Dynamin 2 Potentiates Invasive Migration of Pancreatic Tumor Cells through Stabilization of the Rac1 GEF Vav1 (original) (raw)
. Author manuscript; available in PMC: 2014 Mar 25.
Summary
The large GTPase Dynamin 2 (Dyn2) is markedly upregulated in pancreatic cancer, is a potent activator of metastatic migration, and is required for Rac1-mediated formation of lamellipodia. Here we demonstrate an unexpected mechanism of Dyn2 action in these contexts, via direct binding to the Rac1 guanine nucleotide exchange factor (GEF) Vav1. Surprisingly, disruption of the Dyn2-Vav1 interaction targets Vav1 to the lysosome for degradation, via an interaction with the cytoplasmic chaperone Hsc70, resulting in a dramatic reduction of Vav1 protein stability. Importantly, a specific mutation in Vav1 near its Dyn2-binding C-terminal SH3 domain prevents Hsc70 binding, resulting in a stabilization of Vav1 levels. Dyn2 binding regulates the interaction of Vav1 with Hsc70 to control the stability and subsequent activity of this oncogenic GEF. These findings elucidate how Dyn2 activates Rac1, lamellipod protrusion, and invasive cellular migration and provide insight into how this specific Vav is ectopically expressed in pancreatic tumors.
Introduction
Invasive cell migration is a crucial process required throughout development, and is aberrantly upregulated in tumor cells, promoting cancer metastasis. Pancreatic cancer is the fourth leading cause of cancer death in the U.S., owing primarily to late detection and a high incidence of metastasis. Upon diagnosis these tumors have very often actively disseminated to a variety of different organs, resulting in an exceptionally low 5-year survival rate of approximately 5% (Jemal et al., 2010).
Several cytoskeletal proteins are aberrantly regulated in pancreatic cancers and correlate with increased tumor burden (Kikuchi et al., 2008; Matsuda et al., 2011; Ni et al., 2008; Wang et al., 2010; Welsch et al., 2007). We have reported that the large GTPase Dynamin 2 (Dyn2) is elevated in the majority of human pancreatic adenocarcinomas, and that these elevated levels support increased lamellipodial extension, cell migration, and invasion in vitro, and dissemination to distal organs in vivo using orthotopic mouse models (Eppinga et al., 2012). The conventional dynamins are well-known for a role in the endocytic process and function as pinchases to liberate newly forming endocytic pits (Doherty and McMahon, 2009; Hinshaw, 2000). This versatile family of mechanoenzymes also functions in endosomal trafficking, Golgi dynamics, and cytoskeletal regulation (Gu et al., 2010; Jones et al., 1998; McNiven et al., 2000a; Mooren et al., 2009; Schroeder et al., 2010). How elevated Dyn2 levels could potentiate invasive properties is not understood, although this enzyme has been implicated in regulating focal adhesion dynamics (Ezratty et al., 2005; Wang et al., 2011) and the assembly of branched actin networks that could facilitate lamellipodial extension (Kruchten and McNiven, 2006; Schafer, 2004).
Rac1 is a small GTPase, which cycles between an active, GTP-bound form and an inactive, GDP-bound form. This cycle is regulated by activating GEFs (guanine nucleotide exchange factors) and inactivating GAPs (GTPase activating proteins). Active Rac1 signals downstream to regulate actin dynamics and branching and induce the formation of lamellipodia (Ridley, 2011). Schlunck et al. demonstrated that Dyn2 regulates the localization of active Rac1, without affecting its activation, by regulating its internalization and trafficking to promote the formation of lamellipodia (Schlunck et al., 2004). However, how these distinct GTPases, the large mechanoenzyme Dyn2 and the small regulatory switch Rac1, might interact structurally or functionally remains undefined. One important study has shown that the Rac1 GEF Vav1 interacts directly with Dyn2 in T cells (Gomez et al., 2005).
Vav1 is a 95 kDa exchange factor that is regulated by phosphorylation, and also has proposed adapter functions through conserved SH2 and SH3 domains (Bustelo, 2001; Lazer et al., 2010; Turner and Billadeau, 2002). Vav1 expression is normally restricted to hematopoietic cells, where it is essential for the development and activation of T cells by modulating transcription and the cytoskeleton (Tybulewicz, 2005). However, Vav1 has also been defined as an oncogene, as deletion of the regulatory amino terminal region or mutation of regulatory tyrosine phosphorylation site Tyr174 results in transformation of fibroblasts (Katzav et al., 1989; Lopez-Lago et al., 2000). Critically, more recent findings that Vav1 is ectopically expressed in multiple tumor types and in cancer cell lines strengthens the argument that Vav1 may be a key driver of oncogenic transformation and tumor progression. Vav1 is aberrantly expressed in several human cancer types, including neuroblastoma, melanoma, lung cancer, and breast cancer (Bartolome et al., 2006; Hornstein et al., 2003; Katzav, 2009; Lazer et al., 2009). Most relevant is a report that Vav1 is ectopically expressed in over 50% of pancreatic ductal adenocarcinomas, where it regulates cell cycle progression through cyclin D1 expression to promote cell survival, proliferation, and transformation of cultured pancreatic cancer cells (Fernandez-Zapico et al., 2005), although no link to migratory invasion was examined.
As Dyn2 and Vav1 are both upregulated in human pancreatic cancers, and known to interact in hematopoietic cells, we hypothesized that Vav1 could link Dyn2 to Rac1 activation to promote migration of pancreatic tumor cells. Here, we demonstrate that Dyn2 and Vav1 associate in pancreatic cancer cells and not only promote, but are essential for Rac1 activation, lamellipodial extension, and invasive cell migration. Most interestingly, we have found that Dyn2 binding is essential for Vav1 protein stability, as a reduction in Dyn2 levels is paralleled by a near complete cellular loss of Vav1 and leads to an accumulation in, and subsequent degradation by, the lysosome. We have identified an interaction between Vav1 and the chaperone Hsc70 that directs Vav1 to the lysosome for degradation and is regulated by the direct binding of Dyn2 to Vav1. These findings define a functional relationship between a large contractile GTPase and the activation of the small GTPase Rac1 to potentiate pancreatic tumor cell invasion.
Results
Dyn2 is required for Rac1-dependent pancreatic cancer cell migration
The large GTPase Dyn2 is overexpressed in pancreatic tumors and promotes enhanced cancer cell migration and invasion (Eppinga et al., 2012), though how Dyn2 participates in this invasive process is unknown. In this study, we utilize four different pancreatic adenocarcinoma cell lines that possess distinct features and advantages as experimental models. Panc04.03 cells are highly motile, and provide an excellent model system to study migration. HPAF-II cells form large, stable lamellipodia at a high frequency following acute EGF stimulation so can be quantitatively measured for lamellipod formation. DanG and Panc04.03 are easily manipulated for biochemical studies. Importantly, DanG, Panc04.03, and HPAF-II cells are all positive for expression of the GEF Vav1, as described below. Finally, Panc1 cells do not express Vav1, and are an excellent model system to study ectopic expression of Vav1 on signaling, lamellipod formation, and cell migration.
The role of Dyn2 in cell migration was studied using a transwell chemotactic cell migration assay. Consistent with our previous report, silencing of Dyn2 in Panc04.03 pancreatic adenocarcinoma cells by siRNA results in a significant decrease in transwell cell migration (Fig 1A). As formation of lamellipodia is an important component of migration, we tested if lamellipod formation and extension were defective following Dyn2 knockdown. HPAF-II pancreatic tumor cells were transfected with either control siRNA or siRNA to Dyn2, and the cells were stimulated with EGF (100 ng/ml) for 20 min to induce the formation of lamellipodia. Depletion of Dyn2 resulted in a 50% decrease in the percent of cells forming lamellipodia, consistent with previous reports (Fig 1B) (Schlunck et al., 2004).
Figure 1. Dyn2 promotes pancreatic cancer cell migration through activation of Rac1.
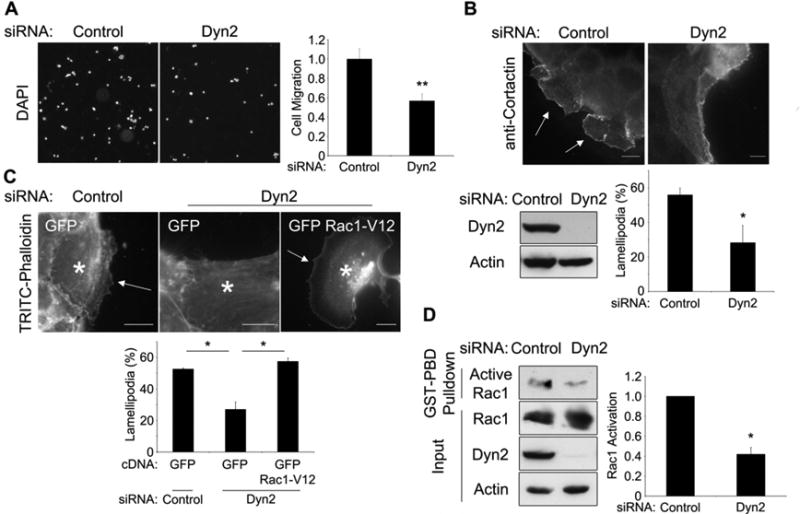
(A-D) Cells were depleted of Dyn2 by siRNA transfection. (A) Panc04.03 cells seeded in a transwell assay were incubated for 16 h, counted by DAPI stain and normalized to siRNA-transfected controls. (B) HPAF-II cells were serum-starved, then stimulated with EGF (50 ng/ml) for 20 min and stained for cortactin to show lamellipodia (arrows). (n > 100 cells/experiment) (C) HPAF-II cells were transfected with GFP-Rac1-V12 or GFP vector alone, then treated as in (B). Asterisks indicate transfected cells. (D) Panc04.03 cells were subjected to a GST-PBD pulldown assay for active Rac1 and immunoblotted for the indicated proteins. Active Rac1 levels were normalized to actin; this ratio was normalized to control siRNA-transfected cells. All scale bars = 10 μm. All graphs represent the mean +/- SEM of at least 3 independent experiments. * indicates p<0.05; ** indicates p<0.01.
