of the Aβ40- and Aβ42 solutions were placed on a formvar-coated grid for 1 min. before wiping off the excess liquid. The protein-coated grids were block-stained with a 2% aqueous solution of uranyl acetate (Merck).
The protein aggregates were also analysed with BN-PAGE and SDS-PAGE as well as subsequent Western blot analysis using the MBC-40 and MBC-42 antibodies to detect Aβ40 and Aβ42, respectively. This experiment was repeated five times.
Results
High-molecular weight Aβ42 aggregates predominate in native protein preparations of the soluble fraction from human brain lysates
BN-PAGE with subsequent Western blot analysis of the soluble fraction of human AD brain lysates revealed a high-molecular weight anti-Aβ42-positive smear >1000 kD in AD cases (Fig. 1A) that was not found in controls. Aβ monomers, dimers, or other loMWAβ species were not observed in AD cases or in controls (Fig. 1A). The high-molecular weight smear was also seen with anti-Aβ1–17 at the level of stacking gel in AD cases (Fig. 1C) but not with anti-Aβ40 (Fig. 1B). However, the detection of synthetic Aβ40 but not Aβ42 indicated specific antibody function (Fig. 1B). Anti-Aβ1–17 stained an additional 150–250 kD band (Fig. 1C) that was also observable with anti-APP antibodies, thereby indicating that this band represents APP-containing material (Fig. 1D). The APP-related band was also present in control cases, whereas the high-molecular weight anti-Aβ1–17 smear was not seen (Fig. 1C). The BN-PAGE blots from brain samples were developed with a chemiluminescence exposure time of 2–3 hrs to detect even very minimal amounts of proteins.
Fig 1.
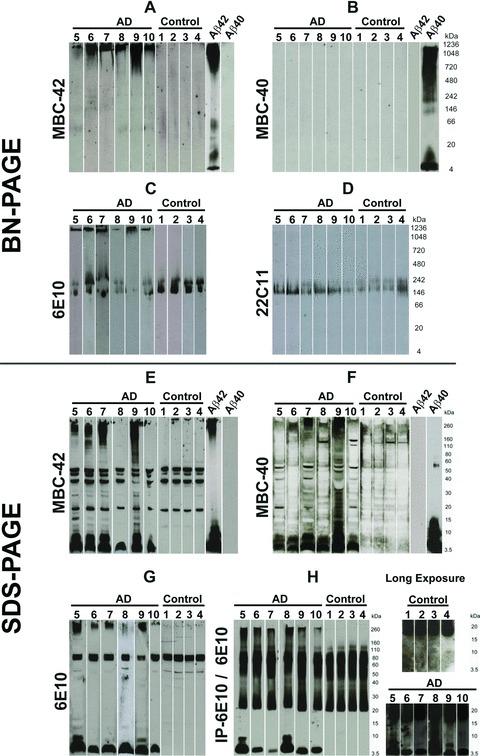
Western blot analysis of sucrose soluble proteins from AD and control brains after BN-PAGE (A–D) and after SDS-PAGE (E–H). All BN-PAGE blots were developed 2–3 hrs for chemiluminescence exposure. SDS-PAGE blots were exposed for 2–5 min. (A) The protein lysates from AD brains (cases no. 5–10) in BN-PAGE showed a high-molecular weight anti-Aβ42-positive smear >1000 kD. Such smears were not observed in controls (cases no. 1–4). Synthetic Aβ42 and Aβ40 were loaded as positive and negative controls, respectively. In Aβ42 preparations, long chemiluminescence exposure led to the detection of additional dimer and ∼50 kD bands that were not observed after 2–5 min. exposures, as shown in Figure 3C. (B) The Aβ42-positive material seen in (A) was not detectable in AD (cases no. 5–10) or in the controls (cases no. 1–4) in the native gel blotted with anti-Aβ40 antibodies. Synthetic Aβ42 and Aβ40 were loaded as positive and negative controls, respectively. After 3 hrs of chemiluminescence exposure, synthetic Aβ40 blots display a dimer band at ∼10 kD in addition to the monomer band and the hiMWAβ smear already detected