From Fear to Safety and Back: Reversal of Fear in the Human Brain (original) (raw)
Abstract
Fear learning is a rapid and persistent process that promotes defense against threats and reduces the need to relearn about danger. However, it is also important to flexibly readjust fear behavior when circumstances change. Indeed, a failure to adjust to changing conditions may contribute to anxiety disorders. A central, yet neglected aspect of fear modulation is the ability to flexibly shift fear responses from one stimulus to another if a once-threatening stimulus becomes safe or a once-safe stimulus becomes threatening. In these situations, the inhibition of fear and the development of fear reactions co-occur but are directed at different targets, requiring accurate responding under continuous stress. To date, research on fear modulation has focused mainly on the shift from fear to safety by using paradigms such as extinction, resulting in a reduction of fear. The aim of the present study was to track the dynamic shifts from fear to safety and from safety to fear when these transitions occur simultaneously. We used functional neuroimaging in conjunction with a fear-conditioning reversal paradigm. Our results reveal a unique dissociation within the ventromedial prefrontal cortex between a safe stimulus that previously predicted danger and a “naive” safe stimulus. We show that amygdala and striatal responses tracked the fear-predictive stimuli, flexibly flipping their responses from one predictive stimulus to another. Moreover, prediction errors associated with reversal learning correlated with striatal activation. These results elucidate how fear is readjusted to appropriately track environmental changes, and the brain mechanisms underlying the flexible control of fear.
Keywords: fear conditioning, prediction error, reversal, amygdala, striatum, vmPFC
Introduction
Fear learning is typically rapid and resistant to modification (LeDoux, 2000). This tendency to persist prevents the need for relearning about danger and can be adaptive in promoting escape and avoidance in the face of threats. However, the ability to flexibly readjust behavior is also advantageous, particularly in an ever-changing environment. This ability may be impaired in anxiety disorders, and patients with such disorders often show fear responses that are inappropriate for current circumstances (Orr et al., 2000; Peri et al., 2000; Shalev et al., 2000; Rauch et al., 2006).
A leading model for studying fear and anxiety in the brain is Pavlovian fear conditioning, a behavioral procedure in which an emotionally neutral conditioned stimulus (CS), such as a tone, is paired with an aversive unconditioned stimulus (US), such as electric shock. Studies over the past several decades have revealed much about the cellular and molecular mechanisms involved in the acquisition and storage of information about fear conditioning (Fendt and Fanselow, 1999; Davis, 2000; LeDoux, 2000; Phelps and LeDoux, 2005). As a result of this work, the mechanisms of fear extinction, whereby fear responses are weakened by presentation of the CS without the US, have also begun to be understood (Paré et al., 2004; Myers and Davis, 2007; Sotres-Bayon et al., 2007; Quirk and Mueller, 2008). However, elucidating how fear responses evolve and weaken through learning provides only partial understanding of how fear is modulated in the brain. To understand emotional control, it is crucial to clarify how fear responses are flexibly maneuvered and readjusted.
One way to study flexibility in fear is through reversal of aversive reinforcement contingencies in a fear conditioning paradigm. In this case, after acquisition of fear to one CS, the fear response is not eliminated as with extinction, but rather is switched to another CS. This is a unique situation in which two processes, the development of a fear reaction and its inhibition, occur in parallel, targeting different stimuli. Fear reversal, therefore, represents a more sophisticated and perhaps more demanding case of fear modulation.
The aim of the present study was to perform a fine-grain analysis of the gradual change in physiological and neural responses to cues that alternate in predicting danger. Specifically, using whole brain functional magnetic resonance imaging (fMRI), we sought to identify the neural mechanisms that underlie the inhibitory control of the fear response while fear is still present but is directed elsewhere. Our second aim was to identify the neural mechanisms tracking the predictive values of the stimuli as they are reversed from fear-inducing to safety-inducing and vice versa. To this end, we also examined the encoding of prediction errors related to such reversals by using a prediction error response pattern generated by the temporal difference reinforcement-learning algorithm (Sutton and Barto, 1990) as a regressor for brain activation.
The experimental procedure (see Fig. 1) consisted of an acquisition stage followed immediately by an unsignaled transition to a reversal stage. During acquisition, subjects were presented with two visual stimuli (faces). One stimulus coterminated with an aversive outcome (US) on one-third of the trials (CS+, face A). The other stimulus was never paired with the US (CS−, face B). The reversal stage was similar to acquisition except that the reinforcement contingency was reversed so that the previously nonreinforced stimulus now sometimes coterminated with the US (new CS+, face B), and the previously reinforced stimulus was now unpaired with the US (new CS−, face A).
Figure 1.
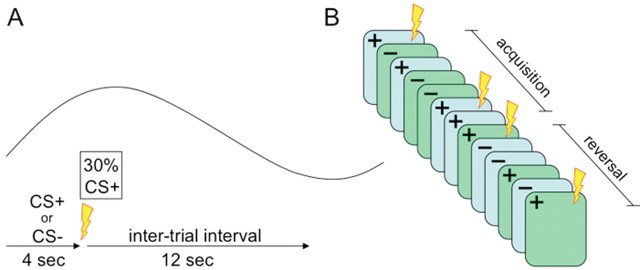
A schematic representation of the procedure. A, Within-trial timeline: stimuli are presented in pseudorandom order for 4 s, and CS+ stimuli terminate with a shock on one-third of the trials. Trials are separated by a 12 s intertrial interval. Above the timeline is a stylized BOLD hemodynamic response in a typical nonreinforced trial. B, Illustration of the overall timeline. Acquisition consisted of presentations of two stimuli, the CS+ and the CS−, on a partial reinforcement schedule. In reversal, the same stimuli were used but the reinforcement contingencies were reversed such that the CS− was paired with the US on about third of the trials. The first trial in which the old CS− terminated with the US (henceforth regarded as “new CS+”) marked the beginning of the reversal stage.
Materials and Methods
Subjects.
Twenty-two healthy right-handed volunteers were recruited for the fMRI reversal task. One subject had excessive head motions during the fMRI scan and was therefore excluded from further analysis. Four subjects had nonmeasurable levels of skin conductance (nonresponders), which did not allow an assessment of fear conditioning. We therefore did not analyze their fMRI data, and they were excluded from the experiment. Thus, the final sample included 17 healthy right-handed volunteers (9 males) between 18 and 31 years of age. The experiment was approved by the University Committee on Activities Involving Human Subjects. All subjects gave informed consent and were paid for their participation.
Conditioning paradigm and physiological assessment.
A fear discrimination and reversal paradigm was used, with delay conditioning and partial reinforcement (Fig. 1). We used partial reinforcement to make learning nontrivial and to slow acquisition and reversal. This allowed us to examine early and late phases in each stage and the gradual development of fear learning and its reversal (for a comparison between a full and partial reinforcement, see Dunsmoor et al., 2007). Subjects were told they would see visual images on a computer screen while receiving shocks. The level of the shocks was set before the experiment, and therefore subjects could experience it beforehand. The instructions were to pay attention to the computer screen and try to figure out the relationship between the stimuli and the shocks. No mention was made of two stages or of a reversal of contingencies.
The CSs were two mildly angry male faces from the Ekman series (Ekman and Friesen, 1976). These stimuli were chosen because they were successful in producing conditioning and amygdala activation in previous studies (Morris et al., 1998; Critchley et al., 2002; Kalisch et al., 2006). Regardless of any a priori emotional saliency of these stimuli, the use of a discrimination procedure allowed us to detect differences in the learned predictive properties of these stimuli. The US was a mild electric shock to the wrist (200 ms duration, 50 pulses/s). The CSs were presented for 4 s, with a 12 s intertrial interval (ITI) in which a fixation point was presented (Fig. 1A).