As Rac1 activation is critical for the extension of lamellipodia, and as both Dyn2 and Rac1 have been implicated in tumor cell migration, we hypothesized that Rac1 activation was impaired as a result of Dyn2 knockdown. Therefore, we predicted that overexpression of constitutively active Rac1 could rescue the defect in lamellipod formation in Dyn2 knockdown cells. Indeed, expression of Rac1-V12 completely restored the formation of lamellipodia in HPAF-II cells depleted of Dyn2 (Fig 1C) suggesting that Rac1 activation is downstream of Dyn2. To test this prediction biochemically, Dyn2 was depleted using siRNA in Panc04.03 cells and Rac1 activation was assessed by a GST pulldown assay using the p21-binding domain (PBD) of PAK1, which binds only GTP-bound, active Rac1. Interestingly, Rac1 activation was decreased by over 50% following siRNA-mediated depletion of Dyn2 (Fig 1D). Together, these data suggest that Dyn2 potentiates lamellipod formation and migration of pancreatic cancer cells by promoting the activation of Rac1.
Dyn2 binds the Rac1 GEF Vav1 in pancreatic cancer cells
From the observations described above, we predicted that Dyn2 could either promote the activity of a Rac1-activating GEF, or inhibit the activity of a Rac1-inactivating GAP. It has been shown in T cells that Dyn2 interacts with the Rac1 GEF Vav1 (Gomez et al., 2005). Vav1 is normally expressed in hematopoietic cells and not normal epithelial cells, but has been shown to be ectopically expressed in over 50% of pancreatic cancers and in multiple pancreatic cancer cell lines (Fernandez-Zapico et al., 2005) (Fig S1A), where it has been implicated in cell cycle regulation. Because Dyn2 is also overexpressed in pancreatic cancers, we predicted that Vav1 could be a link between Dyn2 and Rac1 in pancreatic cancer cells.
Using co-immunoprecipitation, we found that endogenous Vav1 and Dyn2 associate in pancreatic cancer cells (Fig 2A). It was previously reported that the Dyn2/Vav1 interaction is mediated by the proline-rich domain of Dyn2 (PRD), and the C-terminal SH3 domain of Vav1 (Fig 2B) (Gomez et al., 2005). GST pulldown experiments in the tumor cell lines confirmed that the purified Vav1 SH3 domain interacts with full length Dyn2, and this interaction was inhibited by deletion of residues 800-811 within the Dyn2 PRD (Δ800-811, Fig 2C). Disruption of the Vav1 SH3 by mutation (WW820/821YY) blocked the capacity to pull down Dyn2, demonstrating that an intact SH3 domain is required for Vav1/Dyn2 binding (Fig. 2D). To confirm that the Dyn2-Vav1 interaction is direct, the interaction between the purified Vav1 SH3 and the purified, His-tagged Dyn2 PRD was tested. We observed that the Dyn2 PRD bound strongly to GST-Vav1-SH3, and that mutating the Vav1 SH3 domain (WWYY) blocked this interaction (Fig 2E). Similar results were observed using purified full-length Dyn2 (data not shown), and demonstrate that Dyn2 and Vav1 interact directly in pancreatic cancer cells.
Figure 2. Dyn2 binds to Vav1 in the lamellipodia of pancreatic cancer cells.
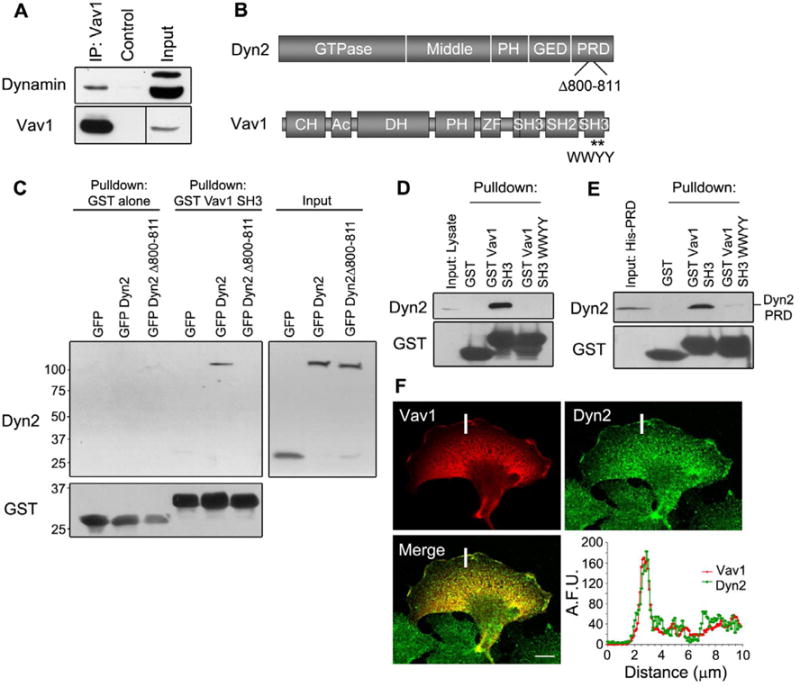
(A) DanG cells were immunoprecipitated for Vav1, and then immunoblotted for Dyn2 or Vav1. A longer exposure was needed to detect Vav1 in the input. (B) Cartoons showing the domain structure of Dyn2 and Vav1. Dyn2 includes a GTPase domain, a middle domain, a plekstrin homology domain (PH), a GTPase effector domain (GED), and a proline and arginine rich domain (PRD). Vav1 contains a calponin homology domain (CH), an acidic domain (Ac), a Dbl homology domain (DH), a plekstrin homology domain (PH), a zinc finger (ZF), two Src homology 3 domains (SH3), and a Src homology 2 domain (SH2). Mutations used throughout the manuscript are indicated (Dyn2 Δ800-811, Vav1 WWYY). (C) MiaPaCa2 cells were transfected with GFP, GFP-Dyn2, or GFP-Dyn2 Δ800-811, and lysates were precipitated using purified GST-Vav1-SH3. (D) Endogenous Dyn2 from MiaPaCa2 cell lysate co-precipitated with purified GST-Vav1-SH3, but not when the SH3 domain was mutated (WWYY). (E) Purified His-Dyn2 PRD was precipitated with purified GST-Vav1-SH3, indicating that Dyn2 and Vav1 interact directly. (F) Panc1 cells were transfected with Vav1, then stained for Vav1 (red) and endogenous Dyn2 (green). The intensity of antibody staining was quantified over the white bar and is plotted in the lower panel. A.F.U. = Arbitrary fluorescence units. All scale bars = 10 μm. See also Table S1.
In independent studies, both Dyn2 and Vav1 proteins have been shown to localize to the leading edge of migrating cells (McNiven et al., 2000b; Miranti et al., 1998). To determine if these interacting proteins colocalize in cells, Panc1 cells were transfected with Vav1, and Vav1 and Dyn2 localization were assessed by immunofluorescence. We observed that both proteins localize to the lamellipodia (Fig 2F) and share similar membrane domains within these extensions in support of the immunoprecipitation data described above.
Expression of Vav1 in pancreatic tumor cells enhances survival, proliferation, and oncogenic transformation (Fernandez-Zapico et al., 2005). In addition, in T cells, Vav1 is a known regulator of spreading and induction of lamellipodia during integrin engagement (del Pozo et al., 2003). As Vav1 is a known activator of Rac1, a central player in cell migration, we hypothesized that Vav1 could also promote the migration of pancreatic tumor cells. To this end, the effects of Vav1 on Rac1 activation, lamellipod formation, and chemotactic cell migration were determined. Depletion of Vav1 expression using siRNA inhibited Rac1 activation in DanG cells by 50% (Fig 3A), a finding that was not necessarily anticipated, as most migratory epithelial cells do not express Vav1. From this observation, as well as the lamellipodial localization of Vav1 in Figure 2F, we predicted that Vav1-dependent Rac1 activation could enhance the formation of lamellipodia in the tumor cells expressing this GEF. Indeed, siRNA-mediated depletion of Vav1 in HPAF-II cells blocked the formation of lamellipodia (Fig 3B), as well as the related process of spreading on a fibronectin substrate (Fig S1B). Knockdown of Vav1 also significantly inhibited chemotactic migration through a transwell chamber by 70% (Fig 3C), and inhibited migration in a wound healing assay (Fig S1C, D). These data indicate that Vav1 is a potent activator of pancreatic tumor cell migration.
Figure 3. Vav1 promotes invasive migration of pancreatic cancer cells.