with shorter exposure times as depicted in Figure 3C. (C) The anti-Aβ1–17 antibody also detected the high-molecular weight protein aggregates >1000 kD in the area of stacking gel in the protein lysate from AD brains (cases no. 5–10), which was not detectable in control brains (cases no. 1–4). In addition, anti-Aβ1–17 also showed APP bands in AD and control cases (140–240 kD). (D) The APP-positive bands were confirmed with an antibody directed against N-terminal epitope of APP (22C11) in control (cases no. 1–4) and AD cases (cases no. 5–10). (E)–(G) SDS-PAGE analysis of AD brain protein lysates from cases no. 5–10 exhibited Aβ monomer and dimer bands with MBC-42 (E), MBC-40 (F) and anti-Aβ1–17 (G) that were not detected in control brains (cases no. 1–4). The MBC-42-dimer (E) and 6E10-dimer bands (G) were not seen in all AD cases, whereas anti-Aβ40 consistently detected dimer bands (F). A high-molecular smear was found in most AD cases with all three antibodies directed against Aβ. Interestingly, cases 6 and 10 exhibited nearly no SDS-stable hiMWAβ42 aggregates (E), whereas both cases showed high-molecular anti-Aβ42-positive material in the BN-PAGE (A). (H) With the help of anti-Aβ1–17 (6E10)-immunoprecipitation, monomer and dimer bands as well as loMWAβ (4–20 kD) smears and hiMWAβ (>160 kD) smears were visible in SDS-PAGE of AD brain lysates (cases no. 5–10) but not in those of controls (cases no. 1–4). The detection of the loMWAβ oligomers required chemiluminescence exposure for 3 hrs (i.e. long exposure times).
SDS-PAGE with subsequent anti-Aβ42, anti-Aβ40 and anti-Aβ1–17 Western blot analysis showed Aβ monomers and dimers in AD cases (Fig. 1E–G). Aβ aggregates with a molecular weight of >160 kD were observed in four of six cases with anti-Aβ42 and a smear >260 kD was observed in all cases with anti-Aβ1–17 (6E10) (Fig. 1E–G). After immunoprecipitation with anti-Aβ1–17, a smear of hiMWAβ- (>160 kD) and loMWAβ aggregates (8–20 kD) as well as dimer bands at ∼10 kD were consistently seen in AD cases (Fig. 1H). LoMWAβ and dimers were detected only after 3 hrs of chemiluminescence exposure but not in controls (Fig. 1H).
A11-antibody and B10AP-antibody fragments precipitate oligomeric and protofibrillar/fibrillar proteins including Aβ oligomers, Aβ protofibrils and Aβ fibrils in AD cases
In controls, immunoelectron microscopy of A11- and B10AP-precipitated proteins revealed a high number of precipitated and aggregated proteins that did not contain Aβ-positive material (Fig. 2A–D). There was no nonspecific labelling with anti-Aβ1–17 in controls. B10AP-precipitated material exhibited a pattern that showed fibril-/ protofibril-like architectures (Fig. 2A, B) whereas the proteins precipitated by A11 displayed a spherical pattern (Fig. 2C, D).
Fig 2.