In the acquisition phase, one face (face A) was paired with the US on one-third of the trials (CS+), and the other (face B) was never paired with the US (CS−). In the reversal stage, these contingencies were reversed such that face B was now paired with the US on approximately one-third of the trials (new CS+) and face A was not paired with the US (new CS−). The order of the different trial types was pseudorandomized (no consecutive reinforced trials and no more than two consecutive trials of each kind), and the designation of faces into CS+ and CS− was counterbalanced across subjects. During acquisition, there were 12 presentations of each of the CSs, intermixed with an additional 6 presentations of the CS+ that coterminated with the US. Reversal immediately followed acquisition, and the transition between the stages was unsignaled. This stage consisted of 16 presentations of each of the CSs, intermixed with 7 additional presentations of the CS+ that coterminated with the US. We considered the first trial in which the previous CS− coterminated with the US as the beginning of the reversal stage (Fig. 1B).
Mild shocks were delivered through a stimulating bar electrode attached with a Velcro strap to the subject's right wrist. A Grass Medical Instruments stimulator charged by a stabilized current was used, with cable leads that were magnetically shielded and grounded through an RF filter. The subjects were asked to set the level of the shock themselves using a work-up procedure before scanning. In this procedure, a subject was first given a very mild shock (10 V, 200 ms, 50 pulses/s), which was gradually increased to a level the subject indicated as “uncomfortable, but not painful” (with a maximum level of 60 V). Skin conductance was assessed with shielded Ag-AgCl electrodes, filled with standard NaCl electrolyte gel, and attached to the middle phalanges of the second and third fingers of the left hand. The electrode cables were grounded through an RF filter panel. The skin conductance signal was amplified and recorded with a BIOPAC Systems skin conductance module connected to a Macintosh computer (Apple Computers). Data were continuously recorded at a rate of 200 samples per second. An off-line analysis of the analog skin conductance waveforms was conducted with AcqKnowledge software (BIOPAC Systems).
The level of skin conductance response was assessed for each trial as the peak-to-peak amplitude difference in skin conductance of the largest deflection (in microsiemens) in the 0.5–4.5 s latency window after stimulus onset. The minimal response criterion was 0.02 μS. Responses below this criterion were encoded as zero. The raw skin conductance scores were square root transformed to normalize the distributions, and scaled according to each subject's mean square-root-transformed US response.
Neuroimaging acquisition and analysis.
A 3T Siemens Allegra head-only scanner and Siemens standard head coil (Siemens) were used for data acquisition. Anatomical images were acquired using a T1-weighted protocol (256 × 256 matrix, 176 1-mm sagittal slices). Functional images were acquired using a single-shot gradient echo EPI sequence (TR = 2000 ms, TE = 25 ms, FOV = 192 cm, flip angle = 75°, bandwidth = 4340 Hz/px, echo spacing = 0.29 ms). Thirty-nine contiguous oblique-axial slices (3 × 3 × 3 mm voxels) parallel to the AC-PC line were obtained. Analysis of the imaging data were conducted using BrainVoyager QX software package (Brain Innovation). Functional imaging data preprocessing included motion correction, slice scan time correction (using sync interpolation), spatial smoothing using a three-dimensional Gaussian filter (4 mm FWHM), and voxelwise linear detrending and high-pass filtering of frequencies above three cycles per time course. One subject with motion >2 mm was not included in the analysis.
A random-effects general linear model analysis was conducted on the fMRI signal during the reversal task with separate predictors for each trial type (face A, face B) at each of four phases: early and late acquisition and early and late reversal (see below). We used separate predictors for trials terminating with a shock. This resulted in 10 box-car predictors corresponding to the length of each trial (4 s), which were convolved with a standard canonical hemodynamic response function. Structural and functional data of each participant were transformed to standard Talairach stereotaxic space (Talairach and Tournoux, 1988). For each region of interest (ROI), we compared the differential mean blood-oxygenation level-dependent (BOLD) responses to the predictive versus nonpredictive stimuli at each phase. These analyses were conducted on the mean percent BOLD signal change at the observed peak activation (4 ± 2 s after stimulus offset) compared with baseline (the mean BOLD response during the last 4 s of the ITI).
In a complementary analysis, a different general linear model design was used to investigate BOLD activation related to errors in fear predictions, in a whole-brain analysis. A temporal difference learning model was used to generate a fear prediction error regressor. For each trial we defined two time points (t), one at the time of the cue (CS+ or CS−) onset and another at the time of its offset. This resulted in four states st (two time points for two cues), each with corresponding predicative value V(st). At each time point, the prediction error (δ_t_) was defined as the difference between two consecutive value predictions: δ_t_ = rt + V(st) − V(st − 1), where rt represents the outcome at every time point, i.e., shock delivery (rt = 1 for shock and rt = 0 for no shock). On the basis of this prediction error, the previous state value predictions were updated according to: V(st − 1) = V(st − 1) + ηδ_t_, where η is the learning rate. The learning rate itself was decreased after every trial according to ηnew = αηold. The parameters of this temporal difference learning model were an initial value Vinit for the two CSs, a learning rate ηacq for the acquisition phase, a learning rate decay term α (which allowed learning to decrease over time), and a learning rate ηrev for the reversal phase (which allowed for the detection of change to again boost up the learning rate that has decayed). To fit these four parameters, we assumed that the skin conductance response at the time of the CS is linearly related to the prediction error at that time (i.e., that it is linearly related to the predictive value of the CS). We thus used linear regression to estimate the scaling of the prediction error for each subject (including in this regression terms for the baseline skin conductance response and a linear drift), and used the residual sum of squared errors from nonreinforced trials only (as in the reinforced trials the skin conductance response was overwhelmed by the response to the shock) as a measure of goodness of fit. Pooling data over subjects, we fit one set of parameters by minimizing the total sum of squared errors. These were: _V_init = 0.69, ηacq = 0.23, ηrev = 0.16 and α = 0.91. The final design matrix for this analysis included, in addition to the prediction error regressor, four additional regressors accounting for the occurrence of CS+ onsets, CS− onsets, trial terminations with US, and trial terminations with no US.
Results
Physiological assessment of fear discrimination and reversal
The results of the skin conductance analysis are presented in Figure 2A. To assess expectations for the aversive outcome separated from unconditioned responses to the shocks themselves, we included only nonreinforced trials of CS+ in this analysis. To assess the development of learning over trials, we defined the first half of the acquisition trials as early acquisition and the second half as late acquisition (six nonreinforced trials each). Similarly, we defined the first half of the reversal trials as early reversal and the last half as late reversal (eight nonreinforced trials each).
Figure 2.

Skin conductance and vmPFC BOLD responses throughout the discrimination and reversal task. A, Mean differential skin conductance responses. The differential responding is calculated as [face A − face B]. Positive scores correspond to stronger responses to face A, which was paired with the shock during acquisition (CS+). Negative scores correspond to stronger responses to face B, which was paired with the shock during reversal (new CS+). B, Statistical activation map depicting the vmPFC revealed by the new CS− > new CS+ in late reversal contrast (false discovery rate <0.05; _x_, _y_, _z_ = 1, 38, −4; BA 32/10). **_C_**, Mean differential percent BOLD signal change for all vmPFC voxels extracted from the new CS− > new CS+ in late reversal contrast (2532 mm3). Error bars indicate SEs. Significant difference from zero: *p < 0.05, **p < 0.01, ***p < 0.001.