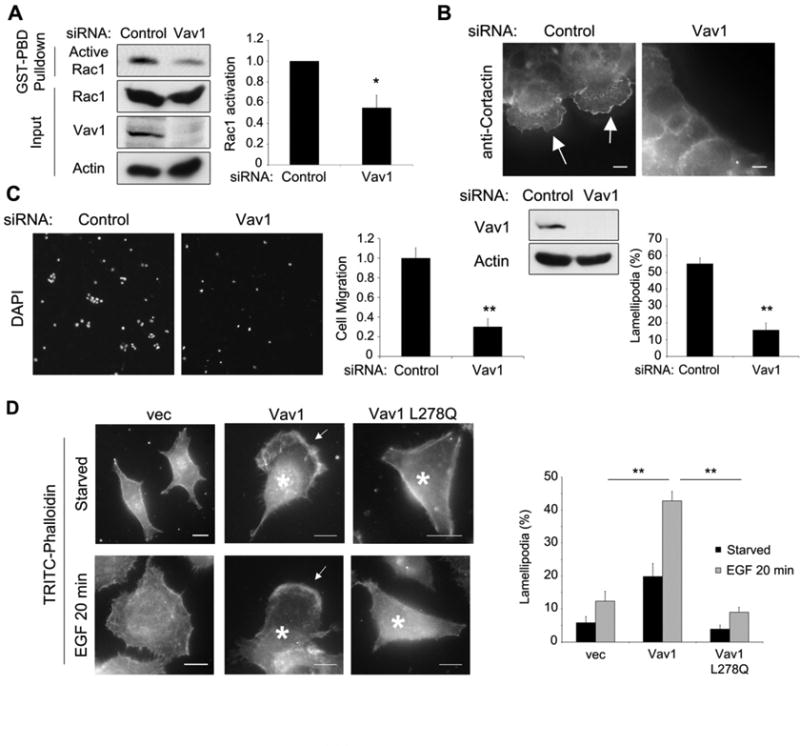
(A-C) Cells were depleted of Vav1 by siRNA transfection. (A) DanG cells were subjected to a GST-PBD pulldown for active Rac1. Active Rac1 was normalized to total Rac1, and ratios were normalized to control cells. (B) HPAF-II cells were serum-starved and stimulated with EGF (50 ng/ml, 20 min) and stained for cortactin to show lamellipodia (arrows). Scale bars = 10 μm. (C) Panc04.03 cells seeded in a transwell assay were incubated for 16 h, counted by DAPI stain and normalized to siRNA-transfected controls. (D) Panc1 cells were transfected with a control vector (vec), WT Vav1, or GEF-inactive Vav1 (L278Q), then serum-starved, and stimulated with EGF (50 ng/ml, 20 min), and stained for actin to show lamellipodia (arrows) and Vav1 (not shown). Asterisks indicate transfected cells. Scale bars = 10 μm. All graphs represent the mean +/- SEM of three independent experiments. * indicates p<0.05; ** indicates p<0.01. See also Fig S1.
As Vav1 is ectopically expressed in many, but not all, pancreatic cancer cell lines (Fig S1A), we tested if Vav1 overexpression in a tumor cell line that does not express this GEF (Panc1 cells) could induce cell migration and lamellipod formation. Panc1 cells were transfected with wild-type Vav1 and stimulated with EGF (50 ng/ml, 20 min) to induce lamellipod formation. Overexpression of wild-type Vav1 markedly increased the percentage of cells capable of forming lamellipodia, but this effect was blocked by expression of a mutated Vav1 lacking GEF activity (L278Q, Fig 3D). Strikingly, high levels of wild-type Vav1 were sufficient to induce massive lamellipodia and induce a motile phenotype in Panc1 cells even in the absence of EGF stimulation (Fig 3D), demonstrating that Vav1 is a potent activator of cell migration in these tumor cells through its exchange factor activity.
A Dyn2/Vav1 interaction promotes Rac1 activation, lamellipod formation, and cell migration
Because Vav1 and Dyn2 are both required for maximal Rac1 activation and the formation of lamellipodia, we tested if the Dyn2-Vav1 interaction was important for Rac1 activation and tumor cell migration. First, Panc04.03 cells were stably transduced with a lentivirus expressing WT Dyn2 or Dyn2 Δ800-811, and Rac1 activation was tested using a biochemical GST-PBD pulldown. Overexpression of WT Dyn2 increased the amount of active Rac1 nearly twofold, whereas Dyn2 Δ800-811, which does not bind Vav1, inhibited Rac1 activation (Fig 4A). Next, Panc1 cells, which do not express endogenous Vav1, were transfected with WT Vav1 or Vav1 WWYY, which does not bind Dyn2 (Fig 2). Importantly, overexpression of Vav1 potently increased levels of active Rac1, and this was completely inhibited by Vav1 WWYY (Fig 4B). These results demonstrate that the Dyn2-Vav1 interaction promotes activation of Rac1.
Figure 4. A Dyn2/Vav1 interaction promotes Rac1 activation and cell migration.
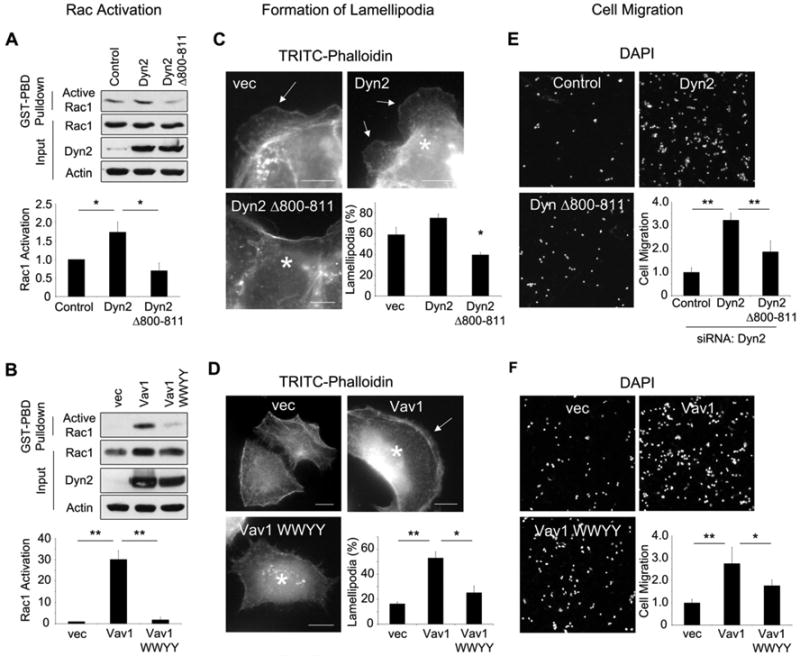
(A) WT Dyn2 or Dyn2 Δ800-811 was stably overexpressed in Panc04.03 cells, or (B) WT Vav1 or Vav1 WWYY were transiently overexpressed in Panc1 cells. For the expression of the Vav1 WWYY mutant, at least 10-fold higher amounts of cDNA were transfected compared to WT Vav1 to obtain comparable protein levels by immunoblot. For both (A) and (B), lysates were subjected to a GST-PBD pulldown for active Rac1. Active Rac1 was normalized to total Rac1, and ratios were normalized to vector-expressing control cells. (C) HPAF-II cells were transfected with empty vector (vec), WT Dyn2, or Dyn2 Δ800-811, or (D) Panc1 cells were transfected with vector, WT Vav1, or Vav1 WWYY. For both (C) and (D), cells were serum-starved, and stimulated with EGF (50 ng/ml, 20 min), and stained for myc-Dyn2 or Vav1 (not shown) and actin to show lamellipodia (arrows). Asterisks indicate transfected cells. Scale bars = 10 μm. (E) Panc04.03 cells stably expressing WT Dyn2 or Dyn2 Δ800-811 were depleted of endogenous Dyn2 by siRNA, or (F) Panc1 cells were transfected with WT Vav1 or WWYY. For both (E) and (F), cells were seeded in transwell chambers and incubated for 16 hours, counted by DAPI staining, and normalized to control cells. Two fields from three independent experiments were scored. All graphs represent the average +/- SEM of three independent experiments. * indicates p<0.05; ** indicates p<0.01. See also Fig S2.
To test if this Rac1 activation translated into lamellipodia formation, wild-type Dyn2 or Dyn2 Δ800-811 was expressed in HPAF-II cells stimulated with EGF. While expression of WT Dyn2 modestly increased the percentage of cells forming lamellipodia, Dyn2 Δ800-811 expression inhibited lamellipod formation (Fig 4C). Similarly, overexpression of WT Vav1 in Panc1 cells increased the percentage of lamellipod-forming cells in response to EGF stimulation, but Vav1 WWYY inhibited lamellipod formation (Fig 4D). These data indicate that the Vav1/Dyn2 interaction promotes the formation of lamellipodia in pancreatic cancer cells.
The biochemical and cell biological observations described above were extended to test the role of this interaction in chemotactic transwell cell migration. Dyn2 was depleted in Panc04.03 cells by siRNA, then WT Dyn2 or Dyn2 Δ800-811 was stably re-expressed using a lentiviral expression system. Consistent with the effects on lamellipod formation, re-expression of WT Dyn2 potently enhanced chemotactic cell migration, but Dyn2 Δ800-811 inhibited this effect by 50% (Fig 4E). Similarly, transient overexpression of Vav1 in Panc1 cells markedly enhanced chemotactic cell migration while this effect was inhibited by Vav1 WWYY (Fig 4F). Together, these data are consistent with the premise that a Dyn2 interaction with the GEF Vav1 is required to promote Rac1 activation and the subsequent formation of lamellipodia to support the migration of pancreatic cancer cells.
Dyn2 binding regulates Vav1 protein stability
Given the potent effects of the Dyn2-Vav1 interaction on Rac1 activity and function, we sought to determine the mechanism by which Dyn2-Vav1 binding regulates Rac1 activation and tumor cell migration. As Dyn2 has been implicated in regulating Rac1 signaling through endocytic trafficking (Kawada et al., 2009; Palamidessi et al., 2008; Schlunck et al., 2004), we first hypothesized that Vav1 binding could regulate Dyn2 enzymatic activity or function in endocytosis. However, no inhibition of endocytosis in cells expressing Dyn2 Δ800-811 was observed as determined by measuring uptake of labeled EGF (Fig S2A). Similarly, expression of Vav1 WWYY did not inhibit EGF uptake compared to cells expressing WT Vav1 (Fig S2B). Dyn2Δ800-811 had no effect on transferrin uptake in Panc1 cells, which do not express Vav1, indicating that this mutant protein can support endocytosis (Fig S2C). These data suggest that the Dyn2-Vav1 interaction does not regulate Dyn2 function in endocytosis, and that Dyn2 regulates Rac1 activation by a distinct mechanism.