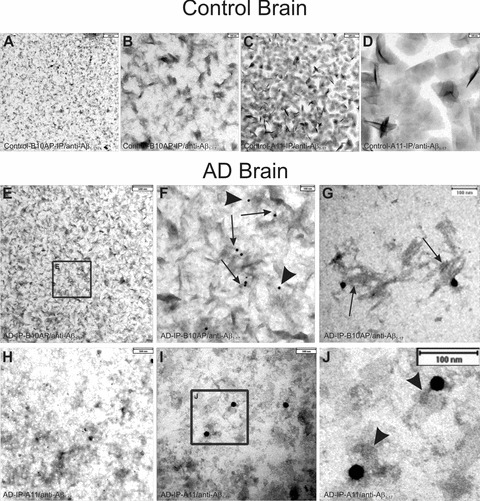
Electron microscopic analysis of immunoprecipitated protein aggregates from AD and control brain as precipitated with B10AP-antibody fragments (B10AP-IP) and A11 antibodies (A11-IP). (A), (B) In the control case no. 3, protein aggregates were precipitated with B10AP, but anti-Aβ1–17 did not show Aβ within these aggregates. There was also no non-specific labelling with anti-Aβ1–17 because no gold particles were observed. The protein aggregates exhibited protofibril-aggregate-like architecture that is more evident at higher magnification (B). This indicates that B10AP does not specifically bind to Aβ protofibrils or fibrils but to proteins with a distinct protofibrillar/fibrillar conformation, as reported previously [2]. (C), (D) A11-IP from control cases resulted in detection of amorphous to spherical presumably oligomeric protein aggregates, as shown in control case no. 4, but did not exhibit Aβ as a component of these protein aggregates. There was also no non-specific labelling with anti-Aβ1–17 because no gold particles were seen. The high magnification demonstrates the spherical shape of the precipitated proteins (D). Thus, A11 also binds spherical protein aggregates other than Aβ oligomers, as reported earlier by others [6]. (E), (F) B10AP-IP from AD brain lysate of case no. 7 showed protein aggregates of protofibril-like morphology. Immunogold labelling indicated Aβ1–17-positive proteins. The frame in E indicates the areas enlarged in (F). At higher magnification, Aβ1–17-positive material following B10AP-IP exhibited protofibril-like morphology (arrows in F) and less frequently amorphous structures (arrowheads in F). These types of Aβ aggregates prevailed in B10AP precipitates. (G) Only a few precipitated Aβ-positive protein aggregates exhibited fibrillar architecture (case no. 10) resembling synthetic Aβ fibrils (Fig. 3A, B). (H) A11-precipitated protein aggregates from AD case no. 7 exhibited spherical to amorphous morphology. Immunogold particles indicate the presence of Aβ1–17-positive aggregates. (I)–(J) Similarly, A11-IP extracted mainly spherical and amorphous protein aggregates from AD case no. 10 shown here at higher magnification. Immunogold labelling indicated Aβ1–17-positive proteins. The frame indicates the area enlarged in (J). The Aβ1–17-positive aggregates observed after A11-IP showed a spherical shape (arrowheads in J).
In B10AP precipitates of the soluble fraction of AD brain homogenates, we observed protein aggregates with a fibril/protofibril-like pattern similar to that seen in controls. However, in AD brains a high number of Aβ-positive protein aggregates were detected with anti-Aβ1–17 (Fig. 2E–G). High-magnification analysis of anti-Aβ1–17-labeled protein aggregates revealed a protofibril-like pattern (Fig. 2F, arrows). However, a few amorphous protein aggregates (Fig. 2F, arrowheads) as well as fibrillar aggregates (Fig. 2G) were seen as well. Spherical Aβ oligomers were not observed following B10AP immunoprecipitation. Amorphous and spherical protein aggregates were observed after A11-immunoprecipitation from AD brain lysates. Anti-Aβ1–17 antibodies detected protein aggregates of spherical and amorphous morphology (Fig. 2H–J). Protofibril-like structures as seen in B10AP precipitates were not observed in A11 precipitates.
SDS treatment destroys native high-molecular weight Aβ42 and Aβ40 aggregates
Synthetic Aβ42 and Aβ40 formed oligomeric and protofibrillar aggregates as detectable by electron microscopy (Fig. 3A, B). With 2–5 min. chemiluminescence exposure time, BN-PAGE blots revealed a monomer band and an additional prominent smear of Aβ42- and Aβ40 aggregates with a molecular weight >700 and 240 kD, respectively (Fig. 3C). However, after longer exposure time of 2–3 hrs, we observed smeary bands at ∼10 and ∼50 kD in Aβ42 preparations, whereas Aβ40 preparations exhibited an additional dimer band that was not detectable in short chemiluminescence exposure blots (Figs 1A, B, 3C). In SDS-PAGE, we observed few high-molecular weight aggregates above 240 kD in Aβ42 preparations but not in Aβ40 preparations, whereas very prominent Aβ monomer, dimer and trimer bands were observed for both synthetic Aβ42- and Aβ40 preparations. In SDS-treated Aβ40 preparations, tetramer and Aβ*56 bands were present that were not seen in SDS-treated Aβ42 preparations (Fig. 3D).