As expected, there was a significantly greater skin conductance response to the CS+ compared with the CS− during both early and late acquisition (paired two-way t tests; t(16) = 3.99, p < 0.001; t(16) = 6.06, p < 0.0001, respectively). When reinforcement contingencies were initially reversed (early reversal), there was a nonsignificant (NS) difference in skin conductance responses to the two stimuli (t(16) = −0.85, NS). However, by late reversal, a significantly greater differential skin conductance response to the new CS+ versus the new CS− was observed (t(16) = −7.23, p < 0.0001). A three-way ANOVA with main factors of stimulus (face A, face B), stage (acquisition, reversal), and phase (early, late) revealed a significant stimulus × stage × phase interaction (F(1,9) = 14.53, p < 0.01). Bonferroni corrected post hoc t tests comparing the difference in skin conductance response between CS+ and CS− at each stage showed a significant difference in all stages (p < 0.001) except for early reversal. These results confirm that fear learning occurred (responses to face A were stronger than to face B during acquisition) and that it was successfully reversed (responses to face B were stronger than to face A during reversal).
Analysis of neuroimaging data
Reversal of fear
Our main objective was to examine neural responses during the reversal stage. Previous fear learning studies have shown that responses to the safe stimuli are stronger in the ventromedial prefrontal cortex (vmPFC) compared with the fear-predictive stimulus (Phelps at al., 2004; Kalisch et al., 2006; Milad et al., 2007). We expected the same pattern to emerge during reversal and therefore used a contrast of new CS− > new CS+ in late reversal. We examined regions on the statistical map showing a significant response (false discovery rate <0.05). Similarly to the physiological analyses, we included only nonreinforced trials of CS+ to assess expectations for the aversive outcome separated from the unconditioned responses. This contrast revealed robust activation in an extensive region of the vmPFC only (Fig. 2B).
To fully characterize the pattern of responding in the vmPFC and perform statistical comparisons on the BOLD signal to the different stimuli, we extracted the mean BOLD responses in all vmPFC voxels (2532 mm3) that emerged on the statistical map. Figure 2C presents the mean differential percent BOLD signal change in response to the CS+ versus CS− in the different phases. Separate examination of early and late phases within acquisition and reversal allowed us to detect gradual changes in BOLD responses. Interestingly, during acquisition, the nonpredictive cue (CS−, face B) elicited stronger responses in the vmPFC compared with the predictive cue (CS+, face A), in both early and late acquisition (t(16) = −2.63, p < 0.05; t(16) = −2.99, p < 0.01, respectively). When reinforcement contingencies were initially reversed (early reversal), there was no significant difference in responding to the two stimuli (t(16) = 0.57, NS). As expected, by late reversal, there was a significantly greater differential BOLD response (t(16) = 5.69, p < 0.001) to the new CS− (face A) versus the new CS+ (face B), which was the criterion for selecting this ROI. A three-way ANOVA with main factors of stimulus (CS+, CS−), stage (acquisition, reversal) and phase (early, late), revealed a significant stimulus × stage × phase interaction (p < 0.01). Bonferroni post hoc t tests comparing the difference in BOLD response between CS+ and CS− at each stage showed a significant difference in early (p < 0.05) and late acquisition (p < 0.01), and late reversal (p < 0.001).
Next, we sought to assess the differences in vmPFC responding during acquisition and reversal. To this aim, we used a conjunction analysis in which the resulting statistical activation map is conditioned on significant responding to two contrasts: CS− > CS+ in late acquisition and new CS− > new CS+ in late reversal. As expected, this analysis revealed activation only in the vmPFC. We extracted the mean BOLD response at peak activation (false discovery rate <0.05; x, y, z = 3, 32, −7) and compared the differential responding between the CS+ and CS− in acquisition to the differential responding between these stimuli in reversal (Fig. 3). This analysis revealed a significantly larger difference in reversal compared with acquisition (t(16) = 1.76, p < 0.05). In addition, responses to the new CS− in reversal were higher, compared not only with the new CS+ at this stage, but also with the old CS− in acquisition (t(16) = 1.97, p < 0.05). In contrast, responses to the old CS+ and the new CS+ were similarly decreased in the two stages (t(16) = 0.67, NS). In other words, the vmPFC dissociated the nonpredictive stimulus in reversal from the nonpredictive stimulus in acquisition, whereas the predictive stimuli in these stages were encoded in a similar manner. These results show that the selective activation of the vmPFC in reversal was driven by responses to the no longer predictive stimulus, that is, the face stimulus that switched from being threatening to being safe.
Figure 3.

Ventromedial prefrontal cortex BOLD responses to the threatening and safe stimuli in the late phase of acquisition and reversal. Mean percent BOLD signal change at peak activation (false discovery rate <0.05; x, y, z = 3, 32, −7) in the vmPFC extracted from the conjunction between the CS− > CS+ in late acquisition and new CS− > new CS+ in late reversal contrasts. The mean BOLD response is presented separately for the predictive (threatening) stimuli and the nonpredictive (safe) stimuli in acquisition and reversal. The differential responding between the CS+ and CS− in reversal was significantly larger than the differential responding between these stimuli in acquisition. In addition, responses to the new CS− in reversal were not only higher compared with the new CS+ at this stage, but also compared with the old CS− in acquisition. In contrast, responses to the old CS+ and the new CS+ were similarly decreased in the two stages. *p < 0.05
Finally, to examine overlap in the neural mechanisms of extinction and reversal, we performed an ROI analysis by applying a vmPFC ROI previously identified in an extinction data set (Phelps et al., 2004), and extracting the BOLD signal during the reversal task. Although these voxels were selected on the basis of a separate extinction data set, the pattern of BOLD response seen in these voxels (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) is consistent with the results reported above (Fig. 3), with stronger responses to the new CS− compared with the old CS−.
Aversive value and prediction error
Our second objective was to explore brain regions tracking the predictive value of the stimuli throughout the task. To this end, we first used a contrast of CS+ > CS− in early acquisition to extract regions of interest, and examined their differential responding to the stimuli in subsequent stages. Again, we excluded CS+ trials coterminating with a US from this analysis. Regions on the statistical map showing a significant response (false discovery rate <0.05) and their differential BOLD response in each stage are summarized in Table 1.
Table 1.