Unexpectedly, when Dyn2 was depleted using siRNA, there was a concomitant and significant (80%) reduction of Vav1 protein (Fig 5A). This effect was observed using multiple siRNAs, using lentiviral-based shRNA depletion of Dyn2, and in multiple cell lines (Fig S3A and data not shown), indicating the loss of Vav1 protein is widespread and not due to an off-target effect of siRNA-mediated knockdown of Dyn2. These findings are exciting as they provide a potential mechanism toward how the Dyn2-Vav1 interaction could regulate Rac1 activity. To define how Dyn2 might affect the levels of Vav1 protein, we first tested if Dyn2 affects Vav1 transcription using RT-PCR. There was no change in Vav1 mRNA levels observed in cells depleted of Dyn2 by siRNA, suggesting that Dyn2 regulates Vav1 post-transcriptionally (Fig S3B). To test if Vav1 protein stability was altered in the absence of Dyn2, a timecourse of cycloheximide treatment was performed to inhibit new protein synthesis and to monitor turnover of endogenous Vav1 in the presence or absence of endogenous Dyn2. Vav1 levels remained stable over the 6-hour timecourse in DanG cells transfected with a control siRNA. However, in the absence of Dyn2, Vav1 protein turned over rapidly, with a half-life of less than four hours (Fig 5B).
Figure 5. Dyn2 interaction mediates Vav1 protein stability.
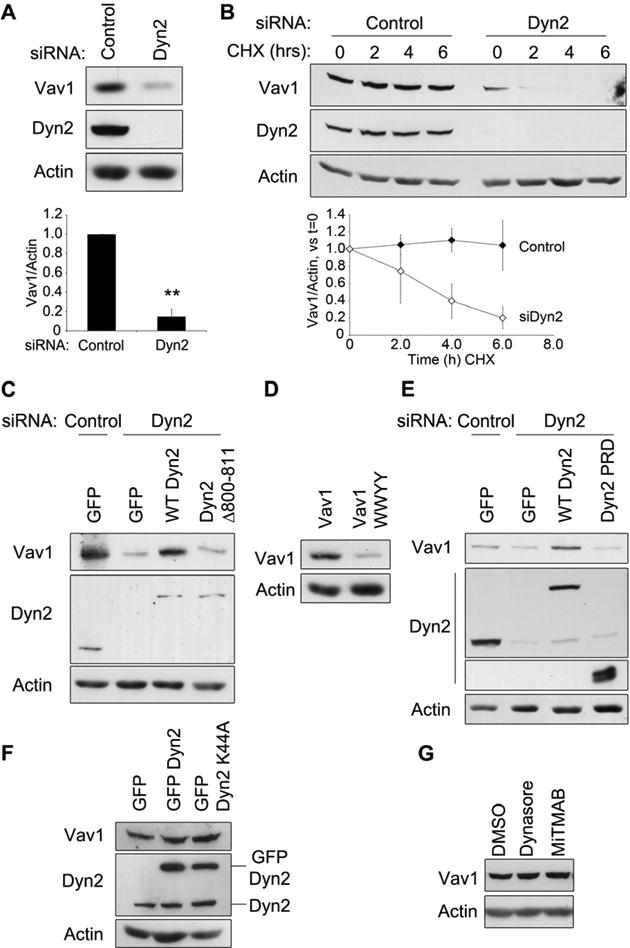
(A) DanG cells were transfected with control siRNA or siRNA targeting Dyn2, and Vav1 protein levels were normalized to actin. (B) DanG cells were transfected with control siRNA (closed symbols) or siRNA targeting Dyn2 (open symbols), then treated with cycloheximide (CHX, 50 μg/ml) for 0-6 hours. Vav1 levels were normalized to actin and compared to t=0. For (A) and (B), graphed values represent the mean +/- SEM of three independent experiments. ** indicates p<0.001. (C-G) Lysates were immunoblotted for the indicated proteins to determine the effects on Vav1 levels. (C) Panc04.03 cells were depleted of Dyn2 using siRNA, then transfected with GFP vector, GFP Dyn2, or GFP Dyn2 Δ800-811. (D) Panc1 cells, which do not express Vav1, were transfected with equal amounts of cDNA for WT Vav1 or Vav1 WWYY. (E) Panc04.03 cells were transfected with siRNA to Dyn2 or control siRNA, and then with either empty GFP vector, WT GFP Dyn2, or GFP-Dyn2-PRD. (F) DanG cells were transfected with GFP, GFP WT Dyn2, or GFP Dyn2 K44A. (G) DanG cells were treated with DMSO, Dynasore (80 μM), or MiTMAB (10 μM) for 24 hours. See also Fig S3.
These findings support the intriguing concept that Dyn2 protein levels affect Vav1 protein stability, possibly by a direct structural interaction. To test this, we assessed Vav1 protein levels upon disruption of Dyn2-Vav1 binding by depleting Dyn2 by siRNA, and then re-expressing either WT Dyn2 or Dyn2 Δ800-811. Vav1 protein levels were completely rescued by expression of GFP-tagged WT Dyn2, but not by re-expression of GFP-Dyn2 Δ800-811 (Fig 5C). Similarly, protein levels of transiently-transfected Vav1 WWYY were also dramatically reduced compared to WT Vav1, even though transcript levels of WT Vav1 and Vav1 WWYY were nearly identical (Fig 5D, Fig S3C). Taken together, these results demonstrate that Dyn2 binding regulates Vav1 protein turnover.
As Dyn2-Vav1 binding is mediated by a PRD/SH3 interaction, and because the PRD is sufficient to bind Vav1 (Fig 2), we tested if re-expression of the Dyn2 PRD alone was sufficient to restore Vav1 protein levels in pancreatic cancer cells depleted of Dyn2. Expression of the Dyn2 PRD did not rescue Vav1 levels in Dyn2 knockdown cells (Fig 5E), indicating that the SH3/PRD interaction alone is not sufficient to stabilize Vav1. Further, the decrease in Vav1 protein stability is not due to a loss of Dyn2 enzymatic activity, as expression of dominant negative Dyn2 (K44A) or treatment with the Dyn2 inhibitors Dynasore or MiTMAB had no effect on Vav1 protein levels (Fig 5F-G, S3D). Taken together, these data indicate that Dyn2 binding is required to maintain Vav1 protein stability.
Dyn2 protects Vav1 from lysosomal degradation
We next sought to determine the mechanism by which Vav1 is degraded in the absence of Dyn2 binding. Previous reports have indicated that Vav1 can be ubiquitinated and degraded by the proteasome (Bustelo et al., 1997; De Sepulveda et al., 2000; Miura-Shimura et al., 2003). However, we were unable to detect any ubiquitination of Vav1 in Dyn2 knockdown cells. Similarly, treatment with the protease inhibitor MG132 did not prevent Vav1 degradation when Dyn2 was depleted (Fig S3E, F). Some cytoplasmic proteins can be targeted to the lysosome for subsequent degradation. To test this possibility, lysosomal degradation was inhibited by treating DanG cells with chloroquine. Surprisingly, chloroquine treatment blocked the degradation of Vav1 when Dyn2 was depleted by siRNA (Fig 6A). Similar results were obtained using NH4Cl to inhibit the lysosome (Fig S3G), suggesting that Dyn2 protects Vav1 from lysosomal degradation.
Figure 6. Dyn2 protects Vav1 from degradation by the lysosome.
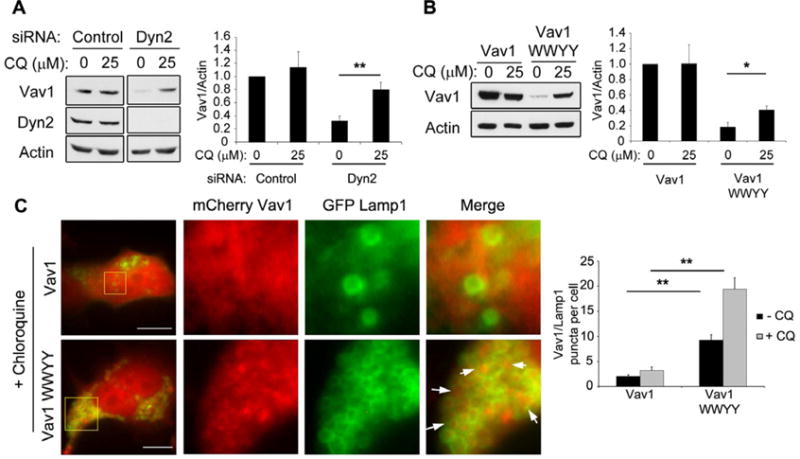
(A) DanG cells were transfected with a control siRNA or an siRNA against Dyn2, and treated with chloroquine (CQ, 25 μM) for 48 hours. Vav1 protein levels were normalized to actin. (B) Panc1 cells were transfected with WT Vav1 or Vav1 WWYY, were treated with 25 μM chloroquine (CQ) for 48 hours, and Vav1 protein levels were normalized to actin. For both (A) and (B), graphs represent the mean +/- SEM of three independent experiments. * indicates p<0.05, ** indicates p<0.001. **(C)** DanG cells were co-transfected with mCherry Vav1 (WT or WWYY) and GFP-Lamp1 and incubated for 48 hours in the presence of CQ (25 μM). Representative images show mCherry Vav1 WWYY accumulating in the lumen of GFP-Lamp1-positive vesicles (arrows). Yellow boxed areas in left images are magnified at right. Scale bar = 10 μm. The number of GFP-Lamp1 positive vesicles per cell containing mCherry Vav1 or mCherry Vav1 WWYY was counted (n>25 cells per experiment). Graphed data represent the mean number of vesicles +/- SEM from three independent experiments; ** indicates p < 0.01.