Fig 3.
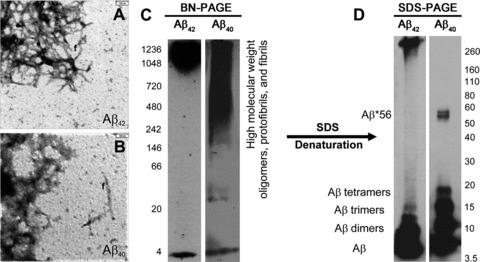
Synthetic Aβ42 (A) and synthetic Aβ40 (B) dissolved in cell culture medium aggregated to amorphous oligomers, protofibrils and fibrils (f), as detectable by electron microscopy. (C) In BN-PAGE, a high-molecular weight smear occurred mainly above 700 kD in Aβ42 preparations in addition to a distinct monomeric band at ∼4 kD. Aβ40 preparations produced a smear of Aβ aggregates with a molecular weight above 242 kD in addition to a clear monomeric band at ∼4 kD. The BN-PAGE blots were developed with standard chemiluminescence exposure time of 2–5 min. Longer chemiluminescence exposure for 3 hrs resulted in additional loMWAβ bands depicted in Figure 1A and B, thereby indicating that these preparations contain low levels of these Aβ species as well. (D) In SDS-PAGE, hiMWAβ42 aggregates were strikingly reduced. Instead, the monomer, dimer and trimer bands displayed strong staining. Other loMWAβ42 oligomers were not evident. No hiMWAβ40 aggregates were seen after denaturing SDS-PAGE, but Aβ40 monomer, dimer, trimer and tetramer bands as well as an Aβ*56 band were detectable at 56 kD.
Discussion
Our results show that hiMWAβ42 oligomers and protofibrils with a molecular weight >1000 kD predominate in the soluble fraction of AD brain homogenates when these samples are analysed under native BN-PAGE conditions. Aβ40 aggregates were not detected following BN-PAGE. Immunoelectron microscopy of immunoprecipitated oligomeric, fibrillar and protofibrillar proteins confirmed the presence of protofibrillar and spherical hiMWAβ aggregates in the soluble fraction of AD brain homogenates. These hiMWAβ aggregates were not seen in controls, whereas analysis of control cases revealed that the A11 and B10AP antibodies/antibody fragments precipitate other proteins of similar morphology, and that only a portion of the precipitated proteins from AD cases were Aβ aggregates. Denaturation of the hiMWAβ aggregates by SDS resulted in the detection of Aβ monomers, dimers, and hiMWAβ with a molecular weight >160 kD in SDS-PAGE analysis of AD cases but not in controls. When using immunoprecipitation with anti-Aβ1–17, a smear of loMWAβ and hiMWAβ was consistently seen following SDS-PAGE and subsequent Western blot analysis, thus indicating that dimers, trimers, tetramers and Aβ*56 are not the only Aβ oligomers that can be detected with SDS-PAGE as shown previously by other groups as well [4, 5]. These results lead us to conclude that, under native conditions, Aβ monomers and loMWAβ aggregates, such as dimers, trimers and Aβ*56, do not represent the major pool of Aβ aggregates in the human AD brain. More likely, loMWAβ aggregates may occur transiently during aggregation or after denaturation of hiMWAβ. The strongest argument in favour of this hypothesis is our finding that hiMWAβ oligomer preparations and Aβ protofibril preparations of synthetic Aβ42- and Aβ40 peptides did not exhibit high levels of loMWAβ oligomers in BN-PAGE but did so in SDS-PAGE. Moreover, subsequent to SDS-induced protein denaturation, hiMWAβ40 aggregates were no longer seen and synthetic hiMWAβ42 aggregates were remarkably reduced. A possible argument against the predominance of hiMWAβ in the soluble fraction is that Aβ monomers tend to aggregate in the presence of oligomers [24] and that this occurs during protein preparation. Nevertheless, in synthetic Aβ preparations with high amounts of aggregated Aβ, we detected a significant monomer band after BN-PAGE. This may indicate that Aβ monomers in the soluble brain lysates remained stable during the process of native protein preparation. As such, it is likely that the hiMWAβ42 aggregates observed in the native soluble fraction indeed represent the major form of soluble Aβ in the human AD brain.