Talairach coordinates of regions extracted from the CS+ > CS− in early acquisition contrast (false discovery rate <0.05) and their differential responding in subsequent stages
| Region | Side | CS+ > CS−a | Coordinates x, y, z | Volume (mm3) | ||
|---|---|---|---|---|---|---|
| Late acquisition | Early reversal | Late reversal | ||||
| Caudate | L | ** | NS | *** | −10, 3, 8 | 210 |
| Caudate | R | *** | NS | ** | 12, 4, 9 | 124 |
| Putamen | L | *** | NS | NS | −26, −3, 7 | 283 |
| Putamen | R | NS | NS | NS | 24, 3, −2 | 291 |
| Insula | L | *** | NS | ** | −44, 6, 6 | 3825 |
| Insula | R | *** | NS | *** | 45, 7, 5 | 3366 |
| Thalamus | R | *** | NS | * | 9, −12, 3 | 310 |
| Thalamus | R | * | NS | ** | 5, −13, 14 | 274 |
| Midbrain | L | * | NS | ** | −7, −12, −7 | 74 |
| Midbrain | L | NS | NS | NS | −7, −23, −10 | 119 |
| dACC | M | *** | NS | *** | 2, 4, 40 (BA32) | 1756 |
| SFG | M | *** | NS | *** | 0, −8, 59 (BA6) | 2782 |
| Precuneus | L | NS | NS | NS | −10, −40, 43 (BA7) | 835 |
| Precuneus | R | NS | NS | NS | 4, −57, 48 (BA7) | 450 |
| Cerebellum | R | NS | NS | NS | 14, −42, −15 | 185 |
Given the prominent role of the striatum and the amygdala in the processing of motivationally significant stimuli (Cardinal et al., 2002; Phelps and LeDoux, 2005; Balleine et al., 2007; Delgado, 2007), we focused on these regions in our subsequent analysis, although additional regions implicated in emotion and arousal might also collaborate (Table 1). Figure 4A presents the mean differential percent BOLD signal change to the CS+ versus CS− in the different phases. Striatal responses (left and right caudate; Fig. 4B) were stronger to the CS+ versus CS− in early acquisition (t(16) = 2.69, p < 0.01), which was the criterion for selecting this ROI. This difference was further seen in late acquisition (t(16) = 2.69, p < 0.01; t(16) = 5.50, p < 0.001; respectively), as well as to the new CS+ versus new CS− in late reversal (t(16) = −3.36, p < 0.01). A three-factor ANOVA (stimulus, phase, stage) revealed a significant stimulus × stage × phase interaction (F(1,9) = 10.35, p < 0.01). Bonferroni post hoc t tests comparing the difference in BOLD response between CS+ and CS− at each stage showed a significant difference in early (p < 0.05) and late acquisition (p < 0.001), and late reversal (p < 0.01).
Figure 4.
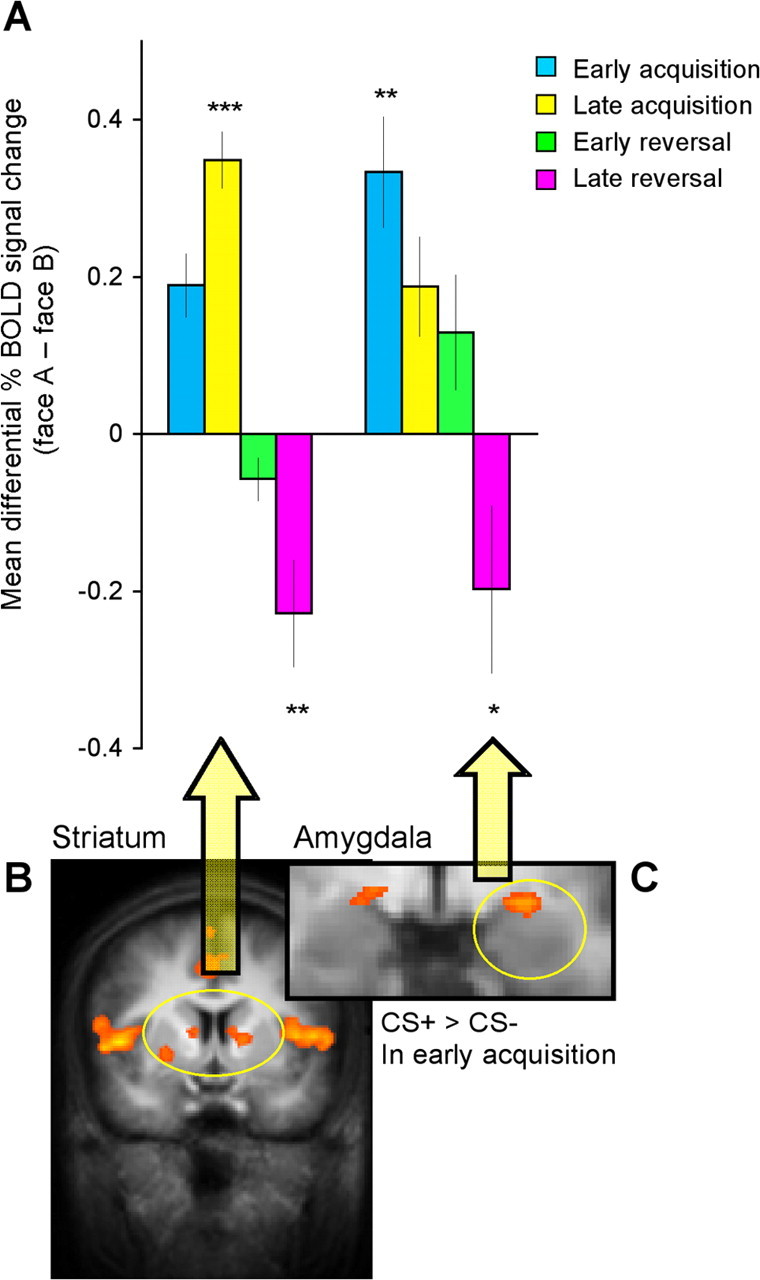
Striatum and amygdala BOLD responses throughout the discrimination and reversal task. A, Mean differential striatal (left and right caudate) and amygdala percent BOLD signal change in the different phases of the task. The differential responding is calculated as [face A − face B]. Positive scores correspond to stronger responses to face A, which was paired with the shock during acquisition (CS+). Negative scores correspond to stronger responses to face B, which was paired with the shock during reversal (new CS+). These BOLD responses were extracted from the CS+ > CS− in early acquisition contrast. B, Striatal activation is denoted by yellow circle (false discovery rate <0.05; R: x, y, z = 12, 4, 9; 124 mm3; L: x, y, z = −10, 3, 8; 210 mm3). C, Left amygdala activation is denoted by yellow circle (p < 0.005, uncorrected; x, y, z = −9, −2, −7; 251 mm3). Error bars indicate SEs. Significant difference from zero: *p < 0.05, **p < 0.01, ***p < 0.001, respectively.
To reveal amygdala BOLD responses (Fig. 4C), we used the contrast of CS+ > CS− in early acquisition with a slightly more liberal threshold (p < 0.005, uncorrected), consistent with previous fear conditioning studies (Büchel et al., 1998; LaBar et al., 1998). Figure 4A presents the mean differential percent BOLD signal change to the CS+ versus CS− in the different phases. In addition to the differential responding to the CS+ versus CS− in early acquisition, amygdala responses were reversed in late reversal, showing stronger responses to the new CS+ versus new CS− (t(16) = −1.85, p < 0.05). A three-factor ANOVA (stimulus, phase, stage) revealed a significant main effect of stimulus (F(1,9) = 5.06, p < 0.05) and a significant stimulus × stage interaction (F(1,9) = 8.04, p < 0.05).