To confirm that lysosomal targeting of Vav1 is regulated by the Dyn2-Vav1 interaction, WT Vav1 or Vav1 WWYY was transfected into Panc1 cells that were then treated with chloroquine. Again, this treatment blocked the degradation of Vav1 WWYY (Fig 6B), indicating that Vav1 WWYY is degraded by the lysosome, consistent with Dyn2 binding protecting Vav1 from lysosomal degradation. To support these biochemical observations, we tested if Vav1 might associate with the lysosomal marker Lamp1. As our antibodies against Vav1 do not recognize endogenous Vav1 by immunofluorescence, we transfected DanG cells with mCherry-tagged WT Vav1 or Vav1 WWYY, along with GFP-tagged Lamp1. While mCherry-WT Vav1 showed minimal colocalization with GFP-Lamp1, the mutant mCherry-Vav1 WWYY protein showed significant accumulation within the lumen of Lamp1-positive vesicles (Fig 6C). Consistent with this finding, the lysosomal localization of Vav1 WWYY was further enhanced by chloroquine treatment to inhibit lysosomal degradation. Taken together, these data support the concept that Dyn2 binding protects Vav1 from targeted degradation by the lysosome.
Cytoplasmic proteins may be targeted for lysosomal degradation via an interaction with the chaperone protein Hsc70. Hsc70 binding directs cytoplasmic proteins to lysosomes via chaperone mediated autophagy (CMA) (Kaushik and Cuervo, 2012), to late endosomes via endosomal microautophagy (Sahu et al., 2011), and to autophagosomes for macroautophagy (Arndt et al., 2010). Therefore, we tested if Vav1 might interact with Hsc70, with the hypothesis that this interaction could be regulated by Dyn2 binding. Indeed, Vav1 co-immunoprecipitated with Hsc70 in DanG cells (Fig 7A). Most striking was the fivefold increase in the interaction between Vav1 and Hsc70 when Dyn2 was depleted by siRNA, suggesting that Dyn2 prevents the binding of Hsc70 to Vav1. To determine if this was an effect of Dyn2 binding, we expressed the Vav1 WWYY mutant that cannot bind Dyn2, or WT Vav1, in Panc1 cells. Again, Vav1 co-immunoprecipitated endogenous Hsc70, and this binding was increased by fivefold to the WWYY mutant (Fig 7B). In both cases, the increase in Hsc70 binding corresponds to the increased lysosomal degradation of Vav1 when Dyn2 association is prevented.
Figure 7. Dyn2 protects Vav1 from Hsc70-mediated degradation.
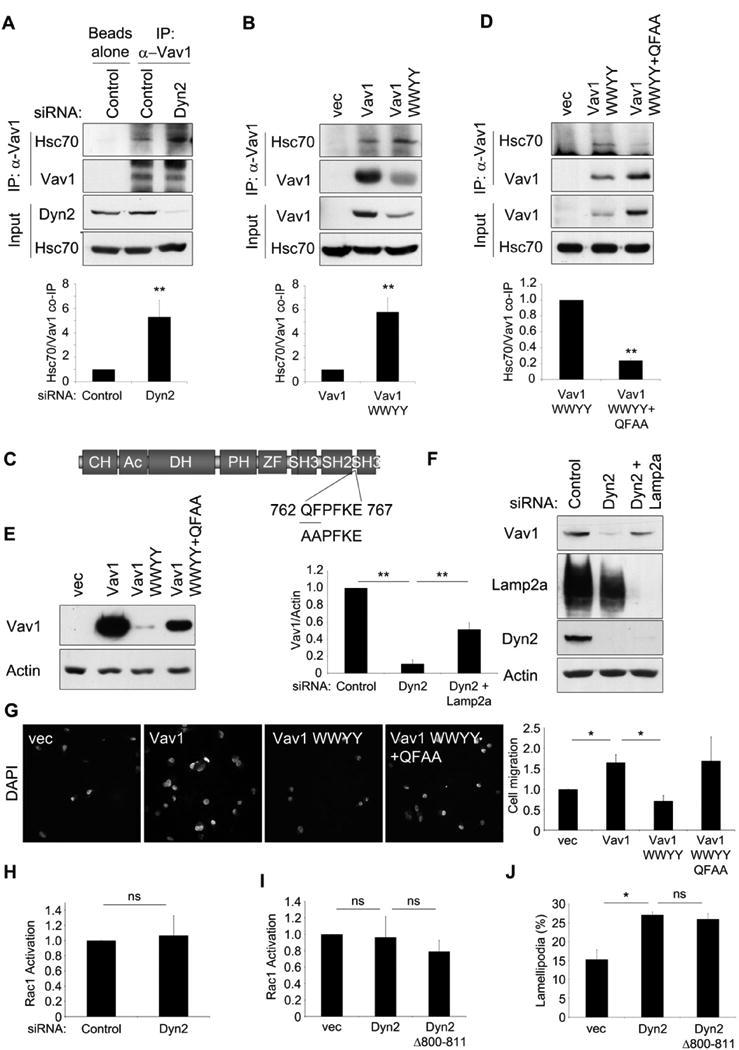
(A) DanG cells were transfected with siRNA targeting Dyn2, or control siRNA. then were immunoprecipitated for Vav1 and blotted for Hsc70. (B) Panc1 cells were transfected with empty vector, WT Vav1, or Vav1 WWYY, and lysates were immunoprecipitated for Vav1 and blotted for Hsc70. (C) Identification of an Hsc70 binding motif in Vav1. Residues QF762/763 were mutated to AA (“QFAA”). (D) Panc1 cells were transfected with empty vector, Vav1 WWYY, or Vav1 WWYY QFAA, and lysates were immunoprecipitated for Vav1 and blotted for Hsc70. For graphs, co-precipitated Hsc70 was normalized to immunoprecipited Vav1 and compared to control cells. (E) Panc1 cells were transfected with equal amounts of cDNA for the indicated constructs, and lysates were immunoblotted for Vav1 or actin. (F) Panc04.03 cells were transfected with siRNAs targeting Dyn2 and Lamp2a to inhibit CMA, and Vav1 levels were normalized to actin. (G) Panc1 cells were transfected with the indicated constructs and were seeded in a transwell migration assay for 16h. The number of migrating cells was determined by DAPI stain and normalized to vector-transfected cells. (H) Panc1 cells were transfected with a control siRNA or siRNA against Dyn2, and Rac1 activation was assessed by GST-PBD pulldown. (I) Panc1 cells were transfected with empty vector (vec), WT Dyn2, or Dyn2 Δ800-811 and subjected to a GST-PBD pulldown to assess Rac1 activation. For both (H) and (I), graphed data represent active Rac1/total Rac1 normalized to control cells. (J) Panc1 cells were transfected with empty vector (vec), WT Dyn2, or Dyn2 Δ800-811, serum-starved, and stimulated with EGF (50 μg/ml) for 20 min. Graph shows the percent of cells forming lamellipodia. All graphs represent the mean +/-SEM of at least three independent experiments. * indicates p<0.05; ** indicates p<0.01; ns= no statistically significant difference. See also Fig S4.
If Hsc70 binding is necessary for targeting Vav1 to the lysosome, then we predicted that disruption of Vav1/Hsc70 binding would block the degradation of Vav1 when it cannot bind Dyn2. Hsc70 targets proteins for lysosomal degradation through recognition of a loosely conserved “KFERQ” motif, consisting of a pentapeptide containing a basic residue, an acidic residue, a bulky hydrophobic residue, one more of these, and a glutamine at the N- or C-terminus (Kaushik and Cuervo, 2012). Vav1 contains multiple potential Hsc70-binding motifs. Using site-directed mutagenesis, we identified one particular motif (762 QFPFKE 767), that, when mutated (QF 762/763 to AA, or “QFAA”), reduced binding to Hsc70 by nearly 80% (Fig 7C, D). While this motif is not a perfect match for the previously described “KFERQ” motif, it does contain all of the essential elements for Hsc70 binding, as described above. Importantly, this mutation also resulted in dramatically increased Vav1 protein levels compared to the Vav1 WWYY mutant, even though the constructs were transcribed at comparable levels (Fig 7E, S3H), and rendered Vav1 WWYY insensitive to chloroquine treatment (Fig S3I). Therefore, blocking Hsc70 binding is sufficient to restore Vav1 stability.
Hsc70 is known to target proteins for lysosomal degradation through CMA, which requires the lysosomal transmembrane protein Lamp2a. To determine if Vav1 degradation is dependent upon Lamp2a and, in turn, CMA, we tested if siRNA-mediated depletion of Lamp2a resulted in elevated levels of Vav1 when Dyn2 is reduced by siRNA. Panc04.03 cells were transfected with siRNAs targeting Dyn2 and Lamp2a, or a control siRNA, and then Vav1 levels were assessed by immunoblotting. Indeed, knockdown of Lamp2a resulted in a marked rescue of Vav1 protein levels (Figure 7F). Taken together, these data strongly suggest that, in the absence of Dyn2 binding, Vav1 is targeted for lysosomal degradation through Hsc70 binding and CMA.