Our finding that Aβ40 was detected in AD cases in SDS-PAGE but not in BN-PAGE could be attributable either to a lower resolution of native gels in comparison to that of denaturing gels or to the fact that a potential smear of Aβ40 aggregates falls far below detectable levels. Presumably, SDS treatment denatures all kinds of Aβ40 oligomers and, in so doing, leads to the accumulation of Aβ40 monomers in a single band. Thus, a non-detectable Aβ40 smear in BN-PAGE might be converted into a detectable well-defined band in the SDS-PAGE. This hypothesis is supported by our finding of a detectable hiMWAβ40 smear in synthetic Aβ40 preparation that disappeared after SDS treatment and converted into strongly stained monomer and loMWAβ oligomer bands. In BN-PAGE, the spectrum of synthetic hiMWAβ40 oligomers was greater (>240 kD) than that of synthetic hiMWAβ42 oligomers (>700 kD). This suggests that the concentrations of distinct hiMWAβ40 oligomers are lower than those of distinct hiMWAβ42 oligomers because of the more widespread distribution of hiMWAβ40 aggregates in the gel. As a result, hiMWAβ40 oligomers and may be less easily detected in native brain lysates. An alternative explanation, on the other hand, could be that Aβ40 interacts with other proteins that hide its C-terminus. In addition, the predominance of hiMWAβ42 in the native soluble fraction of the AD brains investigated here confirms previous reports of a predominant occurrence of Aβ42 in parenchymal soluble and insoluble Aβ aggregates in AD [25, 26].
At first, the results reported here appear to contradict the findings of other authors, who argue that distinct loMWAβ oligomers, such as dimers and Aβ*56, are critical for the development of AD [3, 9, 27]. These authors provide evidence that loMWAβ oligomer preparations received by size-exclusion chromatography are detectable in human as well as transgenic mouse brains, and are capable of inducing cognitive deficits in the rat [9] or altering long-term potentiation [3]. Given our in vitro and in vivo findings, however, one could also speculate that small amounts of loMWAβ oligomers (possibly resulting from the denaturation of hiMWAβ oligomers, protofibrils and fibrils) either are critical for the development of AD or that, upon their administration, loMWAβ oligomers may spontaneously aggregate and form hiMWAβ oligomers, as appears to be the case based upon our native gel analysis of Aβ40- and Aβ42 preparations and the results of Nguyen _et al._[24], who showed that Aβ oligomers accommodate added Aβ monomers. Thus, hiMWAβ aggregates contribute to the pathogenesis of AD either on its own or do so indirectly by providing the reservoir of hiMWAβ aggregates that denature and, in so doing, release loMWAβ oligomers.
That Aβ aggregates, including Aβ plaques, dissociate during the pathogenesis of AD is corroborated by the finding that in late-stage AD cases plaque frequency is lower than in earlier stages [28]. The relevance of soluble hiMWAβ for the pathogenesis of AD may be further supported by the finding of neuritic degeneration near Aβ plaques, i.e. in areas with high levels of hiMWAβ presumably dissolved from Aβ plaques, in the APP transgenic mouse brain [7, 8, 29], in aged rhesus monkeys [30] and in the AD brain [31], and also by the finding that dendritic degeneration in another APP-transgenic mouse model begins at the same time as the deposition of initial diffuse Aβ-plaques, i.e. when hiMWAβ aggregates begin to predominate in the cortex [32].