Thus, both the striatum and the amygdala showed stronger responses to the CS+ versus CS− in acquisition and flipped those responses in reversal. These results suggest that both regions track the predictive aversive value of the stimuli throughout the task. Reinforcement learning theories suggest that learning occurs when outcomes deviate from our expectations. The value of predictive stimuli is continuously updated based on these prediction errors (Rescorla and Wagner, 1972). This was the basis for the temporal difference learning model (Sutton and Barto, 1990) that has been successful in accounting for electrophysiological and imaging data from Pavlovian and instrumental conditioning (McClure et al., 2003; Montague et al., 1996; O'Doherty et al., 2003b; Schultz et al., 1997). Accordingly, in a second analysis targeting regions that track predictive value, we examined BOLD activation related to the errors in fear predictions. For this we used the temporal difference learning model to generate a prediction error regressor. The statistical activation map corresponding to this regressor (false discovery rate <0.05), after accounting for all other events as effects of no interest, revealed the caudate (L: _x_, _y_, _z_ = −7, 3, 9, 217 mm3; R: _x_, _y_, _z_ = 9, 5, 8, 322 mm3), the dorsal anterior cingulate (_x_, _y_, _z_ = −1, 1, 46, BA 32, 3277 mm3), the anterior insula (L: _x_, _y_, _z_ = −34, 14, 9, 1801 mm3; R: _x_, _y_, _z_ = 33, 20, 9, 569 mm3) and the thalamus (_x_, _y_, _z_ = 12, 4, 9, 3874 mm3). Lowering the threshold (_p_ < 0.005 uncorrected; minimal cluster size >100 mm3) did not reveal additional areas. These areas are similar to those that were found in the contrasts examining the differential aversive value of the CS+ and CS− above. However, whereas BOLD responses in both striatum and amygdala corresponded with aversive value in those contrasts (Fig. 4), temporal difference prediction errors were correlated only with striatal BOLD, in accord with previous studies (McClure et al., 2003; O'Doherty et al., 2003b, 2006; Knutson and Wimmer, 2007; Schöenberg et al., 2007; Hare et al., 2008). We note that with this type of model-based analysis, we cannot reliably distinguish between prediction error signals and predicted value signals. Indeed, at the time of the CS the prediction error signal and the predicted value signal are equal and the only difference between them is that the error signal is presumed to be punctate (phasic) whereas the value signal is more sustained for the whole duration of the CS. A recent study (Hare et al., 2008) did try to separate the value signal and the prediction error signal using fMRI, but this was done by using a special experimental design aimed directly at teasing these signals apart. As this is not possible in a standard conditioning design such as ours, here we performed the prediction error analysis in addition to the more conventional “model free” CS+ versus CS− analysis, mainly to verify consistency with previous reports.
Finally, we examined whether, similar to the vmPFC, the striatum and the amygdala dissociated a naive CS− from a CS− that carries conflicting information. We found no difference between these stimuli in the striatum (t(16) = −0.82, NS) or in the amygdala (t(16) = −0.70, NS). However, the amygdala, striatum and vmPFC ROIs were defined on the basis of different contrasts, which might bias a comparison between them. That is, the voxels in the vmPFC were defined as those showing stronger responses to the new CS− in late reversal, whereas the voxels in the amygdala and striatum were defined as those showing weak responses to the CS− in early acquisition. To compare the BOLD responses of these regions under the same conditions, we defined new ROIs in these areas on the basis of their responses to a subset of the trials (all reinforced trials > fixation, false discovery rate <0.05), and then extracted the percent BOLD signal change from each region and examined the differential responding to the nonreinforced trials. Specifically, we subtracted the BOLD response to the safe stimulus in acquisition (CS−) from the responses to the safe stimulus in reversal (new CS−). The differential scores for each region are presented in Figure 5. This analysis confirmed that there was differential responding in the vmPFC, but the striatum and the amygdala did not dissociate these stimuli. Thus, it appears that the selective responding to a stimulus that was once threatening but no longer predicts an aversive outcome is unique to the vmPFC.
Figure 5.

Mean differential percent BOLD signal change to the safe stimuli in reversal versus acquisition. The mean differential percent BOLD signal change, comparing responses to the safe stimuli in the late phase of reversal versus late acquisition, is presented for striatum, amygdala, and vmPFC. These regions of interest were defined by contrasting all reinforced trials with fixation (false discovery rate <0.05). The differential responding is calculated as new CS− minus original CS−. Positive scores correspond to stronger responses to the safe stimulus in reversal (that used to predict danger) compared with the safe stimulus in acquisition (stimulus without previous history). The only area that showed significant (*p < 0.05) differential responding to the safe stimulus between the two stages is the vmPFC. Error bars indicate SEs.
Discussion
The present study provides a detailed analysis of the core processes underlying the reversal of predictive fear and safety reactions. We focused on the gradual development of the reversal, with particular emphasis on safety stimuli (CS−). We found a unique dissociation between a safety stimulus previously predictive of danger and a “naive” safety stimulus, with the former more strongly engaging the vmPFC. The initial fear response and its transference to a new stimulus were mediated through a widespread network, including the amygdala, the striatum, and the vmPFC, that flexibly readjusted fear responses after reversal.
Fear reversal versus fear extinction
Reversal and extinction are two linked interference paradigms of Pavlovian learning. Interference with initial fear learning is introduced by the conflicting information given in the subsequent reversal or extinction phase. In fact, extinction is a component of reversal learning, such that responses to one stimulus are extinguished whereas another stimulus acquires the predictive value (Bouton, 1993; Brooks and Bouton, 1993). Reversal, therefore, is a more demanding process because the extinction association is acquired and retrieved while fear is still present but targeted elsewhere. As such, reversal learning might be based on different causal inference than extinction, and necessitates selective and accurate responding under stressful conditions. Understanding reversal learning is potentially relevant to the treatment of clinical fear disorders such as post-traumatic stress disorder (PTSD), because it may serve as a tool to study the inappropriate control of fear in anxiety disorders. The added value of this paradigm is in allowing an examination not only of how fear responses are diminished, but also of how they are appropriately maneuvered from one predictive stimulus to another without developing either a generalized fear response or perseveration of fear.
Although both reversal and extinction consist of a shift from fear to safety, only the reversal paradigm enables a direct comparison to the opposite shift, from safety to fear. This comparison is of interest because it allows examination of how specific fear responses are decreased while others are acquired, as opposed to an overall reduction in fear. Our results clearly show that under such conditions, neural responses to a safety stimulus learned in acquisition (CS−) are different from responses to a safety stimulus learned in reversal (new CS−). Importantly, such dissociation could not be revealed by extinction because the two stimuli would have been compared under different conditions of fear (present versus not present). We found that these stimuli are uniquely dissociated in the vmPFC, which showed stronger responses to a safety stimulus that previously predicted danger compared with the naive CS−.
Interestingly, vmPFC responses to the fear-predictive stimuli were similar in the two stages, and we could not differentiate a naive CS+ from a CS+ that carried conflicting information (was safe but now predictive of danger). In both cases, the vmPFC showed decreased responding compared with the nonpredictive stimuli. Such decreases to a CS+ versus CS− are typically seen during fear conditioning, and are followed by increased CS+ responses during extinction (Phelps at al., 2004; Kalisch et al., 2006; Milad et al., 2007). The present results provide evidence that these increases are selective to an extinguished CS+, rather than the result of a general reduction in fear arousal. This specificity is indicated by the fact that increased responses to the new CS− (which is equivalent to an extinguished CS+) were accompanied by decreased responses to the new CS+, mirroring acquisition of fear.
We propose two possible roles, which are not mutually exclusive, for the vmPFC in fear reversal. One role might be to provide a selective safety signal while fear responses are still being elicited. By inhibiting fear response to one stimulus, the vmPFC may facilitate the transference of this response to the currently predictive stimulus. In essence, the vmPFC is not generally signaling that it is “safe to let your guard down,” but rather is signaling which particular stimuli in the environment is safe to ignore. Impairments in such selective fear inhibition might lead to a generalized fear response on the one hand, or to preservative fear responses on the other hand (Morgan and LeDoux, 1993).