Re-expression of Vav1 rescues defects in cells depleted for Dyn2
The observations described above underscore the importance of a Dyn2-Vav1 interaction in Vav1 stability (Fig 5), Rac1 activity, lamellipod extension, and cell motility (Fig 4). From these findings we postulated that overexpression of Vav1 in Dyn2 knockdown cells should overcome the loss of Vav1 protein stability and therefore rescue the defects in these processes. To test this, Panc04.03 cells were depleted of Dyn2 using siRNA, and Vav1 was overexpressed using a lentiviral system. As described above, transwell cell migration was reduced by 50% upon Dyn2 knockdown (Fig S4A). Strikingly, re-expression of Vav1 in the Dyn2 knockdown cells completely rescued the defects in cell migration, showing that overcoming the loss of Vav1 protein stability restores Dyn2-dependent migration (Fig S4A). Similarly, stabilization of Vav1 WWYY by inhibiting Hsc70 binding also rescued cell migration (Fig 7G). These data are important because they suggest that Vav1 protein levels are regulated downstream of Dyn2, and indicate that the primary mechanism by which Dyn2 regulates Rac1 activity and invasive cell migration in pancreatic cancer cells is through modulation of Vav1 protein levels.
If this is true, then we predicted that depletion of Dyn2 should have no effect on Rac1 activation in pancreatic cancer cells that do not express Vav1. To test this, Dyn2 was knocked down in Panc1 cells, which do not express Vav1, and Rac1 activation was measured by GST-PBD pulldown. As predicted, Dyn2 depletion did not inhibit Rac1 activation in Panc1 cells, further indicating that the effects of Dyn2 on Rac1 signaling are mediated through Vav1 (Fig 7H, S4B). We also predicted that expression of the Dyn2 Δ800-811 mutant should not affect Rac1 activation in cells that do not express Vav1. To this end Panc1 cells were transfected with WT Dyn2 or Dyn2 Δ800-811 prior to testing for Rac1 activation and lamellipod formation as described above. In Panc1 cells, in contrast to DanG, Panc04.03, or HPAF-II cells that do express endogenous Vav1 (Fig S1A), expression of either WT Dyn2 or Dyn2 Δ800-811 had no effect on Rac1 activation (Fig 7I, S4C). Overexpression of Dyn2 did increase the percentage of Panc1 cells forming lamellipodia (Fig 7J, S4D), but this was not inhibited by Dyn2 Δ800-811. These data show that Dyn2 mediates Rac1 activation through Vav1, and that the inhibitory effects of Dyn2 Δ800-811 are also due to regulation of Vav1. Taken together, these data suggest that a physical interaction with Dyn2 controls Vav1 stability to regulate Rac1-mediated lamellipod formation and tumor cell migration.
Discussion
In this study we provide evidence for the large GTPase Dyn2 regulating the small GTPase Rac1 to potentiate the invasive migration of pancreatic tumor cells. Dyn2 plays an essential role in the regulation of Rac1-mediated pancreatic tumor cell migration through modulation of the Rac1 activator Vav1 via a direct interaction. Surprisingly, this interaction is required to maintain Vav1 protein stability, as this GEF is turned over rapidly by the lysosome in the absence of Dyn2, leading to decreased Rac1 activation, lamellipod formation, and cell migration.
An enzymatic versus adapter role of Dyn2 in regulating tumor cell migration
Dyn2 has been proposed to regulate the activity of Rac1 indirectly by promoting its endocytosis and trafficking. Rac1 is internalized and activated on early endosomes, and is then recycled back to the plasma membrane to ensure its localization to the leading edge, promoting the formation of migratory lamellipodia. This process is disrupted by expression of dominant negative Dyn2 (Dyn2 K44A), which has been shown to inhibit endocytosis, perturb the localization of active Rac1, and attenuate the formation of lamellipodia (Palamidessi et al., 2008; Schlunck et al., 2004)_ENREF 2_. In support of this model, disruption of Dyn2 enzymatic activity using Dynasore also inhibits lamellipod formation and tumor cell migration (Eppinga et al., 2012; Yamada et al., 2009). Interestingly, while expression of Dyn2 K44A disrupts Rac1 localization and downstream effects, it does not inhibit the activation of Rac1 by itself, but only Rac1 activation induced by the overexpression of Rab5 (Palamidessi et al., 2008; Schlunck et al., 2004). In other model systems, RNAi-mediated depletion of Dyn2 inhibits growth factor-induced Rac1 activation (Feng et al., 2011; Singleton et al., 2007). Thus, there is not a clear role for Dyn2 in regulating the biochemical activation of Rac1.
However, we demonstrate here that Dyn2 can directly regulate Rac1 activation through Vav1, and in a manner independent of its enzymatic function, highlighting a role for Dyn2 in regulating cell migration. In Vav1-expressing cells, depletion of Dyn2, or expression of a mutated form of Dyn2 that cannot bind Vav1, reduces Rac1 activation by 50%. We have found that mutations that disrupt Dyn2-Vav1 binding have no effect on endocytosis, indicating that the effects on Rac1 signaling and cell migration are distinct from Dyn2 enzymatic function, but rather are due to regulation of Vav1 protein stability. Further, inhibition of Dyn2 enzymatic activity through a dominant negative mutation (K44A) or by treatment with pharmacological inhibitors of Dyn2 has no effect on Vav1 protein stability, demonstrating that this stabilization is not regulated by Dyn2 enzymatic activity or indirectly through endocytic events. Therefore, this study identifies an additional mechanism by which Dyn2 can regulate Rac1.
The enzymatic activity of Dyn2 requires both binding to phospholipids and assembly into polymers (Hinshaw, 2000). It is not known if the lipid-binding or oligomerization properties of Dyn2 are required for its endocytosis-independent functions, including cytoskeletal remodeling and Vav1 stabilization. Further structural studies of Dyn2 will be useful in determining the role of the multiple functional domains of Dyn2 in tumor cell migration.
Because Vav1 expression is restricted to hematopoietic cells and some tumor cells, the mechanism described here is also specific to Vav1-positive cells. Indeed, this may be one reason why previous studies have not demonstrated a definitive effect of Dyn2 on Rac1 activation, as cells used in those experiments did not express Vav1. As Dyn2 promotes lamellipodia formation and cell migration even in the absence of Vav1 (Eppinga et al., 2012; Schlunck et al., 2004), it is assumed that Dyn2 can enhance migration in both neoplastic and normal cells by Vav1-independent mechanisms as well. In addition to regulating Rac1 localization indirectly through endocytosis, as described above, it is possible that Dyn2 can contribute to tumor cell migration through interactions with other GEF proteins that regulate Rho family GTPases. Alternatively, Dyn2 can promote lamellipod formation and cell migration by an interaction with a variety of actin-binding proteins, including cortactin (Kruchten and McNiven, 2006; Schafer, 2004), or by a direct interaction with actin itself (Gu et al., 2010). Finally, Dyn2 is known to participate in the remodeling of the extracellular matrix, which is also an important component of cell migration (Baldassarre et al., 2003; Ezratty et al., 2005; Wang et al., 2011). Regardless of these additional mechanisms, many pancreatic tumors, and perhaps other tumor types, overexpress both Vav1 and Dyn2, which can synergize to regulate Rac1-mediated cell migration. Indeed, we have found that a significant percentage of human pancreatic cancer tumors are upregulated for both Vav1 and Dyn2 (Table S1).
Regulation of Vav1 function by modulating protein stability
Modulation of Vav1 protein level is a potent mechanism to regulate signaling and cell fate. The data presented here demonstrate the potent effects of Vav1 overexpression or siRNA-mediated knockdown on Rac1 activation and cell migration in tumor cells. To date, several mechanisms regulating Vav1 protein stability have been reported. First, ubiquitination of Vav1 by the E3 ligase Cbl results in degradation of Vav1 by the proteasome, thereby inhibiting Vav1-dependent signaling (Bustelo et al., 1997; De Sepulveda et al., 2000; Miura-Shimura et al., 2003). Second, proteolytic cleavage of Vav1 by caspases occurs both during T cell anergy and apoptosis, thereby regulating downstream signaling during these processes (Hofmann et al., 2000; Puga et al., 2008). Third, calpain cleavage of Vav1 appears to play a role in signaling during platelet aggregation (Miyakawa et al., 1997). In this study we have defined a surprising pathway regulating Vav1 turnover. In the absence of Dyn2 binding, Vav1 is targeted for degradation, not by the proteasome as might be predicted for a cytosolic protein, but by the lysosome, utilized for degradation of endocytosis proteins and in autophagy.
How does Vav1, a cytosolic protein, gain access to the membrane-bound lysosome for degradation? There are several mechanisms that have been implicated in this process including chaperone-associated selective macroautophagy, microautophagy, or chaperone-mediated autophagy (CMA). A factor implicated in all three of these pathways is the chaperone Hsc70 (Arndt et al., 2010; Kaushik and Cuervo, 2012; Sahu et al., 2011). Here we have identified an interaction between Vav1 and Hsc70, and have found that this interaction is required for the degradation of Vav1. Vav1 WWYY is highly unstable, but its protein levels are markedly restored when its putative Hsc70 binding motif is disrupted. This motif appears to reside within the linker situated between the Vav1 SH2 and SH3 domains. We propose that, as Dyn2 binds to Vav1 via the SH3 domain, Dyn2 binding physically restricts the access of Hsc70 to its binding motif, thereby preventing lysosomal targeting and degradation of Vav1. This would explain why expression of the isolated PRD is not sufficient to prevent Vav1 degradation, as it is not bulky enough to block access of Hsc70 to Vav1. As the proto-oncogene Vav1 has potent transforming and migratory effects, regulation of its turnover is highly relevant in both tumor cells and normal hematopoietic cells.