Here, the stability of hiMWAβ42 aggregates was greater than that of Aβ40 aggregates in in vitro experiments, thus confirming previous reports that soluble Aβ42 aggregates are more stable than Aβ40 aggregates [33, 34]. Taken together with our finding that hiMWAβ42 aggregates predominate in the native soluble fraction of the brain, it may be speculated that it is the stability of soluble Aβ42 aggregates in the soluble compartment of the brain that accounts for its predominance in parenchymal Aβ plaque deposition [26].
In conclusion, the results of the present study strongly suggest that hiMWAβ oligomers, protofibrils and fibrils are the predominant soluble Aβ aggregates in the AD brain. loMWAβ oligomers in high concentrations are detectable only after denaturation of hiMWAβ aggregates. In view of the denaturation of hiMWAβ aggregates and fibrils into loMWAβ oligomers, we propose that Aβ plaques consisting of both fibrillar Aβ as well as soluble hiMWAβ aggregates may serve as reservoirs for the release of loMWAβ oligomers.
Acknowledgments
We thank Kelly Del Tredici, M.D., Ph.D. (University of Ulm, Department of Neurology, Center for Clinical Research) for reading the final version of the revised manuscript.
Conflict of interest
D.R.T. received research grants from the Deutsche Forschungsgemeinschaft (DFG-grant TH624/6–1) and from the Alzheimer Forschung Initiative (AFI Grant #10810). M.F. was supported by the Deutsche Forschungsgemeinschaft (SFB 610) and the Landesexzellenz-Netzwerk Biowissenschaften. There are no other conflicts of interest.
References
- 1.Masters CL, Simms G, Weinman NA, et al. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–9. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Habicht G, Haupt C, Friedrich RP, et al. Directed selection of a conformational antibody domain that prevents mature amyloid fibril formation by stabilizing Abeta protofibrils. Proc Natl Acad Sci USA. 2007;104:19232–7. doi: 10.1073/pnas.0703793104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shankar GM, Li S, Mehta TH, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosen RF, Ciliax BJ, Wingo TS, et al. Deficient high-affinity binding of Pittsburgh compound B in a case of Alzheimer’s disease. Acta Neuropathol. 2010;119:221–33. doi: 10.1007/s00401-009-0583-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosen RF, Tomidokoro Y, Ghiso JA, et al. SDS-PAGE/immunoblot detection of Abeta multimers in human cortical tissue homogenates using antigen-epitope retrieval. J Vis Exp. 2010;38 doi: 10.3791/1916. doi: 10.3791/1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–9. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 7.Spires TL, Meyer-Luehmann M, Stern EA, et al. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–87. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai J, Grutzendler J, Duff K, et al. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004;7:1181–3. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- 9.Lesne S, Koh MT, Kotilinek L, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–7. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 10.Thal DR, Papassotiropoulos A, Saido TC, et al. Capillary cerebral amyloid angiopathy identifies a distinct APOE epsilon4-associated subtype of sporadic Alzheimer’s disease. Acta Neuropathol. 2010;120:169–83. doi: 10.1007/s00401-010-0707-9. [DOI] [PubMed] [Google Scholar]
- 11.American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th ed. WashingtonDC: American Psychiatric Association; 1994. [Google Scholar]
- 12.The National Institute on Aging. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging. 1997;18:S1–2. [PubMed] [Google Scholar]
- 13.Insausti R, Amaral DG. Hippocampal Formation. In: Paxinos G, Mai JK, editors. The human nervous system. 2nd ed. London: Elsevier; 2004. pp. 872–914. [Google Scholar]
- 14.Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim KS, Miller DL, Sapienza VJ, et al. Production and characterization of monoclonal antibodies reactive to synthetic cerebrovascular amyloid peptide. Neurosci Res Commun. 1988;2:121–30. [Google Scholar]