Another role might be to provide a reward signal associated with the omission of the aversive outcome to the new CS− in reversal. It could be argued that a naive CS− is encoded as irrelevant, thus not eliciting reward related activation, whereas the omission of an aversive US from the new CS− confers rewarding properties. Consistent with this idea, the vmPFC has been shown to increase activation in response to reward outcomes and reduce activation in reposes to punishment or reward omission (O'Doherty et al., 2001, 2003a; Gottfried et al., 2002; Hampton et al., 2007). An alternative possibility is that any safe stimulus, regardless of its past, might engage inhibitory mechanisms or even be considered rewarding after reversal has occurred. Examining the neural response to a second CS− that does not change roles during the experiment might be informative in this respect: according to this hypothesis, the vmPFC should become more active in response to this stimulus after reversal.
Aversive predictive value and prediction errors
Similar to the vmPFC, the amygdala and the striatum also discriminated the CS+ from CS− throughout the task, albeit in the opposite direction. During acquisition, these areas showed increased responses to the CS+ compared with the CS−. In reversal, these regions increased responding to the new CS+ and reduced their responding to the new CS−. Thus, a complete reversal of neural activation mirrored the reversal in skin conductance responses, our behavioral index of fear. Unlike the vmPFC, these regions did not dissociate a naive CS− from a CS− that carried conflicting information (Fig. 5).
Striatal activation was also correlated with prediction errors in the reversal task. There is accumulated evidence linking striatal BOLD responses with temporal difference prediction errors for rewards (McClure et al., 2003; O'Doherty et al., 2003b, 2006; Knutson and Wimmer, 2007; Schöenberg et al., 2007; Hare et al., 2008). The present finding adds to the growing body of evidence supporting the role of this structure in temporal difference prediction error for aversive outcomes as well (Ploghous et al., 2000; Seymour et al., 2004; Jensen et al., 2007; Menon et al., 2007). Although striatal activation has been observed in aversive learning paradigm in humans (LaBar et al., 1998; Ploghaus et al., 2000; Jensen et al., 2003, 2007; Phelps et al., 2004; Seymour et al., 2004; Menon et al., 2007), and animals (Horvitz 2000; Schoenbaum and Setlow, 2003; Pezze and Feldon, 2004), the role of this region in aversive learning is only beginning to be understood (McNally and Westbrook, 2006). The present study provides robust evidence for the role of the striatum in fear predictions and their associated errors, as well as in the flexible reversal of predictive fear learning.
In addition to the striatum, responses in other regions, including the dorsal anterior cingulate and anterior insula, also correlated with prediction errors. These findings are consistent with previous report using aversive learning (Seymour et al., 2004; Menon et al., 2007) and may point to interesting differences between aversive and appetitive prediction errors. However, amygdala BOLD responses were not significantly correlated with prediction errors in our task. Two recent studies found that the amygdala has a role in signaling appetitive (Seymour et al., 2005) and aversive (money loss) prediction errors (Yacubian et al., 2006). However, a recent study of electrophysiological responses in the primate amygdala could not disentangle prediction error–related signals from a number of other signals such as CS value, stimulus valence, and US-selective responses (Belova et al., 2007). Thus, the exact computation performed by amygdala neurons while learning about aversive consequences is currently unclear.
Nevertheless, the amygdala appears to have an important role in initial acquisition of fear, as seen by the more robust activation in early compared with late acquisition. In the later phase, the differential responding to the CS+ versus CS− was reduced. This finding is consistent with previous reports that CS+ evoked amygdala activation decreases over time (Quirk et al., 1997; Büchel et al., 1998; LaBar et al., 1998; Büchel and Dolan, 2000). It might also be related to the lack of correlation with the prediction error signal, because the temporal difference model predicts increased differentiation between the stimuli over time. Here we show that despite this decrease, the amygdala also flexibly readjusts its responding after reversal, allowing for the opposite differential responding to emerge.
Different types of reversal
Although very little is known about reversal of Pavlovian fear conditioning, the neural mechanisms underlying the reversal of instrumental responses driven by aversive outcomes have been more thoroughly investigated, implicating the lateral region of the ventral PFC (Cools et al., 2002; O'Doherty et al., 2003a; Schoenbaum and Setlow, 2003; Morris and Dolan, 2004; Rolls, 2004; Evers et al., 2005). Increased activation in this area has also been associated with punishment, reward omission, and response switch (Schoenbaum et al., 1998, 1999, 2000; O'Doherty et al., 2001, 2003a). It is possible that aversive instrumental and Pavlovian reversals might be dissociated in the lateral and medial regions of the ventral PFC, respectively. The former may mediate inhibition of instrumental responses, whereas the latter may mediate inhibition of physiological fear reactions. However, there are other fundamental differences between these studies. For example, here, the reversal was between aversive and neutral associations, whereas previous studies shifted between appetitive and aversive associations. Those studies also use serial reversals, which might engage higher order rule learning. Thus, additional studies are required to elucidate the differential contribution of these two regions to reversal learning.
In sum, the present study provides a first detailed analysis of the components of reversal learning in humans, with a particular focus on safety stimuli. We found evidence for the unique contribution of the vmPFC to inhibition of fear under adverse conditions, in which fear is not diminished but rather needs to be properly assigned and controlled. These finding are important for understating the neural dysfunctions leading to the inappropriate control of fear associated with anxiety disorders.
Footnotes
This work was supported by a Seaver Foundation grant to the Center for Brain Imaging (CBI), National Institutes of Health (NIH) Grants R01 K05 MH067048 and P50 MH58911 (J.E.L.), a James S. McDonnell Foundation grant and NIH Grant R21 MH072279 (E.A.P.), a Human Frontiers Science Program fellowship (Y.N.), and a Fulbright award (D.S.). We thank Mauricio Delgado for fruitful discussions and comments, and Keith Sanzenbach and the CBI at New York University for technical assistance. We also thank Kenji Doya and the members of Okinawa Computational Neuroscience Course 2005.