The role of Hsc70 in selective lysosomal degradation is best established in chaperone mediated autophagy. In this process, generally upregulated during cellular stress, Hsc70-bound cargo is imported into the lysosome in an unfolded state via a Lamp2a channel (Kaushik and Cuervo, 2012). Often the “KFERQ” Hsc70-binding motif is masked by the tertiary structure of the protein, and is revealed upon conformation change, posttranslational modification, or misfolding, resulting in increased Hsc70 binding and lysosomal degradation. Our data suggest that Dyn2 may regulate the accessibility of an Hsc70-binding motif on Vav1, and that Vav1 can be degraded via chaperone-mediated autophagy. Interestingly, selective autophagic processes may be aberrantly regulated in cancer cells (Mizushima et al., 2008; Welsch et al., 2010). Thus, even in the absence of activating, oncogenic mutations in Vav1, misregulation of pathways that regulate Vav1 stability may have the same biological consequence, leading to an upregulation of Vav1 expression and subsequent Rac1-mediated invasive cell migration.
Interestingly, the Hsc70-binding motif in Vav1 (QFPFKE) is not a perfect match for the consensus Hsc70 binding motif, as it contains a proline in the middle of the pentapeptide. The presence of a proline has been shown to be disruptive for Hsc70 binding. Our data suggest there may be additional flexibility in the already highly variable Hsc70 binding motif. Alternatively, while our data indicate that disruption of the QFPFKE motif reduces Hsc70 binding, it is possible that this represents a non-conventional Hsc70 interaction motif or that Vav1 interacts with Hsc70 via a modified mechanism. Finally, Vav1 degradation may also be regulated by additional mechanisms. While Vav1 WWYY stabilization is increased significantly upon the QFAA mutation to inhibit Hsc70 binding, it is not restored to the levels of WT Vav1. It is possible that additional “KFERQ”-like motifs on Vav1 are able to compensate for the mutation of the QFPFKE motif, allowing some degradation to continue. In addition, the WWYY mutation represents a disruption of the Vav1 C-terminal SH3 domain, which has also been shown to interact with several proteins including zyxin, RNA binding proteins such as Sam68, and transcriptional regulators (Katzav, 2009). Thus, the stability of this Vav1 construct may also be regulated by other binding partners. Regardless, our data demonstrate a regulation of Vav1 levels by its degradation through an Hsc70 chaperone-mediated targeting to the lysosome.
In pancreatic cancer, aberrant Vav1 expression is due to promoter demethylation and transcriptional upregulation (Fernandez-Zapico et al., 2005). In addition, our data demonstrate that Vav1 interaction with Dyn2 promotes Vav1 stability at the protein level. As Dyn2 is also upregulated in pancreatic cancers, our data raise the intriguing possibility that once Vav1 expression is transcriptionally induced in pancreatic cancer cells, high levels of Dyn2 can further stabilize Vav1 in tumors, thereby potentiating the effects of Vav1 overexpression and further increasing tumor cell survival, proliferation and metastasis. Therefore, pharmacological agents that target Vav1, or its interaction with Dyn2, could be potent inhibitors of tumor cell migration.
Experimental Procedures
Cell Culture and Transfections
DanG, Panc1, and MiaPaCa2 cells were maintained in DMEM + 10% FBS, Panc04.03 cells were maintained in RPMI + 15% FBS, and HPAF-II cells were maintained in MEM + 10% FBS. All cells were grown in the presence of 100 units/ml penicillin and 100 μg/ml streptomycin. Transfections procedures are described in the Supplemental Experimental Procedures.
Immunoprecipitation, GST Pulldowns, and Immunoblotting
Cells were lysed and subject to immunoprecipitation as described in Supplemental Experimental Procedures. For protein turnover experiments, cells were treated with 50 μM cycloheximide for 0-6 hours prior to lysis. For lysosomal inhibition, cells were incubated with 25 μM chloroquine for 48 hours. Primary antibodies were used as indicated in Supplemental Experimental Procedures.
Migration and Lamellipodia Formation Assays
Transwell migration assays were performed as described previously (Eppinga et al., 2012), with modifications as described in Supplemental Experimental Procedures. To measure formation of lamellipodia, cells were serum-starved overnight, then stimulated with EGF (50 ng/ml, Sigma Aldrich, St. Louis, MO) for 20 min, and fixed, stained, and imaged as described in Supplemental Experimental Procedures.
Immunofluoresence imaging
Immunofluorescence was performed as described in Supplemental Experimental Procedures. For lysosomal localization, DanG cells were cotransfected with mCherry-Vav1 (WT or WWYY) and GFP-Lamp1, and were fixed and imaged after 48 hours. The number of GFP-Lamp1 positive vesicles containing mCherry Vav1 or mCherry Vav1 WWYY was counted per cell. At least 25 cells were scored in each of three independent experiments.
Supplementary Material
01
Highlights.
- Dyn2 promotes lamellipodia formation and pancreatic tumor cell migration.
- Direct binding between Dyn2 and Vav1 promotes Rac1 activation and migration.
- Dyn2 binding protects Vav1 from degradation by the lysosome.
- Vav1 is targeted for degradation through an interaction with Hsc70.
Acknowledgments
We gratefully acknowledge the members of the McNiven lab for their contributions, in particular, Shaun Weller and Hong Cao for technical assistance, and Barbara Schroeder, Robbin Eppinga, and Ryan Schulze for critical reading of the manuscript. This work was financially supported by grants from the National Cancer Institute (CA104125 to M.A.M.), the Optical Morphology Core of the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK84567), and Mayo Clinic Pancreatic Cancer SPORE grant CA102701. G.L.R. was supported by T32 CA148073.
List of Abbreviations
CHX
Cycloheximide
CQ
Chloroquine
Dyn2
Dynamin 2
EGF
Epidermal Growth Factor
GAP
GTPase Activating Protein
GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
GEF
Guanine nucleotide Exchange Factor
GST
Glutathione S Transferase
PAK1
p21 Activated Kinase 1
PBD
p21 Binding Domain
PRD
Proline Rich Domain
Rh-EGF
Rhodamine-conjugated Epidermal Growth Factor
RT-PCR
Reverse Transcriptase – Polymerase Chain Reaction
SEM
Standard Error of the Mean
SH3
Src Homology 3 domain
WT
Wild Type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, Hesse M, Furst DO, Saftig P, Saint R, Fleischmann BK, et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. 2010;20:143–148. doi: 10.1016/j.cub.2009.11.022. [DOI] [PubMed] [Google Scholar]
- Baldassarre M, Pompeo A, Beznoussenko G, Castaldi C, Cortellino S, McNiven MA, Luini A, Buccione R. Dynamin participates in focal extracellular matrix degradation by invasive cells. Mol Biol Cell. 2003;14:1074–1084. doi: 10.1091/mbc.E02-05-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolome RA, Molina-Ortiz I, Samaniego R, Sanchez-Mateos P, Bustelo XR, Teixido J. Activation of Vav/Rho GTPase signaling by CXCL12 controls membrane-type matrix metalloproteinase-dependent melanoma cell invasion. Cancer Res. 2006;66:248–258. doi: 10.1158/0008-5472.CAN-05-2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustelo XR. Vav proteins, adaptors and cell signaling. Oncogene. 2001;20:6372–6381. doi: 10.1038/sj.onc.1204780. [DOI] [PubMed] [Google Scholar]
- Bustelo XR, Crespo P, Lopez-Barahona M, Gutkind JS, Barbacid M. Cbl-b, a member of the Sli-1/c-Cbl protein family, inhibits Vav-mediated c-Jun N-terminal kinase activation. Oncogene. 1997;15:2511–2520. doi: 10.1038/sj.onc.1201430. [DOI] [PubMed] [Google Scholar]
- De Sepulveda P, Ilangumaran S, Rottapel R. Suppressor of cytokine signaling-1 inhibits VAV function through protein degradation. J Biol Chem. 2000;275:14005–14008. doi: 10.1074/jbc.c000106200. [DOI] [PubMed] [Google Scholar]
- del Pozo MA, Schwartz MA, Hu J, Kiosses WB, Altman A, Villalba M. Guanine exchange-dependent and -independent effects of Vav1 on integrin-induced T cell spreading. J Immunol. 2003;170:41–47. doi: 10.4049/jimmunol.170.1.41. [DOI] [PubMed] [Google Scholar]
- Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- Eppinga RD, Krueger EW, Weller SG, Zhang L, Cao H, McNiven MA. Increased expression of the large GTPase dynamin 2 potentiates metastatic migration and invasion of pancreatic ductal carcinoma. Oncogene. 2012;31:1228–1241. doi: 10.1038/onc.2011.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezratty EJ, Partridge MA, Gundersen GG. Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat Cell Biol. 2005;7:581–590. doi: 10.1038/ncb1262. [DOI] [PubMed] [Google Scholar]
- Feng H, Liu KW, Guo P, Zhang P, Cheng T, McNiven MA, Johnson GR, Hu B, Cheng SY. Dynamin 2 mediates PDGFRalpha-SHP-2-promoted glioblastoma growth and invasion. Oncogene. 2011 doi: 10.1038/onc.2011.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Zapico ME, Gonzalez-Paz NC, Weiss E, Savoy DN, Molina JR, Fonseca R, Smyrk TC, Chari ST, Urrutia R, Billadeau DD. Ectopic expression of VAV1 reveals an unexpected role in pancreatic cancer tumorigenesis. Cancer Cell. 2005;7:39–49. doi: 10.1016/j.ccr.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Gomez TS, Hamann MJ, McCarney S, Savoy DN, Lubking CM, Heldebrant MP, Labno CM, McKean DJ, McNiven MA, Burkhardt JK, et al. Dynamin 2 regulates T cell activation by controlling actin polymerization at the immunological synapse. Nat Immunol. 2005;6:261–270. doi: 10.1038/ni1168. [DOI] [PubMed] [Google Scholar]