- 16.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 17.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 18.Thal DR, Rüb U, Schultz C, et al. Sequence of Abeta-protein deposition in the human medial temporal lobe. J Neuropathol Exp Neurol. 2000;59:733–48. doi: 10.1093/jnen/59.8.733. [DOI] [PubMed] [Google Scholar]
- 19.Thal DR, Rüb U, Orantes M, et al. Phases of Abeta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 20.Utter S, Tamboli IY, Walter J, et al. Cerebral small vessel disease-induced apolipoprotein E leakage is associated with Alzheimer disease and the accumulation of amyloid beta-protein in perivascular astrocytes. J Neuropathol Exp Neurol. 2008;67:842–56. doi: 10.1097/NEN.0b013e3181836a71. [DOI] [PubMed] [Google Scholar]
- 21.Yamaguchi H, Sugihara S, Ogawa A, et al. Diffuse plaques associated with astroglial amyloid beta protein, possibly showing a disappearing stage of senile plaques. Acta Neuropathol. 1998;95:217–22. doi: 10.1007/s004010050790. [DOI] [PubMed] [Google Scholar]
- 22.Weidemann A, Konig G, Bunke D, et al. Identification, biogenesis, and localization of precursors of Alzheimer’s disease A4 amyloid protein. Cell. 1989;57:115–26. doi: 10.1016/0092-8674(89)90177-3. [DOI] [PubMed] [Google Scholar]
- 23.Huang X, Atwood CS, Moir RD, et al. Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer’s Abeta peptides. J Biol Inorg Chem. 2004;9:954–60. doi: 10.1007/s00775-004-0602-8. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen PH, Li MS, Stock G, et al. Monomer adds to preformed structured oligomers of Abeta-peptides by a two-stage dock-lock mechanism. Proc Natl Acad Sci USA. 2007;104:111–6. doi: 10.1073/pnas.0607440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy MP, Beckett TL, Ding Q, et al. Abeta solubility and deposition during AD progression and in APPxPS-1 knock-in mice. Neurobiol Dis. 2007;27:301–11. doi: 10.1016/j.nbd.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Roher AE, Lowenson JD, Clarke S, et al. beta-Amyloid-(1–42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:10836–40. doi: 10.1073/pnas.90.22.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reed MN, Hofmeister JJ, Jungbauer L, et al. Cognitive effects of cell-derived and synthetically derived Abeta oligomers. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.11.007. doi: 10.1016/j.neurobiolaging.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thal DR, Arendt T, Waldmann G, et al. Progression of neurofibrillary changes and PHF-tau in end-stage Alzheimer’s disease is different from plaque and cortical microglial pathology. Neurobiol Aging. 1998;19:517–25. doi: 10.1016/s0197-4580(98)00090-6. [DOI] [PubMed] [Google Scholar]
- 29.Meyer-Luehmann M, Spires-Jones TL, Prada C, et al. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451:720–4. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah P, Lal N, Leung E, et al. Neuronal and Axonal Loss Are Selectively Linked to Fibrillar Amyloid-{beta} within Plaques of the Aged Primate Cerebral Cortex. Am J Pathol. 2010;177:325–33. doi: 10.2353/ajpath.2010.090937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serrano-Pozo A, William CM, Ferrer I, et al. Beneficial effect of human anti-amyloid-beta active immunization on neurite morphology and tau pathology. Brain. 2010;133:1312–27. doi: 10.1093/brain/awq056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capetillo-Zarate E, Staufenbiel M, Abramowski D, et al. Selective vulnerability of different types of commissural neurons for amyloid beta-protein induced neurodegeneration in APP23 mice correlates with dendritic tree morphology. Brain. 2006;129:2992–3005. doi: 10.1093/brain/awl176. [DOI] [PubMed] [Google Scholar]
- 33.Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–53. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levine H., 3rd Soluble multimeric Alzheimer beta(1–40) pre-amyloid complexes in dilute solution. Neurobiol Aging. 1995;16:755–64. doi: 10.1016/0197-4580(95)00052-g. [DOI] [PubMed] [Google Scholar]