References
- Balleine BW, Delgado MR, Hikosaka O. The role of the dorsal striatum in reward and decision-making. J Neurosci. 2007;27:8161–8165. doi: 10.1523/JNEUROSCI.1554-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belova MA, Paton JJ, Morrison SE, Salzman CD. Expectation modulates neural responses to pleasant and aversive stimuli in primate amygdala. Neuron. 2007;55:970–984. doi: 10.1016/j.neuron.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouton ME. Context, time, and memory retrieval in the interference paradigms of Pavlovian learning. Psychol Bull. 1993;114:80–99. doi: 10.1037/0033-2909.114.1.80. [DOI] [PubMed] [Google Scholar]
- Brooks DC, Bouton ME. A retrieval cue for extinction attenuates spontaneous recovery. J Exp Psychol Anim Behav Process. 1993;19:77–89. doi: 10.1037//0097-7403.19.1.77. [DOI] [PubMed] [Google Scholar]
- Buchel C, Dolan RJ. Classical fear conditioning in functional neuroimaging. Curr Opin Neurobiol. 2000;10:219–223. doi: 10.1016/s0959-4388(00)00078-7. [DOI] [PubMed] [Google Scholar]
- Buchel C, Morris J, Dolan RJ, Friston KJ. Brain systems mediating aversive conditioning: an event-related fMRI study. Neuron. 1998;20:947–957. doi: 10.1016/s0896-6273(00)80476-6. [DOI] [PubMed] [Google Scholar]
- Cardinal RN, Parkinson JA, Hall J, Everitt BJ. Emotion and motivation: the role of the amygdala, ventral striatum, and prefrontal cortex. Neurosci Biobehav Rev. 2002;26:321–352. doi: 10.1016/s0149-7634(02)00007-6. [DOI] [PubMed] [Google Scholar]
- Cools R, Clark L, Owen AM, Robbins TW. Defining the neural mechanisms of probabilistic reversal learning using event-related functional magnetic resonance imaging. J Neurosci. 2002;22:4563–4567. doi: 10.1523/JNEUROSCI.22-11-04563.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchley HD, Mathias CJ, Dolan RJ. Fear conditioning in humans: the influence of awareness and autonomic arousal on functional neuroanatomy. Neuron. 2002;33:653–663. doi: 10.1016/s0896-6273(02)00588-3. [DOI] [PubMed] [Google Scholar]
- Davis M. The role of the amygdala in conditioned and unconditioned fear and anxiety. In: Aggleton JP, editor. The amygdala: a functional analysis. Oxford: Oxford UP; 2000. pp. 213–288. [Google Scholar]
- Delgado MR. Reward-related responses in the human striatum. Ann N Y Acad Sci. 2007;1104:70–88. doi: 10.1196/annals.1390.002. [DOI] [PubMed] [Google Scholar]
- Dunsmoor JE, Bandettini PA, Knight DC. Impact of continuous versus intermittent CS−UCS pairing on human brain activation during Pavlovian fear conditioning. Behav Neurosci. 2007;121:635–642. doi: 10.1037/0735-7044.121.4.635. [DOI] [PubMed] [Google Scholar]
- Ekman P, Friesen W. Palo Alto, CA: Consulting Psychologists; 1976. Pictures of facial affect. [Google Scholar]
- Evers EA, Cools R, Clark L, van der Veen FM, Jolles J, Sahakian BJ, Robbins TW. Serotonergic modulation of prefrontal cortex during negative feedback in probabilistic reversal learning. Neuropsychopharmacology. 2005;30:1138–1147. doi: 10.1038/sj.npp.1300663. [DOI] [PubMed] [Google Scholar]
- Fendt M, Fanselow MS. The neuroanatomical and neurochemical basis of conditioned fear. Neurosci Biobehav Rev. 1999;23:743–760. doi: 10.1016/s0149-7634(99)00016-0. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Tononi G, Reeke GN, Jr, Sporns O, Edelman GM. Value-dependent selection in the brain: simulation in a synthetic neural model. Neuroscience. 1994;59:229–243. doi: 10.1016/0306-4522(94)90592-4. [DOI] [PubMed] [Google Scholar]
- Gottfried JA, O'Doherty J, Dolan RJ. Appetitive and aversive olfactory learning in humans studied using event-related functional magnetic resonance imaging. J Neurosci. 2002;22:10829–10837. doi: 10.1523/JNEUROSCI.22-24-10829.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton AN, Bossaerts P, O'Doherty JP. The role of the ventromedial prefrontal cortex in abstract state-based inference during decision making in humans. J Neurosci. 2006;26:8360–8367. doi: 10.1523/JNEUROSCI.1010-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton AN, Adolphs R, Tyszka JM, O'Doherty JP. Contributions of the amygdala to reward expectancy and choice signals in human prefrontal cortex. Neuron. 2007;55:545–555. doi: 10.1016/j.neuron.2007.07.022. [DOI] [PubMed] [Google Scholar]
- Hare TA, O'Doherty J, Camerer CF, Schultz W, Rangel A. Dissociating the role of he orbitofrontal cortex and the striatum in the computation of goal values and prediction errors. J Neurosci. 2008;28:5623–5630. doi: 10.1523/JNEUROSCI.1309-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvitz JC. Mesolimbocortical and nigrostriatal dopamine responses to salient non-reward events. Neuroscience. 2000;96:651–656. doi: 10.1016/s0306-4522(00)00019-1. [DOI] [PubMed] [Google Scholar]
- Jensen J, McIntosh AR, Crawley AP, Mikulis DJ, Remington G, Kapur S. Direct activation of the ventral striatum in anticipation of aversive stimuli. Neuron. 2003;40:1251–1257. doi: 10.1016/s0896-6273(03)00724-4. [DOI] [PubMed] [Google Scholar]
- Jensen J, Smith AJ, Willeit M, Crawley AP, Mikulis DJ, Vitcu I, Kapur S. Separate brain regions code for salience vs. valence during reward prediction in humans. Hum Brain Mapp. 2007;28:294–302. doi: 10.1002/hbm.20274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalisch R, Korenfeld E, Stephan KE, Weiskopf N, Seymour B, Dolan RJ. Context-dependent human extinction memory is mediated by a ventromedial prefrontal and hippocampal network. J Neurosci. 2006;26:9503–9511. doi: 10.1523/JNEUROSCI.2021-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Shimojo S, O'Doherty JP. Is avoiding an aversive outcome rewarding? Neural substrates of avoidance learning in the human brain. PLoS Biol. 2006;4:e233. doi: 10.1371/journal.pbio.0040233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson B, Wimmer GE. Splitting the difference: how does the brain code reward episodes? Ann N Y Acad Sci. 2007;1104:54–69. doi: 10.1196/annals.1390.020. [DOI] [PubMed] [Google Scholar]
- LaBar KS, Gatenby JC, Gore JC, LeDoux JE, Phelps EA. Human amygdala activation during conditioned fear acquisition and extinction: a mixed-trial fMRI study. Neuron. 1998;20:937–945. doi: 10.1016/s0896-6273(00)80475-4. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- McClure SM, Berns GS, Montague PR. Temporal prediction errors in a passive learning task activate human striatum. Neuron. 2003;38:339–346. doi: 10.1016/s0896-6273(03)00154-5. [DOI] [PubMed] [Google Scholar]
- McNally GP, Westbrook RF. Predicting danger: the nature, consequences, and neural mechanisms of predictive fear learning. Learn Mem. 2006;13:245–253. doi: 10.1101/lm.196606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon M, Jensen J, Vitcu I, Graff-Guerrero A, Crawley A, Smith MA, Kapur S. Temporal difference modeling of the blood-oxygen level dependent response during aversive conditioning in humans: effects of dopaminergic modulation. Biol Psychiatry. 2007;62:765–772. doi: 10.1016/j.biopsych.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Milad MR, Wright CI, Orr SP, Pitman RK, Quirk GJ, Rauch SL. Recall of fear extinction in humans activates the ventromedial prefrontal cortex and hippocampus in concert. Biol Psychiatry. 2007;62:446–454. doi: 10.1016/j.biopsych.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Montague PR, Dayan P, Sejnowski TJ. A framework for mesencephalic dopamine systems based on predictive Hebbian learning. J Neurosci. 1996;16:1936–1947. doi: 10.1523/JNEUROSCI.16-05-01936.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MA, LeDoux JE. Differential contribution of dorsal and ventral medial prefrontal cortex to the acquisition and extinction of conditioned fear in rats. Behav Neurosci. 1993;109:681–688. doi: 10.1037//0735-7044.109.4.681. [DOI] [PubMed] [Google Scholar]