- Gu C, Yaddanapudi S, Weins A, Osborn T, Reiser J, Pollak M, Hartwig J, Sever S. Direct dynamin-actin interactions regulate the actin cytoskeleton. EMBO J. 2010;29:3593–3606. doi: 10.1038/emboj.2010.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinshaw JE. Dynamin and its role in membrane fission. Annu Rev Cell Dev Biol. 2000;16:483–519. doi: 10.1146/annurev.cellbio.16.1.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann TG, Hehner SP, Droge W, Schmitz ML. Caspase-dependent cleavage and inactivation of the Vav1 proto-oncogene product during apoptosis prevents IL-2 transcription. Oncogene. 2000;19:1153–1163. doi: 10.1038/sj.onc.1203406. [DOI] [PubMed] [Google Scholar]
- Hornstein I, Pikarsky E, Groysman M, Amir G, Peylan-Ramu N, Katzav S. The haematopoietic specific signal transducer Vav1 is expressed in a subset of human neuroblastomas. J Pathol. 2003;199:526–533. doi: 10.1002/path.1314. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- Jones SM, Howell KE, Henley JR, Cao H, McNiven MA. Role of dynamin in the formation of transport vesicles from the trans-Golgi network. Science. 1998;279:573–577. doi: 10.1126/science.279.5350.573. [DOI] [PubMed] [Google Scholar]
- Katzav S. Vav1: a hematopoietic signal transduction molecule involved in human malignancies. Int J Biochem Cell Biol. 2009;41:1245–1248. doi: 10.1016/j.biocel.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Katzav S, Martin-Zanca D, Barbacid M. vav, a novel human oncogene derived from a locus ubiquitously expressed in hematopoietic cells. EMBO J. 1989;8:2283–2290. doi: 10.1002/j.1460-2075.1989.tb08354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22:407–417. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawada K, Upadhyay G, Ferandon S, Janarthanan S, Hall M, Vilardaga JP, Yajnik V. Cell migration is regulated by platelet-derived growth factor receptor endocytosis. Mol Cell Biol. 2009;29:4508–4518. doi: 10.1128/MCB.00015-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi S, Honda K, Tsuda H, Hiraoka N, Imoto I, Kosuge T, Umaki T, Onozato K, Shitashige M, Yamaguchi U, et al. Expression and gene amplification of actinin-4 in invasive ductal carcinoma of the pancreas. Clin Cancer Res. 2008;14:5348–5356. doi: 10.1158/1078-0432.CCR-08-0075. [DOI] [PubMed] [Google Scholar]
- Kruchten AE, McNiven MA. Dynamin as a mover and pincher during cell migration and invasion. J Cell Sci. 2006;119:1683–1690. doi: 10.1242/jcs.02963. [DOI] [PubMed] [Google Scholar]
- Lazer G, Idelchuk Y, Schapira V, Pikarsky E, Katzav S. The haematopoietic specific signal transducer Vav1 is aberrantly expressed in lung cancer and plays a role in tumourigenesis. J Pathol. 2009;219:25–34. doi: 10.1002/path.2579. [DOI] [PubMed] [Google Scholar]
- Lazer G, Pe'er L, Farago M, Machida K, Mayer BJ, Katzav S. Tyrosine residues at the carboxyl terminus of Vav1 play an important role in regulation of its biological activity. J Biol Chem. 2010;285:23075–23085. doi: 10.1074/jbc.M109.094508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Lago M, Lee H, Cruz C, Movilla N, Bustelo XR. Tyrosine phosphorylation mediates both activation and downmodulation of the biological activity of Vav. Mol Cell Biol. 2000;20:1678–1691. doi: 10.1128/mcb.20.5.1678-1691.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda Y, Naito Z, Kawahara K, Nakazawa N, Korc M, Ishiwata T. Nestin is a novel target for suppressing pancreatic cancer cell migration, invasion and metastasis. Cancer Biol Ther. 2011;11:512–523. doi: 10.4161/cbt.11.5.14673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNiven MA, Cao H, Pitts KR, Yoon Y. The dynamin family of mechanoenzymes: pinching in new places. Trends Biochem Sci. 2000a;25:115–120. doi: 10.1016/s0968-0004(99)01538-8. [DOI] [PubMed] [Google Scholar]
- McNiven MA, Kim L, Krueger EW, Orth JD, Cao H, Wong TW. Regulated interactions between dynamin and the actin-binding protein cortactin modulate cell shape. J Cell Biol. 2000b;151:187–198. doi: 10.1083/jcb.151.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranti CK, Leng L, Maschberger P, Brugge JS, Shattil SJ. Identification of a novel integrin signaling pathway involving the kinase Syk and the guanine nucleotide exchange factor Vav1. Curr Biol. 1998;8:1289–1299. doi: 10.1016/s0960-9822(07)00559-3. [DOI] [PubMed] [Google Scholar]
- Miura-Shimura Y, Duan L, Rao NL, Reddi AL, Shimura H, Rottapel R, Druker BJ, Tsygankov A, Band V, Band H. Cbl-mediated ubiquitinylation and negative regulation of Vav. J Biol Chem. 2003;278:38495–38504. doi: 10.1074/jbc.M305656200. [DOI] [PubMed] [Google Scholar]
- Miyakawa Y, Oda A, Druker BJ, Ozaki K, Handa M, Ohashi H, Ikeda Y. Thrombopoietin and thrombin induce tyrosine phosphorylation of Vav in human blood platelets. Blood. 1997;89:2789–2798. [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooren OL, Kotova TI, Moore AJ, Schafer DA. Dynamin2 GTPase and cortactin remodel actin filaments. J Biol Chem. 2009;284:23995–24005. doi: 10.1074/jbc.M109.024398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni XG, Zhou L, Wang GQ, Liu SM, Bai XF, Liu F, Peppelenbosch MP, Zhao P. The ubiquitin-proteasome pathway mediates gelsolin protein downregulation in pancreatic cancer. Mol Med. 2008;14:582–589. doi: 10.2119/2008-00020.Ni. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palamidessi A, Frittoli E, Garre M, Faretta M, Mione M, Testa I, Diaspro A, Lanzetti L, Scita G, Di Fiore PP. Endocytic trafficking of Rac is required for the spatial restriction of signaling in cell migration. Cell. 2008;134:135–147. doi: 10.1016/j.cell.2008.05.034. [DOI] [PubMed] [Google Scholar]
- Puga I, Rao A, Macian F. Targeted cleavage of signaling proteins by caspase 3 inhibits T cell receptor signaling in anergic T cells. Immunity. 2008;29:193–204. doi: 10.1016/j.immuni.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ. Life at the leading edge. Cell. 2011;145:1012–1022. doi: 10.1016/j.cell.2011.06.010. [DOI] [PubMed] [Google Scholar]
- Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, Potolicchio I, Nieves E, Cuervo AM, Santambrogio L. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. 2011;20:131–139. doi: 10.1016/j.devcel.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DA. Regulating actin dynamics at membranes: a focus on dynamin. Traffic. 2004;5:463–469. doi: 10.1111/j.1600-0854.2004.00199.x. [DOI] [PubMed] [Google Scholar]
- Schlunck G, Damke H, Kiosses WB, Rusk N, Symons MH, Waterman-Storer CM, Schmid SL, Schwartz MA. Modulation of Rac localization and function by dynamin. Mol Biol Cell. 2004;15:256–267. doi: 10.1091/mbc.E03-01-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder B, Weller SG, Chen J, Billadeau D, McNiven MA. A Dyn2-CIN85 complex mediates degradative traffic of the EGFR by regulation of late endosomal budding. EMBO J. 2010;29:3039–3053. doi: 10.1038/emboj.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton PA, Salgia R, Moreno-Vinasco L, Moitra J, Sammani S, Mirzapoiazova T, Garcia JG. CD44 regulates hepatocyte growth factor-mediated vascular integrity. Role of c-Met, Tiam1/Rac1, dynamin 2, and cortactin. J Biol Chem. 2007;282:30643–30657. doi: 10.1074/jbc.M702573200. [DOI] [PubMed] [Google Scholar]
- Turner M, Billadeau DD. VAV proteins as signal integrators for multi-subunit immune-recognition receptors. Nat Rev Immunol. 2002;2:476–486. doi: 10.1038/nri840. [DOI] [PubMed] [Google Scholar]
- Tybulewicz VL. Vav-family proteins in T-cell signalling. Curr Opin Immunol. 2005;17:267–274. doi: 10.1016/j.coi.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Wang Y, Cao H, Chen J, McNiven MA. A direct interaction between the large GTPase dynamin-2 and FAK regulates focal adhesion dynamics in response to active Src. Mol Biol Cell. 2011;22:1529–1538. doi: 10.1091/mbc.E10-09-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kelber JA, Tran Cao HS, Cantin GT, Lin R, Wang W, Kaushal S, Bristow JM, Edgington TS, Hoffman RM, et al. Pseudopodium-enriched atypical kinase 1 regulates the cytoskeleton and cancer progression [corrected] Proc Natl Acad Sci U S A. 2010;107:10920–10925. doi: 10.1073/pnas.0914776107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsch T, Endlich K, Giese T, Buchler MW, Schmidt J. Eps8 is increased in pancreatic cancer and required for dynamic actin-based cell protrusions and intercellular cytoskeletal organization. Cancer Lett. 2007;255:205–218. doi: 10.1016/j.canlet.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Welsch T, Younsi A, Disanza A, Rodriguez JA, Cuervo AM, Scita G, Schmidt J. Eps8 is recruited to lysosomes and subjected to chaperone-mediated autophagy in cancer cells. Exp Cell Res. 2010;316:1914–1924. doi: 10.1016/j.yexcr.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Abe T, Li SA, Masuoka Y, Isoda M, Watanabe M, Nasu Y, Kumon H, Asai A, Takei K. Dynasore, a dynamin inhibitor, suppresses lamellipodia formation and cancer cell invasion by destabilizing actin filaments. Biochem Biophys Res Commun. 2009;390:1142–1148. doi: 10.1016/j.bbrc.2009.10.105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01