- Morris JS, Dolan RJ. Dissociable amygdala and orbitofrontal responses during reversal fear conditioning. Neuroimage. 2004;22:372–380. doi: 10.1016/j.neuroimage.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Morris JS, Ohman A, Dolan RJ. Conscious and unconscious emotional learning in the human amygdala. Nature. 1998;393:467–470. doi: 10.1038/30976. [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M. Mechanisms of fear extinction. Mol Psychiatry. 2007;12:120–150. doi: 10.1038/sj.mp.4001939. [DOI] [PubMed] [Google Scholar]
- O'Doherty J, Kringelbach ML, Rolls ET, Hornak J, Andrews C. Abstract reward and punishment representations in the human orbitofrontal cortex. Nat Neurosci. 2001;4:95–102. doi: 10.1038/82959. [DOI] [PubMed] [Google Scholar]
- O'Doherty J, Critchley H, Deichmann R, Dolan RJ. Dissociating valence of outcome from behavioral control in human orbital and ventral prefrontal cortices. J Neurosci. 2003a;23:7931–7939. doi: 10.1523/JNEUROSCI.23-21-07931.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Doherty JP, Dayan P, Friston K, Critchley H, Dolan RJ. Temporal difference models and reward-related learning in the human brain. Neuron. 2003b;38:329–337. doi: 10.1016/s0896-6273(03)00169-7. [DOI] [PubMed] [Google Scholar]
- O'Doherty JP, Buchanan TW, Seymour B, Dolan RJ. Predictive neural coding of reward preference involves dissociable responses in human ventral midbrain and ventral striatum. Neuron. 2006;49:157–166. doi: 10.1016/j.neuron.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Orr SP, Metzger LJ, Lasko NB, Macklin ML, Peri T, Pitman RK. De novo conditioning in trauma-exposed individuals with and without posttraumatic stress disorder. J Abnorm Psychol. 2000;109:290–298. [PubMed] [Google Scholar]
- Pare D, Quirk GJ, Ledoux JE. New vistas on amygdala networks in conditioned fear. J Neurophysiol. 2004;92:1–9. doi: 10.1152/jn.00153.2004. [DOI] [PubMed] [Google Scholar]
- Peri T, Ben-Shakhar G, Orr SP, Shalev AY. Psychophysiological assessment of aversive conditioning in posttraumatic stress disorder. Biol Psychiatry. 2000;47:512–519. doi: 10.1016/s0006-3223(99)00144-4. [DOI] [PubMed] [Google Scholar]
- Pezze MA, Feldon J. Mesolimbic dopaminergic pathways in fear conditioning. Prog Neurobiol. 2004;74:301–320. doi: 10.1016/j.pneurobio.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;48:175–187. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: role of the amygdala and vmPFC. Neuron. 2004;43:897–905. doi: 10.1016/j.neuron.2004.08.042. [DOI] [PubMed] [Google Scholar]
- Ploghaus A, Tracey I, Clare S, Gati JS, Rawlins JN, Matthews PM. Learning about pain: the neural substrate of the prediction error for aversive events. Proc Natl Acad Sci U S A. 2000;97:9281–9286. doi: 10.1073/pnas.160266497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Armony JL, LeDoux JE. Fear conditioning enhances different temporal components of tone-evoked spike trains in auditory cortex and lateral amygdala. Neuron. 1997;19:613–624. doi: 10.1016/s0896-6273(00)80375-x. [DOI] [PubMed] [Google Scholar]
- Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research–past, present, and future. Biol Psychiatry. 2006;60:376–382. doi: 10.1016/j.biopsych.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Rescorla RA, Wagner AR. A theory of Pavlovian conditioning: variations in the effectiveness of reinforcement and nonreinforcement. In: Black AH, Prokasy WF, editors. Classical conditioning II: current research and theory. New York: Appleton-Century-Crofts; 1972. pp. 64–99. [Google Scholar]
- Rolls ET. The functions of the orbitofrontal cortex. Brain Cogn. 2004;55:11–29. doi: 10.1016/S0278-2626(03)00277-X. [DOI] [PubMed] [Google Scholar]
- Schoenbaum G, Setlow B. Lesions of nucleus accumbens disrupt learning about aversive outcomes. J Neurosci. 2003;23:9833–9841. doi: 10.1523/JNEUROSCI.23-30-09833.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenbaum G, Chiba AA, Gallagher M. Orbitofrontal cortex and basolateral amygdala encode expected outcomes during learning. Nat Neurosci. 1998;1:155–159. doi: 10.1038/407. [DOI] [PubMed] [Google Scholar]
- Schoenbaum G, Chiba AA, Gallagher M. Neural encoding in orbitofrontal cortex and basolateral amygdala during olfactory discrimination learning. J Neurosci. 1999;19:1876–1884. doi: 10.1523/JNEUROSCI.19-05-01876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenbaum G, Chiba AA, Gallagher M. Changes in functional connectivity in orbitofrontal cortex and basolateral amygdala during learning and reversal training. J Neurosci. 2000;20:5179–5189. doi: 10.1523/JNEUROSCI.20-13-05179.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöenberg T, Daw ND, Joel D, O'Doherty JP. Reinforcement learning signals in the human striatum distinguish learners from nonlearners during reward-based decision making. J Neurosci. 2007;21:12860–12867. doi: 10.1523/JNEUROSCI.2496-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599. doi: 10.1126/science.275.5306.1593. [DOI] [PubMed] [Google Scholar]
- Seymour B, O'Doherty JP, Dayan P, Koltzenburg M, Jones AK, Dolan RJ, Friston KJ, Frackowiak RS. Temporal difference models describe higher-order learning in humans. Nature. 2004;429:664–667. doi: 10.1038/nature02581. [DOI] [PubMed] [Google Scholar]
- Seymour B, O'Doherty JP, Koltzenburg M, Wiech K, Frackowiak R, Friston K, Dolan R. Opponent appetitive-aversive neural processes underlie predictive learning of pain relief. Nat Neurosci. 2005;8:1234–1240. doi: 10.1038/nn1527. [DOI] [PubMed] [Google Scholar]
- Shalev AY, Peri T, Brandes D, Freedman S, Orr SP, Pitman RK. Auditory startle response in trauma survivors with posttraumatic stress disorder: a prospective study. Am J Psychiatry. 2000;157:255–261. doi: 10.1176/appi.ajp.157.2.255. [DOI] [PubMed] [Google Scholar]
- Sotres-Bayon F, Bush DE, LeDoux JE. Acquisition of fear extinction requires activation of NR2B-containing NMDA receptors in the lateral amygdala. Neuropsychopharmacology. 2007;32:1929–1940. doi: 10.1038/sj.npp.1301316. [DOI] [PubMed] [Google Scholar]
- Suri RE, Schultz W. Temporal difference model reproduces anticipatory neural activity. Neural Comput. 2001;13:841–862. doi: 10.1162/089976601300014376. [DOI] [PubMed] [Google Scholar]
- Sutton RS, Barto AG. Time-derivative models of Pavlovian reinforcement. In: Gabriel MJ, Moore J, editors. Learning and computational neuroscience: foundations of adaptive networks. Boston: MIT; 1990. pp. 497–537. [Google Scholar]
- Talairach J, Tournoux P. New York: Thieme; 1998. Co-planar stereotaxic atlas of the human brain: an approach to medical cerebral imaging. [Google Scholar]
- Yacubian J, Glascher J, Schroeder K, Sommer T, Braus DF, Buchel C. Dissociable systems for gain- and loss-related value predictions and errors of prediction in the human brain. J Neurosci. 2006;26:9530–9537. doi: 10.1523/JNEUROSCI.2915-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]